
Superior performance enabled by supramolecular interactions in metal−organic cathode: the power of weak bonds†
An-Che
Li‡
ab,
Cheng-Han
Chang‡
c,
Alexander S.
Ivanov
 d,
Yi-An
Lo
ab,
Ilja
Popovs
d,
Jeng-Lung
Chen
e,
Yu-Chun
Chuang
e,
Yu-Chung
Chang
e,
Bo-Hao
Chen
e,
Jui-Chin
Lee
f,
Teng-Hao
Chen
*g and
Watchareeya
Kaveevivitchai
*ab
d,
Yi-An
Lo
ab,
Ilja
Popovs
d,
Jeng-Lung
Chen
e,
Yu-Chun
Chuang
e,
Yu-Chung
Chang
e,
Bo-Hao
Chen
e,
Jui-Chin
Lee
f,
Teng-Hao
Chen
*g and
Watchareeya
Kaveevivitchai
*ab
aDepartment of Chemical Engineering, National Cheng Kung University, Tainan City 70101, Taiwan. E-mail: wkaveechai@mail.ncku.edu.tw
bHierarchical Green-Energy Materials Research Centre, National Cheng Kung University, Tainan City 70101, Taiwan
cDepartment of Chemistry, Tamkang University, New Taipei City 25137, Taiwan
dChemical Sciences Division, Oak Ridge National Laboratory, Oak Ridge, P.O. Box 2008, TN 37831, USA
eNational Synchrotron Radiation Research Centre, Hsinchu 30076, Taiwan
fCore Facility Centre, National Cheng Kung University, Tainan City 70101, Taiwan
gSchool of Pharmacy, National Cheng Kung University, Tainan City 70101, Taiwan,. E-mail: thchen@gs.ncku.edu.tw
First published on 15th April 2022
Abstract
Metal–organic-based electrode materials are increasingly appealing because both metal and organic linker are capable of undergoing redox processes, thus offering a high specific capacity. High porosity which can be achieved by the rational design of these materials is generally perceived as one of the major criteria for high rate performance. However, while Li-ion transport may be possible, oftentimes the counteranions (e.g., PF6−) and solvent molecules are co-inserted into the porous host lattice, potentially hindering Li-ion diffusion pathways. Here we propose the concept of close-packed metal–organic cathode stabilized by multiple supramolecular interactions as a viable solution for exceptional electrochemical performance. This is illustrated in a modularly designed redox-active [CuL(Py)2]n (LH4 = 1,4-dicyano-2,3,5,6-tetrahydroxybenzene, Py = pyridine). The mechanistic studies and DFT calculations confirm that the supramolecular interactions between its close-packed chains are the key to the flexible host lattice which only allows desolvated Li+ to intercalate, while these weak bonds also stabilize the inserted Li in the preferred hopping sites, creating optimal diffusion paths. The performance observed in this work is found to be among the best ever reported for metal–organic cathodes with a capacity as high as 255 mA h g−1 at 65 mA g−1 (0.25C) and a reversible capacity of 59 mA h g−1 at ∼26 A g−1 (100C) corresponding to 81% retention after 1000 cycles. These findings reveal a potential new strategy towards metal–organic-based electrodes with high performance and enhanced cycling stability.
 Watchareeya Kaveevivitchai | Dr Watchareeya Kaveevivitchai is an Assistant Professor in Chemical Engineering at National Cheng Kung University, Taiwan. She earned a PhD in Chemistry from the University of Houston (2014) and pursued postdoctoral research in Texas Materials Institute at the University of Texas at Austin (2015–2017). Her research group focuses on a variety of investigative studies in the general areas of solid-state chemistry, Li-ion batteries, post-Li-ion batteries, energy storage technologies, functional redox-active materials, and advanced porous compounds for gas storage and separations, carbon capture, and biomedical applications. |
Introduction
Due to the increasing demand for large-scale energy storage systems in electric vehicles and renewable energy smart grids, superior Li-ion batteries (LIBs) in terms of energy/power density are now required at a substantially lower cost.1,2 The next-generation LIBs need to have fast-charge ability, high safety and, most importantly, environmental friendliness. The inorganic intercalation compounds commonly used as cathode materials in conventional LIBs usually contain expensive and/or toxic transition metals, such as cobalt and nickel.2 To increase the limited practical capacity of these inorganic cathodes, efforts have been made such as coating and doping with other metals; however, further enhancement is quite challenging and it appears that traditional inorganic electrodes have reached critical limits in their performance in terms of energy and power density.3–5Organic-based electrodes are considered to be more eco-friendly and sustainable than their inorganic counterparts. Organic materials can be engineered at the molecular level to incorporate reversible redox-active functional groups, which allow multi-electron transfer process leading to a high theoretical capacity.6 The redox potential of organic moieties can also be fine-tuned by introducing electron-withdrawing or electron-donating functional groups for cathode and anode, respectively.7–9 Some of the organic electrode materials including carbonyls,10–12 quinones,13 Schiff bases,14 quinoxaline-based heteroaromatic,15 porous organic polymers16,17 and covalent organic frameworks18,19 exhibit excellent specific capacity and cycling stability, while many of them suffer from issues associated with material dissolution in typical organic solvents and low electronic conductivity, resulting in poor cycling performance. Up until now, the improvement of these organic electrodes has been obstructed by the lack of understanding on the relationship between structure and battery performance due to their poor crystallinity.20 Instead, metal–organic coordination polymers (CPs) are formed by polymerizing organic building blocks via metal–ligand coordination, usually resulting in crystalline frameworks.21 More importantly, the characteristic feature of CPs is modular design, which allows the chemical/physical properties as well as the crystal structure to be fine-tuned simply by modifying the organic or inorganic modules of the CP, therefore leading to large diversity and endless possibility for desired applications.22
