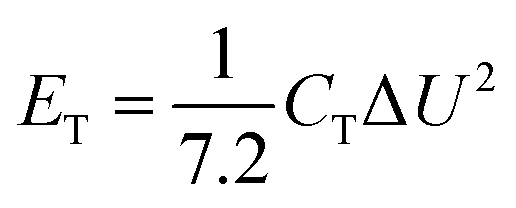DOI:
10.1039/D2TA00957A
(Paper)
J. Mater. Chem. A, 2022,
10, 9932-9940
Synthesis of TiC nanotube arrays and their excellent supercapacitor performance†
Received
5th February 2022
, Accepted 22nd March 2022
First published on 22nd March 2022
Abstract
Nanostructured metal carbides have numerous applications in catalysis and energy storage. However, directional construction remains a significant challenge. In this work, a novel strategy for the direct synthesis of nanostructured metal carbides using nanostructured metal oxides as the precursor is developed. TiO2 nanotube arrays (TiO2 NTAs) can be successfully transformed into TiC nanotube arrays (TiC NTAs) through electro-deoxidation and carbonization reactions in a low-temperature molten salt. TiC NTAs have a highly oriented and ordered array structure, which shows the advantages of large specific surface area, direct electron transport, and good chemical stability. Here, TiC NTA electrodes and PVA-H3PO4 electrolyte gel were assembled into a flexible quasi-solid-state supercapacitor to characterize their energy storage performance. The results show that the TiC NTA electrodes exhibit a high areal capacitance of 53.3 mF cm−2, excellent cycling stability, and mechanical flexibility. Moreover, the energy densities can reach 4.6 μW h cm−2 at a power density of 78.9 μW cm−2. This work provides a new strategy for the directed synthesis of nanostructured metal carbides and demonstrates the energy storage application potential of TiC NTAs. It is expected that this work will contribute to the development of the synthesis and application of nanostructured metal carbides.
1. Introduction
With the rapid depletion of fossil fuels and the worsening of environmental pollution, advanced renewable energy conversion and storage technologies have become increasingly urgent. In recent decades, supercapacitors (SCs) have been considered as a new type of energy storage device with great development potential.1,2 SCs have several advantages, such as high power density, fast charge/discharge rates, long cycle life, and better security than batteries. It is well known that the energy density of SCs is much lower than that of batteries, which is also a major challenge for the application and development of SCs. The overall performance of SCs depends mainly on the electrode material. The ideal electrode should have an abundant nanostructure, which can achieve higher kinetics due to the reduced transport/diffusion path lengths of ions and electrons. Moreover, a large specific surface area can provide more active sites for ion adsorption and redox reactions, thus greatly improving the storage capacity.1 Therefore, it is crucial to investigate and design nanostructured electrode materials.
Recently, transition metal carbides have been considered excellent energy storage electrode materials owing to their good chemical stability and electrical conductivity.3–5 Among them, titanium carbide (TiC) has attracted special attention because of its outstanding characteristics, such as good thermal stability, strong corrosion resistance, oxidation resistance, and low electrical resistivity.6,7 Various nanostructured TiCs have been reported, such as nanotubes,3 nanowires,8 nanoflakes6 and nanospheres,9 which have shown excellent performance in electrochemical energy storage. However, traditional fabrication methods are generally the template method and chemical vapor deposition method (CVD), which require high temperatures (>1100 °C) and multistep processes. Moreover, most electrode materials exist as powders and require additional current collectors and binders for assembly. When the high-quality active materials are loaded, it causes the generation of the charge storage “dead area” and unsatisfactory connection between the active materials and the current collector.10 An ordered nanostructure array is considered to be an ideal electrode structure, which has a highly ordered and oriented structural feature, which can obtain a large electrode–electrolyte interface and low ion transport barrier.11,12 Therefore, nanostructure arrays are expected to overcome the limitations of traditional electrode structures and improve the overall performance of SCs.
In the present work, a new process is proposed to synthesize TiC nanotube arrays (TiC NTAs) by electrochemical techniques, which perfectly maintain the nanoscale morphology during the transformation from oxide to carbide. To the best of our knowledge, it has never been reported to date. In this process, TiO2 nanotube arrays (TiO2 NTAs) were first grown on a titanium substrate by anodization, and then the TiO2 NTAs were transformed into TiC NTAs through electro-deoxidation and carbonization reactions in a low-temperature molten salt (≤600 °C). These TiC NTAs have attractive structural features such as highly oriented and arranged array structures, adjustable apertures and tube lengths, high surface areas, and unobstructed ion transport channels. Furthermore, they are vertically and uniformly arranged on the titanium matrix without additional binders, which provides a straight pathway for electron transport, so the integrated electrode can achieve high performance. To verify their energy storage application, TiC NTA electrodes and PVA-H3PO4 electrolytes were assembled into flexible quasi-solid SCs. The results showed that the SCs have high areal specific capacitance, excellent cycling stability, and remarkable energy and power densities. This work highlights a novel strategy for the synthesis of nanostructured transition metal carbides and demonstrates the application potential of TiC NTAs in the field of energy storage.
2. Experiment
2.1 Preparation of TiO2 nanotube arrays (TiO2 NTAs)
TiO2 NTAs were prepared using a two-step anodic oxidation process. The composition of the electrolyte was ethylene glycol-0.6 wt% NH4F-2 vol% H2O; titanium foil (thickness: 0.1 mm) was used as the anode, and the counter electrode was a platinum sheet. First, the titanium foil was polished with sandpaper and then ultrasonically cleaned in ethanol and deionized water. Subsequently, the titanium foil was chemically etched in a solution of HF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HNO3:H2O = 1:1:4 (volume ratio) to remove the surface oxide film. In the anodic oxidation process, the titanium foil was first oxidized at 50 V for 1 h, after which the nanotubes were removed by ultrasonic treatment and oxidized again for 1 h under the same conditions. Finally, the TiC NTAs were annealed at 400 °C for 1 h.
:HNO3:H2O = 1:1:4 (volume ratio) to remove the surface oxide film. In the anodic oxidation process, the titanium foil was first oxidized at 50 V for 1 h, after which the nanotubes were removed by ultrasonic treatment and oxidized again for 1 h under the same conditions. Finally, the TiC NTAs were annealed at 400 °C for 1 h.
