
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhanced oxygen redox reversibility and capacity retention of titanium-substituted Na4/7[□1/7Ti1/7Mn5/7]O2 in sodium-ion batteries†
Stephanie F.
Linnell
 ab,
Eun Jeong
Kim
ab,
Yong-Seok
Choi
bcd,
Moritz
Hirsbrunner
e,
Saki
Imada
f,
Atin
Pramanik
a,
Aida Fuente
Cuesta
a,
David N.
Miller
a,
Edoardo
Fusco
a,
Bela E.
Bode
a,
John T. S.
Irvine
ab,
Laurent C.
Duda
e,
David O.
Scanlon
bcd and
A. Robert
Armstrong
*ab
ab,
Eun Jeong
Kim
ab,
Yong-Seok
Choi
bcd,
Moritz
Hirsbrunner
e,
Saki
Imada
f,
Atin
Pramanik
a,
Aida Fuente
Cuesta
a,
David N.
Miller
a,
Edoardo
Fusco
a,
Bela E.
Bode
a,
John T. S.
Irvine
ab,
Laurent C.
Duda
e,
David O.
Scanlon
bcd and
A. Robert
Armstrong
*ab
aSchool of Chemistry, University of St Andrews, St Andrews, Fife, KY16 9ST, UK. E-mail: ara@st-andrews.ac.uk
bThe Faraday Institution, Quad One, Harwell Science and Innovation Campus, Didcot, OX11 0RA, UK
cDepartment of Chemistry, University College London, 20 Gordon Street, London, WC1H 0AJ, UK
dThomas Young Centre, University College London, Gower Street, London WC1E 6BT, UK
eDepartment of Physics and Astronomy, Division of Molecular and Condensed Matter Physics, Uppsala University, Uppsala, S-75120, Sweden
fFaculty of Electrical Engineering and Electronics, Kyoto Institute of Technology, Sakyo, Kyoto 606-8585, Japan
First published on 30th March 2022
Abstract
Anion redox reactions offer a means of enhancing the capacity of layered sodium transition metal oxide positive electrode materials. However, oxygen redox reactions typically show limited reversibility and irreversible structural changes upon cycling, resulting in rapid capacity loss. Here, the Ti-substituted Na4/7[□1/7Ti1/7Mn5/7]O2 (where □ represents a transition metal vacancy) is presented as a positive electrode material for sodium-ion batteries. Na4/7[□1/7Ti1/7Mn5/7]O2 delivers a reversible capacity of 167 mA h g−1 after 25 cycles at 10 mA g−1 within the voltage range of 1.6–4.4 V and presents enhanced stability compared with Na4/7[□1/7Mn6/7]O2 over the voltage range 3.0–4.4 V. The structural and electronic structural changes of this Ti4+ substituted phase are investigated by powder X-ray diffraction, X-ray absorption spectroscopy, electron paramagnetic resonance and Raman spectroscopy, supported by density functional theory calculations. These results show that the Na4/7[□1/7Mn6/7]O2 structure is maintained between 3.0 and 4.4 V, and the presence of TiO6 octahedra in Na4/7[□1/7Ti1/7Mn5/7]O2 relieves structural distortions from Jahn–Teller distorted Mn3+O6 between 1.6 and 4.4 V. Furthermore, Ti4+ substitution stabilises the adjacent O 2p orbitals and raises the ionicity of the Mn–O bonds, increasing the operating potential of Na4/7[□1/7Ti1/7Mn5/7]O2. Thereby providing evidence that the improved electrochemical performance of Na4/7[□1/7Ti1/7Mn5/7]O2 can be attributed to Ti4+ substitution. This work provides insight and strategies for improving the structural stability and electrochemical performance of sodium layered oxides.
1. Introduction
The limited availability and hence the high-cost of raw materials used in lithium-ion batteries (LIBs) has driven research towards alternative rechargeable battery technologies. Among these, sodium-ion batteries (SIBs) have attracted considerable attention as a viable replacement to LIBs for large-scale applications, such as grid energy storage because of the low-cost, high-abundance and uniform geographical distribution of sodium.1,2 However, one of the main challenges to overcome is the development of high energy density positive electrode materials for the practical application of SIBs.3–6The use of both cation and anion redox couples has become a promising method to enhance the capacity of positive electrode materials, thereby increasing the energy density of batteries. Lithium-rich layered oxides (Li1+xM1−xO2, M = transition metal), have been widely-studied and are promising positive electrode materials for lithium-ion batteries (LIBs).7–9 For instance, Li1.20Mn0.54Co0.13Ni0.13O2 in which lithium atoms are situated in the transition metal layer, delivers an extraordinarily high capacity, attributed to the cumulative use of transition metal and oxygen redox couples.10–12 However, materials that exhibit oxygen redox generally show large voltage hysteresis during the initial charge/discharge cycles, because of irreversible structural changes including cation migration from the transition metal layers, resulting in the release of molecular O2 and thus, rapid capacity fade.7,13–15 Alternatively, some sodium-based transition metal layered oxides, NaxMyO2, have shown suppressed cation migration owing to the increased ionic size difference between sodium and the transition metal.16–20 One sodium-based layered material that exhibits stable oxygen anion redox is Na4/7[□1/7Mn6/7]O2. The transition metal vacancies in Na4/7[□1/7Mn6/7]O2 (where □ represents a transition metal vacancy) and the unique ordering between vacancies and manganese, generates O 2p nonbonding orbitals, promoting reversible oxygen redox activity.21 Na4/7[□1/7Mn6/7]O2 maintains the layered structure over several cycles without migration of Mn cations and preserves the ABBCCA oxygen stacking sequence.22,23 As a result, Na4/7[□1/7Mn6/7]O2 exhibits more reversible anion redox compared with other sodium-based oxygen redox active materials with negligible voltage hysteresis and can deliver a high specific capacity of up to ∼200 mA h g−1, thereby demonstrating the importance of vacancies in stabilising anion redox.21,24,25
Despite the good reversibility of Na4/7[□1/7Mn6/7]O2 reported to date, its long-term cycle life remains questionable. This is especially true when both cation and anion redox reactions are utilised, where Na4/7[□1/7Mn6/7]O2 undergoes a continuous capacity fade from ∼200 to ∼125 mA h g−1 over 60 cycles.21,25 To promote reversible redox reactions, an effective strategy is through doping with transition metals. Among the dopants studied, Ti-substituted materials have drawn attention in recent years due to the low-cost, chemical stability and wide-spread distribution in the earth's crust. Yoshida et al. revealed that substituting Mn with Ti in P2-type Na2/3Ni1/3Mn2/3−xTixO2, enhanced the cycling performance and increased average discharge voltage to 3.7 V. Moreover, increasing the amount of Ti-substitution resulted in a smoother voltage profile, possibly because of the suppression of the P2 → O2 phase transition.26 Li et al. demonstrated that Ti4+ substitution reduced the Li+/Mn4+ ordering for Na0.72Li0.24Ti0.10Mn0.66O2 which enhanced the reversibility of the oxygen redox process and resulted in a superior reversible capacity of 165 mA h g−1 over 80 cycles.27 These results show that Ti-substitution is a proven method to stabilise the structure, smooth the charge/discharge profile, elevate the working potential, and improve the cycling stability.26–28
Liu et al., have recently reported a superior cycling stability of 79% for Na4/7[□1/7Ti1/7Mn5/7]O2 compared with 17% for Na4/7[□1/7Mn6/7]O2 within 1.5–4.6 V at 50 mA g−1, negligible volume change (0.11%) during charge/discharge and no irreversible O2 evolution upon charge to 4.6 V.29 The present study aims to understand the role of Ti-substitution on the charge compensation mechanism of Na4/7[□1/7Mn6/7]O2. Herein, we report that Ti-substituted Na4/7[□1/7Ti1/7Mn5/7]O2 shows a remarkable improvement over Na4/7[□1/7Mn6/7]O2 for both cation and anion redox stability: for a wide voltage range (1.6–4.4 V) over which both cation (Mn3+/Mn4+) and anion (O2−/O2n−) redox reactions are activated, Na4/7[□1/7Ti1/7Mn5/7]O2 delivers higher operating potential, reversible capacity (167 mA h g−1), and greater capacity retention of 88% than Na4/7[□1/7Mn6/7]O2 with a lower reversible capacity of 143 mA h g−1 and capacity retention of 76% after 25 cycles at 10 mA g−1. Additional electrochemical tests performed for Na4/7[□1/7Ti1/7Mn5/7]O2 over the voltage range 3.0–4.4 V revealed a highly reversible oxygen redox process with no capacity loss after 50 cycles at 10 mA g−1. We evaluated the changes in the crystal structure and the electronic structure upon cycling using a combination of spectroscopic techniques, together with DFT calculations to determine the redox mechanism and the effects of Ti-substitution on the electrochemical performance. This work on Na4/7[□1/7Ti1/7Mn5/7]O2, which has the oxygen stacking (ABBCCA) characteristic of P3-type structures extends our survey of P3-type sodium-based layered oxides, and our understanding of the importance of transition metal dopants on stabilising oxygen anion redox.30–32
2. Results and discussion
2.1. Structure and electrochemical properties
Na4/7[□1/7Mn6/7]O2 and Na4/7[□1/7Ti1/7Mn5/7]O2 were synthesised by a solid-state method.25 The synchrotron PXRD pattern of as-synthesised Na4/7[□1/7Ti1/7Mn5/7]O2 can be indexed to a triclinic lattice with space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . The Rietveld refinement of the Na4/7[□1/7Mn6/7]O2 model using synchrotron PXRD data for Na4/7[□1/7Ti1/7Mn5/7]O2 is shown in Fig. 1(a) and the results are given in Table S1.† Similarly, the diffraction peaks of the laboratory PXRD pattern of as-synthesised Na4/7[□1/7Mn6/7]O2 can be indexed using the same model (P), as shown in Fig. S1(a),† consistent with previous reports.21,22,25 The structure of Na4/7[□1/7Mn6/7]O2 consists of one Na+ ion (Na1) which occupies distorted prismatic sites above and below the vacant Mn sites, and the other Na+ ion (Na2) occupies distorted octahedral sites, the Mn4+ ions occupy octahedral sites with distorted P3-type oxygen stacking (ABBCCA), as shown in Fig. 1(b). As for Ti-substituted Na4/7[□1/7Ti1/7Mn5/7]O2, the Ti4+ ions can occupy any one of the three Mn sites, as confirmed by calculations which found negligible energy difference between the three Mn sites (see ESI for more details, Fig. S2†). Furthermore, the position of the (001) diffraction peak which relates to the interlayer distance between the transition metal oxide layers is compared in Fig. S3.† The (001) diffraction peak for Na4/7[□1/7Ti1/7Mn5/7]O2 shifts towards a lower angle. This corresponds to a greater interlayer separation of 5.569 Å for Na4/7[□1/7Ti1/7Mn5/7]O2, compared with 5.534 Å for Na4/7[□1/7Mn6/7]O2, as a result of the partial substitution of Ti4+ for Mn4+ ions, because of the larger ionic radius of Ti4+ (0.605 Å) compared with Mn4+ (0.53 Å).33
