
Promoting oxygen reduction via coordination environment modulation through secondary metal-atom incorporation†‡
Haozhou
Yang
 a,
Tianyu
Zhang
a,
Xiao
Chi
b,
Xiaojiang
Yu
b,
Junmei
Chen
a,
Jiayi
Chen
a,
Chunfeng
Li
a,
Shengdong
Tan
c,
Qian
He
c,
Xun
Wang
d and
Lei
Wang
*a
a,
Tianyu
Zhang
a,
Xiao
Chi
b,
Xiaojiang
Yu
b,
Junmei
Chen
a,
Jiayi
Chen
a,
Chunfeng
Li
a,
Shengdong
Tan
c,
Qian
He
c,
Xun
Wang
d and
Lei
Wang
*a
aDepartment of Chemical and Biomolecular Engineering, National University of Singapore, 117585, Singapore. E-mail: wanglei8@nus.edu.sg
bDepartment of Physics and Singapore Synchrotron Light Source, National University of Singapore, 119077, Singapore
cDepartment of Material Science Engineering, National University of Singapore, 117575, Singapore
dKey Lab of Organic Optoelectronics and Molecular Engineering, Department of Chemistry, Tsinghua University, Beijing 100084, China
First published on 25th May 2022
Abstract
Fe-based single-atom catalysts (SACs) have been recognized as attractive candidates for the oxygen reduction reaction (ORR). However, several limitations such as unsatisfactory intrinsic activity and poor stability hinder their practical applications, motivating further optimization of Fe-SACs which requires precise control of their composition and coordination environment. Herein, we establish a facile solvothermal method to incorporate Cu into Fe-SACs to form heterometallic SACs. We observe that the adjacent Cu atoms can regulate the coordination environment of FeN4 sites and significantly enhance the ORR activity. As a result, the heterometallic SACs show superior ORR catalytic performance compared to the mono-Fe-SAC counterpart (an enhancement factor of ∼5 times), delivering a half-wave potential (E1/2) of 0.889 V vs. RHE and a high kinetic current density (jk) of 4.77 mA cm−2 at 0.9 V vs. RHE, and also outperform the state-of-the-art Pt/C catalyst under identical testing conditions.
 Lei Wang | Lei Wang completed his doctorate at the KTH Royal Institute of Technology in 2015, where he studied water oxidation based on molecular catalysts. After receiving his PhD, he undertook a short period of research on catalyst discovery for light-induced hydrogen evolution at Uppsala University. In 2016, he joined Stanford University as a Wallenberg-Stanford postdoc fellow and worked on electrochemical CO2 reduction. He assumed his current position at the National University of Singapore at the end of 2020, where he is part of the NUS Green Energy Programme. His current research focuses on catalyst discovery and understanding reaction mechanisms for electrochemical CO2 reduction. |
The oxygen reduction reaction (ORR) plays a key role in many crucial energy conversion devices including fuel cells and metal–air batteries, which are highly promising technologies for clean energy usage to realize global carbon neutralization and are regarded as next-generation power supply systems.1–5 However, occurring at the air cathode, the four-electron/proton transfer ORR process is kinetically sluggish and usually requires precious metal catalysts especially Pt.6–9 To tackle this challenge, many efforts have been made to develop high-performance and low-cost ORR catalysts. First-row transition metals are broadly applied in diverse electrocatalytic fields owing to their versatile oxidation states, comparable durability to noble metals under certain conditions, and low cost.9–12 Specifically, transition-metal-based single-atom catalysts (SACs) composed of atomically dispersed metal atoms embedded on nitrogen-doped carbon matrixes, especially Fe SACs, have been developed towards cost-effective ORR.13–23 While tremendous success has been achieved, greater efforts on reducing the overpotential (half-wave potential) and enhancing the intrinsic activity (kinetic current density) for the ORR are still desirable to bring these SACs into practical applications. The key to further improving the performance of SACs for the ORR is the precise control of their metal composition and coordination environment, which still remains a challenge.
Although diverse methods have been developed to fabricate SACs on carbon matrixes, pyrolysis remains one of the most promising ways to synthesize well-defined SACs with a definite structure and composition.24,25 Separately, 2-methylimidazole (2-Hmim) based zeolitic imidazolate frameworks (ZIF-8) with zinc as metal nodes have demonstrated the ability to afford diverse structural and compositional variations.26,27 Thus, ZIF-8 analogues have been extensively employed as a template/precursor for preparing different SACs, including Fe-based SACs for the ORR, and Zn is usually chosen as the co-incorporated precursor since the reduced metallic Zn is volatile at a pyrolysis temperature of ∼900 °C, and the loss of Zn atoms can further provide additional N-coordination sites for the binding of desired single atoms.28 Several publications have shown that the coordination type of the FeN4 modification (typical active sites within Fe-based SACs) greatly impacts the activity of the ORR. More specifically, the D1 type FeN4 site with high-spin ferric FeN4 sites exhibits better ORR activity compared with D2 sites (medium-spin ferrous FeN4 sites) due to enhanced adsorption of oxygen molecules.29–31 Thus, fabricating Fe-based SACs with dominant D1 sites is preferred. On the other hand, the adjacent “non-bonding” coordination sites can facilitate electronic interactions and regulate the electronic and coordination environment of the single-atom active sites, e.g., FeN4 sites.32 A recent study suggests that the adjacent FeN4 sites tend to have electronic interactions with each other, and this interaction will promote the formation of D1 sites and further facilitate the ORR.33 Taking the above into consideration, we believe that the introduction of secondary metal single sites can further optimize the electronic interactions and modulate the formation of desirable FeN4 sites. Considering the formation ability of MN4 sites, structural compatibility with ZIF assembly and electronegativity compared with Fe, we chose Cu as the secondary component to prepare the target heterometallic SACs aiming to achieve improved ORR performance.34,35
Herein, we developed a facile two-step method to synthesize the aforementioned heterometallic SACs with controllable metal loadings as illustrated in Fig. 1a. First, trimetallic ions and 2-Hmim ligands were added together and assembled into the uniform ZnCuFe-ZIF precursor under solvothermal conditions, while the Fe content within the precursor was modulated by varying the amount of Fe salt added. Then, a well-defined SAC was obtained after thermal treatment. The as-fabricated heterometallic SAC exhibited high surface area and porosity, which is beneficial for the gas-involved catalysis.36,37 ORR tests indicated that the Cu,Fe co-doped SAC with 10 mg of Fe(NO3)3 precursor obtained via 900 °C calcination (denoted as CuFe10-900) exhibited the best performance. The spectroscopic characterization studies reveal that the incorporation of Cu can tune the coordination environment of the FeN4 sites and significantly enhance the ORR catalytic activity.38 Unary Cu- and Fe-based SACs and CuFe SACs with different Fe loadings and pyrolysis temperatures were then studied to elucidate the effects of different factors. Finally, CuFe10-900 was fabricated as an air cathode for a Zn–air battery which outperformed the commercial Pt/C counterpart. Overall, this work provides guidance in designing, preparing and utilizing heterometallic SACs towards diverse energy-related applications.