2.2 Preparation of TiC nanotube arrays (TiC NTAs)
The electrolysis process was performed in a tubular electrolysis furnace. 450 g analytical grade LiCl–CaCl2–KCl mixed molten salt (50:40:10 mol%) was dried at 300 °C for 12 h, then added into a corundum crucible and placed in an electrolytic furnace. During electrolysis, high-purity argon gas was continuously injected into the furnace to ensure an inert atmosphere. After the furnace was heated from room temperature to 600 °C, a graphite rod was used as the anode and a stainless-steel rod was used as the cathode; the impurities in the molten salt were removed by pre-electrolysis for 12 h at a voltage of 3 V. Then, Li2O (0.1 mol%) was added to the molten salt and 300 mL CO2 gas was continuously introduced into the molten salt at a rate of 2 mL min−1, and the molten salt system was stabilized for 30 min. Subsequently, TiO2 NTAs were used as the cathode, and electrolysis was performed for 90 min at 3.3 V. The resulting cathode products were cleaned in dilute hydrochloric acid (0.5 mol L−1) and deionized water to remove the residual molten salt on the sample surface, and then vacuum dried.
2.3 Preparation of the quasi-solid-state SC
Two identical TiC NTA electrodes with a working area of 0.9 cm2 (0.5 cm × 1.8 cm) were used to prepare the quasi-solid flexible SC. The SC is composed of two identical TiC NTA electrode sheets and a PVA-H3PO4 polymer electrolyte sandwiched in the middle. The PVA polymer acted as a separator and solid electrolyte. PVA-H3PO4 polymer electrolytes were prepared using a solution-casting method. PVA (2 g) was dissolved in 20 mL deionized water and stirred at 80 °C for 1 h until it was completely dissolved. Then, 2 g of H3PO4 was added dropwise and stirred for 2 h until a uniform gel was formed, and finally dried at room temperature for 30 h to obtain a flexible electrolyte thin film.
2.4 Characterization
The products were characterized by field-emission scanning electron microscopy (FE-SEM, Thermo Fisher Scientific, Quattro S), transmission electron microscopy (TEM, Thermo Fisher Scientific, Talos F200S), X-ray diffraction (XRD, PANalytical X'PertPowder with Cu Kα radiation, Panalytical B.V.), and X-ray photoelectron spectroscopy (XPS, Thermo Fisher Scientific, ESCALAB 250Xi).
The cathodic reaction in the electrolysis process was analyzed by cyclic voltammetry (CV, 100 mV s−1) on a CHI1140C electrochemical workstation (CH Instruments, China). A Mo wire or TiO2 NTA-coated Mo wire (1 mm in diameter) is used as the working electrode, a spectrally pure graphite rod is used as the counter electrode (4 mm in diameter), and Ag/AgCl is used as the reference electrode. The potentials will be measured with respect to this reference electrode and then converted to Cl−/Cl2.
The electrochemical performance of the TiC NTA electrodes was investigated using a CHI1140C electrochemical workstation (CH Instruments, China). The electrochemical performance of the quasi-solid flexible SC was evaluated in a voltage window of 0.8 V. Cyclic voltammetry (CV, 10 to 100 mV s−1), galvanostatic charge/discharge (GCD, 0.2 mA cm−2 to 3 mA cm−2), cycling stability (10000 cycles at a current density of 3 mA cm−2) were assessed. The electrochemical impedance spectroscopy (EIS) measurements were conducted with a superimposed 5 mV sinusoidal voltage in the frequency range of 100 kHz to 0.1 Hz using a DH7002 electrochemical workstation (DH instrument, China). The total capacitance (CT) of the full SC was calculated from the GCD curves based on eqn (1):
| |  | (1) |
where
CT (mF cm
−2) is the area specific capacitance of the full SC; and
I (mA), Δ
t (s), Δ
U (V), and
S (cm
2) denote the discharge current through the full SC, discharge time, potential drop during discharge, and total area of the positive and negative electrodes, respectively.
The energy density (ET) and power density (PT) of the full SC were calculated based on eqn (2) and (3):
| | 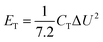 | (2) |
| |  | (3) |
where
ET (μWh cm
−2) is the energy density,
PT (μW cm
−2) is the power density,
CT (mF cm
−2), Δ
t (s), and Δ
U (V) are the area specific capacitance of the full SC, discharge time, and potential drop during discharge, respectively.
3. Results and discussion
3.1 Fabrication and characterization of TiC NTAs
The manufacturing process of the TiC NTAs is shown in Fig. 1. First, ordered TiO2 NTAs are formed on a titanium plate through an anodic oxidation process,13,14 as shown in Fig. 1a. These nanotubes were vertically arranged on the titanium substrate, and the diameters were mostly concentrated in the range of 110–130 nm (Fig. 1b). After high-temperature annealing, the amorphous TiO2 NTAs obtained by anodic oxidation were converted into the anatase phase, which improves the conductivity of the TiO2 NTA film.15 In addition, the annealing treatment can improve the adhesion of the TiO2 NTA film to the titanium substrate16 and prevent the film from falling off during the subsequent electrolysis process. As shown in Fig. 1c, the local area of the TiO2 NTAs cracked after annealing, but the overall array structure remained intact, and the TiO2 nanotube morphology remained unchanged. Finally, the annealed TiO2 NTAs were used as the precursor for electro-deoxidation and carbonization, and the resulting TiC NTAs still maintain the array structure of the TiO2 nanotubes (Fig. 1d) with a tube length of approximately 5 μm (Fig. S1†). The detailed microstructure of the TiC NTAs was further analyzed using TEM. The hollow tubular structure of TiC can be clearly distinguished in the TEM images (Fig. 2a and d), and these highly oriented nanotubes are closely arranged. Elemental mapping analysis of Ti and C further confirmed the nanotube composition (Fig. 2c). Furthermore, the SAED pattern (inset of Fig. 2b) reveals typical crystalline rings characteristic of TiC and the measured spacing of two adjacent lattices is 0.22 nm, which corresponds well to the (200) crystal plane of cubic TiC (JCPDS 65-0242).