. The Rietveld refinement of the Na4/7[□1/7Mn6/7]O2 model using synchrotron PXRD data for Na4/7[□1/7Ti1/7Mn5/7]O2 is shown in Fig. 1(a) and the results are given in Table S1.† Similarly, the diffraction peaks of the laboratory PXRD pattern of as-synthesised Na4/7[□1/7Mn6/7]O2 can be indexed using the same model (P), as shown in Fig. S1(a),† consistent with previous reports.21,22,25 The structure of Na4/7[□1/7Mn6/7]O2 consists of one Na+ ion (Na1) which occupies distorted prismatic sites above and below the vacant Mn sites, and the other Na+ ion (Na2) occupies distorted octahedral sites, the Mn4+ ions occupy octahedral sites with distorted P3-type oxygen stacking (ABBCCA), as shown in Fig. 1(b). As for Ti-substituted Na4/7[□1/7Ti1/7Mn5/7]O2, the Ti4+ ions can occupy any one of the three Mn sites, as confirmed by calculations which found negligible energy difference between the three Mn sites (see ESI for more details, Fig. S2†). Furthermore, the position of the (001) diffraction peak which relates to the interlayer distance between the transition metal oxide layers is compared in Fig. S3.† The (001) diffraction peak for Na4/7[□1/7Ti1/7Mn5/7]O2 shifts towards a lower angle. This corresponds to a greater interlayer separation of 5.569 Å for Na4/7[□1/7Ti1/7Mn5/7]O2, compared with 5.534 Å for Na4/7[□1/7Mn6/7]O2, as a result of the partial substitution of Ti4+ for Mn4+ ions, because of the larger ionic radius of Ti4+ (0.605 Å) compared with Mn4+ (0.53 Å).33
 | ||
| Fig. 1 (a) Synchrotron X-ray Rietveld fit of as-synthesised Na4/7[□1/7Ti1/7Mn5/7]O2. Observed data points are shown in blue, with fitted profile in black. Tick marks indicate allowed reflections. (b) Refined structural model of Na4/7[□1/7Ti1/7Mn5/7]O2 with Na+ ions shown as yellow spheres, Mn as purple polyhedra, Ti shown in white and O as red spheres. The top image shows the alternating layers of Na+ ions and [□1/7Ti4+1/7Mn4+5/7] layers. The Na1 ions occupy distorted prismatic sites and Na2 ions occupy distorted octahedral sites. The bottom image shows the Mn vacancies in the [□1/7Ti4+1/7Mn4+5/7] layer. (c) SEM micrograph and (d) EDS mapping of as-synthesised Na4/7[□1/7Ti1/7Mn5/7]O2. | ||
The morphology of Na4/7[□1/7Ti1/7Mn5/7]O2 was examined by SEM. Fig. 1(b) is representative of Na4/7[□1/7Ti1/7Mn5/7]O2 and shows a homogeneous morphology with agglomerated primary particle size of roughly 100 nm. As-synthesised Na4/7[□1/7Mn6/7]O2 displays a similar morphology to Na4/7[□1/7Ti1/7Mn5/7]O2 (Fig. S1(b)†), showing that there is no obvious change in the morphology for the Ti-substituted sample compared with the parent material. EDS elemental maps of Na4/7[□1/7Ti1/7Mn5/7]O2 are given in Fig. 1(c) and show uniform distribution of Na, Mn, Ti and O. Furthermore, the stoichiometries of Mn and Ti (normalised to Mn + Ti = 6/7) were determined by EDS analysis averaged across several regions to be 0.72 and 0.14, respectively, and close to the expected stoichiometry. This stoichiometry ratio of Mn and Ti corresponds to the solubility limit of Ti in Na4/7[□1/7TixMn6/7−x]O2 at the synthesis temperature of 600 °C, indicating that the maximum amount of Ti (x) is substituted in Na4/7[□1/7Mn6/7]O2 (see Fig. S4–S6 and Tables S2–S4 in ESI† for the detailed procedures for the Ti solubility limit calculations).
The electrochemical performance of Na4/7[□1/7Ti1/7Mn5/7]O2 and Na4/7[□1/7Mn6/7]O2 was evaluated in sodium-half cells, cycled galvanostatically between 1.6 and 4.4 V at a current rate of 10 mA g−1. Fig. 2(a) and (b) presents the galvanostatic charge/discharge profiles and the corresponding differential capacity versus voltage (dQ/dV) plots, respectively, collected for the first cycle of Na4/7[□1/7Ti1/7Mn5/7]O2 and Na4/7[□1/7Mn6/7]O2. The materials exhibit similar charge/discharge profiles (Fig. 2(a)) for the first cycle. Na4/7[□1/7Mn6/7]O2 delivers an initial charge capacity of 63.7 mA h g−1, corresponding to the removal of 0.22 Na+ ions. It shows a reversible plateau at around 4.20 V, observed in the dQ/dV plot (Fig. 2(b)) as sharp oxidation and reduction peaks at 4.22 and 4.18 V, respectively, based on oxygen redox, which is consistent with data previously reported for Na4/7[□1/7Mn6/7]O2.21,22 On discharge to 1.6 V, Na4/7[□1/7Mn6/7]O2 delivers a discharge capacity of 189.9 mA h g−1 equivalent to the insertion of 0.65 Na+ ions, and exhibits a long, flat discharge plateau at 2.10 V associated with the Mn4+/Mn3+ redox couple.21
 | ||
| Fig. 2 (a) Galvanostatic charge/discharge curves of the first cycle for Na4/7[□1/7Mn6/7]O2 (black) and Na4/7[□1/7Ti1/7Mn5/7]O2 (blue) cycled within the voltage range 1.6–4.4 V at a current rate of 10 mA g−1 at 30 °C and (b) the corresponding differential capacity versus voltage dQ/dV plots. (c) Voltammetric analysis of Na4/7[□1/7Mn6/7]O2 (black) and Na4/7[□1/7Ti1/7Mn5/7]O2 (blue) collected at a scan rate of 30 μV s−1 at 30 °C using 1 M NaClO4 in propylene carbonate containing 3% fluoroethylene carbonate by weight as electrolyte. (d) Galvanostatic cycling performance of Na4/7[□1/7Mn6/7]O2 (black) and Na4/7[□1/7Ti1/7Mn5/7]O2 (blue) cycled within the voltage range 1.6–4.4 V at 30 °C. Square-shaped symbols represent discharge capacities of cells cycled at a rate of 10 mA g−1 and triangle-shaped symbols represent discharge capacities of cells cycled at a rate of 100 mA g−1. | ||
As for Na4/7[□1/7Ti1/7Mn5/7]O2, Fig. 2(b) shows it delivers an initial charge capacity of 94.9 mA h g−1, equivalent to the extraction of 0.32 Na+, and exhibits a discharge capacity of 189.9 mA h g−1 corresponding to the insertion of 0.65 Na+. Fig. 2(b) reveals oxidation and reduction peaks, at higher potentials of 4.28 and 4.24 V, respectively, for the oxygen redox process, and a reduction peak at a higher potential of 2.12 V for the Mn4+/Mn3+ redox couple for Na4/7[□1/7Ti1/7Mn5/7]O2. To confirm the higher working potential of Na4/7[□1/7Ti1/7Mn5/7]O2, cyclic voltammograms were collected for both materials between 1.6 and 4.4 V at a scan rate of 30 μV s−1 and these data are compared in Fig. 2(c). The cyclic voltammograms are consistent with the dQ/dV plots (Fig. 2(b)), showing Na4/7[□1/7Ti1/7Mn5/7]O2 operates at a higher potential (4.31 and 4.23 V) to Na4/7[□1/7Mn6/7]O2 (4.28 and 4.16 V). Furthermore, Fig. 2(d) compares the galvanostatic cycling performance of Na4/7[□1/7Mn6/7]O2 and Na4/7[□1/7Ti1/7Mn5/7]O2, cycled between 1.6 and 4.4 V at rates of 10 and 100 mA g−1. The discharge capacity of Na4/7[□1/7Mn6/7]O2 fades rapidly on cycling with only 62 and 44% of the initial discharge capacity retained at 10 and 100 mA g−1, respectively, after 50 cycles. This agrees with earlier work on Na4/7[□1/7Mn6/7]O2 by de Boisse et al. which reported similar cycling performance.21 By contrast, Na4/7[□1/7Ti1/7Mn5/7]O2 reveals superior cycling stability at 10 and 100 mA g−1, maintaining 77% and 65%, respectively, of its initial discharge capacity after 50 cycles, showing that Ti-substitution improves the reversibility and cycling performance, consistent with data reported for Na4/7[□1/7Ti1/7Mn5/7]O2 by Liu et al. and other Ti-substituted materials.26,27,29,34–41
In order to separate the effect of Ti-substitution on oxygen anionic redox from the combined cation and anion redox reactions, cells of Na4/7[□1/7Mn6/7]O2 and Na4/7[□1/7Ti1/7Mn5/7]O2 were cycled galvanostatically between 3.0 and 4.4 V where only oxygen redox couples become active. Fig. 3(a) and (b) shows the differential capacity versus voltage for Na4/7[□1/7Mn6/7]O2 and Na4/7[□1/7Ti1/7Mn5/7]O2, respectively, cycled between 3.0 and 4.4 V at a current rate of 10 mA g−1 over 50 cycles. Fig. 3(a) shows that on cycling Na4/7[□1/7Mn6/7]O2 the redox couple at 4.2 V, associated with oxygen redox, is lost after about 30 cycles. Whereas, Fig. 3(b) shows that on cycling Na4/7[□1/7Ti1/7Mn5/7]O2 the redox couple centred about 4.25 V persists after 50 cycles, thereby demonstrating enhanced oxygen redox stability due to the partial substitution of Ti. Additionally, Fig. 3(c) presents the galvanostatic cycling performance of Na4/7[□1/7Mn6/7]O2 and Na4/7[□1/7Ti1/7Mn5/7]O2 cycled between 3.0 and 4.4 V at 10 mA g−1. Na4/7[□1/7Ti1/7Mn5/7]O2 exhibits excellent cycling performance, showing no capacity loss. Whereas, Na4/7[□1/7Mn6/7]O2 only retains 82% of its initial discharge capacity. The aforementioned tests show that Ti4+ has a remarkable effect on the cycling stability, especially for the oxygen redox reversibility. In order to study the role of Ti4+ in improving the reversibility of the insertion and extraction of Na+ ions in Na4/7[□1/7Ti1/7Mn5/7]O2 a computational study was performed (see Section 2.4). The rate capability of Na4/7[□1/7Mn6/7]O2 and Na4/7[□1/7Ti1/7Mn5/7]O2 cycled between 3.0 and 4.4 V at 50, 100, 200 mA g−1 has also been tested. Fig. 3(d) reveals that both materials exhibit similar rate performance in this voltage window.