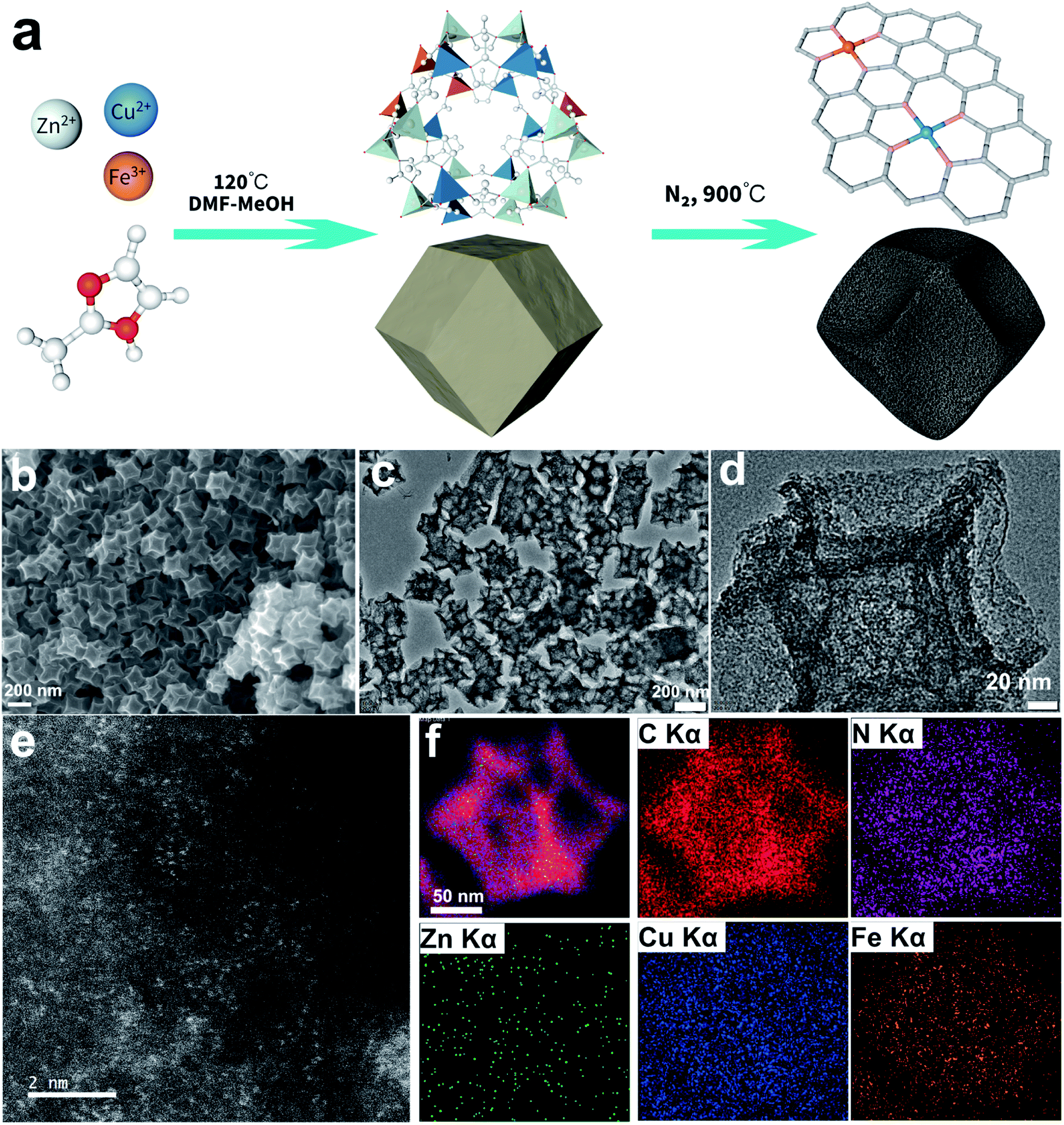 | ||
| Fig. 1 (a) Schematic illustration of the catalyst synthetic route, (b) SEM secondary electron image, (c and d) TEM bright-field images, (e) HAADF-STEM image and (f) representative HAADF-STEM image with sum spectra and the corresponding EDS elemental maps taken from the CuFe10-900 sample. | ||
The morphology of the ZnCuFe10-ZIF precursor was first characterized with secondary electron imaging using a scanning electron microscope (SEM). As shown in Fig. S1a and b,‡ it can be clearly seen that the samples are rhombododecahedra with an average size of about 220 nm. Energy dispersive spectroscopy (EDS) elemental mapping images (Fig. S1c‡) show that all three kinds of metal elements are uniformly distributed, indicating that these different kinds of metal–ligand moieties are evenly distributed in the ZIF precursor. Bright field transmission electron microscopy (BF-TEM) imaging (Fig. S1d‡) further confirms the morphology. The powder X-ray diffraction (PXRD) pattern of ZIF is shown in Fig. S2,‡ from which we can determine that the measured patterns match well with that of the simulated ZIF-8, indicating the successful formation of the ZIF-8 topological skeleton. After pyrolysis at 900 °C in Ar, ZIF transformed into concave mesoporous nanoparticles while the rhombododecahedral mesostructure was retained, as shown in Fig. 1b and c. No observable metal particles or clusters can be found in the carbon matrix (Fig. 1d), suggesting that metal ions were mainly converted into single sites. High angle annular dark-field imaging from scanning transmission electron microscopes (HAADF-STEM) was used to directly visualize the single-atom species in the catalyst. In Fig. 1e, the bright spots in the HAADF-STEM image represent the monodisperse metal single atoms. EDS elemental mapping (Fig. 1f) suggests that all elements are uniformly distributed, and the negligible zinc signals reveal that the majority of zinc species were vaporized after high-temperature pyrolysis. The EDS elemental line scanning profiles (Fig. S3a and b‡) provide further support. It should be noted that the relatively higher signal intensity of Cu results from the Cu-based TEM grid. The PXRD pattern of CuFe10-900 is presented in Fig. S3c.‡ The broad peaks located at 26° and 42° can be indexed to C (002) and (100), respectively.39 Moreover, Brunauer–Emmett–Teller (BET) specific surface area and pore size distribution evaluations were carried out to prove the mesoporous nature of CuFe10-900. In Fig. S3d‡ we can find a typical type-IV N2 adsorption–desorption isotherm, which strongly confirms its mesoporous nature. Finally, the BET surface area was calculated to be 797.4 m2 g−1, and a pore volume of 0.962 cm3 g−1 was also estimated. Overall, the above morphological characterization studies suggest that CuFe10-900 exhibits a large surface area and high pore volume, which is expected to be beneficial for the absorption of oxygen gas and further promote the overall ORR process.