 |
| | Fig. 1 Fabrication process and FE-SEM characterization. (a) Schematic diagram of TiO2 NTA formation; (b) SEM images of the TiO2 NTAs obtained by two-step anodic anodization; (c) SEM images of annealed TiO2 NTAs; (d) SEM images of TiC NTAs obtained by electrolysis; (e) deoxidation and carbonization process of TiO2 NTAs in the molten salt. | |
 |
| | Fig. 2 TEM characterization of TiC NTAs. (a) TEM images of the TiC NTAs; (b) the corresponding the SAED pattern and HRTEM image; (c) elemental mapping of Ti and C; (d) TEM images of a single TiC nanotube. | |
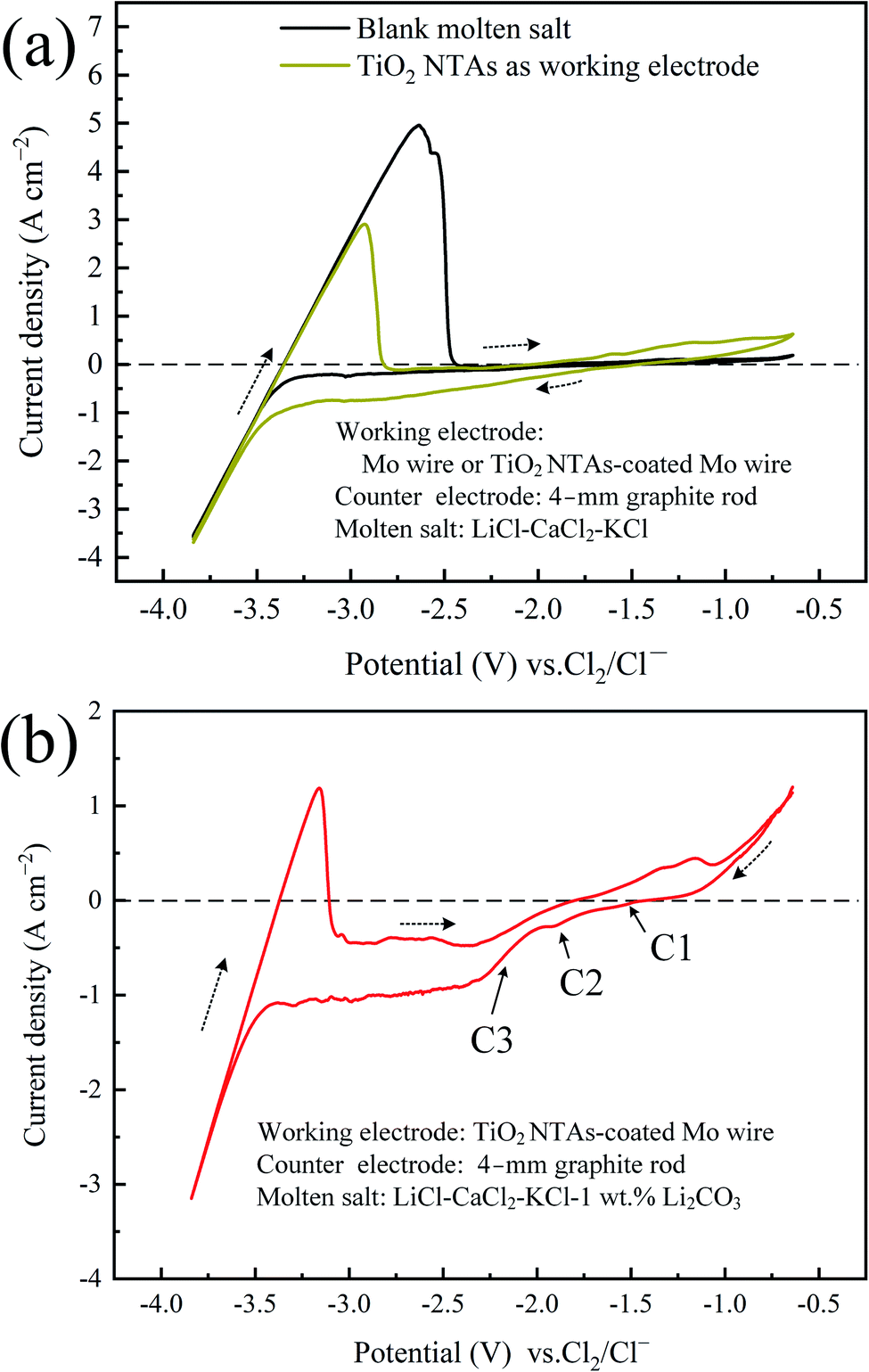
The reaction mechanism of the cathode region in the electrolysis process was analyzed by the cyclic voltammetry. As shown in Fig. 3a, during the scan toward negative potentials, there was no obvious reduction peak in the potential window before the decomposition of the molten salt, and the current remained at almost 0 A. When TiO2 NTAs were used as the working electrode, the current gradually increased due to the stepwise deoxidation of TiO2 (Fig. S2†). When CO32− was added to the molten salt, the CV curve (Fig. 3b) exhibited an additional small peak at C2 (−1.8 V vs. Cl2/Cl−), which may be attributed to the reduction reaction of CO32− (reaction (5)). As the oxygen content of titanium oxide decreases, the difficulty of deoxidation gradually increases,17,18 and further deoxidation of low-oxygen titanium can only be achieved at a higher decomposition potential. Thus, the increase in current at C1 (−1.5 V vs. Cl2/Cl−) corresponds to the deoxidation of TiO2 to form suboxides (reaction (4)), and the increase in current at C3 (−2.1 V vs. Cl2/Cl−) corresponds to the deoxidation of titanium suboxides (reaction (6)). The current in Fig. 3b increases significantly at C3 when CO32− is added, possibly due to the suppressed deoxygenation reaction. When the potential reached C2, the reduction reaction of CO32− occurred mainly at the cathode, and the deoxidation reaction was inhibited. When the CO32− near the cathode was depleted, the oxides that were too late to deoxidize in the early stage were rapidly deoxidized at high voltages, and thus the current increased sharply. Finally, the titanium suboxides were reduced to metallic titanium, which reacted with the deposited carbon to form TiC (reaction (7)).