In order to obtain higher energy density, more accessible redox-active sites per unit volume or weight of active material are required. To achieve high rate capability and power density, electrode materials need sufficiently rapid Li-ion and electron transport in the structure, which are governed by their intrinsic ionic and electronic conductivity.23 In the past decade, metal–organic frameworks (MOFs), a porous subclass of CPs well-known for their high porosity and structural tunability, have been widely studied as battery electrodes due to their multiple redox centres from both metal and organic moieties, rigid frameworks and structural diversity.24 The high surface areas and large pores of these emerging materials are perceived to be essential for accessible redox-active sites and rapid Li-ion transport, which are required for high-capacity and high-rate battery operation.25 Among these materials, two-dimensional (2D) layer-stacked MOFs with 1D channels demonstrate intriguing properties such as high intrinsic electronic conductivity and structural stability.23 Cu-THQ26 (THQ = tetrahydroxy-1,4-quinone) and Cu-BHT27 (BHT = benzenehexathiolate) were capable of undergoing reversible three-electron and four-electron redox reactions with high initial specific capacities of 387 and 232 mA h g−1, respectively, at a current density of 50 mA g−1. For high rates, these materials were able to be cycled up to 1000 and 2000 mA g−1 delivering capacities of 93 and 100 mA h g−1, respectively; however, only a few cycles were reported. Although various porous, conductive and structurally stable 2D MOFs have been investigated as electrode materials, they barely provide satisfactory rate capability, energy density and long-term cycling performance needed for emerging practical applications.28,29 One possible reason is that MOFs with large voids usually have a low density of redox-active sites and their pores and channels tend to also be filled by anions and solvent molecules from the organic electrolyte, significantly hindering the Li-ion diffusion pathways during the battery operation.30,31 In our previous work, a 1D CP [CuL(DMF)2]n (DMF = N,N-dimethylformamide) was prepared from Cu(II) and redox-active organic ligand 1,4-dicyano-2,3,5,6-tetrahydroxybenzene (LH4) and used as cathode material for LIBs.32 It was found that even though multi-electron redox reaction was possible, the compound suffered from capacity loss and poor rate capability, as observed in many other metal–organic electrodes mainly due to unstable structures and low ionic and electronic conductivity.24
Supramolecular interactions are commonly observed in self-assembled crystalline organic materials. They are a driving force allowing stable packing of small discrete molecules and dynamic assembly of large frameworks.33,34 These weak interactions are also responsible for the flexibility and self-healing properties of polymeric materials.35,36 Despite these useful aspects, supramolecular interactions have rarely been mentioned in the context of energy storage. Recently, a few small-molecule-based cathodes have been reported to utilize weak intermolecular interactions such as H-bonding and π-interactions to stabilize the structures, therefore improving the cycling performance.37–39 A supramolecular self-assembled organic small molecule stitched together by multiple π⋯π and unconventional C–H⋯O interactions has been found to deliver unparalleled electrochemical performance and ranked among the best organic molecules for LIBs.37 Inspired by the remarkable electrochemical properties enhanced by these noncovalent interactions, we propose the concept of close-packed crystalline metal–organic cathode stabilized by multiple supramolecular interactions as a viable solution for exceptional electrochemical performance. This is demonstrated in a novel CP [CuL(Py)2]n (Fig. 1), which has been prepared by modular synthesis. Pyridine (Py) was our module of choice to replace the coordinating DMF molecules in [CuL(DMF)2]n. The conjugation in Py introduces additional π-interactions between the close-packed 1D chains; as a result, supramolecular interactions are maximized between the CP chains (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N⋯π, CN⋯H, C–H⋯π and π⋯π). Our mechanistic studies, by comprehensive spectroscopic, diffraction and simulation techniques, reveal the importance of the supramolecular interactions between the close-packed chains being responsible for the flexible CP structure which only allows desolvated Li+ to insert/extract during discharge–charge processes. These weak interactions also further stabilize the inserted Li ions in their preferred hopping sites and create optimal diffusion paths, therefore ultimately leading to excellent electrochemical performance especially at high rates. The CP electrode can undergo a current density as high as 100C (∼26
N⋯π, CN⋯H, C–H⋯π and π⋯π). Our mechanistic studies, by comprehensive spectroscopic, diffraction and simulation techniques, reveal the importance of the supramolecular interactions between the close-packed chains being responsible for the flexible CP structure which only allows desolvated Li+ to insert/extract during discharge–charge processes. These weak interactions also further stabilize the inserted Li ions in their preferred hopping sites and create optimal diffusion paths, therefore ultimately leading to excellent electrochemical performance especially at high rates. The CP electrode can undergo a current density as high as 100C (∼26![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 162 mA g−1) providing a reversible capacity of 59 mA h g−1 with an 81% retention after 1000 cycles, which is found to be among the best metal–organic cathode materials ever reported to date. The lithiation dynamics and mechanistic approach to high electrode performance of metal–organic-based materials have, for the first time, been revealed. This work undoubtedly sheds light on the new concept and provides a profound understanding on design strategy of metal–organic electrode materials with high performance and cycling stability.