In order to find out the roles of heterometallic ions and calcination conditions for the ORR, a series of controllable experiments were conducted. We first synthesized two kinds of bimetallic ZIF-8 analogues: ZnCu-ZIF and ZnFe10-ZIF. As shown in Fig. S4a and b,‡ the morphology and size of similar rhombododecahedral ZIFs were maintained, and PXRD patterns (Fig. S5‡) confirm the ZIF-8 topological structure. Cu-900 and Fe10-900 samples were obtained after pyrolysis under the same conditions. As shown in Fig. S6a and b,‡ Fe10-900 retained a well-defined rhombododecahedral shape, while Cu-900 encountered structural degradation, and this might be attributed to the relatively easier formation of Fe–N and therefore a better-retained matrix. The XRD patterns of the above two samples (Fig. S7a and b‡) resemble that of CuFe10-900 owing to the same carbon skeleton. Secondly, the amount of Fe(NO3)3 precursor added when synthesizing the heterometallic SACs was changed to 5 mg and 20 mg, and the obtained ZIFs were denoted as ZnCuFe5-ZIF and ZnCuFe20-ZIF, respectively. SEM images (Fig. S4c and d‡) and PXRD patterns (Fig. S5‡) confirm that a similar morphology was obtained for these samples. Subsequently, CuFe5-900 and CuFe20-900 were prepared as well through calcination under the same conditions. SEM and TEM images (Fig. S8a–d‡) indicate that the Fe20 analogue showed a better-maintained morphology compared with the Fe5 analogue. Based on the morphological changes of Cu-900, we can infer that the increase of Fe loading is favorable for retaining the morphology of the pristine ZIF. Finally, we studied the effect of pyrolysis temperature. Fig. S9a–d‡ present the morphology of SACs obtained at 800 °C and 1000 °C, respectively (denoted as CuFe10-800 and CuFe10-1000). It can be clearly seen that the polyhedron shape gradually transformed into more concave particles with larger hollow interior structures with the increase of calcination temperature, which can be attributed to the thermally induced mass diffusion. Inductively coupled plasma optical emission spectrometry (ICP-OES) was employed to precisely determine the elemental compositions of the above samples (Table S1‡). It was found that all the samples obtained with a calcination temperature of 900 °C or via 1000 °C pyrolysis have very low Zn content (typically 0.01%); we therefore believe that the trace amount of Zn will likely make no appreciable contribution to the ORR activity. In addition, CuFe10-900 and Fe10-900 show similar Fe contents, which is consistent with the amount of Fe salt added during the precursor synthesis. Overall, the Fe loading in CuFex-900 still matches well with those in ZIF precursors. Specifically, the loading weight percentages of Fe are 1.54%, 2.42% and 4.57% for x = 5, 10 and 20 respectively. In CuFe10-800, the Zn weight percentage is high, while in CuFe10-1000, Zn is utterly evaporated leaving a more open C–N matrix, and therefore the Fe loading amount is slightly lower in the former and higher in the latter. Based on the above demonstration, we can conclude that the strategy of one-pot incorporation of multimetallic ZIF precursors developed here is highly promising for preparing SACs with precisely controlled multi-metallic compositions.
We then utilized the synchrotron-based X-ray absorption fine spectroscopy (XAFS) technique to ascertain the chemical state and coordination environment of the metal single atoms in our samples. Fig. 2a shows the X-ray absorption near edge structure (XANES) of the Fe K-edge. It can be seen that the Fe K-pre-edges of both CuFe10-900 and Fe10-900 lie between those of Fe2O3 and FeO and are close to that of Fe2O3, indicating that the valence states of Fe in these samples are positive and close to +3. Specifically, the spectra of CuFe10-900 and Fe10-900 show different characteristics in the pre-edge and white-lines, which can be indexed to the discrepancy of D1 and D2 types of FeN4 moieties as demonstrated in the literature. The XANES spectrum of CuFe10-900 accords well with the non-planar FeN4 coordination reported in the literature, suggesting the existence of more D1 type FeN4 in CuFe10-900.33,38,40 Then, we employed the extended X-ray absorption fine structure (EXAFS) characterization to elucidate the coordination structures of Fe atoms in SACs. Fig. 2b presents the Fourier-transformed (FT)-EXAFS curves of four samples; the Fe–Fe interaction in Fe foil is absent in CuFe10-900, indicating that no Fe metal particles exist. The minor peaks at around 2.5 Å represent Fe–C interactions, which also appear in the reference FePc spectrum. The dominant peak of CuFe10-900 located at around 1.47 Å can be attributed to the Fe–N single scattering path in the FeN4 moiety, as fitted in Fig. 2c. The FT-EXAFS spectrum and first-shell fitting curve of Fe10-900 are shown in Fig. S13a and b.‡ The slightly longer Fe–N distance of CuFe10-900 (Table S2‡) further supports the previous report that D1 type FeN4 sites are more distorted from the ideal planar coordination.37,39 Besides, XANES and FT-EXAFS spectra of the Cu K-edge are shown in Fig. S10.‡ It can be seen that the oxidation state of Cu in CuFe10-900 and Cu-900 lies between those in Cu foil and CuPc, slightly less than +2. Similarly, no Cu–Cu interaction is seen in Fig. S10b,‡ suggesting that the Cu is also present in single-atom sites. R space fitting of the Cu EXAFS spectra is shown in Fig. S13c–f,‡ and both Cu-900 and CuFe10-900 exhibit a typical CuN4 coordination environment. According to the fitted data, the coordination number (CN) of both Cu and Fe in CuFe10-900 decreased compared with those in Cu-900 and Fe10-900 (Table S3‡), likely resulting from the increased defect densities caused by the additional metal introduction.41