| | | TiO2 + (4 − 2x)e− = TiOx + (2 − x)O2− (x ≤ 2) | (4) |
| | | CO32− + 4e− = C + 3O2− | (5) |
| | | TiOx + 2xe− = Ti + xO2− (x ≤ 2) | (6) |
 |
| | Fig. 3 (a) CV curve in LiCl–CaCl2–KCl at 600 °C with the Mo wire as the working electrode (black line) and TiO2 NTA-coated Mo wire as the working electrode (yellow line); (b) CV curve in LiCl–CaCl2–KCl-1 wt% Li2CO3 (TiO2 NTA-coated Mo wire as the working electrode). | |
Electrochemical fixation of CO2 to prepare advanced functional materials is a promising solution to promote carbon neutrality.19–21 Notably, the carbon source for the electrolysis process is the pre-injected CO2, which can be captured by O2− in the molten salt to form soluble CO32−.22–25 During electrolysis, the nanostructure can be maintained during the transformation from oxide to carbide, which is attributed to the high reactivity of the electrodeposited carbon and the lower reaction temperature. When a voltage is applied between the anode and cathode, the TiO2 nanotubes begin to deoxidize rapidly, and the conductivity is improved. Meanwhile, CO32− migrates from the molten salt to the nanotube interface and rapidly reduces to form carbon, and this carbon is composed of sp2 hybridized amorphous carbon, which has a high reaction activity.26 When TiO2 is reduced to Ti, the carbon deposited at the interface quickly reacts with Ti to form high melting point TiC, thereby avoiding morphological damage caused by sintering. In addition, the low reaction temperature of this process is also the key to maintaining the tubular morphology of TiC. Due to the limitations of kinetics, the synthesis of TiC usually requires a reaction temperature above 1000 °C and a long holding time.27 In this case, it is challenging to obtain nanoscale TiC (nanomaterials have stronger sintering kinetics, and the sintering temperature is approximately 20–40% of the melting point temperature of the material).28 However, in our fabrication process, the oriented pore structure of the TiO2 nanotubes provides an unhindered channel for CO32− transportation, and CO32− can be directly reduced to form carbon on the tube wall, which can expand the carbonization contact area and shorten the diffusion distance of carbon atoms, thus greatly improving the carbonization reaction kinetics. Therefore, the process can achieve TiC synthesis and maintain the nanostructure at low temperatures (≤600 °C).
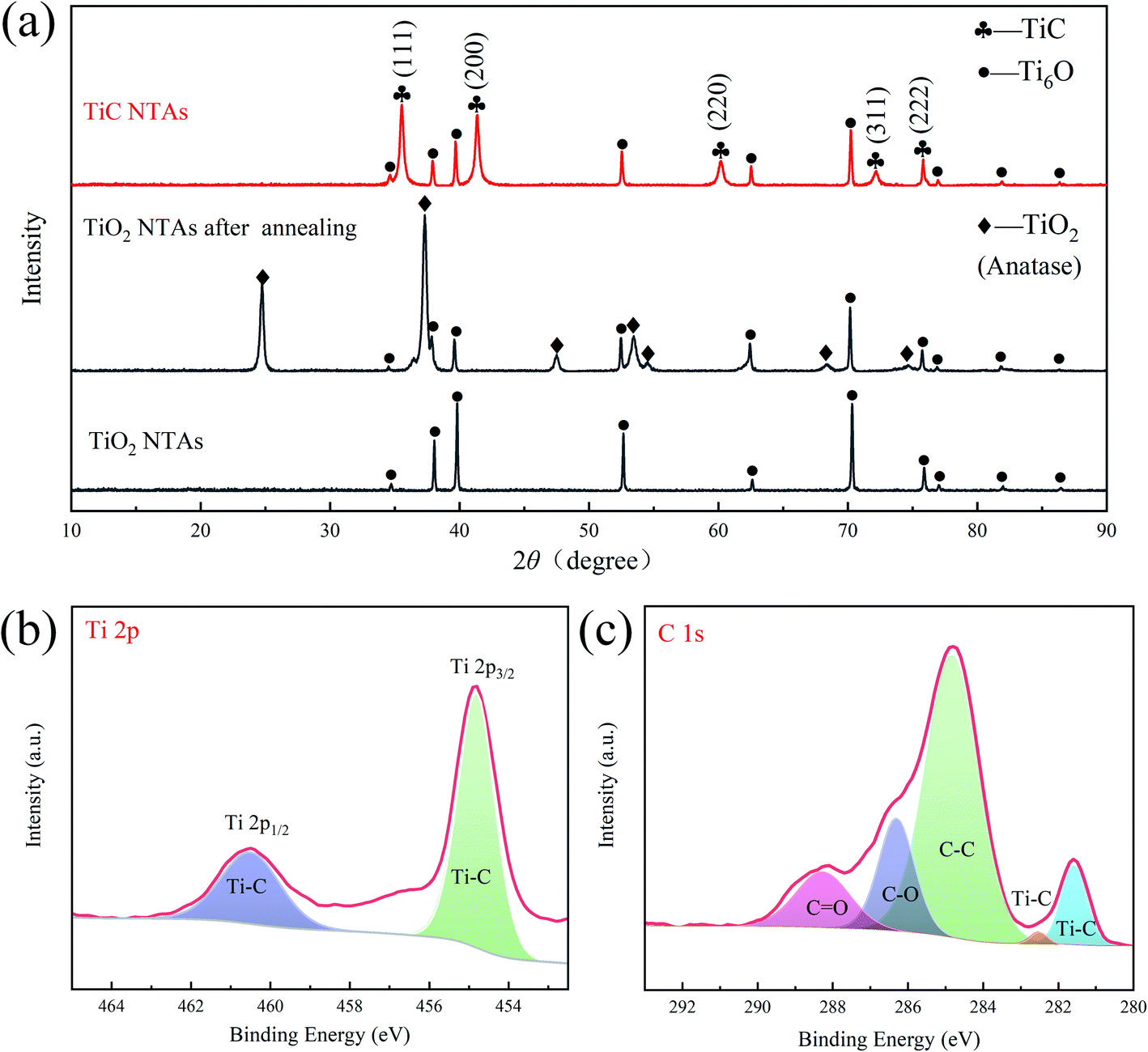
The formation and composition of the TiC phase were determined using XRD and XPS. The XRD patterns (Fig. 4a) indicate that the as-anodized TiO2 NTAs were amorphous because there were only peaks from the metal Ti substrate and no crystalline TiO2 peaks were identified. After the annealing treatment, anatase TiO2 diffraction peaks were detected, indicating that the amorphous TiO2 was converted into anatase. Finally, TiO2 NTAs were transformed into TiC NTAs through electro-deoxidation and carbonization, and the XRD pattern exhibited the characteristic diffraction peaks of the cubic TiC phase (JCPDS 65-0242). These diffraction peaks correspond to the (111), (200), (220), (311), and (222) planes, respectively, which is consistent with the results of the diffraction rings (Fig. 2b). In addition, XPS was used to analyze the elemental composition of the TiC NTAs. In the Ti 2p spectra (Fig. 4b), two obvious peaks at Ti 2p1/2 (460.5 eV) and Ti 2p3/2 (454.8 eV) correspond to the Ti–C bond.29 In addition, two typical peaks of TiC at 281.6 and 282.5 eV were observed in the C 1s spectrum (Fig. 4c).30 The XPS results further confirmed the successful synthesis of TiC by an electrolytic process.