162 mA g−1) providing a reversible capacity of 59 mA h g−1 with an 81% retention after 1000 cycles, which is found to be among the best metal–organic cathode materials ever reported to date. The lithiation dynamics and mechanistic approach to high electrode performance of metal–organic-based materials have, for the first time, been revealed. This work undoubtedly sheds light on the new concept and provides a profound understanding on design strategy of metal–organic electrode materials with high performance and cycling stability.
 | ||
| Fig. 1 (a) and (b) The single crystal structure of [CuL(Py)2]n viewed along the x-axis and y-axis, respectively. The hydrogen atoms are omitted for clarity. (c) Packing of [CuL(Py)2]n by supramolecular CN⋯π (green dotted line), CN⋯H (red dotted lines), C–H⋯π (blue dotted lines) and π⋯π (magenta dotted line) interactions. Colours of atoms: blue: Cu(II); grey: C; red: O; cyan: N; white: H. (d) The perspective view of supramolecular network in [CuL(Py)2]n. The structure of [CuL(Py)2]n is greyed for clarity. | ||
Results and discussion
Material synthesis and crystal structure
By reacting LH4 and Py with Cu(NO3)2·3H2O in DMF at 100 °C for 48 h, orange crystals were obtained. The single crystal X-ray analysis reveals 1D metal–ligand chains with a chemical formula [CuL(Py)2]n (Fig. 1a). The octahedral Cu(II) metal centre is coordinated by two pairs of oxygen atoms from two deprotonated ligands L at its equatorial positions, and capped with two pyridine molecules at its axial positions. The Fourier-transform infrared (FT-IR) spectrum of [CuL(Py)2]n shows the stretching band at 1636 cm−1 assigned as C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of the benzoquinoid ligand, while the spectrum of LH4 shows no sign of the carbonyl groups (Fig. S2†).32 This indicates that when deprotonation and oxidation take place, the two adjacent hydroxyl groups on LH4 become CO and C–O−. The resulting p-quinoid structure with a total of two negative charges balances Cu(II) to yield a neutral CP. One Cu(II) is bridged by two ligands L and the coordinating CO (or C–O−) groups are at trans-positions, which allow the formation of 1D linear metal–organic chain. The 1D chains pack closely to form a layered structure without any solvent molecules in between (Fig. 1b). Multiple supramolecular interactions such as CN⋯π, CN⋯H, C–H⋯π and π⋯π are observed. The measured distances from the nitrogen atom (nitrile group) to the centre of the adjacent ligand L (CN⋯π, Fig. 1c, green dotted line) and the hydrogen atoms of the pyridine molecule (CN⋯H, Fig. 1c, red dotted lines) are 3.59 and 2.54 Å, respectively. The distances between the centroid of ligand L and the closest hydrogens of pyridine molecules (C–H⋯π, Fig. 1c, blue dotted lines) are 3.65–3.85 Å. The distance of π⋯π interaction between pyridine molecules is 4.07 Å (Fig. 1c, magenta dotted lines). These weak bonds, whose distances are similar to those of π-stacking observed in graphite, a common anode material for LIBs, potentially provide a flexible network for Li+ to be transported three-dimensionally (Fig. 1d).38,39
O of the benzoquinoid ligand, while the spectrum of LH4 shows no sign of the carbonyl groups (Fig. S2†).32 This indicates that when deprotonation and oxidation take place, the two adjacent hydroxyl groups on LH4 become CO and C–O−. The resulting p-quinoid structure with a total of two negative charges balances Cu(II) to yield a neutral CP. One Cu(II) is bridged by two ligands L and the coordinating CO (or C–O−) groups are at trans-positions, which allow the formation of 1D linear metal–organic chain. The 1D chains pack closely to form a layered structure without any solvent molecules in between (Fig. 1b). Multiple supramolecular interactions such as CN⋯π, CN⋯H, C–H⋯π and π⋯π are observed. The measured distances from the nitrogen atom (nitrile group) to the centre of the adjacent ligand L (CN⋯π, Fig. 1c, green dotted line) and the hydrogen atoms of the pyridine molecule (CN⋯H, Fig. 1c, red dotted lines) are 3.59 and 2.54 Å, respectively. The distances between the centroid of ligand L and the closest hydrogens of pyridine molecules (C–H⋯π, Fig. 1c, blue dotted lines) are 3.65–3.85 Å. The distance of π⋯π interaction between pyridine molecules is 4.07 Å (Fig. 1c, magenta dotted lines). These weak bonds, whose distances are similar to those of π-stacking observed in graphite, a common anode material for LIBs, potentially provide a flexible network for Li+ to be transported three-dimensionally (Fig. 1d).38,39
Electrochemical performance
A few redox-active benzoquinoid-based CPs or MOFs have been investigated as LIB cathode materials; however, their battery performance are still unsatisfactory.26,31,32 In this work, the two electron-withdrawing nitrile groups in the redox-active benzoquinoid ligand L can enhance the charge delocalization, leading to a relatively high redox potential required for good cathodes.40 The benzoquinoid ligand L in [CuL(Py)2]n can reversibly undergo a two-electron transfer redox reaction.32,41 Nitrile-based compounds have also been demonstrated as high-performance battery electrolyte due to their unique properties, such as high anodic oxidization potential, high dielectric constant and favorable interactions with Li ions.42 Combining the abovementioned features with the flexible structure linked by multiple supramolecular interactions, [CuL(Py)2]n has great potential as efficient platform for Li-ion storage and transport.[CuL(Py)2]n was used as cathode material with Li metal as anode and 1 M LiPF6 in 1:1 v/v ethylene carbonate (EC)/dimethyl carbonate (DMC) as electrolyte. Fig. 2a displays the discharge–charge profile of [CuL(Py)2]n (active material:Ketjen black:polyvinylidene difluoride (PVDF) = 3:6:1; active mass loading = ∼1 mg cm−2) at 0.25C (reaction with 4 Li in 4 h, ∼65 mA g−1). During discharge, a very short plateau is observed at ∼3.2 V vs. Li/Li+, followed by two more plateaus at 2.4 and 1.9 V with a discharge capacity of 255 mA h g−1. During charge, three plateaus can be observed at 2.1, 2.8 and 3.3 V. The positions of these plateaus are in good agreement with cyclic voltammogram (CV) of the CP in Fig. 2b. The Cu redox couple is at 3.1/3.3 V and the redox centres in the quinone-based organic linker are responsible for the other two major pairs of anodic and cathodic peaks in the CV.32 This reveals the redox reactions on both the metal centre and organic moiety of the CP, which enable a multi-electron process, leading to a high specific capacity (one electron transfer corresponds to 65.4 mA h g−1).