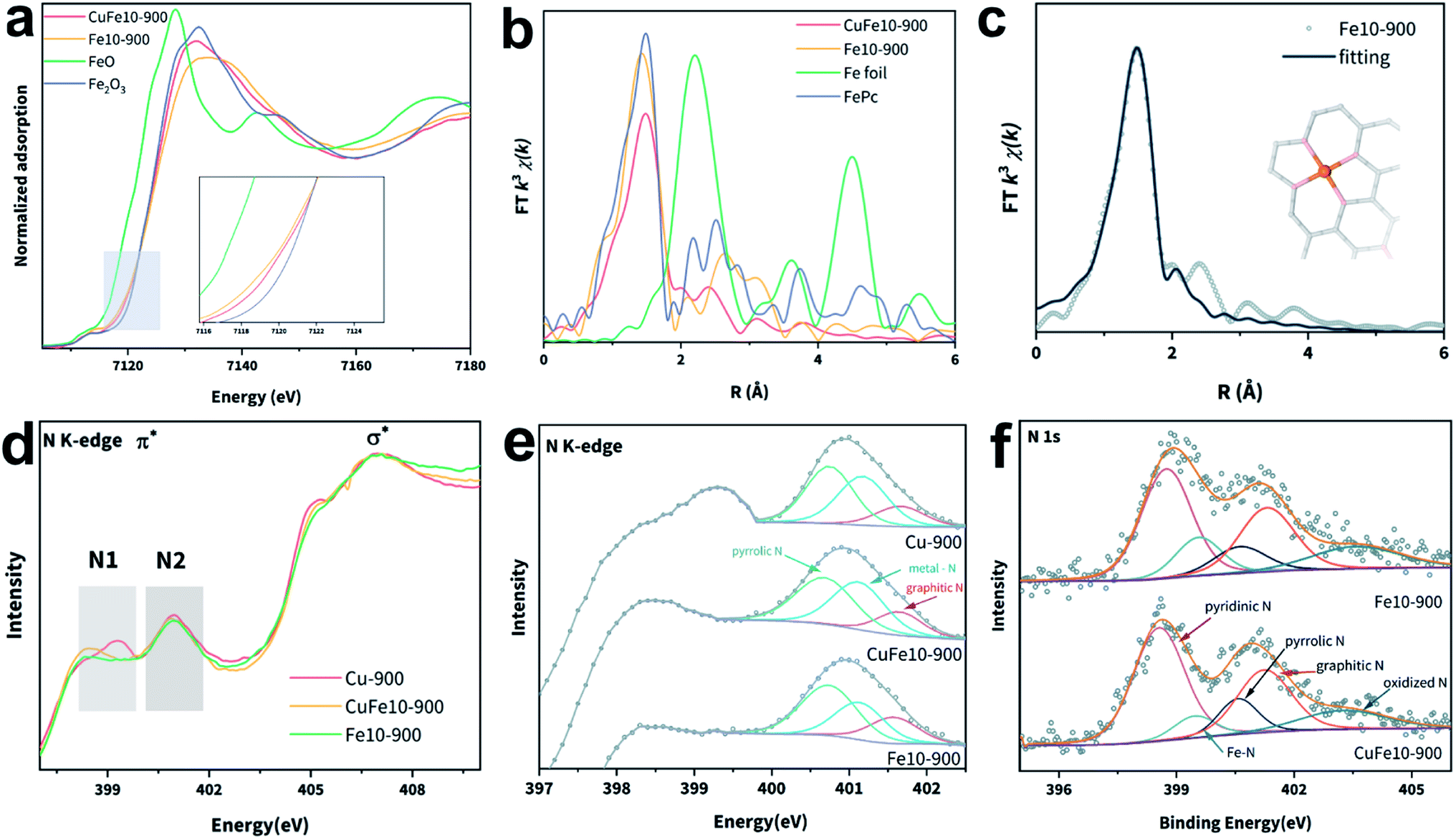 | ||
| Fig. 2 (a) Fe K-edge XANES spectra, inset: magnified spectra, (b) FT-EXAFS spectra in the R space of CuFe10-900, Fe10-900 and standards (FeO and Fe2O3), (c) first-shell fitting of the FT-EXAFS spectrum of CuFe10-900, (d) N K-edge XANES spectra and (e) magnified and deconvoluted N1 + N2 regions of the three SACs, and (f) deconvoluted N 1s XPS spectra of CuFe10-900 and Fe10-900. | ||
Metal–N coordination sites were further studied by the soft X-ray absorption spectroscopy (sXAS) technique. As shown in Fig. S14,‡ there are two dominant C K-edge absorptions located at 285.3 and 288.3 eV, consistent with previous reports of C–C π* and C–N–C, respectively.39,42–44 All three samples present the same C K-edge absorption positions, indicating that the carbon matrix environments are similar under 900 °C pyrolysis conditions. The deconvoluted C 1s XPS spectrum of CuFe10-900 is shown in Fig. S15a,‡ and four peaks are observed and can be attributed to C–C, C–N, C–O and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, respectively. In contrast, the N K-edge spectra differ from each other (Fig. 2d). The highlighted N1 and N2 peaks can be assigned to N π* transitions, and the broad peaks at 407 eV match the N σ* transition.45,46 The N1 domain represents the π* transitions of pyridinic N.39 As the deconvoluted N2 spectra (Fig. 2e) show, the two peaks at around 400.7 eV and 401.6 eV can be indexed to pyrrolic N and graphitic N, respectively. The peak between these two peaks corresponds to the metal–N species.33,47 Compared with CuFe10-900, Fe10-900 presents a stronger pyrrolic N peak intensity and a lower metal–N peak intensity. The deconvoluted N 1s XPS spectra of CuFe10-900 and Fe10-900 are shown in Fig. 2f. The peaks located at 398.6 eV, 399.5 eV, 400.5 eV, and 401.7 eV can be assigned to pyridinic N, metal–N, pyrrolic N, graphitic N and oxidized N, respectively.48 In addition, the Cu and Fe 2p XPS spectra are shown in Fig. S15b and c.‡ It should be noted that the poor Fe XPS signals likely resulted from the unsatisfactory detection limit of the conventional XPS instrument,48 a common challenge encountered elsewhere for analyzing Fe-based SACs with low Fe loadings especially for definite models.23,49,50
O, respectively. In contrast, the N K-edge spectra differ from each other (Fig. 2d). The highlighted N1 and N2 peaks can be assigned to N π* transitions, and the broad peaks at 407 eV match the N σ* transition.45,46 The N1 domain represents the π* transitions of pyridinic N.39 As the deconvoluted N2 spectra (Fig. 2e) show, the two peaks at around 400.7 eV and 401.6 eV can be indexed to pyrrolic N and graphitic N, respectively. The peak between these two peaks corresponds to the metal–N species.33,47 Compared with CuFe10-900, Fe10-900 presents a stronger pyrrolic N peak intensity and a lower metal–N peak intensity. The deconvoluted N 1s XPS spectra of CuFe10-900 and Fe10-900 are shown in Fig. 2f. The peaks located at 398.6 eV, 399.5 eV, 400.5 eV, and 401.7 eV can be assigned to pyridinic N, metal–N, pyrrolic N, graphitic N and oxidized N, respectively.48 In addition, the Cu and Fe 2p XPS spectra are shown in Fig. S15b and c.‡ It should be noted that the poor Fe XPS signals likely resulted from the unsatisfactory detection limit of the conventional XPS instrument,48 a common challenge encountered elsewhere for analyzing Fe-based SACs with low Fe loadings especially for definite models.23,49,50
Based on the spectroscopic characterization results, we believe that CuFe10-900 with dominant D1 sites can deliver the optimal ORR electrocatalytic activity. The ORR performance evaluation was carried out using a rotating ring-disk electrode (RRDE) system in O2-saturated 0.1 M KOH as the electrolyte. The cyclic voltammetry (CV) curves of the different samples in O2-saturated electrolyte are shown in Fig. S17.