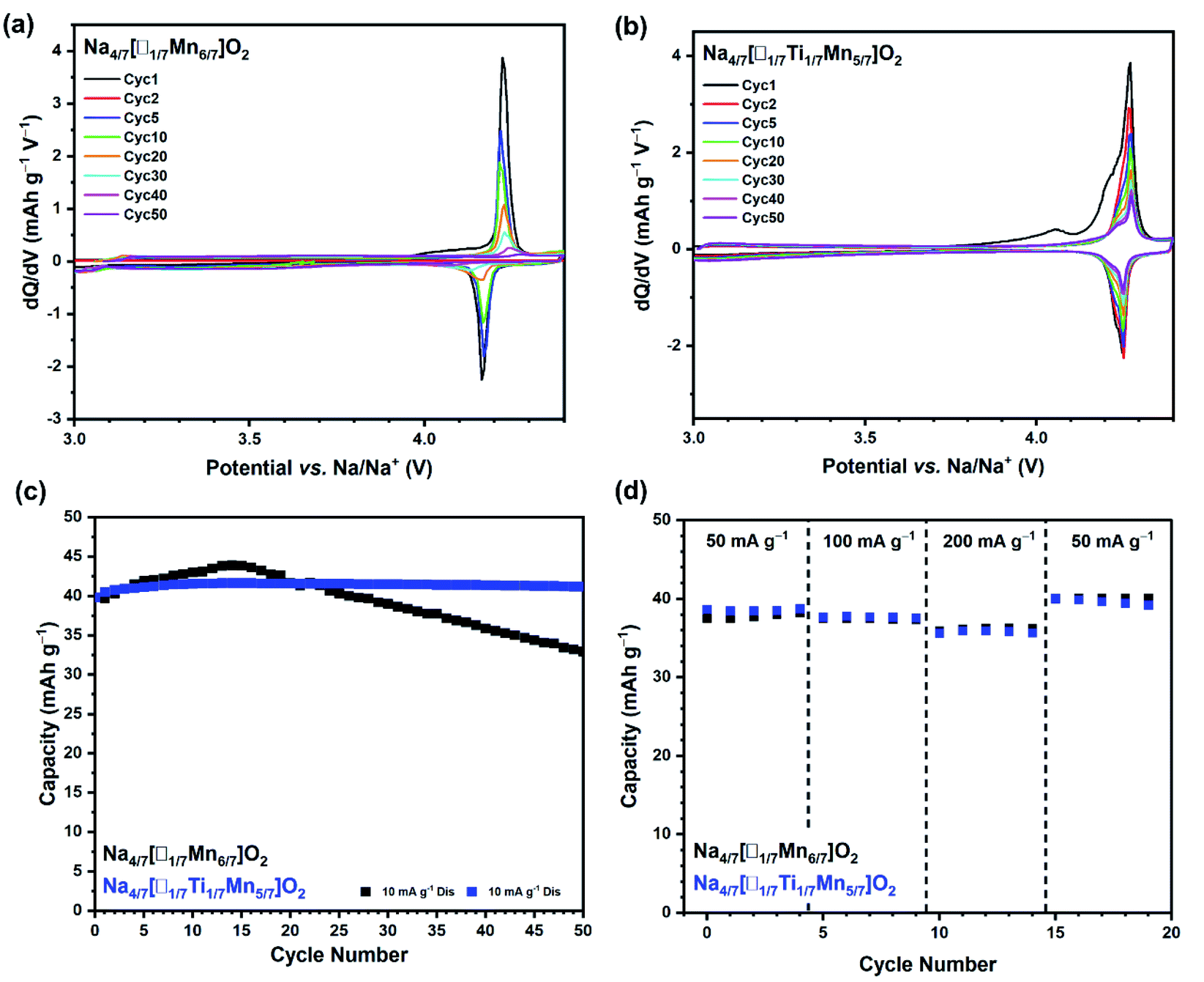 | ||
| Fig. 3 Derivative dQ/dV plots of (a) Na4/7[□1/7Mn6/7]O2 and (b) Na4/7[□1/7Ti1/7Mn5/7]O2, collected at 30 °C between 3.0 and 4.4 V at a rate of 10 mA g−1 showing the first (black), second (red), fifth (blue), tenth (green), twentieth (orange), thirtieth (cyan), fortieth (pink) and fiftieth (purple) cycles. (c) Galvanostatic cycling performance of Na4/7[□1/7Mn6/7]O2 (black) and Na4/7[□1/7Ti1/7Mn5/7]O2 (blue) cycled between 3.0 and 4.4 V at a current rate of 10 mA g−1 at 30 °C, showing the discharge capacities. (d) Rate capability of Na4/7[□1/7Mn6/7]O2 (black) and Na4/7[□1/7Ti1/7Mn5/7]O2 (blue) cycled between 3.0 and 4.4 V at a current rate of 50, 100, and 200 mA g−1 at 30 °C, showing the discharge capacities. | ||
2.2. Structural evolution and charge compensation mechanism
The changes in structure of Na4/7[□1/7Ti1/7Mn5/7]O2 upon Na+ ion extraction and insertion were investigated using powder X-ray diffraction (PXRD). Ex situ PXRD patterns were collected over the initial charge/discharge cycle between 1.6 and 4.4 V (Fig. 4(a)). Rietveld refinements were performed, and the fits obtained for data from the electrodes charged to 4.4 V (CH4.4V), and then subsequently discharged to 3.0 V (DIS3.0V) and 1.6 V (CYC1.6V) are shown in Fig. 4(b)–(d), respectively. The PXRD pattern for Na4/7[□1/7Ti1/7Mn5/7]O2 after charge to 4.4 V (Fig. 4(b)), can be fitted to a simple P3-type model with the R3m space group, with refined unit cell parameters a = 2.849(1), c = 16.637(14) Å. This shows that upon Na+ ion removal there is an increase in the crystallographic symmetry, with a transition from the Na4/7[□1/7Mn6/7]O2 structure (P space group) for pristine Na4/7[□1/7Ti1/7Mn5/7]O2 (Fig. 1(a)) to a structure which can be well-described in R3m symmetry, demonstrating that the P3 oxygen stacking sequence is preserved (Table S5†), consistent with the structural evolution observed for Na4/7[□1/7Mn6/7]O2.22,29 Furthermore, Na4/7[□1/7Ti1/7Mn5/7]O2 shows a minimal volume contraction of ∼0.6%. Upon subsequent Na+ ion insertion, the PXRD pattern after discharge to 3.0 V can effectively be modelled using the P space group (Fig. 4(c)), showing that the Na4/7[□1/7Mn6/7]O2 structure is restored and reveals a small volume contraction of ∼0.3% relative to the pristine phase. Moreover, upon charge to 4.4 V and subsequent discharge to 3.0 V, the shift in the position of the 001 diffraction peak (7.39° 2θ), corresponding to the interlayer distance between the [□1/7Ti1/7Mn5/7]O2 layers is negligible (0.01° 2θ), suggesting that the interlayer distance is preserved at 5.57 Å which agrees with data reported for Na4/7[□1/7Mn6/7]O2.21,23 Additionally, the superstructure is restored on discharge and it is likely that the superstructure is preserved after charge to 4.4 V, but cannot be resolved in ex situ measurements. Upon further Na+ ion insertion, the PXRD pattern of Na4/7[□1/7Ti1/7Mn5/7]O2 discharged to 1.6 V after charge to 4.4 V (Fig. 4(d)), shows partial transformation of 15% to an O3-type phase which shows an expansion of 1.5% relative to the volume of the pristine phase. This is reasonable since the insertion of additional Na+ ions increases the coulombic repulsion between the Na and Mn cations and the P3 structure undergoes a structural transformation to the O3-type phase to relieve this repulsion.30,42 Since 85% of the Na4/7[□1/7Mn6/7]O2 structure is retained and shows essentially no volume change, there is an average overall expansion of ∼0.2% relative to the pristine phase. Na4/7[□1/7Ti1/7Mn5/7]O2 undergoes minimal volume changes upon cycling, agreeing with in situ PXRD measurements previously reported by Liu et al. who also showed a larger volume change of 6.8% for Na4/7[□1/7Mn6/7]O2,29 correlating enhanced structural stability with superior cycle life.