 |
| | Fig. 4 (a) XRD pattern of TiC NTAs and TiO2 NTAs; XPS spectra: (b) Ti 2p spectrum and (c) C 1s spectrum. | |
3.2 Supercapacitor performance
The SC performance of the TiC NTAs was studied to evaluate their application potential in energy storage. A quasi-solid-state SC was assembled with TiC NTAs as the cathode and anode, and a PVA-H3PO4 polymer electrolyte was used as the electrolyte and diaphragm. Solid electrolytes can avoid the leakage and corrosion of liquid electrolytes, improve the safety of SCs, and easily realize the lightweight, high flexibility, and portability of energy storage devices.31,32 As shown in Fig. 5a, cyclic voltammetry was carried out at different scanning rates of 10–100 mV s−1 in the voltage range of 0–0.8 V. The CV curve exhibited an ideally quasi-rectangular shape without an obvious redox peak, indicating that the charge storage of the TiC NTA electrode originated from typical double-layer capacitor (EDLC) characteristics. Moreover, the curve maintained a good quasi-rectangular shape at all scan rates, which illustrates the outstanding rate capability of the TiC NTA electrode. The GCD curves of the TiC NTA electrode at different current densities are shown in Fig. 5b. The charge curve and the corresponding discharge curve were symmetrical, indicating good electrochemical capacitive characteristics and reversibility. In addition, the slope of the curve maintained a constant value at a specific current density, showing a typical linear charge–discharge characteristic, which further demonstrates the EDLC behavior of the TiC NTA electrode. Fig. 5c shows the area specific capacitance of the TiC NTA electrode at different current densities, with a maximum specific capacitance of 53.3 mF cm−2 obtained at a current density of 0.2 mA cm−2. Even when the current density increased to 3 mA cm−2, the capacitance could still be maintained at 35.3 mF cm−2 (retention rate of 66%). These values are obviously higher than those reported in the literature (Table S1†). Fig. 5 shows the EIS of the TiC NTA electrode. It can be seen from the Nyquist diagram that the capacitor demonstrates ideal electrochemical capacitor behavior, that is, the imaginary part of the impedance in the low-frequency region is approximately perpendicular to the real part. The approximate semicircle in the high to medium frequency range corresponds to the charge transfer through the electrode/electrolyte interface, and the charge transfer resistance can be estimated from the diameter of the semicircle.3 However, TiC NTAs have small impedance arcs in the high-frequency region, indicating low charge-transfer resistance (Rct). The excellent performance of TiC NTAs depends on their unique structural characteristics. The highly oriented tubular nanostructure not only provides a large specific surface area for ion adsorption, but also improves the diffusion channels of ions and electrolytes. Meanwhile, the ordered array structure can effectively avoid the agglomeration and folding of electrode materials and greatly reduce the generation of “dead zones” for charge storage; compared with independent and disordered nanoparticles, the ordered array structure can achieve more active material loading per unit area. Furthermore, TiC NTAs directly connect to the conductive substrate without additional binders or conductive additives, which can reduce the self-weight of the electrode and provide a straight pathway for electron transport, thereby ensuring low electrode interface resistance.