‡ We observed that CuFe10-900 shows a more positive oxygen reduction peak position and larger current density compared with the state-of-the-art 20% Pt/C catalyst, suggesting a higher half-wave potential (E1/2) and improved intrinsic activity, which are further confirmed by linear sweep voltammetry (LSV) curves shown in Fig. 3b. Specifically, CuFe10-900 delivered an impressive E1/2 of 0.889 V vs. RHE and a large diffusion-limited current density (j0) of around −6.1 mA cm−2. For comparison, the above two activity indicators of commercial 20% Pt/C are 0.89 V vs. RHE and −5.5 mA cm−2, respectively. We also tested two monometallic SACs for comparison. Fe10-900 also shows a decent E1/2 of 0.855 V vs. RHE, however with a much lower j0 (−5.6 mA cm−2). While Cu-900 shows a relatively large j0 (−6.2 mA cm−2), its E1/2 is as low as 0.798 V vs. RHE. This result verifies that our design strategy of incorporation of copper can enhance the intrinsic activity of Fe-based SACs towards the ORR. A comparison of the kinetic current densities (jk) of different catalysts at 0.9 V vs. RHE is provided in Fig. 3b. As expected, we found that CuFe10-900 presented the largest jk of around 4.77 mA cm−2, which is significantly larger than those of the other three counterparts, including Pt/C. In order to find out the contribution of Cu single sites in CuFe10-900, potassium thiocyanate (KSCN) was employed to conduct the poisoning test.51 As shown in Fig. S18,‡ CuFe10-900, CuFe20-900 and Fe10-900 presented nearly identical degraded ORR performance after the addition of KSCN, while Cu-900 exhibited almost no change, suggesting that FeN4 instead of CuN4 serves as the dominant ORR active site in CuFe10-900. We further conducted ORR tests using CuX-900 (X = Mn, Co and Ni) to support this assumption. As shown in Fig. S19,‡ the ORR polarization curves of these samples diverge from each other, indicating that X single-sites should play a dominant role in the enhanced ORR activity. The electron transfer number (n) and peroxide production yield were evaluated using a RRDE. According to Fig. 3c, n of CuFe10-900 is higher than 3.9 in an extended potential window from 0.2 to 0.8 V vs. RHE, and only minimal H2O2 is produced (less than 5%), indicating a very high selectivity towards the four-electron transfer process in CuFe10-900 based ORR. In comparison, all other samples exhibit an obviously reduced n as well as increased H2O2 yields, especially at low potentials. We also used the Koutecky–Levich (K–L) equation to estimate n using the rotating disk electrode (RDE) test.52 As shown in the K–L plot (Fig. 3d), the obtained n at different potentials remains close to 4, which is consistent with the above RRDE result. Interestingly, all four samples exhibit similar Tafel slopes of around 60 mV dec−1, indicating that the cleavage of the oxygen–oxygen bond on the catalyst surface is the kinetic rate determining step for the ORR under the testing conditions.53 Specifically, CuFe10-900 displays the most positive onset potential, which is close to that of Pt/C, indicating the best intrinsic activity towards the ORR. Furthermore, the stability of CuFe10-900 was evaluated using a chronoamperometry method and compared with that of commercial Pt/C using a chronoamperometric method performed at 0.85 V vs. RHE. As shown in Fig. 3f, in clear contrast to Pt/C, CuFe10-900 not only delivered a clearly higher current density at the same potential, but also presented a less than 10% drop in activity after 30 h electrolysis, while the activity of Pt/C dropped by one-fourth in 12 h. The morphology of CuFe10-900 was characterized after the stability test. As shown in Fig. S21,‡ the well-defined rhombododecahedral morphology remained after catalysis with the appearance of slightly roughened surfaces.
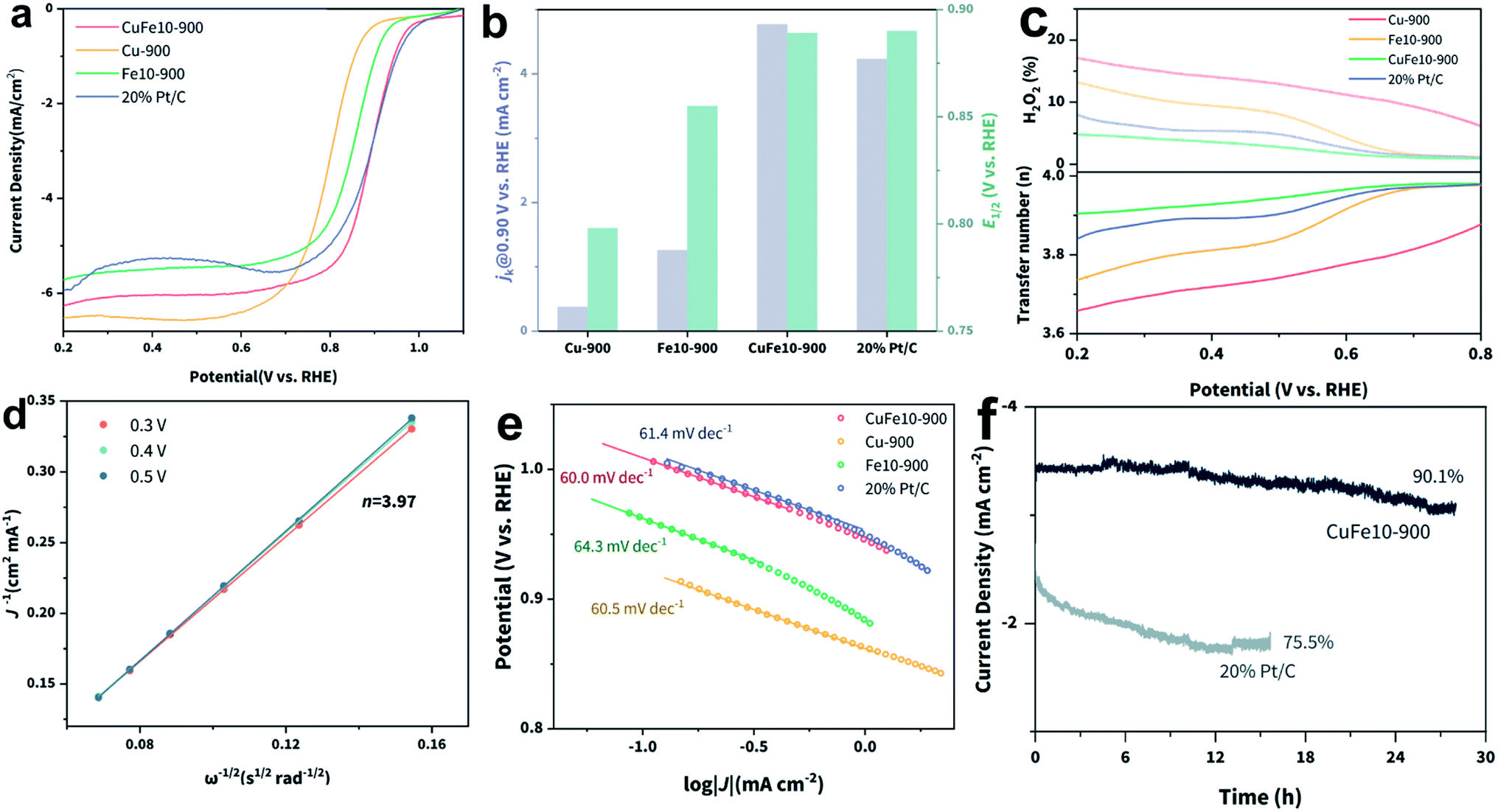 | ||
| Fig. 3 (a) LSV curves, (b) jk @ 0.9 V vs. RHE and E1/2 comparison, (c) H2O2 selectivity and electron transfer number (n) of Cu-900, Fe10-900, CuFe10-900 and 20% Pt/C, (d) K–L plots of CuFe10-900, (e) Tafel plots of the four tested samples and (f) chronoamperometric tests of CuFe10-900 and Pt/C at 0.85 V vs. RHE. | ||
Moreover, we examined the ORR activity of CuFe-SACs with different Fe loadings and pyrolysis temperatures. As can be seen in Fig. S22,‡ all four samples present an E1/2 value similar to that of Fe10-900, which is much smaller than that of CuFe10-900, indicating that only appropriate modulation of Fe loading and pyrolysis temperature can enhance the intrinsic activity of FeN4 sites and thus facilitate the ORR process. Moreover, both CuFe20-900 and CuFe10-1000 show obvious decreases in j0, indicating that a high density of FeN4 sites alone or very high temperature will hamper the intrinsic activity and mass transport. Fig. S23‡ reveals that the above four samples show jk values that are close to that of Fe10-900, but much lower than that of CuFe10-900. As shown in Fig. S22c,‡ all these samples have similar Tafel slopes of around 60 mV dec−1, suggesting that these catalysts share the same rate limiting step for the ORR. In addition, CuFe5-900 shows the largest n among the four samples, as well as a similar n–potential relation to that of CuFe10-900. Taken together, we can conclude that both suitable Fe loading amount and pyrolysis temperature play important roles in optimizing the ORR catalytic activity and selectivity. Specifically, too low Fe loading will not provide sufficient active sites, thus leading to low intrinsic activity, while on the other hand, too high Fe loading will weaken the interactions between Cu and Fe and further lead to reduced ORR activity. Separately, low pyrolysis temperature (<900 °C) is not enough for sufficient removal of Zn atoms which will likely hinder the formation of neighboring Fe and Cu single sites and further inhibit the ORR kinetics, while very high pyrolysis temperature can damage the highly porous carbon skeleton and is thus unfavorable for gas-involved catalysis.