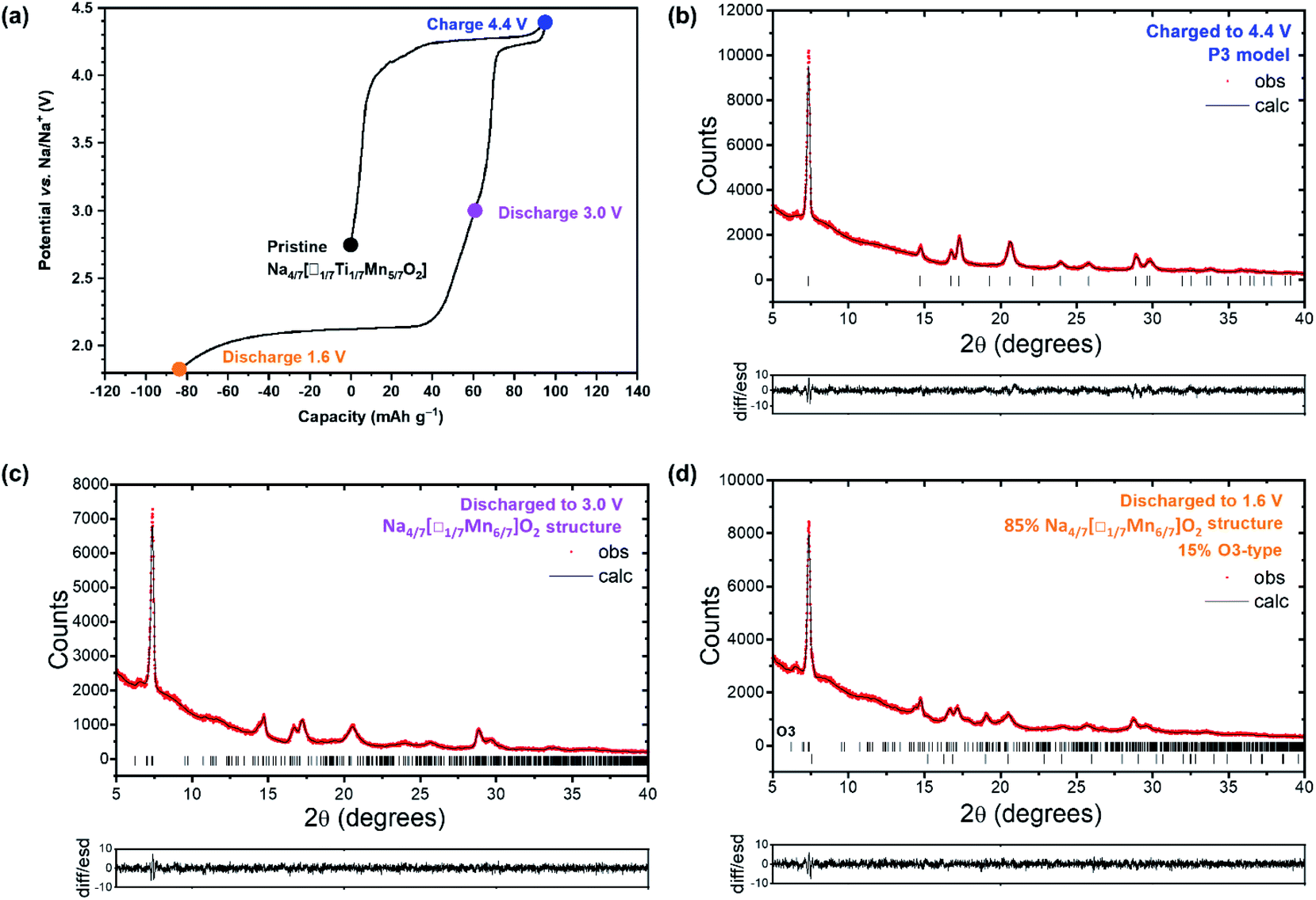 | ||
| Fig. 4 (a) Galvanostatic charge/discharge profile for Na4/7[□1/7Ti1/7Mn5/7]O2 cycled at 30 °C between 1.6 and 4.4 V at a rate of 10 mA g−1, showing the voltages for the ex situ PXRD measurements. (b) Laboratory X-ray Rietveld fits for Na4/7[□1/7Ti1/7Mn5/7]O2 charged to 4.4 V, (c) cycled between 3.0 and 4.4 V, and (d) cycled between 1.6 and 4.4 V. The observed data points are shown in red, with the fitted profiles in black. Tick marks indicate allowed reflections. | ||
To follow changes in the oxidation states of Mn, Ti and O during the first cycle, ex situ SXAS spectra were collected at different states of charge and discharge over the first cycle between 3.0 and 4.4 V. SXAS measurements were collected at the Mn L2,3-edge (Fig. 5(a)), Ti L2,3-edge (Fig. 5(b)) and O K-edge (Fig. S7(a)†) using the partial fluorescence yield (PFY) and total electron yield (TEY) modes. During cycling the Mn L2,3-edge spectrum of pristine Na4/7[□1/7Ti1/7Mn5/7]O2 shows absorption peaks at 643.4 and 653.4 eV, which correspond to the L3 and L2 edges, respectively, and is consistent with Mn4+.43 The spectra show no shift in energy upon cycling (Fig. 5(c)), indicating that Mn remains tetravalent and does not participate in the charge transfer mechanism between 3.0 and 4.4 V. The Ti L2,3-edge SXAS spectrum of the pristine electrode shows that Ti exists as Ti4+ and similarly reveals no changes upon charge/discharge (Fig. 5(c)), confirming that Ti does not contribute towards the capacity between 3.0 and 4.4 V.44 Therefore, these data suggest that lattice oxygen participates in the charge compensation mechanism.
 | ||
| Fig. 5 Electronic structure changes of Na4/7[□1/7Ti1/7Mn5/7]O2 during charge and discharge. (a) Ex situ soft X-ray absorption spectra for Mn L2,3-edge in partial fluorescence yield mode (PFY). (b) Ex situ soft X-ray absorption spectra for Ti L2,3-edge in PFY. (c) First-cycle charge–discharge load curve and the points indicate the states of charge and discharge at which ex situ measurements were collected. Variations in the Mn L2-peak energy and Ti L2 eg peak energies. | ||
The O K-edge SXAS spectrum of pristine Na4/7[□1/7Ti1/7Mn5/7]O2 shows two pre-edge peaks which correspond to the excitation from the O 1s to unoccupied O 2p states hybridised with the transition metal 3d t2g (ca. 530 eV) and eg states (532 eV).12,45,46 The broad peaks above 535 eV are characteristic of transitions to the O 2p states hybridised with transition metal 4s and 4p states. The area under the pre-edge peak (between 525 and 533 eV) relates to changes in the density of empty states above the Fermi level. The relative intensity of the pre-edge peaks is shown in Fig. S7(b).† This reveals minimal intensity changes and does not provide sufficient evidence of oxygen redox activity. These metastable On− species where 3 ≥ n ≥ 1 relax over time and it is reasonable to assume that they relaxed prior to data acquisition. Therefore, electron paramagnetic resonance (EPR) spectroscopy (Fig. 6) and Raman spectroscopy (Fig. S10†) were used to investigate the oxygen redox activity. By contrast EPR spectroscopy demonstrate stronger indications of oxygen anion redox activity, consistent with the Mn and Ti L-edge SXAS spectra (Fig. 5), and these findings are discussed in more detail below.
 | ||
| Fig. 6 (a) EPR spectra of pristine Na4/7[□1/7Ti1/7Mn5/7]O2 and ex situ electrodes of Na4/7[□1/7Ti1/7Mn5/7]O2 cycled between 3.0 and 4.4 V at 10 mA g−1, mixed with conductive carbon C65 additive. EPR spectra were collected on the same day after cycling ceased and normalised based on the mass of the sample. The sharp, intense peak is assigned to conductive carbon C65 additive. (b) The corresponding galvanostatic charge/discharge profile with the voltages highlighted where the ex situ EPR measurements were conducted. | ||
Ex situ EPR measurements were collected for Na4/7[□1/7Mn6/7]O2 and Na4/7[□1/7Ti1/7Mn5/7]O2 at different states of charge and discharge over the first cycle between 3.0 and 4.4 V. EPR spectroscopy can identify the formation and disappearance of radical oxygen species and therefore, provide mechanistic insights into oxygen anion redox.8,47,48Fig. 6 presents the evolution of the EPR spectra for Na4/7[□1/7Ti1/7Mn5/7]O2 at various states of charge and discharge. Pristine Na4/7[□1/7Ti1/7Mn5/7]O2, without conductive carbon additive, shows an intense broad symmetric signal, attributed to the antiferromagnetic Mn4+–O. The Mn4+–O EPR signal presents a single Lorentzian line shape centred at g ≈ 2.00.49 The addition of conductive carbon C65 to Na4/7[□1/7Ti1/7Mn5/7]O2 gives rise to a sharp peak, as shown for the Na4/7[□1/7Ti1/7Mn5/7]O2 electrodes, due to delocalised electrons within C65. After charge to 4.1 V, the intensity of the EPR spectrum decreases relative to that of pristine Na4/7[□1/7Ti1/7Mn5/7]O2. Upon further charge to 4.4 V, the intensity of the EPR signal reduces further. Based on the SXAS data (Fig. 5), Mn and Ti do not participate in the charge compensation mechanism between 3.0 and 4.4 V. Therefore, the extraction of Na+ ions upon charge to 4.4 V is associated with O2− oxidation to O2n− where 3 ≥ n ≥ 1. Of these anion redox species, only O23− and superoxo O2− species have a single unpaired electron (S = ½) and are therefore detectable by EPR. Whereas peroxo O22− has an even number of paired electrons (S = 0) and is EPR-silent. Upon discharge, the EPR signal intensity increases with decreasing potential, demonstrating the reversibility of the O2n− redox reaction.
As for Na4/7[□1/7Mn6/7]O2, Fig. S8† shows the EPR spectra for Na4/7[□1/7Mn6/7]O2 cycled between 3.0 and 4.4 V. Na4/7[□1/7Mn6/7]O2 presents the same trend as Na4/7[□1/7Ti1/7Mn5/7]O2 upon charge/discharge. The intensity of the EPR signal similarly decreases on charge to 4.4 V and increases again on discharge. These data also agree well with EPR data previously reported for Na4/7[□1/7Mn6/7]O2 by Song et al.22
Further analysis of the same samples after 5 days (Fig. S9†) shows a change in the EPR signals of the electrodes after charge to 4.4 V. The Na4/7[□1/7Mn6/7]O2 electrode after charge to 4.4 V reveals an increase in intensity after 5 days, suggesting that the O2n− species formed at the end of charge is unstable and relaxes over 5 days. The Na4/7[□1/7Ti1/7Mn5/7]O2 system shows a similar relaxation of the EPR signal after five days but to a lesser extent to that of the Na4/7[□1/7Mn6/7]O2 system. Thereby, implying that the presence of Ti4+ increases the stability of the O2n− species somewhat. Based on these data, the O2n− species decays within days of being charge/discharged and therefore explains the minimal changes observed for the SXAS O K-edge spectra (Fig. S7†), as these data were collected roughly four weeks after preparation.