 |
| | Fig. 5 Electrochemical characterization of the SC based on TiC NTA electrode. (a) CV curves at different scan rates ranging from 10 to 100 mV s−1; (b) GCD curves at various current densities ranging from 0.2 to 3 mA cm−2; (c) specific capacitances at various current densities; (d) Nyquist plots of the flexible SC, the inset shows the details in the high-frequency range. | |
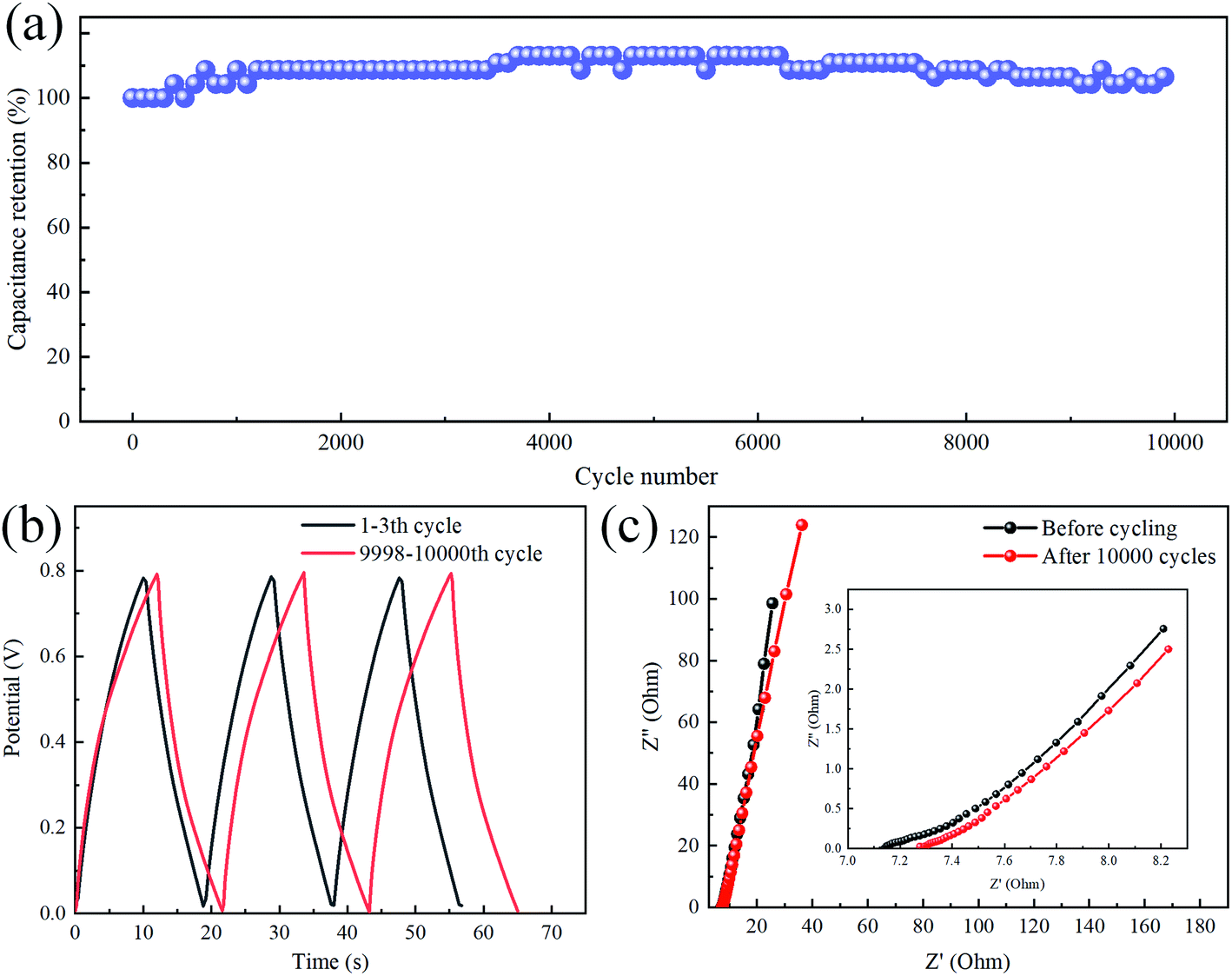
A durable and stable cycle life is also an essential parameter for evaluating the performance of the SC. As shown in Fig. 6a, the quasi-solid flexible SC was charged and discharged at a current density of 3 mA cm−2 for 10000 cycles. The capacitance of the TiC NTA electrode gradually increased due to the self-activation process at the beginning of the cycle,33,34 and the capacitance gradually decreased after 4000 cycles. Finally, the capacitance retention reached 106% (compared with the initial capacitance) after 10000 cycles. The GCD curves before and after the cycle tests are shown in Fig. 6b. The GCD curve after the cycle was still very symmetrical, reflecting good charge and discharge stability. The excellent cycle life of TiC NTA-based supercapacitors is caused by two factors: (1) TiC has good oxidation resistance, which is conducive to long-term operation at high current density.3 (2) The solid electrolyte can stabilize the nanostructure during the cycle, and its low water content also inhibits the electrochemical oxidation of TiC NTAs.33 In addition, the change in the Nyquist diagram (Fig. 6c) in the high-frequency region is negligible, indicating that the Rct of the electrode remains low after cycling. The curve intercept on the Z′ axis reflects the electrolyte and electrode resistance. It can be seen that the inner SC resistance increases slightly after 10000 cycles, which is caused by the decrease in the polymer electrolyte conductivity.35
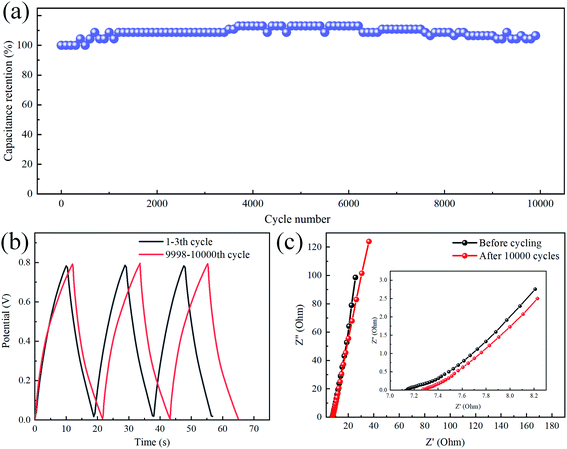 |
| | Fig. 6 (a) Cycling stability of the TiC NTAs at a current density of 3.0 mA cm−2 for 10000 cycles; (b) GCD curve comparison before and after 10000 cycles; (c) the Nyquist plots of the flexible SC before and after 10000 cycles, and the inset shows the details in the high-frequency range. | |
The energy and power densities of the full SC are shown in the Ragone plot (Fig. 7). The results showed that the energy density of the device was as high as 4.6 μW h cm−2 at a power density of 78.9 μW cm−2. This is significantly higher than other electrode materials reported in the literature in recent years, such as graphene thin films (0.235 μW h cm−2),36 MXene/Fe3O4/MXene thin films (0.97 μW h cm−2),34 rGO/Ti3C2 (2.1 μW h cm−2),37 graphene/carbon nanotubes (1.36 μW h cm−2),38 porous SiC/C (1.64 μW h cm−2),39 and other electrode materials;40–45 the detailed information is shown in Table S2.† Although the energy density of TiC NTAs is better than that of many nanostructured electrodes, it can be further improved through a series of methods: (1) increasing the nanotube length or optimizing the TiO2 nanotube structure; (2) TiC NTAs can act as a support matrix for the deposition of some electroactive species or build asymmetric supercapacitors.46–49
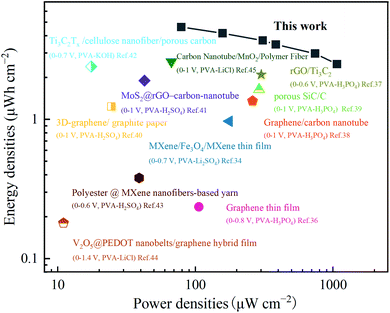 |
| | Fig. 7 Ragone plot of TiC NTAs compared with other representative nanostructure-based quasi-solid flexible SCs. | |
Fig. 8 demonstrates the application potential of the TiC NTA electrode in flexible and portable electronic products. Fig. 8a and b are schematic diagrams and optical photographs of a flexible quasi-solid SC with good flexibility. Furthermore, CV measurements were performed for the flexible device with different curvatures at a scan rate of 100 mV s−1. Fig. 8c and d show the excellent flexibility and mechanical stability of the SC device. The CV curves at different bending angles almost overlap. There is no obvious distortion even at a large bending angle of 180°, and the capacitance can be maintained at 96%.