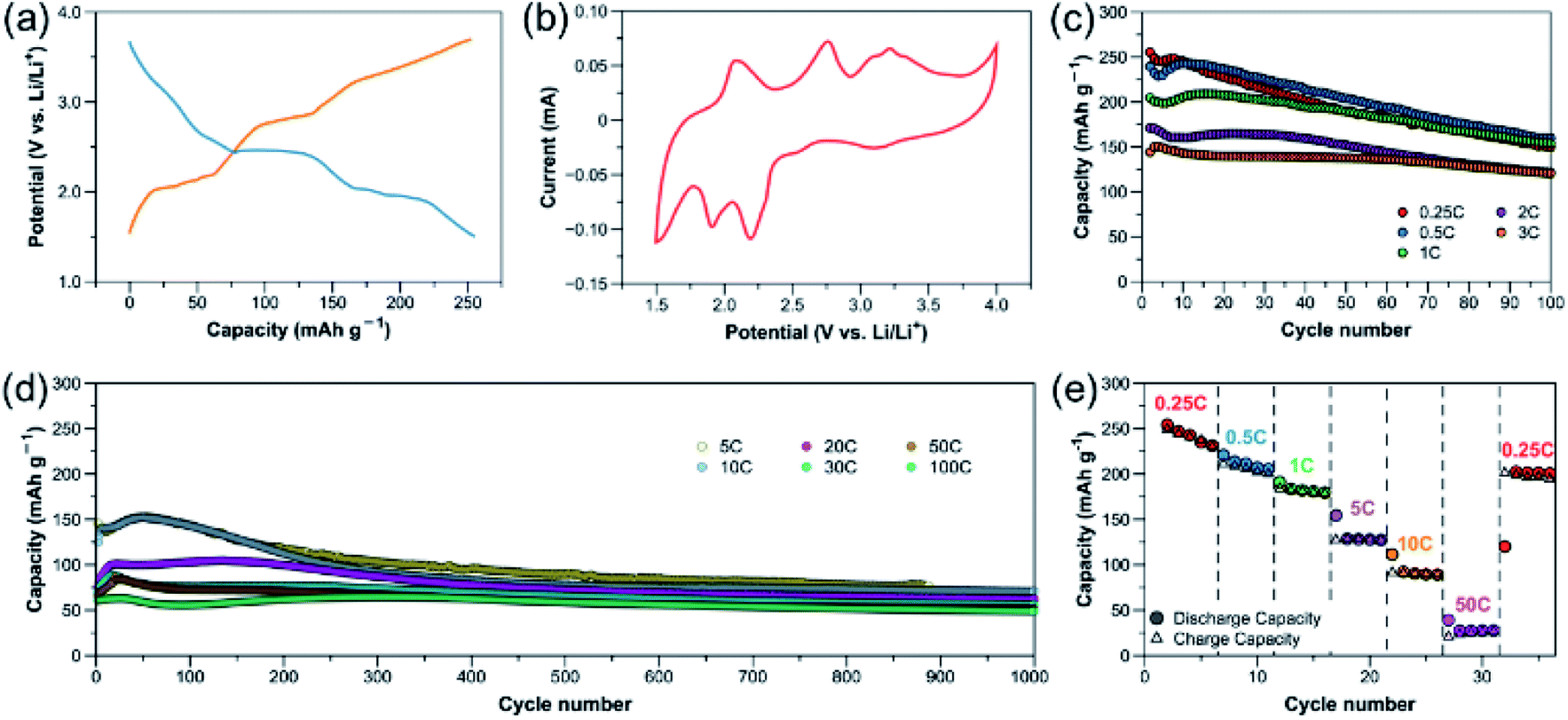 | ||
| Fig. 2 Electrochemical properties of [CuL(Py)2]n cathode: (a) discharge–charge profile at 0.25C (∼65 mA g−1). (b) CV plot with a scan rate of 0.5 mV s−1. (c) Capacity retention at rates of 0.25C to 3C (∼785 mA g−1). (d) Capacity retention at rates of 5C (∼1308 mA g−1) to 100C (∼26162 mA g−1). (e) Rate capability at various current densities. | ||
The capacity retention at different rates is shown in Fig. 2c and d. At lower rates of 0.25C and 0.5C (∼131 mA g−1), the initial capacities obtained are close to the theoretical capacity of 262 mA h g−1; however, within 100 cycles, an obvious capacity drop is observed. This phenomenon was also reported on [CuL(DMF)2]n (with the highest reported current density of 120 mA g−1) as a result of structural decomposition with Cu(II) being reduced to Cu(0).32 At higher rates, the initial capacities decrease due to kinetic effects. At 3C (∼785 mA g−1), the compound delivers an initial capacity of 145 mA h g−1, with a slight drop within 100 cycles. This capacity is comparable to those of the high-performance MOF electrodes (Cu-THQ and Cu-BHT) previously reported at a similar current density.26,27
To further test the rate performance and long-term stability of the CP, the cells were cycled at even higher rates, as shown in Fig. 2d. At 5C (∼1308 mA g−1) and 10C (∼2616 mA g−1), the retention trends are similar, starting with the capacities of initial cycles at ∼145 mA h g−1 which, after 100 cycles, slightly drops to 144 and 143 mA h g−1 before stabilizing at ∼90 and 80 mA h g−1 for up to 1000 cycles, respectively. Surprisingly, the close-packed [CuL(Py)2]n cathode can further undergo extremely high rates of up to 100C (∼26162 mA g−1). This result is contradicting to the conventional perception on electrode design that the high porosity of active material is essential for efficient Li-ion transport and high rate capability.21,24,43 At higher rates of 20C to 100C, the CP apparently shows excellent capacity retention compared to the lower rates, delivering initial specific capacities of 75, 70, 66 and 61 mA h g−1, with retention values of 82.2, 80.3, 78.1 and 80.5% after 1000 cycles for 20C (∼5232 mA g−1), 30C (∼7848 mA g−1), 50C (∼13081 mA g−1) and 100C (∼26162 mA g−1), respectively. It is worth noting that at such high cycling rates of 20–100C, the capacity contribution from the conducting additive is small (∼10–20 mA h g−1, Fig. S7†). Based on these results, it is clear that [CuL(Py)2]n electrodes can undergo significantly higher current densities and exhibit much better capacity retention than [CuL(DMF)2]n, which indicates superior structural stability due to more supramolecular interactions and better packing of [CuL(Py)2]n. This CP is found to be remarkably stable for over 1000 cycles, providing a power density as high as 60171 W kg−1 at 100C, which is, to the best of our knowledge, among the highest ever reported for metal–organic-based cathode materials. The rate capability was also tested by varying the current density from 0.25C to 50C, and back to 0.25C. A relatively reversible rate capability curve is obtained as shown in Fig. 2e. These results indicate that the [CuL(Py)2]n electrodes feature exceptional electrochemical performance, with high cycling stability especially at high rates. Table S2† summarizes the electrochemical properties of the CP in comparison with the other metal–organic cathodes reported for LIBs. The CV measurements (Fig. S27†) reveal that as the rate is higher, a higher proportion of capacitive contribution is observed on the overall capacity, which confirms an excellent rate performance of [CuL(Py)2]n cathode.
Redox mechanism at various rates
To elucidate the change in the redox centres of [CuL(Py)2]n during discharge–charge cycles, X-ray photoelectron spectroscopy (XPS) was carried out on the CP electrodes (7:2:1 electrode ratio used for all mechanistic studies). At 20 mA g−1 (∼0.08C), the deconvolution of the high-resolution Cu 2p spectra of electrodes (Fig. S10†) shows gradual reduction of Cu species to Cu(I) and Cu(0) during discharge and oxidation during charge.32,44 The CP electrodes cycled at three additional rates, 0.5C, 5C and 50C, were also analyzed. The XPS spectra in Fig. 3 reveal that at the end of discharge (1.5 V), both Cu(0) and Cu(I) are still present; however, going from low (0.5C), to moderate (5C) and high rate (50C), less and less Cu(0) signal is observed with more and more being Cu(I). Interestingly, only a very small amount of Cu(0) appears at 50C. These results imply that a full reduction of Cu(II) to Cu(0) tends to take place only at a low cycling rate, which accounts for the higher number of electron transfers observed in the electrochemical study. Subsequently, on charge up to 4.0 V, the amount of Cu(0) significantly decreases with Cu(I) being the main component for all the rates. These XPS spectra reveal the different levels of Cu reduction at various discharge/charge rates. The increase in formation of Cu(0) leads to decomposition of the material, which is responsible for more severe capacity decay at low rates.
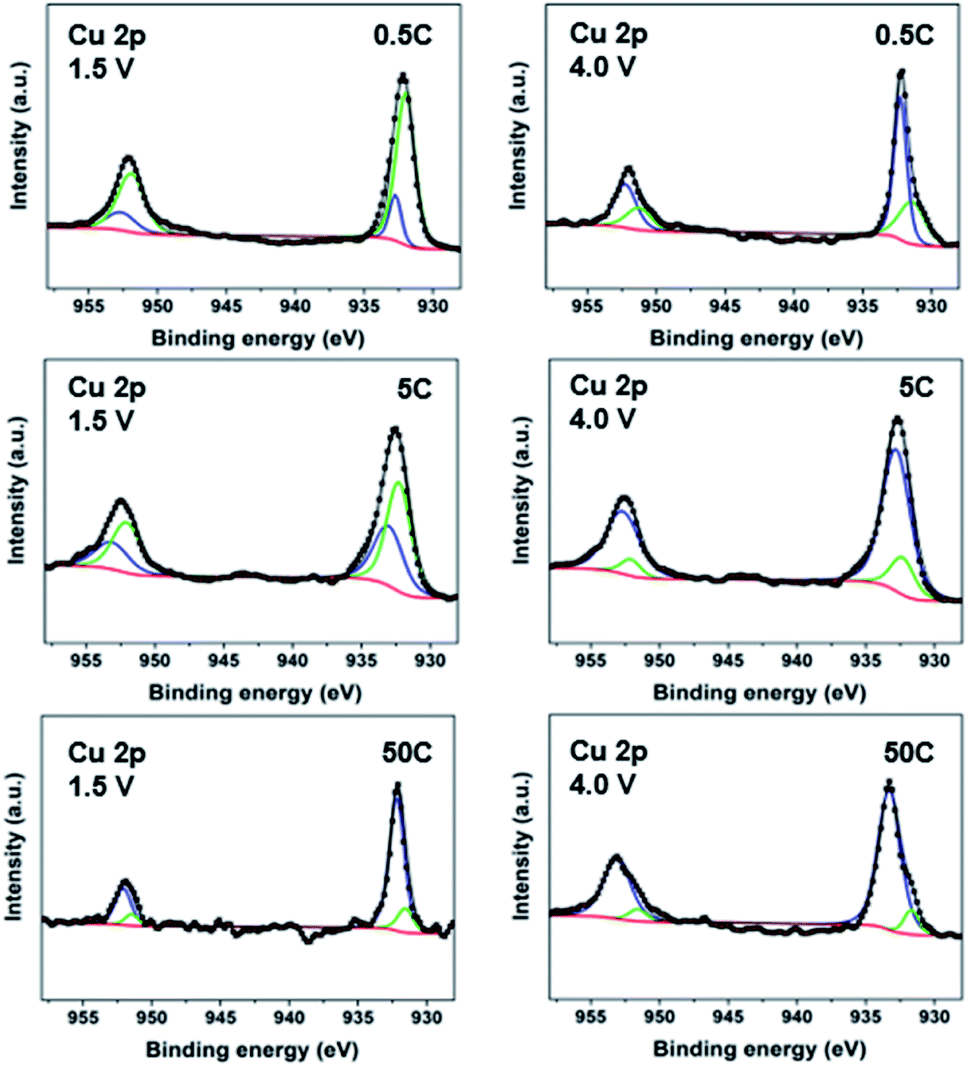 | ||
| Fig. 3 Deconvolution of Cu 2p3/2 and 2p1/2 XPS spectra of [CuL(Py)2]n electrodes at various rates. 0.5C (top), 5C (middle) and 50C (bottom) at end of discharge (1.5 V, left) and end of charge (4.0 V, right) (green: Cu(0); blue: Cu(I)). | ||
The deconvolution of the C 1s XPS spectra also reveals the redox process of the organic moieties in [CuL(Py)2]n (Fig. S11 and S12†). The peaks attributed to C–C, C–O and CO bonds,32,45 reveal that on discharge, the carbonyl group is transformed into an enol structure, while the reverse process occurs on charge. This behaviour confirms the reversible redox reaction of the organic moieties. Ex situ FT-IR spectra (Fig. S13†) were also collected to further confirm the change at the redox centres of the organic moieties during cycling. The data are in good agreement with the C 1s XPS mentioned above.