Raman spectroscopy was used to evaluate the structural evolution of Na4/7[□1/7Ti1/7Mn5/7]O2 upon cycling and examine if peroxide species, O22−, form. Pristine Na4/7[□1/7Ti1/7Mn5/7]O2 and Na4/7[□1/7Ti1/7Mn5/7]O2 mixed with carbon C65 (Fig. S10(a)†) show an intense band at ∼640 cm−1 and a broad shoulder at ∼582 cm−1 which probably correspond to the O–TM–O bending and TM–O stretching modes (TM = Mn, Ti) of the transition metal TMO6 layer, respectively. Li et al. examined the structural evolution of Na4/7[□1/7Mn5/7]O2 upon cycling and likewise observed bands at 638 and 580 cm−1 for the pristine material.23 Fig. S10(b)† presents the ex situ Raman spectra recorded for Na4/7[□1/7Ti1/7Mn5/7]O2 at different states of charge and discharge over the first cycle between 3.0 and 4.4 V. These spectra show that the position of the TM–O (TM = Mn, Ti) bands remain constant upon cycling which agrees with the PXRD results (Fig. 4, i.e., no structural transformation between 3.0 and 4.4 V) and Raman data previously presented for Na4/7[□1/7Mn5/7]O2.23 Essentially the spectra do not reveal any clear additional bands or changes in intensity in the region where O–O stretches (700–900 cm−1) have been observed for peroxide-containing species (e.g., Li2O2, Na2O2, H2O2 and ZnO2).12 Unfortunately, contributions from the EL-Cell holder/separator (∼790–1090 cm−1) are within the same region for O–O stretches and therefore hinder definitive detection of O–O bonds. This can be seen in Fig. S10(a),† where the bands within the ∼790–1090 cm−1 region are only observed for the measurements carried out inside the EL-Cell for both the pristine Na4/7[□1/7Ti1/7Mn5/7]O2 and Na4/7[□1/7Ti1/7Mn5/7]O2 mixed with carbon C65. Similar to this work, ex situ Raman spectroscopic studies performed on Na4/7[□1/7Mn6/7]O2 could not confirm the presence of peroxide species O22− upon charging.23 However, in situ Raman spectroscopic experiments on related systems have revealed bands between 700 and 900 cm−1, corresponding to O–O stretches of peroxo-species.50–52
2.3. Interpretation on the increased voltages after Ti substitution
To gain insight into the origin of the higher potentials observed for Na4/7[□1/7Ti1/7Mn5/7]O2 (Fig. 2), theoretical voltage profiles were constructed, and the associated electronic structures were analysed. We first constructed model atomic structures for Nax[□1/7Mn6/7]O2 and Nax[□1/7Ti1/7Mn5/7]O2, with Na content being 0.0 ≤ x ≤ 6/7, based on the x values (∼0.25 to 1.0 Na) from the galvanostatic charge/discharge curves over the voltage range 1.6–4.4 V (see Fig. 2). In this regard, partially desodiated phases with x < 4/7 were generated by removing Na+ ions from primitive Na4/7[□1/7Mn6/7]O2 and Na4/7[□1/7Ti1/7Mn5/7]O2, whereas phases after discharging with x > 4/7 were created by adding Na+ ions into stable interstitial sites (see ESI† for more details). Tables S6 and S7† show the constructed ground state structures of fully discharged Na6/7[□1/7Mn6/7]O2 and Na6/7[□1/7Ti1/7Mn5/7]O2.After generating intermediate Nax[□1/7Mn6/7]O2 and Nax[□1/7Ti1/7Mn5/7]O2 phases, with Na contents being 0.0 ≤ x ≤ 6/7, we next searched the stable low-energy structures that primarily determine the shape of theoretical voltage curves. This was attained by calculating total energies of all symmetry unique atomic arrangements, followed by plotting their convex-hull diagrams as a function of Na content (Fig. 7(a)). For both Nax[□1/7Mn6/7]O2 and Nax[□1/7Ti1/7Mn5/7]O2, the convex-hull diagrams show that there are six stable structures, with Na concentrations of x = 1/7, 2/7, 3/7, 4/7 and 6/7. This suggests that voltage profiles upon charging from x = 4/7 to 2/7 are characterised by two voltage plateaux, whereas those upon discharge from x = 4/7 to 6/7 will have a single voltage plateau.
 | ||
| Fig. 7 (a) Calculated formation energies of all intermediate phases constructed from Nax[□1/7Mn6/7]O2 and Nax[□1/7Ti1/7Mn5/7]O2. Convex hull diagrams were plotted by connecting ground state structures (empty circles). Unstable atomic configurations are denoted as horizontal lines for reference. (b) Theoretical and experimental voltage curves of Nax[□1/7Mn6/7]O2 and Nax[□1/7Ti1/7Mn5/7]O2. Among all symmetrically unique Na and Ti arrangements, those that can form under thermal vibration energy (0.0259 eV) at room temperature are considered. Mean and standard deviation of voltages calculated from the selected atomic arrangements are denoted as dotted and vertical black lines, respectively. Mean theoretical voltages are denoted above each plateau for better visualisation. | ||
Close comparison of the formation energies of two convex-hull diagrams of Nax[□1/7Mn6/7]O2 and Nax[□1/7Ti1/7Mn5/7]O2 show that, upon charging, Ti-doped materials are relatively unstable, as can be confirmed by the higher formation energy of Na2/7[□1/7Ti1/7Mn5/7]O2 than Na2/7[□1/7Mn6/7]O2 (see inset of Fig. 7(a)). This suggests that the addition of Ti destabilises the desodiated materials, resulting in the increase of charging voltage. By incorporating the stable phases predicted from the convex-hull diagrams and other unstable Na arrangements that can form under thermal vibration at room temperature, we calculated theoretical voltage profiles of Nax[□1/7Mn6/7]O2 and Nax[□1/7Ti1/7Mn5/7]O2 (Fig. 7(b) and (c)). In this calculation, the mean and standard deviations of voltages that arise from differing Na arrangements are considered to reflect the potential errors that occur during Na diffusion (see ESI†). The predicted voltage profiles are in good agreement with experimental data, especially for the Ti-substituted material which shows slightly higher voltages over Na4/7[□1/7Mn6/7]O2. The voltages required to remove and insert Na from Ti-substituted Nax[□1/7Ti1/7Mn5/7]O2 are greater by 0.08–0.15 V and 0.09 V, respectively, which is consistent with the capacity versus voltage and dQ/dV plots shown in Fig. 2.
Having reproduced the higher voltages of Ti-substituted Na4/7[□1/7Mn6/7]O2 (Fig. 7), we proceeded to analyse the redox mechanisms of voltage plateaux to identify the origin of the increased voltages. To this end, projected densities of states were calculated for differing Na concentrations (x) of Nax[□1/7Mn6/7]O2 and Nax[□1/7Ti1/7Mn5/7]O2 (Fig. 9). For both materials, Na extraction during charging oxidises non-bonding oxygens adjacent to the unoccupied Mn sites, causing the oxygens to have localised excess holes. On the other hand, adding excess Na into Na4/7[□1/7Mn6/7]O2 and Na4/7[□1/7Ti1/7Mn5/7]O2 accompanies the localisation of excess electrons on Mn4+, reducing it to Mn3+. The above results indicate that the high potentials (4.3 V) during charging (x < 4/7) arise from anion redox, as revealed by EPR measurements (Fig. 6), whereas the low potentials (2.0 V) during discharging (4/7 < x < 6/7) are associated with the Mn3+/Mn4+ redox couple. Unlike Mn and O, Ti does not participate in the redox reaction (between 1.6 and 4.4 V), as none of the excess electrons/holes are centred near Ti upon charge or discharge.