The electrochemical active surface areas (ECSAs) of Cu-900, Fe10-900 and CuFe10-900 were estimated by measuring the electrochemical double-layer capacitance (Cdl) in non-faradaic regions (Fig. S24‡). As plotted in Fig. 4a, while CuFe10-900 exhibits the largest Cdl, it is very close to those of the other two SACs. Taking electrochemically accessible active sites into consideration, we calculated the corresponding ECSA normalized jk as shown in Fig. S25.‡ As can be seen, CuFe10-900 still presents the highest ECSA normalized jk, which is 3.5 and 11.9 fold higher than those of Fe10-900 and Cu-900, respectively.
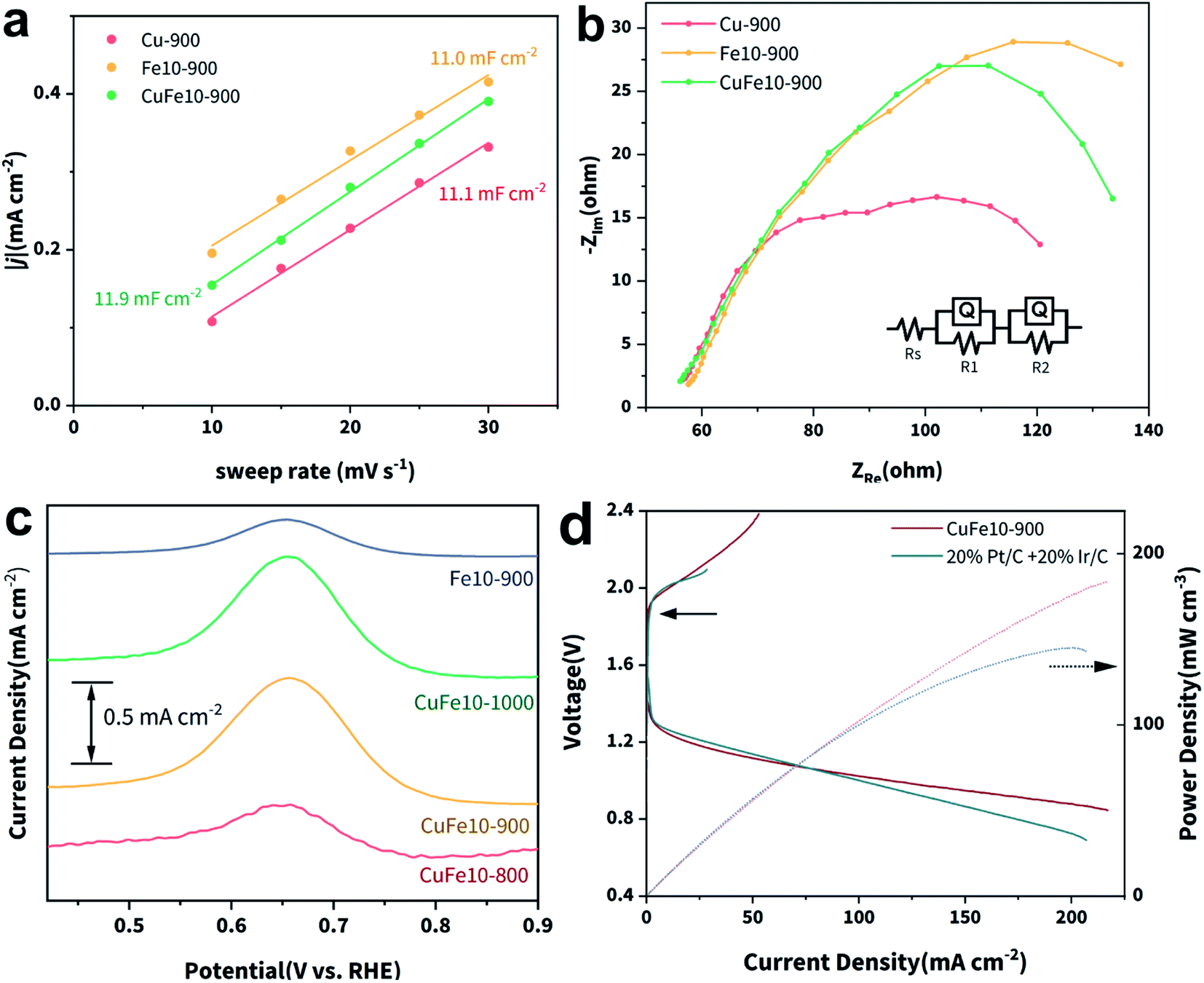 | ||
| Fig. 4 (a) Cdl fitting plots of Cu-900, Fe10-900 and CuFe10-900, (b) Nyquist plots of the three samples and equivalent circuit, (c) SWV curves of the four SACs and (d) charge–discharge and power density curves of CuFe10-900 and Pt/C for the Zn–air battery. | ||
Electrochemical impedance spectroscopy (EIS) was used to analyze the intrinsic charge transfer kinetics for the above SACs. Fig. 4b presents the Nyquist plots of the three SACs and the equivalent circuit used for the curve fittings. It can be simulated that the charge transfer resistance (R1) of CuFe10-900 (6.631 Ω) is much smaller than those of Cu-900 (83.44 Ω) and Fe10-900 (127.4 Ω), resulting in an enhanced electron transfer rate during the ORR on CuFe10-900. Moreover, square-wave voltammetry (SWV) was employed to investigate the Fe coordination environments.54,55 As shown in Fig. 4c, there is a peak located at around 0.65 V vs. RHE, which can be attributed to the redox signal of D1 type FeN4.38 Clearly, the D1 peak intensities of CuFe10-900 and CuFe10-1000 are much stronger than those of Fe10-900 and CuFe10-800. Since these four samples have a similar Fe content based on the ICP-OES data, we can conclude that the incorporation of Cu and complete removal of Zn are crucial to obtain SACs with a high density of D1 type active sites. Fig. S26‡ shows the SWV curves of CuFex-900, and it can be found that the intensity of the redox peak is in good accordance with the density of FeN4 sites. Overall, the SWV results are in agreement with the XAFS results, further confirming that increased densities of D1 type FeN4 sites can be ascribed to the incorporation of Cu SACs and concomitant electronic interactions. Finally, we studied the ORR catalytic activity of the SACs in acidic media. As shown in Fig. S27,‡E1/2 of CuFe10-900 is 0.78 V vs. RHE, similar to that of Fe10-900 and is comparable with that of Pt/C. Besides, the n of the acidic ORR on CuFe10-900 is still close to 4. Based on all of this, we can conclude that the design strategy of incorporating secondary metal atoms is beneficial for the ORR activity in both alkaline and acidic media (Table S4‡).