To closely monitor the valence state change and the local environment of Cu during battery cycling, Cu K-edge X-ray absorption spectroscopy (XAS) were performed on the CP electrodes. In situ X-ray absorption near edge structure (XANES) measurement was carried out during discharge (Fig. 4a) and charge (Fig. 4b) at 20 mA g−1 (∼0.08C). The signal of the pristine electrode confirms the Cu(II) valence state. Interestingly, as soon as the cell is discharged, the characteristic Cu(II) signal immediately disappears (within seconds) even when the amount of charge transfer is less than one-electron reduction. This phenomenon reflects that the electrons are delocalized in the coordination networks of the CP presumably due to the nature of the non-innocent ligand L.46,47 During discharge to 1.5 V (Fig. 4a), the pre-edge peak at 8982 eV gradually emerges and the edge peak slowly shifts to a lower energy indicating the reduction of Cu(II) species.32,48 On charge to 4.0 V (Fig. 4b), a reverse process occurs with the curves shifting back very close to the original position of pristine electrode. Ex situ Cu K-edge extended X-ray absorption fine structure (EXAFS) patterns are shown in Fig. S15.† It is obvious that when the pristine CP electrode is discharged down to 1.5 V, the intensity of Cu–O bond becomes weaker while the Cu–Cu bond becomes stronger. The curve fitting of the EXAFS patterns in Fig. S16† shows the change in coordination number of Cu–O and Cu–Cu, confirming Cu–O bond dissociation and formation during discharge and charge, respectively.32 The incomplete oxidation of Cu back to its original valence of 2+ observed in both ex situ and in situ XAS measurements is in good agreement with the Cu XPS data mentioned above. This incomplete oxidation of Cu redox centres during charge may be responsible for the capacity loss of CP electrodes at low rates.
 | ||
| Fig. 4 XAS of [CuL(Py)2]n electrodes: (a) and (b) in situ Cu K-edge XANES patterns during discharge and charge at 20 mA g−1 (∼0.08C), respectively. (c) and (d) Ex situ Cu K-edge EXAFS patterns at three different rates (0.5C, 5C and 50C) at end of discharge (1.5 V) and end of charge (4.0 V), respectively (with Cu foil, Cu2O and CuO as references). | ||
Ex situ XAS experiments were also performed on the CP electrodes at different rates. EXAFS patterns of the electrodes cycled at 0.5C, 5C and 50C at the end of discharge, 1.5 V, and the end of charge, 4.0 V, are shown in Fig. 4c and d, respectively. It is clear that the rate plays a major role on the degree of redox activity at the metal centres. On discharge (Fig. 4c), the lower the rate, the higher the intensity of Cu–Cu bond. It is worth noting that at the rate of 50C, Cu–Cu bond is barely observed in the EXAFS pattern. On charge to 4.0 V, the intensity of Cu–Cu bond is found to be lower than that at the end of discharge (1.5 V) at all rates, which suggests that some Cu–Cu bonds are dissociated with an oxidation of Cu(0) returning to Cu–O bonds, especially at lower rates. Comparing the pattern at 50C (Fig. 4d) to that of pristine, it is apparent that there is a high degree of similarity between the two, suggesting that the 1D chains of [CuL(Py)2]n are likely to remain intact at such high rate of 50C. It is obvious that less formation of Cu–Cu bonds allows better recovery of Cu–O bonds at the end of charge. Thus, the metal–ligand coordination of [CuL(Py)2]n is more likely to be maintained. Based on our results, at high discharge/charge rate with a small contribution from Cu, the organic redox centres which are believed to have better kinetics and thus high reversibility will then have a larger contribution to the capacity of the CP, allowing the material to have high rate performance and excellent capacity retention. At a low cycling rate, the full redox reactions at both the metal and organic ligand take place, resulting in a high specific capacity from [CuL(Py)2]n. However, a deep reduction of Cu species which may be sluggish and cause deformation of the CP chains, leads to poor reversibility. Overall, structural integrity is the main factor allowing better capacity retention of the CP.
Role of supramolecular interactions on rate performance
The abovementioned mechanistic studies provide insight into why [CuL(Py)2]n displays better long-term cycling stability at high rates than at low rates, but its ability to undergo cycling rate as high as 100C or ∼26162 mA g−1 which is much higher than that of [CuL(DMF)2]n is still highly intriguing. It is, therefore, essential to elucidate the distinct structure–performance relationship of [CuL(Py)2]n during electrochemical cycling. The current trend of research on metal–organic electrode materials focuses on high porosity for faster ion diffusion.21 However, those large pores usually allow PF6−, the counteranion of Li+ in LiPF6, together with the solvent molecules to co-insert into the pores, which may affect Li-ion diffusion during cell cycling. This phenomenon has been observed in some MOF electrodes reported previously.26,46 In this work, due to the close-packing of 1D [CuL(Py)2]n chains (according to the single-crystal XRD), there is not enough space for PF6− co-insertion. In order to confirm this, we performed the ex situ FT-IR experiments to verify the absence of PF6− at different cycling rates and discharge–charge states.26 The disassembled electrodes were washed with DMC and then dried in vacuum to remove residual solvents. The results (Fig. S23†) show that PF6− signal (∼840 cm−1) can only be observed in electrodes that were wetted with the LiPF6 electrolyte.26 Once the electrodes were washed and the electrolyte was removed from the electrode surface, PF6− signal can no longer be observed at any cycling rate and voltage position.26 This provides evidence for the absence of PF6− anions in the CP structure during electrochemical cycling. Due to the limited space between the 1D chains, the insertion of bulky solvated Li ions and electrolyte solvent molecules into the CP can also be ruled out. It has been indicated that the discrepancy between the kinetics and diffusion rates of cations and anions usually hampers the battery performance of porous electrode materials at high rates, as they allow both cations and anions to diffuse into the channels.31,46 The supramolecular networks between close-packed chains of [CuL(Py)2]n can provide flexible yet minimal space for efficient Li+ transport while excluding PF6− anions and solvent molecules, allowing good rate performance. This phenomenon is similar to that found in the traditional inorganic-based cathodes currently used in commercial LIBs (e.g., LiCoO2, LiNi1/3Mn1/3Co1/3O2, LiNi0.8Co0.15Al0.05O2, and LiFePO4) which also possess close-packed structures.4,49 Without co-intercalation of anions and solvent molecules, Li ions are able to transport freely in the structure. Li-ion diffusion rate which is known to be much slower than the electron transfer rate generally determines the rate capability of electrode materials.40 Therefore, to achieve high rate capability, efficient Li+ transport within the structure is found to be a more important factor than intrinsic electronic conductivity. It is worth noting that the absence of large anions and solvent molecules co-inserted into the structure also helps with overall structural integrity during long-term cycling.