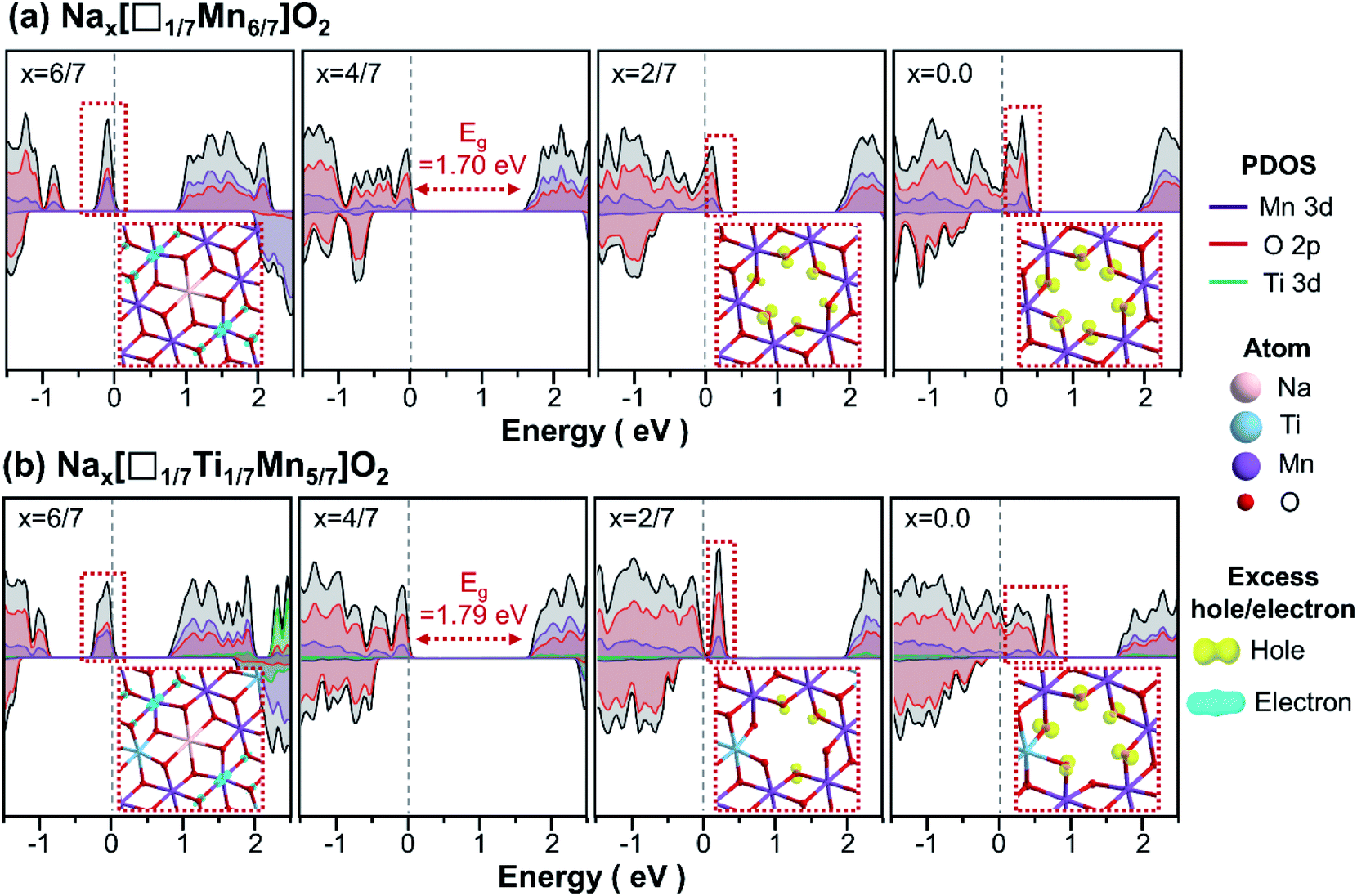 | ||
| Fig. 8 Projected density of states of primitive (a) Nax[□1/7Mn6/7]O2 and (b) Nax[□1/7Ti1/7Mn5/7]O2 calculated from the most stable atomic arrangement at each Na composition of x = 6/7, 4/7, 2/7, and 0.0. The insets show the partial charge densities of excess holes (electrons) created after desodiation (sodiation) of Na4/7[□1/7Mn6/7]O2 or Na4/7[□1/7Ti1/7Mn5/7]O2. | ||
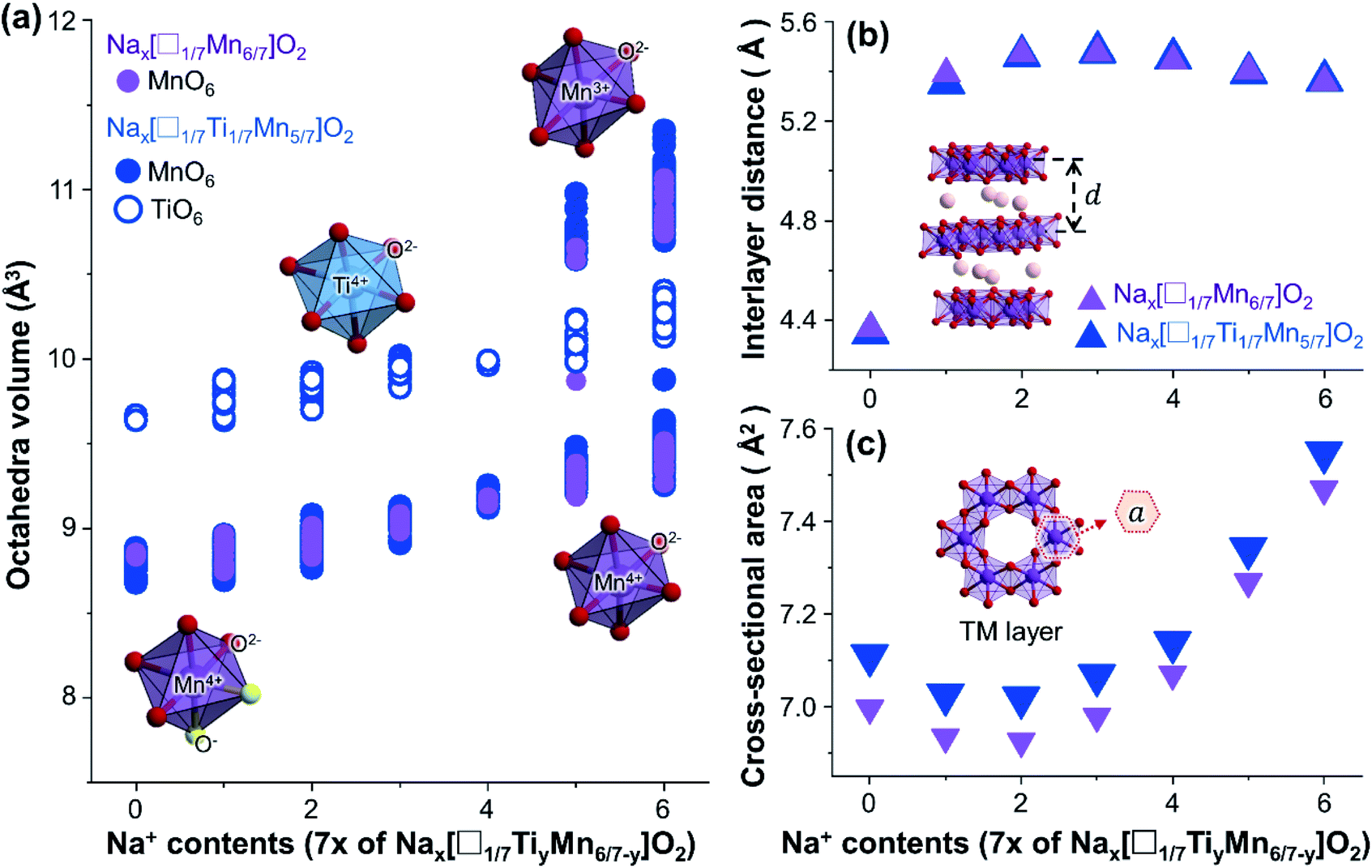 | ||
| Fig. 9 (a) Volumes of MnO6 and TiO6 octahedra of all Nax[□1/7Mn6/7]O2 and Nax[□1/7Ti1/7Mn5/7]O2 structures optimised in this study. Representative oxidation states of octahedra at each Na composition are represented in the insets. Changes in the (b) average interlayer distance (d) and (c) average cross-sectional area (a) of TMO6 octahedra calculated for Nax[□1/7Mn6/7]O2 and Nax[□1/7Ti1/7Mn5/7]O2 structures, plotted with respect to their Na contents. | ||
Despite Ti being redox-inactive upon cycling, it influences the bond ionicity and associated electronic structures. Ti, with slightly lower electronegativity than Mn, tends to make stronger ionic Ti–O bonds, as revealed by the greater band gap of Na4/7[□1/7Ti1/7Mn5/7]O2 compared to that of Na4/7[□1/7Mn6/7]O2. From an atomistic perspective, Ti makes adjacent O 2p orbitals more stable and therefore, harder to remove. Upon Na extraction, this causes the electron holes to be localised away from Ti, as shown by the partial charge density of Na2/7[□1/7Ti1/7Mn5/7]O2 in Fig. 8(b). Additional calculations of the local Madelung potentials (Fig. S11†),53,54 further support this argument, that oxygens positioned further away from Ti atoms have lower Madelung potential and can readily be oxidised upon charging. Such hindrance of Ti on anion redox increases the energy required to remove Na, resulting in greater charge voltages.
The stronger ionicity of Ti–O bonds also contributes to the increase in the discharge voltage: when a less electronegative Ti atom bonds with an oxygen, the oxygen has a greater share of electrons, thereby making the nearby Mn–O bonding more ionic (Fig. S12†). The increased ionicity of Mn–O bonds reduces the repulsion between the bonding and antibonding orbitals, pulling the antibonding orbitals away from the Na+/Na redox couple.55 These changes are observed as higher discharge voltages for Na4/7[□1/7Mn6/7]O2 and Na4/7[□1/7Ti1/7Mn5/7]O2, as their discharge voltages are related to the location of the antibonding Mn–O hybrid orbitals with respect to the Na+/Na redox couple.
2.4. Interpretation of enhanced cyclability after Ti substitution
The cation and anion redox couples have their own respective mechanisms that can reduce the cycling stability of Mn-based materials. Cation redox reactions can cause Jahn–Teller distortion of MnO6 octahedra upon reduction of Mn4+ to Mn3+, reducing the structural stability of Mn-based materials.56 To better understand the effect of Ti-substitution on the structural stability and associated cyclability of Na4/7[□1/7Mn6/7]O2 upon cation redox, we compared the structural changes upon cycling before and after Ti-substitution (Fig. 9). The volumes of the MnO6 octahedra for Na4/7[□1/7Mn6/7]O2 and Na4/7[□1/7Ti1/7Mn5/7]O2 (Fig. 9(a)), tend to expand with increasing Na content. The greatest volumetric changes occur during discharge from x = 4/7 to 6/7, where MnO6 octahedra volumes are enlarged by up to roughly 20%. Such large volumetric change can be attributed to excess electrons supplied from inserted Na+ ions, which reduces Mn4+ to Mn3+ and form Jahn–Teller distorted Mn3+O6 octahedra, with larger volumes. On the other hand, the charging process from x = 4/7 to 0.0 is governed by anion redox reactions and compresses the octahedra by oxidation of O2− to O− with smaller ionic radius. Compared to the volume changes from the cation redox couples, the volume changes from this anion redox reaction have a minor effect, which can enlarge/compress octahedra volumes by up to only ∼5%. Therefore, between 1.6 and 4.4 V, where both cation and anion redox reactions occur, the major structural degradation arises from cation redox reactions. From this perspective, one can improve the structural stability of positive electrodes by reducing the voltage window above cation redox voltage of ∼2.1 V. However, this method compromises cation-redox capacities, as revealed by the galvanostatic cycling performance in Fig. 2.Another important finding from Fig. 9(a) is that the volumes of TiO6 octahedra are located in between those of Mn3+O6 and Mn4+O6 during cation redox reactions (see Na contents range of 4/7 < x < 6/7). This is because of the ionic radius of Ti4+ (0.605 Å), is close to the average value of those of Mn3+ (0.645 Å) and Mn4+ (0.53 Å).33 Moreover, unlike MnO6 octahedra, these TiO6 octahedra do not undergo cation redox reactions and are immune to the associated volumetric changes. This suggests that TiO6 octahedra are mechanically stable and act as a buffer that mediates the structural changes of MnO6 octahedra upon cation redox reactions. In this regard, Ti-substitution can be an effective method to relieve the structural degradation and improve cyclability under the wide voltage windows, while exploiting the large capacities from Mn3+/Mn4+ redox couples.