 |
| | Fig. 8 (a) Schematic diagram of a flexible quasi-solid SC; (b) optical photograph of a flexible quasi-solid SC; (c) cyclic voltammetry curves for the SC with different curvatures with a scan rate of 100 mV s−1; (d) capacitance retention at different bending angles. | |
4. Conclusion
In this study, a new method to synthesize nanostructured metal carbides using the corresponding metal oxides as precursors was developed. As a preliminary demonstration, TiC NTAs were successfully prepared in a low-temperature molten salt with TiO2 NTAs as structural templates and CO2 as a carbon source, which perfectly maintained the nanoscale morphology during the transformation from oxide to carbide. TiC NTAs exhibit a highly oriented and ordered array structure, which has the advantages of a large specific surface area, direct electron transport, and good chemical stability. Moreover, TiC NTAs directly connect to the conductive substrate without extra binders or conductive additives, which ensures low electrode interface resistance and a rapid electrochemical reaction rate. We assembled them with a PVA-H3PO4 polymer electrolyte to obtain a flexible quasi-solid SC, and its electrochemical energy storage performance was tested. The results show that TiC NTAs have outstanding EDLC performance, including high areal specific capacitance, excellent cycling stability, good mechanical flexibility, and remarkable energy and power density. In summary, we provide an unprecedented strategy for the directional synthesis of nanostructured metal carbides, which can convert metal oxides into the corresponding carbides while maintaining the original nanostructure. And further demonstrates the excellent performance and application potential of TiC NTAs in the field of energy storage. Moreover, the process also achieves electrochemical fixation of CO2 and the conversion of advanced energy materials.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Funding: this work was financially supported by the National Natural Science Foundation of China (No. 52174299) and supported by the Chongqing Key Laboratory of Vanadium–Titanium Metallurgy and New Materials, Chongqing University, Chongqing 400044, PR China.
References
- G. Wang, L. Zhang and J. Zhang, Chem. Soc. Rev., 2012, 41, 797–828 RSC.
- M. Salanne, B. Rotenberg, K. Naoi, K. Kaneko, P. L. Taberna, C. P. Grey, B. Dunn and P. Simon, Nat. Energy, 2016, 1, 16070 CrossRef CAS.
- X. Xia, Y. Zhang, D. Chao, Q. Xiong, Z. Fan, X. Tong, J. Tu, H. Zhang and H. J. Fan, Energy Environ. Sci., 2015, 8, 1559–1568 RSC.
- Y. Zhong, X. H. Xia, F. Shi, J. Y. Zhan, J. P. Tu and H. J. Fan, Adv. Sci., 2015, 3, 1500286 CrossRef PubMed.
- K. N. Dinh, Q. H. Liang, C. F. Du, J. Zhao, A. L. Y. Tok, H. Mao and Q. Y. Yan, Nano Today, 2019, 25, 99–121 CrossRef CAS.
- T. Chen, M. Li, S. Song, P. Kim and J. Bae, Nano Energy, 2020, 71, 104549 CrossRef CAS.
- B. X. Dong, F. Qiu, Q. Li, S. L. Shu, H. Y. Yang and Q. C. Jiang, Nanomaterials, 2019, 9, 1152 CrossRef CAS PubMed.
- X. Xia, J. Zhan, Y. Zhong, X. Wang, J. Tu and H. J. Fan, Small, 2017, 13, 1602742 CrossRef PubMed.
- Y. Zhong, X. Xia, J. Zhan, Y. Wang, X. Wang and J. Tu, J. Mater. Chem. A, 2016, 4, 18717–18722 RSC.
- W. Guo, C. Yu, S. Li and J. Qiu, Energy Environ. Sci., 2021, 14, 576–601 RSC.
- H. Zhao, M. Zhou, L. Wen and Y. Lei, Nano Energy, 2015, 13, 790–813 CrossRef CAS.
- J. Liu, G. Cao, Z. Yang, D. Wang, D. Dubois, X. Zhou, G. L. Graff, L. R. Pederson and J. G. Zhang, ChemSusChem, 2008, 1, 676–697 CrossRef CAS PubMed.
- K. Huo, B. Gao, J. Fu, L. Zhao and P. K. Chu, RSC Adv., 2014, 4, 17300–17324 RSC.
- P. Roy, S. Berger and P. Schmuki, Angew. Chem., Int. Ed., 2011, 50, 2904–2939 CrossRef CAS PubMed.
- H. T. Fang, M. Liu, D. W. Wang, T. Sun, D. S. Guan, F. Li, J. Zhou, T. K. Sham and H. M. Cheng, Nanotechnology, 2009, 20, 225701 CrossRef PubMed.
- J. Xiong, X. Wang, Y. Li and P. D. Hodgson, J. Phys. Chem. C, 2011, 115, 4768–4772 CrossRef CAS.
- Y. Zhang, Z. Z. Fang, P. Sun, T. Zhang, Y. Xia, C. Zhou and Z. Huang, J. Am. Chem. Soc., 2016, 138, 6916–6919 CrossRef CAS PubMed.
- Y. Zhang, Z. Z. Fang, Y. Xia, P. Sun, B. Van Devener, M. Free, H. Lefler and S. Zheng, Chem. Eng. J., 2017, 308, 299–310 CrossRef CAS.
- X. Liang, J. Xiao, W. Weng and W. Xiao, Angew. Chem., Int. Ed., 2021, 60, 2120–2124 CrossRef CAS PubMed.