Li-ion insertion and optimal diffusion pathways stabilized by supramolecular interactions
To gain insight into the significance of supramolecular interactions, we conducted the density functional theory (DFT) simulation on the crystal structure of [CuL(Py)2]n. As mentioned above, at a low cycling rate, a high capacity close to four-electron transfer (two from the metal centre and two from the organic ligand L) may be achieved in the CP. However, the full reduction of Cu(II) to Cu(0) has been found not to be completely reversible, which causes structural decomposition and capacity decay. According to high-resolution transmission electron microscopy (HR-TEM), at the rate of 5C, there are no Cu(0) particles formed in the electrode during cycling (Fig. S20c†), suggesting that at this rate, the overall lowest oxidation state of Cu is 1+. The average reversible capacity within the first 100 cycles at 5C is ∼150 mA h g−1 (Fig. 2d), corresponding to over two-electron transfer after subtracting the capacity from the conductive carbon. As a result, we chose to simulate the insertion of three Li ions into the CP and an optimized structure of [CuL(Py)2Li3]n was obtained as shown in Fig. 5a.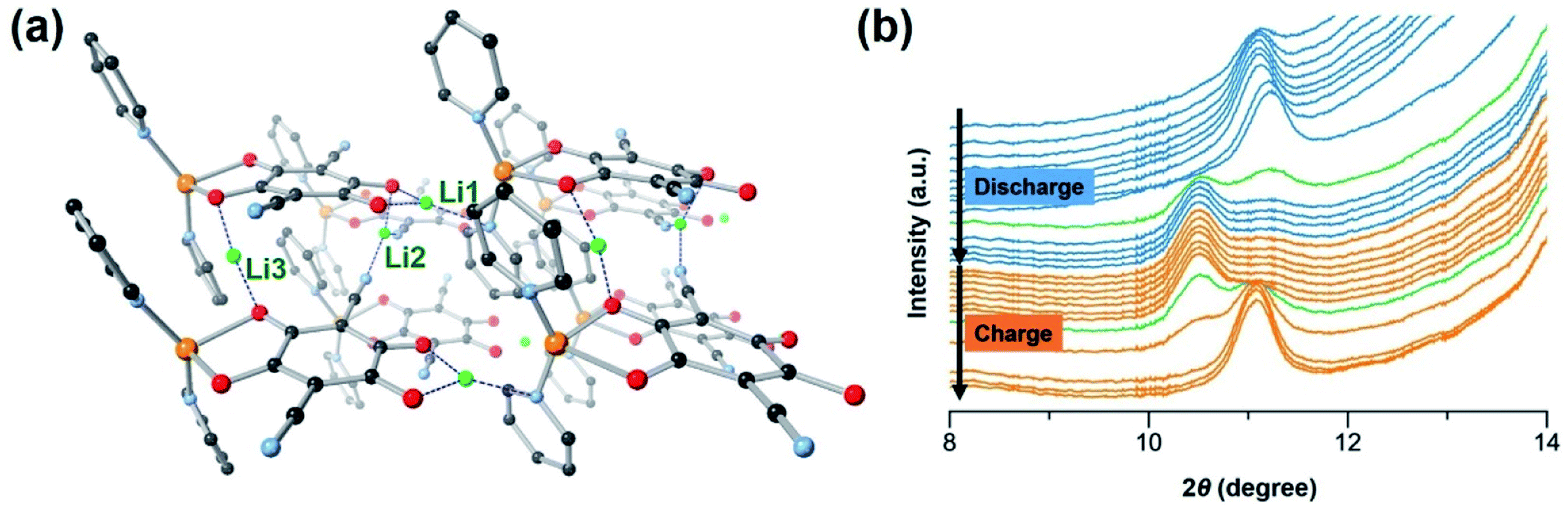 | ||
| Fig. 5 (a) Computational simulation of lithiated CP [CuL(Py)2Li3]n showing the DFT optimized structure. Colours of atoms: orange: Cu(I); grey: C; red: O; cyan: N; green: Li; H is omitted for clarity. (b) In situ synchrotron PXRD patterns of CP electrode collected during the first discharge–charge cycle at current density of 120 mA g−1 at a voltage between 1.5 V and 4.0 V. | ||
Starting from pristine material, Li insertion is accompanied by the reduction of the CP. The octahedrally coordinated Cu(II) centre is reduced to tetrahedral Cu(I), with one Li+ (denoted as Li1 in Fig. 5a) inserted right between ligand L and Cu(I) of the 1D chain. Li1 is stabilized by the surrounding oxygen and nitrogen atoms from L and pyridine which is axially coordinated to the Cu(I) (blue dashed lines in Fig. 5a), respectively.50 At high cycling rates, the four-coordinated Cu(I) may not be easily accessible by another Li+. Thus, further reduction of Cu(I) to Cu(0) is believed to have poor kinetics and is unlikely to take place, especially at a high discharge rate. As a consequence, the structural integrity of CP and capacity retention are better at high rates than those at low rates. Reduction of the CP during discharge also takes place at the organic ligand L. The first reduction of ligand L occurs on the CO redox centre next to Li1, with an insertion of another Li+ (denoted as Li2 in Fig. 5a) into the space between the CP chains. Li2 is stabilized by one oxygen atom from the ligand and two nitrogen atoms from the cyano groups of two neighbouring ligands. This simulation result is not surprising because cyano-based compounds have been widely studied as battery electrolytes due to their favorable interactions with Li ions.42 The second reduction of ligand L takes place on the other CO which is at the para-position of the first CO being reduced; as a result, another Li+ (denoted as Li3 in Fig. 5a) inserts into the space between the CP chains. Li3 is stabilized by two oxygen atoms, one from CO being reduced and one from the neighboring ligand L. The distances between any two inserted Li ions in the simulated [CuL(Py)2Li3]n structure are within 2.99–5.84 Å, ideal for Li+ hopping (Fig. S24†).51 Thus, the 1D metal–ligand chains of [CuL(Py)2]n with supramolecular interactions (CN⋯π, CN⋯H, C–H⋯π and π⋯π) provide an excellent platform with preferred hopping sites for Li ions to transport efficiently in three dimensions (Fig. 1d).
To investigate the change of the CP structure during the discharge–charge cycle, in situ synchrotron powder X-ray diffraction (PXRD) was performed at a current density of 120 mA g−1 between 1.5 V and 4.0 V (Fig. 5b). During cell cycling, the diffraction patterns show an obvious change in the position of the first peak. As the discharge progresses, the peak (2θ = 11.1°) which corresponds to the Miller index plane (hkl) = (001) of [CuL(Py)2]n slightly shifts to a lower angle when compared to that of the pristine material, indicating an expansion of the 1D structure due to the interaction with Li. Upon charging, the peak shifts back close to the original position when Li is deintercalated.28 These results are in agreement with our simulations, indicating the appearance of the first peak (010) at a lower angle for the lithiated [CuL(Py)2Li3]n structure than the first peak of the parent material (Fig. S25†). The peak shifting observed during cycling confirms the reversible lithiation processes and structural flexibility of [CuL(Py)2]n caused by the supramolecular interactions.