Finally, a Zn–air battery was fabricated to evaluate the potential of the practical application of the SAC catalyst developed here. As can be seen in Fig. 4d, the CuFe10-900 + Ir/C and 20% Pt/C + Ir/C couples show comparable charge–discharge voltage gaps, suggesting the intriguing performance of CuFe10-900 as an air cathode in the Zn–air battery. Notably, the maximum power density of CuFe10-900 reaches 200 mW cm−2, higher than that of the state-of-the-art Pt/C–Ir/C couple. These results provide persuasive verification of our effective catalyst design principle and CuFe10-900 as a potential ORR catalyst candidate for future applications.
In conclusion, we have successfully demonstrated an effective catalyst design principle for the ORR that consists in incorporating secondary metal atoms into single-atom catalysts. Based on this, we developed a facile two-step method to synthesize a well-defined CuFe heterometallic single-atom catalyst. The method is highly tunable for achieving diverse atom kinds and loadings. Through spectroscopic characterization and performance evaluation, we reached the conclusion that the incorporated Cu single sites promote the formation of D1 type FeN4 sites, leading to a significantly enhanced electrocatalytic ORR activity. We believe that this strategy and the obtained SACs can be further applied to versatile energy and environment related applications such as CO2 reduction, and pave a new way for the precisely controlled synthesis of single-atom catalysts.
Author contributions
L. Wang led the whole project. H. Yang synthesized the catalysts, performed SEM, TEM, XRD, BET, XPS, XAFS, ICP, and electrochemical measurements, and data analysis, and wrote the manuscript. X. Wang guided the synthesis of catalysts. X. Chi and X. Yu provided assistance with soft X-ray absorption characterization. S. Tan and Q. He recorded the HAADF-STEM images. T. Zhang assisted in the analysis of the XAFS data. Junmei Chen, Jiayi Chen and C. Li provided help with characterization and manuscript writing. Q. He and L. Wang revised the manuscript.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
We acknowledge the National University of Singapore, Ministry of Education and National Research Foundation Singapore for their financial support, through grants R-279-000-622-133, R-279-000-553-646 and A-0009195-00-00. Q. He would also like to acknowledge the support by the National Research Foundation (NRF) Singapore, under its NRF Fellowship (NRF-NRFF11-2019-0002).References
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- S. Chu, Y. Cui and N. Liu, Nat. Mater., 2016, 16, 16–22 CrossRef PubMed.
- F. Cheng and J. Chen, Chem. Soc. Rev., 2012, 41, 2172–2192 RSC.
- M. Shao, Q. Chang, J.-P. Dodelet and R. Chenitz, Chem. Rev., 2016, 116, 3594–3657 CrossRef CAS PubMed.
- X. X. Wang, M. T. Swihart and G. Wu, Nat. Catal., 2019, 2, 578–589 CrossRef CAS.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Norskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- I. Katsounaros, S. Cherevko, A. R. Zeradjanin and K. J. Mayrhofer, Angew. Chem., Int. Ed., 2014, 53, 102–121 CrossRef CAS.
- Z.-F. Huang, J. Wang, Y. Peng, C.-Y. Jung, A. Fisher and X. Wang, Adv. Energy Mater., 2017, 7, 1700544 CrossRef.
- Y. Xue, S. Sun, Q. Wang, Z. Dong and Z. Liu, J. Mater. Chem. A, 2018, 6, 10595–10626 RSC.
- S. Zheng, X. Li, B. Yan, Q. Hu, Y. Xu, X. Xiao, H. Xue and H. Pang, Adv. Energy Mater., 2017, 7, 1602733 CrossRef.
- C. X. Zhao, J. N. Liu, J. Wang, D. Ren, B. Q. Li and Q. Zhang, Chem. Soc. Rev., 2021, 50, 7745–7778 RSC.
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2015, 44, 2060–2086 RSC.
- D. Zhao, Z. Zhuang, X. Cao, C. Zhang, Q. Peng, C. Chen and Y. Li, Chem. Soc. Rev., 2020, 49, 2215–2264 RSC.
- H. Xu, D. Wang, P. Yang, A. Liu, R. Li, Y. Li, L. Xiao, X. Ren, J. Zhang and M. An, J. Mater. Chem. A, 2020, 8, 23187–23201 RSC.
- Y. Li, N. Wang, H. Lei, X. Li, H. Zheng, H. Wang, W. Zhang and R. Cao, Coord. Chem. Rev., 2021, 442, 213996 CrossRef CAS.
- Y. Chen, S. Ji, C. Chen, Q. Peng, D. Wang and Y. Li, Joule, 2018, 2, 1242–1264 CrossRef CAS.
- Y. Chen, S. Ji, Y. Wang, J. Dong, W. Chen, Z. Li, R. Shen, L. Zheng, Z. Zhuang, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2017, 56, 6937–6941 CrossRef CAS PubMed.
- H. T. Chung, D. A. Cullen, D. Higgins, B. T. Sneed, E. F. Holby, K. L. More and P. Zelenay, Science, 2017, 357, 479–484 CrossRef CAS PubMed.
- Z. Zhang, J. Sun, F. Wang and L. Dai, Angew. Chem., Int. Ed., 2018, 57, 9038–9043 CrossRef CAS PubMed.
- R. Jiang, L. Li, T. Sheng, G. Hu, Y. Chen and L. Wang, J. Am. Chem. Soc., 2018, 140, 11594–11598 CrossRef CAS PubMed.
- Y. Chen, S. Ji, S. Zhao, W. Chen, J. Dong, W. C. Cheong, R. Shen, X. Wen, L. Zheng, A. I. Rykov, S. Cai, H. Tang, Z. Zhuang, C. Chen, Q. Peng, D. Wang and Y. Li, Nat. Commun., 2018, 9, 5422 CrossRef CAS PubMed.
- C. C. Hou, L. Zou, L. Sun, K. Zhang, Z. Liu, Y. Li, C. Li, R. Zou, J. Yu and Q. Xu, Angew. Chem., Int. Ed., 2020, 59, 7384–7389 CrossRef CAS PubMed.
- K. Yuan, D. Lutzenkirchen-Hecht, L. Li, L. Shuai, Y. Li, R. Cao, M. Qiu, X. Zhuang, M. K. H. Leung, Y. Chen and U. Scherf, J. Am. Chem. Soc., 2020, 142, 2404–2412 CrossRef CAS PubMed.
- E. Luo, Y. Chu, J. Liu, Z. Shi, S. Zhu, L. Gong, J. Ge, C. H. Choi, C. Liu and W. Xing, Energy Environ. Sci., 2021, 14, 2158–2185 RSC.
- C. Hu, R. Paul, Q. Dai and L. Dai, Chem. Soc. Rev., 2021, 50, 11785–11843 RSC.
- B. You, N. Jiang, M. Sheng, W. S. Drisdell, J. Yano and Y. Sun, ACS Catal., 2015, 5, 7068–7076 CrossRef CAS.
- P. Yin, T. Yao, Y. Wu, L. Zheng, Y. Lin, W. Liu, H. Ju, J. Zhu, X. Hong, Z. Deng, G. Zhou, S. Wei and Y. Li, Angew. Chem., Int. Ed., 2016, 55, 10800–10805 CrossRef CAS PubMed.
- H. Yang and X. Wang, Adv. Mater., 2019, 31, 1800743 CrossRef PubMed.
- U. I. Kramm, M. Lefevre, N. Larouche, D. Schmeisser and J. P. Dodelet, J. Am. Chem. Soc., 2014, 136, 978–985 CrossRef CAS PubMed.
- N. D. Leonard, S. Wagner, F. Luo, J. Steinberg, W. Ju, N. Weidler, H. Wang, U. I. Kramm and P. Strasser, ACS Catal., 2018, 8, 1640–1647 CrossRef CAS.
- H. Zhang, H. T. Chung, D. A. Cullen, S. Wagner, U. I. Kramm, K. L. More, P. Zelenay and G. Wu, Energy Environ. Sci., 2019, 12, 2548–2558 RSC.
- L. Jiao, J. Zhu, Y. Zhang, W. Yang, S. Zhou, A. Li, C. Xie, X. Zheng, W. Zhou, S. H. Yu and H. L. Jiang, J. Am. Chem. Soc., 2021, 143, 19417–19424 CrossRef CAS PubMed.
- Z. Jin, P. Li, Y. Meng, Z. Fang, D. Xiao and G. Yu, Nat. Catal., 2021, 4, 615–622 CrossRef CAS.
- Y. Qu, Z. Li, W. Chen, Y. Lin, T. Yuan, Z. Yang, C. Zhao, J. Wang, C. Zhao, X. Wang, F. Zhou, Z. Zhuang, Y. Wu and Y. Li, Nat. Catal., 2018, 1, 781–786 CrossRef CAS.
- F. Li, G.-F. Han, H.-J. Noh, S.-J. Kim, Y. Lu, H. Y. Jeong, Z. Fu and J.-B. Baek, Energy Environ. Sci., 2018, 11, 2263–2269 RSC.
- X. Xu, Z. Zhang and X. Wang, Adv. Mater., 2015, 27, 5365–5371 CrossRef CAS PubMed.
- H. Yang and X. Wang, Adv. Mater., 2019, 31, e1800743 CrossRef PubMed.
- Q. Jia, N. Ramaswamy, H. Hafiz, U. Tylus, K. Strickland, G. Wu, B. Barbiellini, A. Bansil, E. F. Holby, P. Zelenay and S. Mukerjee, ACS Nano, 2015, 9, 12496–12505 CrossRef CAS PubMed.
- N. Zhang, T. Zhou, M. Chen, H. Feng, R. Yuan, C. a. Zhong, W. Yan, Y. Tian, X. Wu, W. Chu, C. Wu and Y. Xie, Energy Environ. Sci., 2020, 13, 111–118 RSC.
- J. Li, S. Ghoshal, W. Liang, M.-T. Sougrati, F. Jaouen, B. Halevi, S. McKinney, G. McCool, C. Ma, X. Yuan, Z.-F. Ma, S. Mukerjee and Q. Jia, Energy Environ. Sci., 2016, 9, 2418–2432 RSC.
- M. Zhao, H. Liu, H. Zhang, W. Chen, H. Sun, Z. Wang, B. Zhang, L. Song, Y. Yang, C. Ma, Y. Han and W. Huang, Energy Environ. Sci., 2021, 14, 6455–6463 RSC.
- Y. Liang, Y. Li, H. Wang, J. Zhou, J. Wang, T. Regier and H. Dai, Nat. Mater., 2011, 10, 780–786 CrossRef CAS.
- X. Wang, W. Chen, L. Zhang, T. Yao, W. Liu, Y. Lin, H. Ju, J. Dong, L. Zheng, W. Yan, X. Zheng, Z. Li, X. Wang, J. Yang, D. He, Y. Wang, Z. Deng, Y. Wu and Y. Li, J. Am. Chem. Soc., 2017, 139, 9419–9422 CrossRef CAS PubMed.
- H. Jiang, J. Gu, X. Zheng, M. Liu, X. Qiu, L. Wang, W. Li, Z. Chen, X. Ji and J. Li, Energy Environ. Sci., 2019, 12, 322–333 RSC.
- Y. Liang, H. Wang, J. Zhou, Y. Li, J. Wang, T. Regier and H. Dai, J. Am. Chem. Soc., 2012, 134, 3517–3523 CrossRef CAS PubMed.
- P. Leinweber, J. Kruse, F. L. Walley, A. Gillespie, K. U. Eckhardt, R. I. Blyth and T. Regier, J. Synchrotron Radiat., 2007, 14, 500–511 CrossRef CAS PubMed.
- D. N. Kelly, C. P. Schwartz, J. S. Uejio, A. M. Duffin, A. H. England and R. J. Saykally, J. Chem. Phys., 2010, 133, 101103 CrossRef PubMed.
- M. Primbs, Y. Sun, A. Roy, D. Malko, A. Mehmood, M.-T. Sougrati, P.-Y. Blanchard, G. Granozzi, T. Kosmala, G. Daniel, P. Atanassov, J. Sharman, C. Durante, A. Kucernak, D. Jones, F. Jaouen and P. Strasser, Energy Environ. Sci., 2020, 13, 2480–2500 RSC.
- Y. Li, W. Shan, M. J. Zachman, M. Wang, S. Hwang, H. Tabassum, J. Yang, X. Yang, S. Karakalos, Z. Feng, G. Wang and G. Wu, Angew. Chem., Int. Ed., 2022, e202205632 Search PubMed.
- Z. Wang, X. Jin, C. Zhu, Y. Liu, H. Tan, R. Ku, Y. Zhang, L. Zhou, Z. Liu, S.-J. Hwang and H. J. Fan, Adv. Mater., 2021, 33, 2104718 CrossRef CAS PubMed.
- M. S. Thorum, J. M. Hankett and A. A. Gewirth, J. Phys. Chem. Lett., 2011, 2, 295–298 CrossRef CAS.
- S. Guo, S. Zhang and S. Sun, Angew. Chem., Int. Ed., 2013, 52, 8526–8544 CrossRef CAS PubMed.
- T. Shinagawa, A. T. Garcia-Esparza and K. Takanabe, Sci. Rep., 2015, 5, 13801 CrossRef PubMed.
- N. Ramaswamy, U. Tylus, Q. Jia and S. Mukerjee, J. Am. Chem. Soc., 2013, 135, 15443–15449 CrossRef CAS PubMed.
- B. Huang, M. Wang, C. Wu and L. Guan, CCS Chem., 2021, 3, 3473–3484 CrossRef.
Footnotes |
| † This paper is dedicated to Prof. Licheng Sun on the occasion of his 60th birthday. |
| ‡ Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ta01962k. |
| This journal is © The Royal Society of Chemistry 2022 |