Owing to the larger ionic radius of Ti4+ compared to Mn4+, the addition of Ti increases the size of the TMO2 layer and thereby the size of the unit cell. Fig. 9(b) and (c) shows the effect of Ti on the interlayer distance (d) and the area of octahedra cross-sectioned by a plane parallel to TMO2 layer. The interlayer distance is slightly greater for Na4/7[□1/7Ti1/7Mn5/7]O2 (5.45 Å) than Na4/7[□1/7Mn6/7]O2 (5.44 Å), which is consistent with (001) diffraction peaks from PXRD results. However, the broadened interlayer spacing may not persist upon cycling, as the interlayer spacing becomes greater for Na4/7[□1/7Mn6/7]O2 for Na composition ranges (x = 0.0, 1/7, 2/7, and 5/7). Unlike the interlayer spacing, the cross-sectional area of TMO6 octahedra is greater for the Ti-substituted material over the full range of Na compositions (Fig. 9(c)).
The above results suggest that Ti-substitution in Na4/7[□1/7Mn6/7]O2 primarily enlarges the TMO2 layer parallel to the (001) planes, while having only minor influences on the interlayer spacing. In practical experiments, such structural changes may cause negative effects on the rate performances of the positive electrode materials, as they will lengthen the pathways for Na diffusion along (001) planes. Further examination of Fig. 9(b) and (c) shows that, upon discharge from x = 2/7 to 6/7, both materials show decrease in their interlayer distances, accompanied by a widening of the cross-sectional area. These structural changes can be explained based on the type of Na sites: when examining the calculated (see ESI†) and observed structures (see Rietveld refinement results in Table S1†) of Nax[□1/7Mn6/7]O2 and Nax[□1/7Ti1/7Mn5/7]O2, Na atoms prefer to be in prismatic sites under Na-poor conditions (x = 2/6) and gradually transform to octahedral sites as the Na content increases to x = 6/7, as confirmed by ex situ PXRD results (Fig. 4). This is common for transition metal oxides, as prismatic and octahedral stackings are the optimal configurations to minimise face-sharing between cations and the associated electrostatic repulsions under Na-poor and Na-rich conditions, respectively.57,58 Among these two stackings, prismatic sites are characterised by greater interlayer distance and smaller cross-sectional TM layer area compared to octahedra,57,59 resulting in wider and shorter Na diffusion channels for prismatic stackings. Both Nax[□1/7Mn6/7]O2 and Nax[□1/7Ti1/7Mn5/7]O2 undergo transition from prismatic to octahedral stacking, which can cause the cyclability to deteriorate.
3. Conclusions
Partial substitution of Mn by Ti has been shown to enhance the electrochemical performance of Na4/7[□1/7Mn6/7]O2. Ti-substituted Na4/7[□1/7Ti1/7Mn5/7]O2 delivers a highly reversible capacity of 146 mA h g−1 after 50 cycles with 77% capacity retention within the voltage range 1.6–4.4 V at 10 mA g−1. In the voltage range of 3.0–4.4 V, Na4/7[□1/7Ti1/7Mn5/7]O2 shows no capacity fade after 50 cycles, based on oxygen redox reactions. The improved cyclability is likely to be rooted in the presence of TiO6 octahedra that maintain their volume upon cycling, thus relieving structural distortions arising from Jahn–Teller distorted Mn3+O6 octahedra below 3.0 V. The stable anion redox activity above 3.0 V, on the other hand, is believed to arise from the low electronegativity of Ti that enhances bond ionicity and thereby bond strength of Mn–O bonds. The low electronegativity of Ti also stabilises the adjacent O 2p orbitals, increasing the charge/discharge voltages of Na4/7[□1/7Ti1/7Mn5/7]O2. Finally, despite the larger ionic radius of the Ti4+, its effect on the interlayer separation is insignificant, as the so-formed TiO6 octahedra distort the TMO2 layer to be stretched mostly along the TM layer direction, as the TiO6 octahedra distort rather than widening interlayer distances. This work provides new methods to preserve the structural integrity in oxygen redox active sodium layered oxides which can achieve high energy densities and show enhanced oxygen redox reversibility.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for the provision of beam time and assistance from instrument scientists at beamline I11 at Diamond (Rapid Access experiment CY26699). We gratefully acknowledge Spring 8 for beamtime (Proposal No. 2021A1425) and technical support, particularly beamline scientist Kiyofumi Nitta. L. D. gratefully acknowledges financial support from the Swedish Energy Agency (contract 2020-005246). Y. C. and D. O. S. are grateful to the Faraday Institution for funding the MICHAEL computing cluster hosted at University College London (UCL). The calculations have been also carried out on the Thomas (Thomas@UCL) and Young (Young@UCL) High Performance Computing Facility provisioned by UCL. Via our membership of the UKs HEC Materials Chemistry Consortium, which is funded by EPSRC (EP/L000202, EP/R029431), this work used the ARCHER2 UK National Supercomputing Service (http://www.archer2.ac.uk). We are also grateful to the UK Materials and Molecular Modelling Hub for computational resources, which is partially funded by EPSRC (EP/P020194/1 and EP/T022213/1). We are grateful to the Engineering and Physical Sciences Research Council (EPSRC) Light Element Facility Grant (EP/T019298/1) for funding the acquisition of the Raman spectrometer. EF is grateful for an EaStCHEM studentship. This work was supported by the Faraday Institution (grant number FIRG018).References
- J. Y. Hwang, S. T. Myung and Y. K. Sun, Chem. Soc. Rev., 2017, 46, 3529–3614 RSC.
- N. Yabuuchi, K. Kubota, M. Dahbi and S. Komaba, Chem. Rev., 2014, 114, 11636–11682 CrossRef CAS PubMed.
- Y. Li, Y. Lu, C. Zhao, Y. S. Hu, M. M. Titirici, H. Li, X. Huang and L. Chen, Energy Storage Mater., 2017, 7, 130–151 CrossRef.
- C. Fang, Y. Huang, W. Zhang, J. Han, Z. Deng, Y. Cao and H. Yang, Adv. Energy Mater., 2016, 6, 1–18 Search PubMed.
- J. Do, I. Kim, H. Kim and Y. Jung, Energy Storage Mater., 2020, 25, 62–69 CrossRef.
- N. Tapia-ruiz, A. R. Armstrong, H. Alptekin, M. A. Amores, H. Au, J. Barker, R. Boston, W. R. Brant, J. M. Brittain, Y. Chen, M. Chhowalla, Y. Choi, S. I. R. Costa, M. C. Ribadeneyra, S. A. M. Dickson, E. I. Eweka, J. D. Forero-saboya, C. P. Grey, Z. Li, S. F. L. Mertens, R. Mogensen, L. Monconduit, D. M. C. Ould, R. G. Palgrave, P. Poizot, A. Ponrouch, S. Renault, E. M. Reynolds, A. Rudola, R. Sayers, D. O. Scanlon, S. Sen, V. R. Seymour, B. Silv, G. S. Stone, C. I. Thomas, M. Titirici, J. Tong, T. J. Wood, D. S. Wright and R. Younesi, J. Phys.: Energy, 2021, 3, 031503 CAS.
- G. Assat and J. M. Tarascon, Nat. Energy, 2018, 3, 373–386 CrossRef CAS.
- M. Sathiya, G. Rousse, K. Ramesha, C. P. Laisa, H. Vezin, M. T. Sougrati, M. L. Doublet, D. Foix, D. Gonbeau, W. Walker, A. S. Prakash, M. Ben Hassine, L. Dupont and J. M. Tarascon, Nat. Mater., 2013, 12, 827–835 CrossRef CAS PubMed.
- N. Yabuuchi, M. Takeuchi, M. Nakayama, H. Shiiba, M. Ogawa, K. Nakayama, T. Ohta, D. Endo, T. Ozaki, T. Inamasu, K. Sato and S. Komaba, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 7650–7655 CrossRef CAS PubMed.
- H. Koga, L. Croguennec, M. Ménétrier, K. Douhil, S. Belin, L. Bourgeois, E. Suard, F. Weill and C. Delmas, J. Electrochem. Soc., 2013, 160, A786–A792 CrossRef CAS.
- H. Koga, L. Croguennec, M. Ménétrier, P. Mannessiez, F. Weill and C. Delmas, J. Power Sources, 2013, 236, 250–258 CrossRef CAS.
- K. Luo, M. R. Roberts, R. Hao, N. Guerrini, D. M. Pickup, Y. S. Liu, K. Edström, J. Guo, A. V. Chadwick, L. C. Duda and P. G. Bruce, Nat. Chem., 2016, 8, 684–691 CrossRef CAS PubMed.
- C. J. Chen, W. K. Pang, T. Mori, V. K. Peterson, N. Sharma, P. H. Lee, S. H. Wu, C. C. Wang, Y. F. Song and R. S. Liu, J. Am. Chem. Soc., 2016, 138, 8824–8833 CrossRef CAS PubMed.
- B. Xu, C. R. Fell, M. Chi and Y. S. Meng, Energy Environ. Sci., 2011, 4, 2223–2233 RSC.
- W. E. Gent, K. Lim, Y. Liang, Q. Li, T. Barnes, S. J. Ahn, K. H. Stone, M. McIntire, J. Hong, J. H. Song, Y. Li, A. Mehta, S. Ermon, T. Tyliszczak, D. Kilcoyne, D. Vine, J. H. Park, S. K. Doo, M. F. Toney, W. Yang, D. Prendergast and W. C. Chueh, Nat. Commun., 2017, 8, 1–12 CrossRef CAS PubMed.