Conclusions
High porosity has long been utilized as a strategy for facile Li-ion transport. While this may be possible, co-insertion of bulky and slow-diffusing counteranions and solvent molecules also tends to take place, potentially hampering Li-ion diffusion. Herein, we propose the concept of close-packed metal–organic cathode stabilized by multiple supramolecular interactions as a viable solution for exceptional electrochemical performance. Via modular design, changing only one structural component of the CP (DMF replaced by Py), which introduces more weak interactions and more stable packing, can significantly improve electrochemical performance especially at high rates. Our studies reveal that such weak bonds are the key to flexible host lattice which only allows desolvated Li+ to intercalate, while they also stabilize the inserted Li in the preferred hopping sites, creating optimal diffusion paths. To the best of our knowledge, the performance of CP is found to be among the best for all metal–organic-based cathode materials ever reported and also comparable to that of inorganic cathodes currently used in commercial LIBs (Fig. S28†). Although the CP demonstrated here still has an issue with Cu deep reduction affecting cycling stability at low rates, our concept has been proved excellent at high rates where the issue from the metal no longer exists. Our design strategy combined with mechanistic and structural insights is now being used to expand the series of high-performance metal–organic-based cathodes (with more stable metal–ligand coordination), which will be reported in due course. With this, next-generation energy storage systems with unprecedentedly high energy/power density can certainly be achieved by crystal engineering.Author contributions
C.-H. C. designed and synthesized [CuL(Py)2]n, and performed all the material characterization. A.-C. L. performed all the electrochemical and mechanistic studies, with intellectual input from I. P., W. K. and T.-H. C. I. P. synthesized the compound LH4. J.-L. C. and Y.-C. C. collected and analyzed the XAS data, Y.-C. C. and B.-H. C. designed and performed the in situ synchrotron PXRD experiments, and J.-C. L. conducted the XPS experiments. Y.-A. L. performed part of the electrochemical testing and surface characterization. The computational simulation was performed by A. S. I. The interpretation of experiments was done by W. K. and T.-H. C. W. K. and T.-H. C. designed the study and wrote the manuscript, incorporating the input from all the other authors.Conflicts of interest
Patents related to this research have been filed by the authors (W. K., T.-H. C., A.-C. L. and C.-H. C.) and National Cheng Kung University. Taiwan patent entitled “Cathode Material and Fabricating Method Thereof, and Lithium-Ion Battery”, No. I 738593, dated 1 September 2021. US Application entitled “Coordination-Polymer Cathode Material and Fabricating Method Thereof, and Lithium-Ion Battery”, No. 17233486, filed on 18 April 2021.Acknowledgements
This work was supported by the Ministry of Science and Technology (MOST) of Taiwan under grant MOST 108-2113-M-006-016 (to T.-H. C.) and the Young Scholar Fellowship Program MOST 108-2636-E-006-001 (to W. K.). This work was also financially supported by the Hierarchical Green-Energy Materials (Hi-GEM) Research Center, from the Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education (MOE) and MOST (MOST 109-2634-F-006-020) in Taiwan (to W. K.). This research was supported in part by High Education Sprout Project, Ministry of Education of the Headquarters of University Advancement at National Cheng Kung University (NCKU) (to T.-H. C. and W. K.). Simulation work by A. I. was supported as part of the Fluid Interface Reactions, Structures and Transport (FIRST) Center, an Energy Frontier Research Center funded by the U.S. Department of Energy (DOE), Office of Science and Office of Basic Energy Sciences. I. P. was supported by the U.S. DOE, Office of Science, Office of Basic Energy Sciences, Materials Sciences and Engineering Division under contract number DE-AC05-00OR22725. This research used resources of the Computer and Data Environment for Science (CADES) at Oak Ridge National Laboratory, managed by UT-Battelle, LLC for the U.S. DOE under contract DE-AC05-00OR22725. The authors gratefully acknowledge the use of ESCA000200 and EM000800 of MOST 108-2731-M-006-001 belonging to the Core Facility Center of NCKU. The authors are indebted to Dr Ting-Shen Kuo (National Taiwan Normal University) for the collection and the refinement of crystallographic data.Notes and references
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- A. Manthiram, ACS Cent. Sci., 2017, 3, 1063–1069 CrossRef CAS PubMed.
- A. Manthiram, Nat. Commun., 2020, 11, 1550 CrossRef CAS PubMed.
- F. Wu, J. Maier and Y. Yu, Chem. Soc. Rev., 2020, 49, 1569–1614 RSC.
- M. H. Braga, N. S. Grundish, A. J. Murchison and J. B. Goodenough, Energy Environ. Sci., 2017, 10, 331–336 RSC.
- P. Poizot, J. Gaubicher, S. Renault, L. Dubois, Y. Liang and Y. Yao, Chem. Rev., 2020, 120, 6490–6557 CrossRef CAS PubMed.
- M. E. Bhosale, S. Chae, J. M. Kim and J.-Y. Choi, J. Mater. Chem. A, 2018, 6, 19885–19911 RSC.
- G. S. Vadehra, R. P. Maloney, M. A. Garcia-Garibay and B. Dunn, Chem. Mater., 2014, 26, 7151–7157 CrossRef CAS.
- J. Lee, H. Kim and M. J. Park, Chem. Mater., 2016, 28, 2408–2416 CrossRef CAS.
- H. Wang, C.-J. Yao, H.-J. Nie, K.-Z. Wang, Y.-W. Zhong, P. Chen, S. Mei and Q. Zhang, J. Mater. Chem. A, 2020, 8, 11906–11922 RSC.
- Y. Liang, P. Zhang, S. Yang, Z. Tao and J. Chen, Adv. Energy Mater., 2013, 3, 600–605 CrossRef CAS.
- Y. Lu, X. Hou, L. Miao, L. Li, R. Shi, L. Liu and J. Chen, Angew. Chem., Int. Ed., 2019, 58, 7020–7024 CrossRef CAS PubMed.
- C. Cui, X. Ji, P.-F. Wang, G.-L. Xu, L. Chen, J. Chen, H. Kim, Y. Ren, F. Chen, C. Yang, X. Fan, C. Luo, K. Amine and C. Wang, ACS Energy Lett., 2020, 5, 224–231 CrossRef CAS.
- Z. Lei, X. Chen, W. Sun, Y. Zhang and Y. Wang, Adv. Energy Mater., 2019, 9, 1801010 CrossRef.
- C. Peng, G.-H. Ning, J. Su, G. Zhong, W. Tang, B. Tian, C. Su, D. Yu, L. Zu, J. Yang, M.-F. Ng, Y.-S. Hu, Y. Yang, M. Armand and K. P. Loh, Nat. Energy, 2017, 2, 17074 CrossRef CAS.
- S. Xu, G. Wang, B. P. Biswal, M. Addicoat, S. Paasch, W. Sheng, X. Zhuang, E. Brunner, T. Heine, R. Berger and X. Feng, Angew. Chem., Int. Ed., 2019, 58, 849–853 CrossRef CAS PubMed.
- F.-Y. Chou, J.-C. Tang, H.-Y. Lee, J.-C. Lee, S. Ratchahat, T.-H. Chen and W. Kaveevivitchai, ACS Appl. Energy Mater., 2020, 3, 11300–11306 CrossRef CAS.