- T. Lv, J. Xiao, W. Weng and W. Xiao, Adv. Energy Mater., 2020, 10, 2002241 CrossRef CAS.
- J. Wang, J. Xiao, Y. Shen, X. Liang, T. Lv and W. Xiao, J. Mater. Chem. A, 2021, 9, 27442–27447 RSC.
- W. Weng, B. M. Jiang, Z. Wang and W. Xiao, Sci. Adv., 2020, 6, eaay9278 CrossRef CAS PubMed.
- W. Weng, L. Z. Tang and W. Xiao, J. Energy Chem., 2019, 28, 128–143 CrossRef.
- L. Li, Z. Shi, B. Gao, X. Hu and Z. Wang, Electrochim. Acta, 2016, 190, 655–658 CrossRef CAS.
- Z. Fan and W. Xiao, Angew. Chem., Int. Ed., 2021, 60, 7664–7668 CrossRef CAS PubMed.
- L. Massot, P. Chamelot, F. Bouyer and P. Taxill, Electrochim. Acta, 2002, 47, 1949–1957 CrossRef CAS.
- H. Nadimi, H. Sarpoolaky and M. Soltanieh, Ceram. Int., 2021, 47, 12859–12869 CrossRef CAS.
- J. P. Ramos, A. M. R. Senos, T. Stora, C. M. Fernandes and P. Bowen, J. Eur. Ceram. Soc., 2017, 37, 3899–3908 CrossRef CAS.
- Y. Cheng and Y. F. Zheng, Surf. Coat. Technol., 2007, 201, 4909–4912 CrossRef CAS.
- E. Lewin, P. O. Å. Persson, M. Lattemann, M. Stüber, M. Gorgoi, A. Sandell, C. Ziebert, F. Schäfers, W. Braun, J. Halbritter, S. Ulrich, W. Eberhardt, L. Hultman, H. Siegbahn, S. Svensson and U. Jansson, Surf. Coat. Technol., 2008, 202, 3563–3570 CrossRef CAS.
- T. Lv, M. Liu, D. Zhu, L. Gan and T. Chen, Adv. Mater., 2018, 30, 1705489 CrossRef PubMed.
- P. Yang and W. Mai, Nano Energy, 2014, 8, 274–290 CrossRef CAS.
- X. Lu, G. Wang, T. Zhai, M. Yu, S. Xie, Y. Ling, C. Liang, Y. Tong and Y. Li, Nano Lett., 2012, 12, 5376–5381 CrossRef CAS PubMed.
- H. Li, Y. Liu, S. Lin, H. Li, Z. Wu, L. Zhu, C. Li, X. Wang, X. Zhu and Y. Sun, J. Power Sources, 2021, 497, 229882 CrossRef CAS.
- H. Yu, J. Wu, L. Fan, K. Xu, X. Zhong, Y. Lin and J. Lin, Electrochim. Acta, 2011, 56, 6881–6886 CrossRef CAS.
- Q. Chen, X. Li, X. Zang, Y. Cao, Y. He, P. Li, K. Wang, J. Wei, D. Wu and H. Zhu, RSC Adv., 2014, 4, 36253–36256 RSC.
- N. Radha, A. Kanakaraj, H. M. Manohar, M. R. Nidhi, D. Mondal, S. K. Nataraj and D. Ghosh, Appl. Surf. Sci., 2019, 481, 892–899 CrossRef.
- Y. Wang, Y. Zhang, G. Wang, X. Shi, Y. Qiao, J. Liu, H. Liu, A. Ganesh and L. Li, Adv. Funct. Mater., 2020, 30, 1907284 CrossRef CAS.
- B. Yang, R. Sun, X. Li, M. Ma, X. Zhang, Z. Wang, W. Yi, Z. Zhang, R. Yang, H. Sun, G. Gao, Y. Chu, Z. Zhao and X. Liu, J. Mater. Sci., 2021, 56, 16068–16081 CrossRef CAS.
- A. Ramadoss, K. Y. Yoon, M. J. Kwak, S. I. Kim, S. T. Ryu and J. H. Jang, J. Power Sources, 2017, 337, 159–165 CrossRef CAS.
- W. Yang, L. He, X. Tian, M. Yan, H. Yuan, X. Liao, J. Meng, Z. Hao and L. Mai, Small, 2017, 13, 1700639 CrossRef PubMed.
- W. Chen, D. Zhang, K. Yang, M. Luo, P. Yang and X. Zhou, Chem. Eng. J., 2021, 413, 127524 CrossRef CAS.
- W. Shao, M. Tebyetekerwa, I. Marriam, W. Li, Y. Wu, S. Peng, S. Ramakrishna, S. Yang and M. Zhu, J. Power Sources, 2018, 396, 683–690 CrossRef CAS.
- L. Wang, T. Shu, S. Guo, Y. Lu, M. Li, J. Nzabahimana and X. Hu, Energy Storage Mater., 2020, 27, 150–158 CrossRef.
- C. Choi, S. H. Kim, H. J. Sim, J. A. Lee, A. Y. Choi, Y. T. Kim, X. Lepró, G. M. Spinks, R. H. Baughman and S. J. Kim, Sci. Rep., 2015, 5, 9387 CrossRef CAS PubMed.
- C. Wan, Y. Jiao and J. Li, J. Mater. Chem. A, 2017, 5, 3819–3831 RSC.
- C. Wan, Y. Jiao, W. Bao, H. Gao, Y. Wu and J. Li, J. Mater. Chem. A, 2019, 7, 9556–9564 RSC.
- C. Wan, Y. Jiao, D. Liang, Y. Wu and J. Li, Adv. Energy Mater., 2018, 8, 1802388 CrossRef.
- Y. Tian, C. Yang, W. Que, X. Liu, X. Yin and L. B. Kong, J. Power Sources, 2017, 359, 332–339 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2ta00957a |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b,
Buxin
Chen
a,
Lang
Zhao
a,
Liwen
Hu
a,
Liangying
Wen
a and
Meilong
Hu
*a
*b,
Buxin
Chen
a,
Lang
Zhao
a,
Liwen
Hu
a,
Liangying
Wen
a and
Meilong
Hu
*a