- P. Rozier, M. Sathiya, A. R. Paulraj, D. Foix, T. Desaunay, P. L. Taberna, P. Simon and J. M. Tarascon, Electrochem. Commun., 2015, 53, 29–32 CrossRef CAS.
- B. Mortemard De Boisse, G. Liu, J. Ma, S. I. Nishimura, S. C. Chung, H. Kiuchi, Y. Harada, J. Kikkawa, Y. Kobayashi, M. Okubo and A. Yamada, Nat. Commun., 2016, 7, 1–9 Search PubMed.
- B. Mortemard de Boisse, M. Reynaud, J. Ma, J. Kikkawa, S.-i. Nishimura, M. Casas-Cabanas, C. Delmas, M. Okubo and A. Yamada, Nat. Commun., 2019, 10, 2185 CrossRef PubMed.
- U. Maitra, R. A. House, J. W. Somerville, N. Tapia-Ruiz, J. G. Lozano, N. Guerrini, R. Hao, K. Luo, L. Jin, M. A. Pérez-Osorio, F. Massel, D. M. Pickup, S. Ramos, X. Lu, D. E. McNally, A. V. Chadwick, F. Giustino, T. Schmitt, L. C. Duda, M. R. Roberts and P. G. Bruce, Nat. Chem., 2018, 10, 288–295 CrossRef CAS PubMed.
- R. A. House, U. Maitra, M. A. Pérez-Osorio, J. G. Lozano, L. Jin, J. W. Somerville, L. C. Duda, A. Nag, A. Walters, K. J. Zhou, M. R. Roberts and P. G. Bruce, Nature, 2020, 577, 502–508 CrossRef CAS PubMed.
- B. Mortemard de Boisse, S.-i. Nishimura, E. Watanabe, L. Lander, A. Tsuchimoto, J. Kikkawa, E. Kobayashi, D. Asakura, M. Okubo and A. Yamada, Adv. Energy Mater., 2018, 8, S1–S7 Search PubMed.
- B. Song, M. Tang, E. Hu, O. J. Borkiewicz, K. M. Wiaderek, Y. Zhang, N. D. Phillip, X. Liu, Z. Shadike, C. Li, L. Song, Y. Y. Hu, M. Chi, G. M. Veith, X. Q. Yang, J. Liu, J. Nanda, K. Page and A. Huq, Chem. Mater., 2019, 31, 3756–3765 CrossRef CAS.
- Y. Li, X. Wang, Y. Gao, Q. Zhang, G. Tan, Q. Kong, S. Bak, G. Lu, X. Q. Yang, L. Gu, J. Lu, K. Amine, Z. Wang and L. Chen, Adv. Energy Mater., 2019, 9, 1–9 Search PubMed.
- A. Tsuchimoto, X. Shi, K. Kawai, B. M. De Boisse, J. Kikkawa, D. Asakura, M. Okubo and A. Yamada, Nat. Commun., 2021, 12, 1–7 CrossRef PubMed.
- E. Adamczyk and V. Pralong, Chem. Mater., 2017, 29, 4645–4648 CrossRef CAS.
- H. Yoshida, N. Yabuuchi, K. Kubota, I. Ikeuchi, A. Garsuch, M. Schulz-Dobrick and S. Komaba, Chem. Commun., 2014, 50, 3677–3680 RSC.
- C. Li, C. Zhao, B. Hu, W. Tong, M. Shen and B. Hu, Chem. Mater., 2020, 32, 1054–1063 CrossRef CAS.
- S. Guo, J. Yi, Y. Sun and H. Zhou, Energy Environ. Sci., 2016, 9, 2978–3006 RSC.
- Y. Liu, C. Wang, M. Ren, H. Fang, Z. Jiang and F. Li, J. Energy Chem., 2021, 63, 351–357 CrossRef.
- E. J. Kim, L. A. Ma, D. M. Pickup, A. V. Chadwick, R. Younesi, P. Maughan, J. T. S. Irvine and A. R. Armstrong, ACS Appl. Energy Mater., 2020, 3, 10423–10434 CrossRef CAS.
- E. J. Kim, L. A. Ma, L. C. Duda, D. M. Pickup, A. V. Chadwick, R. Younesi, J. T. S. Irvine and A. Robert Armstrong, ACS Appl. Energy Mater., 2020, 3, 184–191 CrossRef CAS.
- E. J. Kim, K. Mofredj, D. M. Pickup, A. V. Chadwick, J. T. S. Irvine and A. R. Armstrong, J. Power Sources, 2021, 481, 229010 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- W. Zhao, A. Tanaka, K. Momosaki, S. Yamamoto, F. Zhang, Q. Guo and H. Noguchi, Electrochim. Acta, 2015, 170, 171–181 CrossRef CAS.
- X. Cao, X. Li, Y. Qiao, M. Jia, F. Qiu, Y. He, P. He and H. Zhou, ACS Energy Lett., 2019, 4, 2409–2417 CrossRef CAS.
- K. Tang, Y. Huang, X. Xie, S. Cao, L. Liu, H. Liu, Z. Luo, Y. Wang, B. Chang, H. Shu and X. Wang, Chem. Eng. J., 2020, 399, 125725 CrossRef CAS.
- B. Zhang, B. Zhang, L. Wang, X. Chen, Y. Lu, B. Xu and W. Yang, J. Alloys Compd., 2020, 824, 153938 CrossRef CAS.
- J. H. Stansby, W. M. Dose, N. Sharma, J. A. Kimpton, J. M. López del Amo, E. Gonzalo and T. Rojo, Electrochim. Acta, 2020, 341, 1–9 CrossRef.
- D. Pahari and S. Puravankara, J. Power Sources, 2020, 455, 227957 CrossRef CAS.
- S. Bao, S.-h. Luo, Z.-y. Wang, S.-x. Yan and Q. Wang, J. Colloid Interface Sci., 2019, 544, 164–171 CrossRef CAS PubMed.
- J. K. Park, G. G. Park, H. H. Kwak, S. T. Hong and J. W. Lee, ACS Omega, 2017, 3, 361–368 CrossRef PubMed.
- J.-P. Parant, R. Olazcuaga, M. Devalette, C. Fouassier and P. Hagenmuller, J. Solid State Chem., 1971, 3, 1–11 CrossRef CAS.
- D. Asakura, E. Hosono, Y. Nanba, H. Zhou, J. Okabayashi, C. Ban, P. A. Glans, J. Guo, T. Mizokawa, G. Chen, A. J. Achkar, D. G. Hawthron, T. Z. Regier and H. Wadati, AIP Adv., 2016, 6, 035105 CrossRef.
- J. Lee, J. K. Papp, R. J. Clément, S. Sallis, W. Yang, B. D. Mccloskey, G. Ceder, D. Kwon and T. Shi, Nat. Commun., 2017, 8, 1–10 CrossRef PubMed.
- K. Luo, M. R. Roberts, N. Guerrini, N. Tapia-Ruiz, R. Hao, F. Massel, D. M. Pickup, S. Ramos, Y. S. Liu, J. Guo, A. V. Chadwick, L. C. Duda and P. G. Bruce, J. Am. Chem. Soc., 2016, 138, 11211–11218 CrossRef CAS PubMed.
- M. Okubo and A. Yamada, ACS Appl. Mater. Interfaces, 2017, 9, 36463–36472 CrossRef CAS PubMed.
- M. Tang, A. Dalzini, X. Li, X. Feng, P. H. Chien, L. Song and Y. Y. Hu, J. Phys. Chem. Lett., 2017, 8, 4009–4016 CrossRef CAS PubMed.
- M. Sathiya, J. B. Leriche, E. Salager, D. Gourier, J. M. Tarascon and H. Vezin, Nat. Commun., 2015, 6, 1–7 Search PubMed.
- R. Stoyanova, M. Gorova and E. Zhecheva, J. Phys. Chem. Solids, 2000, 61, 615–620 CrossRef CAS.
- Q. Li, Y. Qiao, S. Guo, K. Jiang, Q. Li, J. Wu and H. Zhou, Joule, 2018, 2, 1134–1145 CrossRef CAS.
- X. Li, Y. Qiao, S. Guo, Z. Xu, H. Zhu, X. Zhang, Y. Yuan, P. He, M. Ishida and H. Zhou, Adv. Mater., 2018, 30, 2–7 Search PubMed.
- Y. Qiao, S. Guo, K. Zhu, P. Liu, X. Li, K. Jiang, C. J. Sun, M. Chen and H. Zhou, Energy Environ. Sci., 2018, 11, 299–305 RSC.
- D. O. Scanlon, C. W. Dunnill, J. Buckeridge, S. A. Shevlin, A. J. Logsdail, S. M. Woodley, C. R. A. Catlow, M. J. Powell, R. G. Palgrave, I. P. Parkin, G. W. Watson, T. W. Keal, P. Sherwood, A. Walsh and A. A. Sokol, Nat. Mater., 2013, 12, 798–801 CrossRef CAS PubMed.
- J. Buckeridge, K. T. Butler, C. R. A. Catlow, A. J. Logsdail, D. O. Scanlon, S. A. Shevlin, S. M. Woodley, A. A. Sokol and A. Walsh, Chem. Mater., 2015, 27, 3844–3851 CrossRef CAS.
- J. Vinckeviciute, D. A. Kitchaev and A. Van Der Ven, Chem. Mater., 2021, 33, 1625–1636 CrossRef CAS.
- E. Gonzalo, M. Zarrabeitia, N. E. Drewett, J. M. López del Amo and T. Rojo, Energy Storage Mater., 2021, 34, 682–707 CrossRef.
- M. D. Radin and A. Van Der Ven, Chem. Mater., 2016, 28, 7898–7904 CrossRef CAS.
- K. S. Pitzer, J. Am. Chem. Soc., 1960, 82, 4121 CrossRef.
- C. Zhao, Q. Wang, Z. Yao, J. Wang, B. Sánchez-Lengeling, F. Ding, X. Qi, Y. Lu, X. Bai, B. Li, H. Li, A. Aspuru-Guzik, X. Huang, C. Delmas, M. Wagemaker, L. Chen and Y. S. Hu, Science, 2020, 370, 708–712 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2ta01485h |
| This journal is © The Royal Society of Chemistry 2022 |