- T. Sun, J. Xie, W. Guo, D.-S. Li and Q. Zhang, Adv. Energy Mater., 2020, 10, 1904199 CrossRef CAS.
- X. Chen, Y. Li, L. Wang, Y. Xu, A. Nie, Q. Li, F. Wu, W. Sun, X. Zhang, R. Vajtai, P. M. Ajayan, L. Chen and Y. Wang, Adv. Mater., 2019, 31, 1901640 CrossRef PubMed.
- Y. Liang and Y. Yao, Joule, 2018, 2, 1690–1706 CrossRef CAS.
- Z. Wu, J. Xie, Z. J. Xu, S. Zhang and Q. Zhang, J. Mater. Chem. A, 2019, 7, 4259–4290 RSC.
- S. Kitagawa, R. Kitaura and S.-i. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed.
- J. Liu, X. Song, T. Zhang, S. Liu, H. Wen and L. Chen, Angew. Chem., Int. Ed., 2021, 60, 5612–5624 CrossRef CAS PubMed.
- S. Gu, Z. Bai, S. Majumder, B. Huang and G. Chen, J. Power Sources, 2019, 429, 22–29 CrossRef CAS.
- L. Kong, M. Zhong, W. Shuang, Y. Xu and X.-H. Bu, Chem. Soc. Rev., 2020, 49, 2378–2407 RSC.
- Q. Jiang, P. Xiong, J. Liu, Z. Xie, Q. Wang, X.-Q. Yang, E. Hu, Y. Cao, J. Sun, Y. Xu and L. Chen, Angew. Chem., Int. Ed., 2020, 59, 5273–5277 CrossRef CAS PubMed.
- Z. Wu, D. Adekoya, X. Huang, M. J. Kiefel, J. Xie, W. Xu, Q. Zhang, D. Zhu and S. Zhang, ACS Nano, 2020, 14, 12016–12026 CrossRef CAS PubMed.
- Z. Zhang, H. Yoshikawa and K. Awaga, J. Am. Chem. Soc., 2014, 136, 16112–16115 CrossRef CAS PubMed.
- W. Kaveevivitchai and A. J. Jacobson, J. Power Sources, 2015, 278, 265–273 CrossRef CAS.
- L. Liu, X. Chen, W. Lu, A. Han and Y. Qiao, Phys. Rev. Lett., 2009, 102, 184501 CrossRef PubMed.
- M. E. Ziebel, C. A. Gaggioli, A. B. Turkiewicz, W. Ryu, L. Gagliardi and J. R. Long, J. Am. Chem. Soc., 2020, 142, 2653–2664 CrossRef CAS PubMed.
- C.-H. Chang, A.-C. Li, I. Popovs, W. Kaveevivitchai, J.-L. Chen, K.-C. Chou, T.-S. Kuo and T.-H. Chen, J. Mater. Chem. A, 2019, 7, 23770–23774 RSC.
- T.-H. Chen, I. Popov, W. Kaveevivitchai, Y.-C. Chuang, Y.-S. Chen, O. Daugulis, A. J. Jacobson and O. Š. Miljanić, Nat. Commun., 2014, 5, 5131 CrossRef CAS PubMed.
- M. I. Hashim, H. T. M. Le, T.-H. Chen, Y.-S. Chen, O. Daugulis, C.-W. Hsu, A. J. Jacobson, W. Kaveevivitchai, X. Liang, T. Makarenko, O. Š. Miljanić, I. Popovs, H. V. Tran, X. Wang, C.-H. Wu and J. I. Wu, J. Am. Chem. Soc., 2018, 140, 6014–6026 CrossRef CAS PubMed.
- E. Vorobyeva, F. Lissel, M. Salanne and M. R. Lukatskaya, ACS Nano, 2021, 15, 15422–15428 CrossRef CAS PubMed.
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS PubMed.
- M.-S. Wu, N. T. H. Luu, T.-H. Chen, H. Lyu, T.-W. Huang, S. Dai, X.-G. Sun, A. S. Ivanov, J.-C. Lee, I. Popovs and W. Kaveevivitchai, Adv. Energy Mater., 2021, 11, 2100330 CrossRef CAS.
- K. Nakashima, T. Shimizu, Y. Kamakura, A. Hinokimoto, Y. Kitagawa, H. Yoshikawa and D. Tanaka, Chem. Sci., 2020, 11, 37–43 RSC.
- L. Sieuw, A. Jouhara, É. Quarez, C. Auger, J.-F. Gohy, P. Poizot and A. Vlad, Chem. Sci., 2019, 10, 418–426 RSC.
- M. Amores, K. Wada, K. Sakaushi and H. Nishihara, J. Phys. Chem. C, 2020, 124, 9215–9224 CrossRef CAS.
- W. Kaveevivitchai, X. Wang, L. Liu and A. J. Jacobson, Inorg. Chem., 2015, 54, 1822–1828 CrossRef CAS PubMed.
- P. Hu, J. Chai, Y. Duan, Z. Liu, G. Cui and L. Chen, J. Mater. Chem. A, 2016, 4, 10070–10083 RSC.
- R. Du, Y. Wu, Y. Yang, T. Zhai, T. Zhou, Q. Shang, L. Zhu, C. Shang and Z. Guo, Adv. Energy Mater., 2021, 11, 2100154 CrossRef CAS.
- Z. Jin, C. Liu, K. Qi and X. Cui, Sci. Rep., 2017, 7, 39695 CrossRef CAS PubMed.
- J. Wang, Q. Deng, M. Li, K. Jiang, J. Zhang, Z. Hu and J. Chu, Sci. Rep., 2017, 7, 8903 CrossRef PubMed.
- K. Wada, K. Sakaushi, S. Sasaki and H. Nishihara, Angew. Chem., Int. Ed., 2018, 57, 8886–8890 CrossRef CAS PubMed.
- W. Kaim, Inorg. Chem., 2011, 50, 9752–9765 CrossRef CAS PubMed.
- H. H. Lee, J. B. Lee, Y. Park, K. H. Park, M. S. Okyay, D.-S. Shin, S. Kim, J. Park, N. Park, B.-K. An, Y. S. Jung, H.-W. Lee, K. T. Lee and S. Y. Hong, ACS Appl. Mater. Interfaces, 2018, 10, 22110–22118 CrossRef CAS PubMed.
- H. Ji, J. Wu, Z. Cai, J. Liu, D.-H. Kwon, H. Kim, A. Urban, J. K. Papp, E. Foley, Y. Tian, M. Balasubramanian, H. Kim, R. J. Clément, B. D. McCloskey, W. Yang and G. Ceder, Nat. Energy, 2020, 5, 213–221 CrossRef CAS.
- J. Du, Y. Li, H. Liu, W. Shi, L. V. Moskaleva and P. Cheng, ACS Appl. Mater. Interfaces, 2019, 11, 25863–25869 CrossRef CAS.
- K. Dokko, D. Watanabe, Y. Ugata, M. L. Thomas, S. Tsuzuki, W. Shinoda, K. Hashimoto, K. Ueno, Y. Umebayashi and M. Watanabe, J. Phys. Chem. B, 2018, 122, 10736–10745 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Details on the synthesis, characterisation, and electrochemistry. CCDC 2089952. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2ta01463g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |