
An efficient screening strategy towards multifunctional catalysts for the simultaneous electroreduction of NO3−, NO2− and NO to NH3†
Peng
Lv
ae,
Donghai
Wu
a,
Bingling
He
a,
Xue
Li
a,
Rui
Zhu
a,
Gang
Tang
 b,
Zhansheng
Lu
c,
Dongwei
Ma
*ae and
Yu
Jia
*ade
b,
Zhansheng
Lu
c,
Dongwei
Ma
*ae and
Yu
Jia
*ade
aKey Laboratory for Special Functional Materials of Ministry of Education, School of Materials Science and Engineering, Henan University, Kaifeng 475004, China. E-mail: madw@henu.edu.cn; jiayu@henu.edu.cn
bAdvanced Research Institute of Multidisciplinary Science, Beijing Institute of Technology, Beijing 100081, China
cSchool of Physics, Henan Normal University, Xinxiang 453007, China
dInternational Laboratory for Quantum Functional Materials of Henan, School of Physics, Zhengzhou University, Zhengzhou 450001, China
eJoint Center for Theoretical Physics, Center for Topological Functional Materials, Henan University, Kaifeng 475004, China
First published on 21st March 2022
Abstract
Multifunctional electrocatalysts that simultaneously reduce all NOx species (NO3−, NO2− and NO) to NH3 can deliver superior efficiency compared with that for individual species. Herein, by using the first-principles method, an efficient strategy was proposed to screen NOxRR multifunctional electrocatalysts, which focuses on the key protonation process of the essential NORR for the NOxRR. Taking double-atom catalysts (DACs) with transition-metal dimer (M1/M2 = V, Cr, Mn, Fe, Co, Ni, and Cu) embedded N-doped graphene as examples, we showed that the proposed strategy was effective for electrocatalyst screening, and we identified Cu2@NG as the best one, with rather low limiting potentials of −0.36, −0.32, and −0.30 V for the NO3RR, NO2RR, and NORR, respectively. Furthermore, a simple descriptor for evaluating electrocatalytic activity was established. And the physical mechanism in terms of the electronic structures was analyzed for the Cu2 DAC case in view of NO activation. Finally, from the screening test in some 4d and 5d late transition-metal based DACs, the proposed strategy was confirmed to be feasible to find another NOxRR multifunctional electrocatalyst. Our work provides a procedure for efficiently screening multifunctional electrocatalysts to simultaneously reduce all NOx species to synthesize NH3 and sheds light on the atomic mechanisms for NOx electroreduction as well.
1. Introduction
For a long time, the production of valuable ammonia (NH3) mainly has depended on the traditional Haber–Bosch process that occurs at high temperature and pressure and is extremely economically and environmentally unfriendly. Yet, researchers turned to the electrochemical N2 reduction reaction (NRR) mimicking biological nitrogen fixation towards NH3.1–6 Due to the poor NRR performance resulting from the extremely inert N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond, alternatively the electrochemical conversion of other important nitrogenous species, NOx (NO3−, NO2− and NO), came into focus very recently. It turns out that this not only produces NH3 beyond NRR under ambient conditions, but also removes the waste NOx species.7,8 In contrast, this denitrification process in nature reduces the NOx species to N2O by fungi and N2 by bacteria.9
N bond, alternatively the electrochemical conversion of other important nitrogenous species, NOx (NO3−, NO2− and NO), came into focus very recently. It turns out that this not only produces NH3 beyond NRR under ambient conditions, but also removes the waste NOx species.7,8 In contrast, this denitrification process in nature reduces the NOx species to N2O by fungi and N2 by bacteria.9
It should be noted that nitrate and nitrite ions usually coexist in waste water, such as contaminated groundwater, lakes, and coastal water, which also includes NO from biological denitrification.9 In addition, the NOx species can be easily obtained by the plasma activation of air.10,11 To this end, developing multifunctional electrocatalysts that simultaneously reduce all the NOx species promises higher conversion efficiency for NH3 synthesis compared with monofunctional ones.10,12,13 Up to now, existing research has mainly focused on monofunctional systems for individual species, while the design strategy of multifunctional catalysts for the NOxRR has been lacking. As we all know, the NORR is an essential reaction for determining the performance of electrocatalysts for the electroreduction of NO3− and NO2− to NH3.14,15 Therefore, for the NOxRR towards NH3 instead of other by-products (H2, N2, and N2O), the premise is to design an efficient NORR electrocatalyst with high activity and NH3 selectivity.
Since the proposal of single-atom catalysts (SACs) by Zhang et al. in 2011,16 they have also been confirmed to show high activity for NH3 synthesis from the NORR, NO2RR or NO3RR (i.e., Mg/Al/Ga/Co@NG,17–19 Zr–C2N,20 Ru@C3N4,21 and Ti/Zr@CN22). Unfortunately, the individual active site gives rise to their deficiency in multifunctional electrocatalytic ability to reduce all the NOx species, which motivates us to design multifunctional electrocatalysts. Given the flexibility of dual active sites in double-atom catalysts (DACs),23–26 there are more possibilities to adjust their geometrical bridge configurations,27 offering more opportunities to modulate the binding strength of reaction intermediates and then optimize the catalytic activity, selectivity and multifunctional ability.28 For instance, compared with atomically dispersed single Fe and Ni catalysts, with the help of the synergetic effect of dual active sites, a bimetallic FeNi DAC exhibits outstanding multifunctional performance for the reversible oxygen evolution reaction and oxygen reduction reaction.29 Furthermore, various DACs have been theoretically and experimentally confirmed to show high activity and selectivity for the NRR.23,30–35 In view of these advantages, it is expected that DACs would be multifunctional electrocatalysts with the potential ability for reducing all NOx species, which has not yet been reported.
Here, by using the first-principles method, we proposed an optimized strategy for screening multifunctional electrocatalysts to reduce all NOx species. The screening strategy consists of the steps of “stability of catalyst”, “NO adsorption”, “NORR activity”, and “NH3 selectivity”. We used the selection metric of ‘‘two steps of key hydrogenation process’’ to measure the “NORR activity”, which is confirmed to be very effective. According to some late transition-metal based SACs with good electrocatalytic ability for the NO2RR or NO3RR in the experiment,18,36–38 together with recently synthesized transition-metal dimer embedded N-doped graphene (M1M2@NG),32,39–43 28 DAC candidates were built (M1/M2 = V, Cr, Mn, Fe, Co, Ni, and Cu) for screening multifunctional electrocatalysts for the NOxRR. Through firstly systematic screening for NORR electrocatalysts, the DAC of Cu2@NG was identified as the best one due to its smallest limiting potential (UL, −0.30 V) and high NH3 selectivity. As expected, the catalytic merits of Cu2@NG can be extended to the NO2RR (UL, −0.36 V) and NO3RR (UL, −0.32 V). Moreover, the electroreduction activity descriptor and physical mechanism in terms of electronic structures were analyzed. Our work provides an effective strategy to screen multifunctional electrocatalysts for the NOxRR, by which a cost-efficient and stable candidate with desirable NH3 activity and selectivity was identified.
2. Methods
All spin-polarized computations were carried out by the generalized gradient approximation (GGA) method with the Perdew–Burke–Ernzerhof (PBE) functional44 based on density functional theory (DFT) implemented in the Vienna ab initio simulation package (VASP),45,46 in which van der Waals (vdW) correction proposed by Grimme (DFT+D3) was chosen.47 The plane-wave basis set with a cut-off energy of 450 eV was employed. The convergence thresholds of the total energy and the Hellmann–Feynman force are 10−5 eV and 0.02 eV Å−1, respectively. A 7 × 7 supercell of graphene with a vacuum layer of ∼18 Å is used to avoid the interactions between periodic images for all calculations. Monkhorst–Pack meshes of 3 × 3 × 1 and 7 × 7 × 1 are adopted for structural optimization and calculation of densities of states (DOS), respectively.The free energy change (ΔG) under the acidic medium (pH = 0) for each elementary reaction step was calculated based on the computational hydrogen electrode (CHE) model,48–50 according to the following equation:
| ΔG = ΔE + ΔEZPE − TΔS |
The hybrid model,53,54i.e., the combination of explicit solvation model with two water molecules near the active site and continuum solvation model,55 was used to investigate the solvation effect on the NOxRR. The solvation effect doesn't change our conclusions about the screening of multifunctional electrocatalysts for the NOxRR (for the details and results, see Note S1 in the ESI†). The pH effect was also considered from the viewpoints of thermodynamics and dynamics. The results show that Cu2 and Ag2 DACs are excellent acidic electrocatalysts to multifunctionally reduce all the NOx species (for the details and results, see Note S2 in the ESI†). The thermal stability of Cu2 and Ag2 DACs was examined by ab initio molecular dynamics simulations56 for 10 ps with the current 7 × 7 supercells of graphene (for the details and results, see Note S3 in the ESI†).
3. Results and discussion
For DACs, graphene containing nitrogen (NG) is an excellent substrate with anchoring sites for metal dimer deposition. Some DACs of Fe2@NG,39 FeCo@NG,40,41 FeNi@NG,24 and CoNi@NG42 have been definitely determined experimentally and exhibit excellent catalytic performances for the O2 reduction reaction (ORR). In addition, a few M1M2@NG DACs also possess a great N2 reduction ability with NH3 selectivity.5,32,43 In this regard, M1M2@NG DACs may be promising electrocatalyst candidates for the NOxRR including both the hydrogenation of N and O atoms in the NOx species. Given that the NORR is the essential reaction of the NOxRR, we will firstly consider the design of a NORR electrocatalyst from the M1M2@NG system. The atomic structure of M1M2@NG is presented in Fig. 1a, where each metal atom is tetra-coordinated with three N atoms and another metal atom. Both the homonuclear and heteronuclear cases were considered for 3d metals (M1/M2 = V, Cr, Mn, Fe, Co, Ni, and Cu), leading to 28 kinds of DAC candidates. The deposited metal dimers are well within the atomic plane of graphene, probably thanks to the large size of the pore decorated with the edge N atoms. Our recent studies5,34 showed that the metal dimer in DACs can provide a bridge active site for the adsorption of N2 and relevant intermediates, which is beneficial for molecular activation and its further hydrogenation.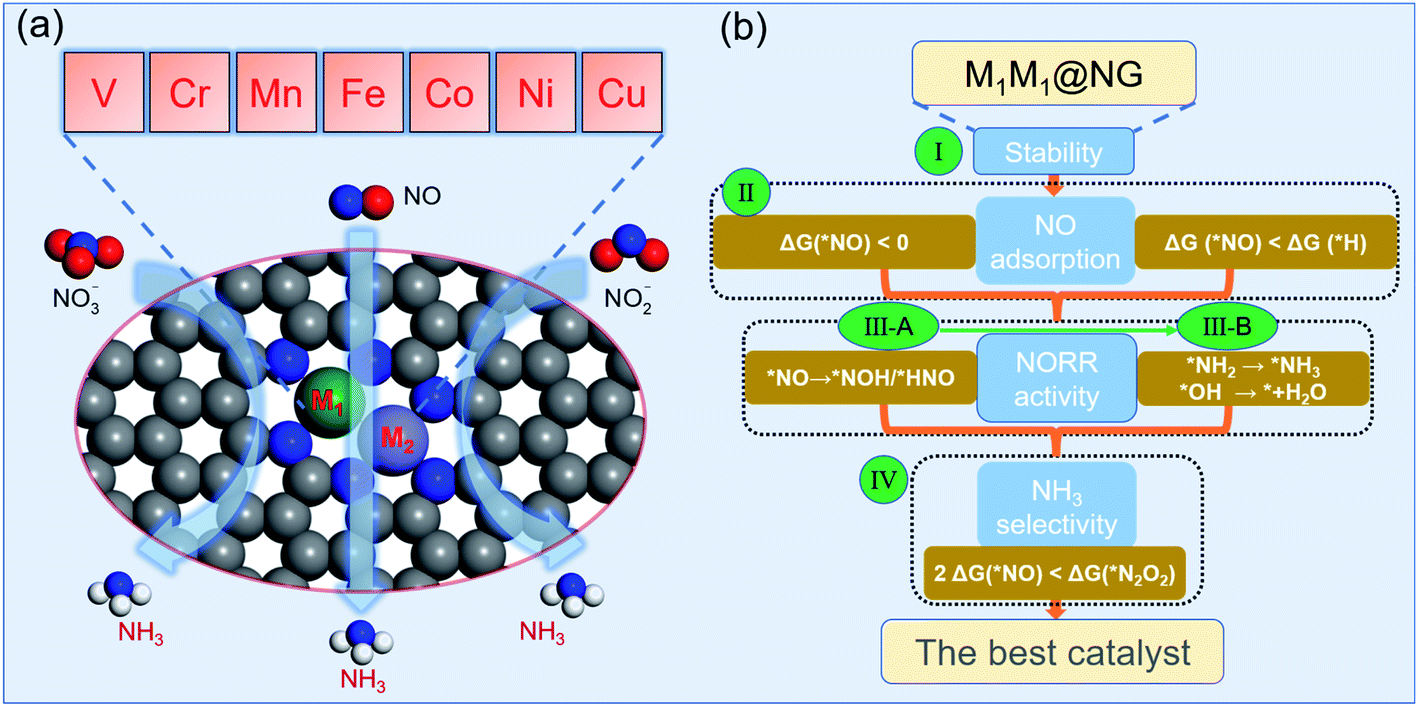 | ||
| Fig. 1 (a) The atomic structure of M1M2@NG DACs (M1/M2 = V, Cr, Mn, Fe, Co, Ni, and Cu). (b) The optimized strategy for screening NORR electrocatalysts including four stepwise selection principles. The third criterion consists of ‘‘two steps of key hydrogenation process’’. | ||
According to the NORR characteristics,12,18,57–59 we comprehensively proposed four stepwise selection principles (Fig. 1b) for the best NORR electrocatalysts: (I) the thermodynamic stability of the catalyst for the feasibility of experimental realization, (II) stable adsorption of molecular NO for activation, and superiority to H, (III) high NORR activity to NH3 with low ΔGmax (here the threshold value (GT) was set as 0.4 eV, i.e., UL ≥ 0.4 V), and (IV) great NH3 selectivity with inhibitive ability of other products (N2 and N2O).14 Particularly, for the third criterion, we used the selection metric of ‘‘two steps of key hydrogenation process’’ to measure the catalytic activity for the NORR, rather than calculating the overall reaction pathway of numerous candidates.
We have investigated the feasibility of experimental realization of these 28 DACs based on the calculated formation energies and binding energies.5 The results showed that their formation energies (about 2.8–4.3 eV) are lower than or comparable with those of several synthesized ones (i.e. FeNi@NG,24 Fe2@NG,39 FeCo@NG,40,41 and CoNi@NG42). And their more negative binding energies (about −9.5 to −13.5 eV) suggest that highly stable complexes can be formed by depositing metal atoms onto an N-doped porous graphene support. As the first step of the NORR, the adsorption and activation of NO on M1M2@NG DACs were investigated. Various kinds of NO adsorption configurations were initially studied to determine the favorable one. The most stable adsorption configurations are summarized in Fig. S1.† It was found that most stable species over M1M2@NG prefer to stay over the metal atoms with three representative configurations (Fig. 2a): (1) the side-on pattern with the N atom binding with both metal atoms and the O atom with one metal atom (NOα), and (2) the end-on pattern with the N atom binding with both metal atoms (NOβM1M2) or one metal atom (NOβM1).
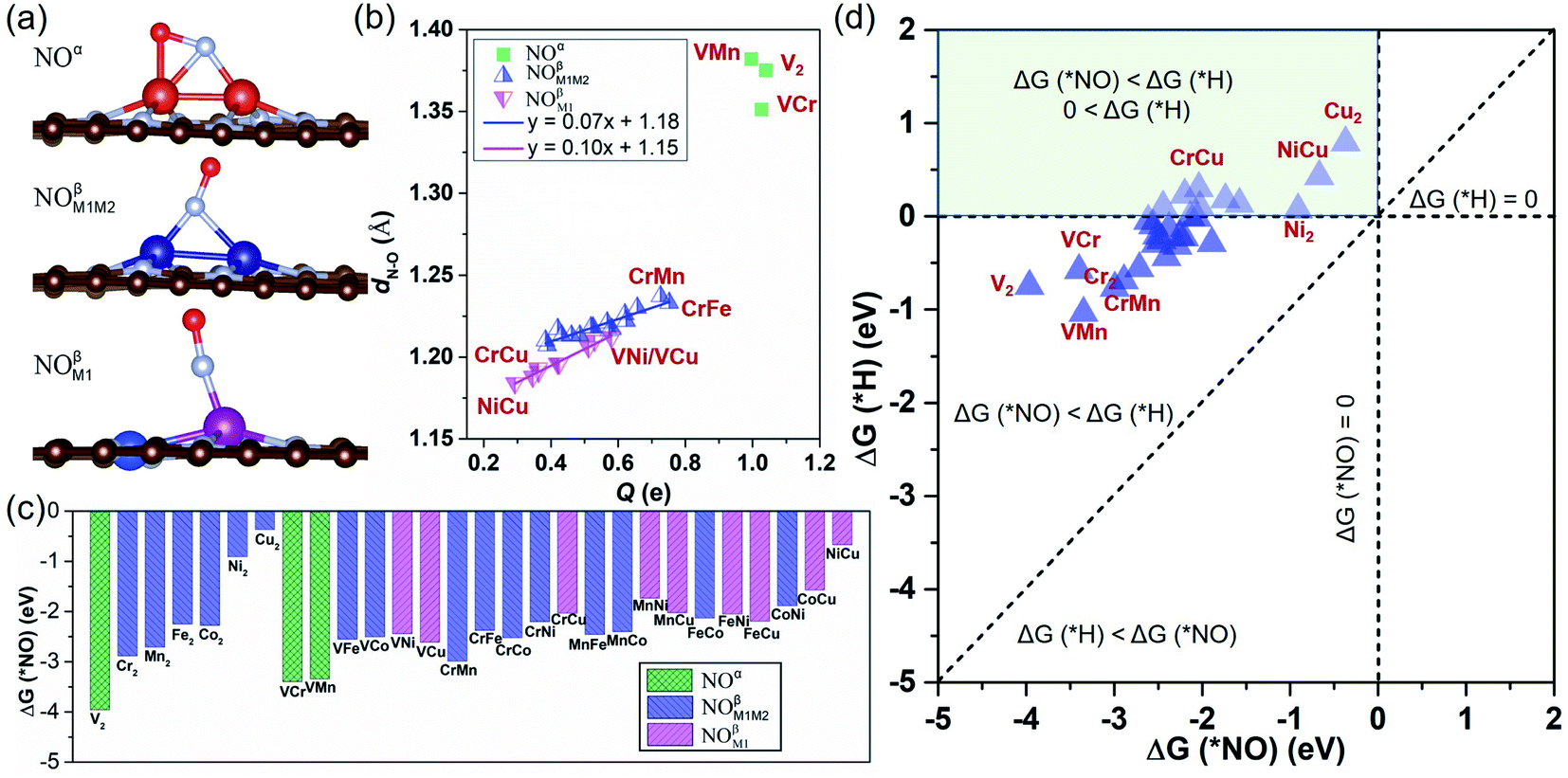 | ||
| Fig. 2 (a) Side views of the three most stable adsorption configurations of NO over M1M2@NG DACs. (b) The relationship between the charge transfer (Q, e) and the N–O bond length (dN–O (*NO), Å) of the most favorable *NO configurations on each M1M2@NG. Here, the positive values of Q indicate electron transfer from the catalyst to the adsorbed NO. (c) The free energy change (ΔG (*NO), eV) of the most stable *NO configuration over various DACs. (d) The ΔG for the *NO and *H over various DACs. | ||
Interestingly, most DACs adopt an end-on configuration for *NO (NOβM1 mainly for Ni and Cu-alloy DACs), while only V2, VCr, and VMn@NG DACs prefer to adopt the side-on NOα configuration (Fig. S1†), which may result from the fact that V2, VCr, and VMn dimers have fewer d-orbital electrons. From the relationship between the charge transfer (Q, e) and N–O bond length in Fig. 2b, the side-on NOα configurations transfer more electrons (1.00–1.04 e) from the DAC support to the *NO and make the length of the N–O bond longer (1.35–1.38 Å from 1.15 Å in the free NO molecule). For each NOβM1 and NOβM1M2 end-on configuration, there is a nearly linear correlation between the electron transfer and the length of the N–O bond. Correspondingly, the NOβM1 configurations have relatively less electron transfer between the *NO and DACs than NOβM1M2 configurations, resulting in the difference of N–O bond lengths. From the free energy change for *NO (Fig. 2c), we can find that *NO over V2, VCr, and VMn DACs with a side-on configuration has smaller ΔG values due to the higher activation of *NO with a large N–O bond length than that with an end-on configuration. Moreover, all the DACs have negative ΔG values for *NO with enlarging N–O bond, indicating the chemical adsorption and activation of *NO. The negative ΔG has a linear relationship with the total number of d-orbital valence electrons (Nd-orbital) of free metal-dimer atoms (Fig. S2†): more Nd-orbital corresponds to less negative ΔG. Thus, the Cu2 DAC has the least negative ΔG for the NO configuration among these DAC systems, while it still possesses the capture ability for NO molecules under ambient conditions.
Since H adsorption is generally strong on most bulk metal surfaces, the active site of metal surfaces can be easily blocked by the covered *H to inhibit electrocatalytic reduction, such as for the NRR.60 Here also for the NORR, NO adsorption has to compete with the H adsorption over active sites. Based on this, we calculated the H adsorption on various DACs and the comparison results of ΔG (*H) and ΔG (*NO) are organized in Fig. 2d. We found that all the electrocatalysts have more negative ΔG values for *NO than that for *H over the corresponding DACs, indicating that they have a strong binding capability for NO to suppress the competing HER.
We then investigated the catalytic performances of the NORR over all DACs as most catalysts have a chemical adsorption ability for NO molecules. However, it's complex, cumbersome and resource-consuming work to consider all the reaction steps of the NORR for the 28 DACs that meet the first two rules (catalyst's stability and NO adsorption), which usually also occurred for other catalyst screening.5,34,43 Thus, a one time and labor saving, and convenient method is an urgent need, which is significant for screening outstanding electrocatalysts for the NORR or multifunctional electrocatalysts for the NOxRR.
From here obtained *NO configurations and previous studies,19,57,61,62 we summarized the detailed reaction pathways for the NORR towards NH3, as shown in Fig. 3. Given that the NO molecule contains N and O atoms, the proton–electron pairs can attack the N atom and also the O atom to produce NH3 and H2O, respectively. The sequential order of two product formation strongly depends on the NO adsorption configuration. Based on *NO with an end-on configuration, the reaction can follow O/N-distal and -alternating mechanisms to firstly form the product H2O. Correspondingly, the NO reduction follows O/N-consecutive and -enzymatic mechanisms to firstly form either H2O or NH3. In particular, under the side-on *NO induced N-consecutive mechanism, protonation can lead to progressive hydrogenation of N atoms until NH3 is preferentially formed with O atoms left over active sites. Moreover, the different reaction mechanisms can convert or hybridize with each other. We can find that no matter what mechanism the reaction follows, the first hydrogenation step is the *NOH/*HNO formation from *NO and the last hydrogenation step is the *NH3 or H2O formation, indicating that the NORR includes both the NRR and ORR features.
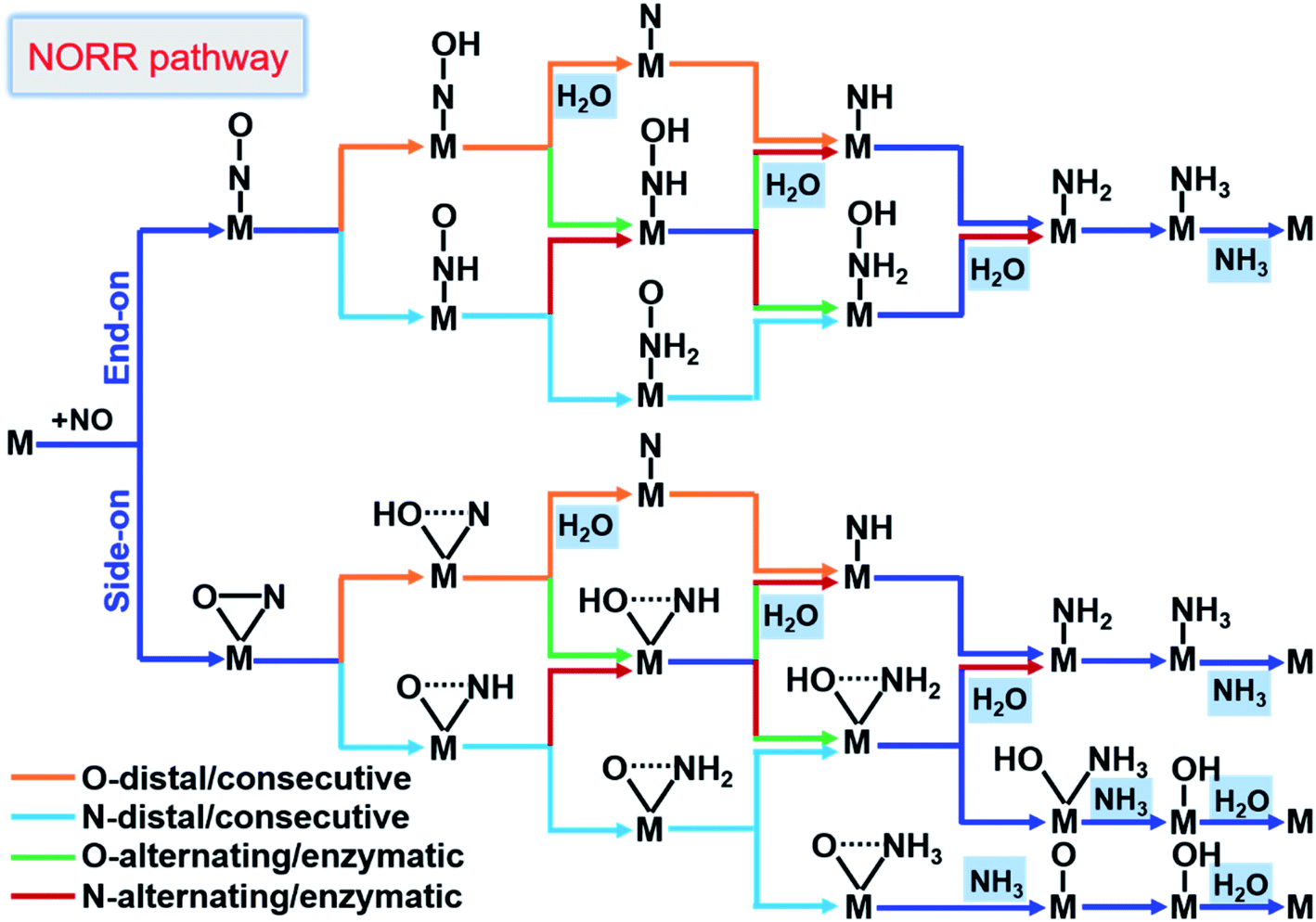 | ||
| Fig. 3 Schematic depiction of the various pathways for the NORR towards NH3. The dotted lines in the adsorbates indicate that bonds may be broken. | ||
According to the results from calculating the entire reaction steps in recent studies about the NORR19,61,62 and the similarity to the NRR,32,63 the PDS for the NORR towards NH3 may be the stabilization of *NOH/*HNO species from NO adsorption (*NO → *NOH/*HNO), or destabilization of *NH2 or *OH species to form the products (*NH2 → *NH3, or *OH → * + H2O). Thus, the third screening criterion (Fig. 1b) for NORR activity consists of ‘‘two steps of key hydrogenation process’’, i.e., (1) *NO → *NOH/*HNO as the first procedure to select the catalyst candidates that meet the free energy threshold requirements (smaller than GT of 0.40 eV) and (2) *NH2 → *NH3 or *OH → * + H2O as the final procedure to confirm the NORR electrocatalyst with high activity. It is worth emphasizing that this reaction process always involves *NH3 and H2O formation, whether in the middle or at the last step. We thus simplified the final screening procedure for NORR activity to a combination of *NH2 → *NH3 and *OH → * + H2O.
From the calculated free energy change as depicted in Fig. 4a, we can find that the first hydrogenation step (*NO → *NOH/*HNO) requires ΔG smaller than GT (0.40 eV) over eight DACs (V2, Cr2, Cu2, VCr, VMn, VFe, VCu, and CrMn), which are mostly V and Cr-based DACs (except for Cu2). Among these DACs, the chemical adsorption of *NO upon hydrogenation forms stable intermediate *HNO or *NOH species with side-on configurations, except for VCu with an end-on configuration (Fig. S3†). The strong stabilization of *HNO or *NOH (only one for *NOH over VCr DAC) is responsible for the high activity for the *NO hydrogenation process. Here most of the DACs with small ΔG give rise to the more favorable *HNO than *NOH, different from that over transition-metal borides with more favorable *NOH.61 Moreover, among these results, the N–O bond in *NO can break spontaneously by hydrogenation under ambient conditions, causing the smallest ΔG of the first hydrogenation step over V2 (−2.03 eV) and VCr (−1.68 eV) DACs.
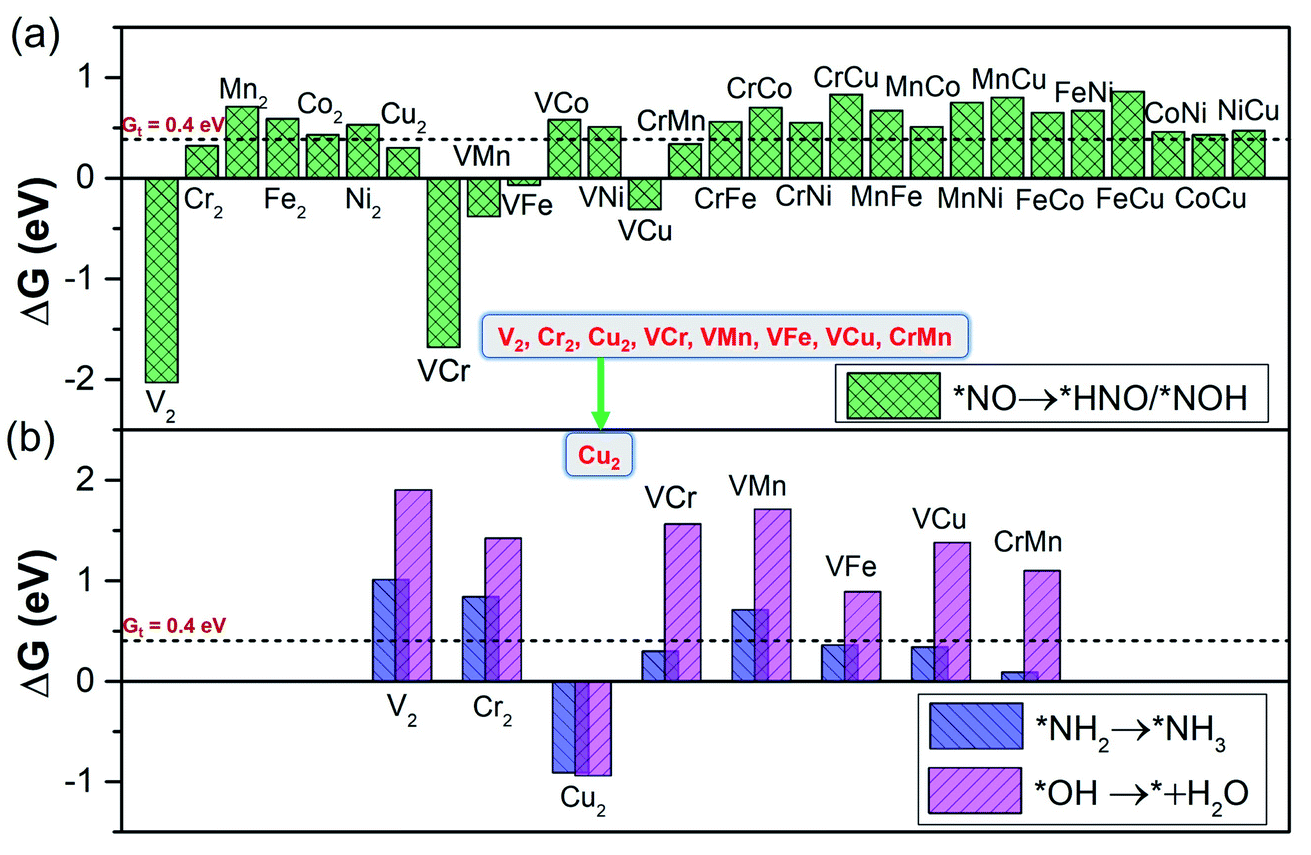 | ||
| Fig. 4 The screening criterion of ‘‘two steps of key hydrogenation process’’ for NORR activity (step III in Fig. 1b), with the threshold value (GT = 0.40 eV). (a) The ΔG for stabilization of *NOH/*HNO species (*NO → *NOH/*HNO) over the 28 DACs. (b) The ΔG for destabilization of *NH2 and *OH species (*NH2 → *NH3 and *OH → * + H2O) over the 28 DACs. | ||
Through the first round of preliminary screening for NORR activity, we thus calculated the free energy change of *NH2 → *NH3 and *OH → * + H2O over the above eight DAC candidates (Fig. 4b). It was found that only the Cu2 DAC meets the screening metric of ΔG smaller than 0.4 eV, indicating that the Cu2 DAC may show the greatest electroactivity for the NORR towards NH3. The other seven DACs have a larger ΔG of H2O formation (1.90 eV for V2, 1.42 eV for Cr2, 1.56 eV for VCr, 1.71 eV for VMn, 0.89 eV for VFe, 1.38 eV for VCu, and 1.10 eV for VMn), which is greater than that for *NH3 formation and leads to the poisoning of active sites of DACs by *OH species. A similar poisoning phenomenon also occurs for CO2 reduction within low limiting potential for the Pd-based catalyst.64
A promising NORR electrocatalyst also requires good NH3 selectivity, which suggests that it should have the ability to suppress the competing reaction to form the by-product. For example, at high potential, the NORR will lead to by-product (N2O or N2) formation on some metal surfaces.14 For this reaction to occur, N–N coupling in the formation of N2O2 is required from two NO co-adsorbed on one active site. In consequence, as the first step in initiating the whole competing reaction, the affinity to *NO (towards NH3) and *N2O2 (towards N2O and N2) on catalysts will give rise to their reduction selectivity performance. We then compared their free energy changes for adsorption over the Cu2 DAC, i.e., ΔG (*N2O2) vs. ΔG (*2NO). It was found that the *N2O2 formation from the NO dimer by N–N bond coupling (Fig. S4†) is unfavorable due to its less negative ΔG (−0.42 eV) than that for two separated *NO (−0.74 eV). This indicates that the two *NO prefer to separately adsorb on different active sites rather than on one site, which can suppress N2O and N2 formation.
To elucidate the results and verify our screening rule for NORR activity, we obtained the free energy diagrams of the most relevant pathway (the other pathways are also considered) for the NORR under zero and applied potential over Cu2, VCr, VMn, VFe, VCu, and CrMn DACs, all of which have ΔG values smaller than 0.4 eV from *NH2 to *NH3. We can see that, over the Cu2 DAC, the protonation of *H2NO can firstly follow the N-consecutive mechanism to form *H2NOH (Fig. 5a) and then transforms into an O-enzymatic mechanism to form the first product H2O. Interestingly, *H2NO species can also continuously follow the N-consecutive mechanism to form *ONH3 that binds on the Cu site by an O–Cu bond, which firstly forms the product NH3 and leaves the *O on the active site (Fig. 5b). The left *O over the Cu2 DAC can be easily removed with small ΔG under ambient conditions via the protonation process: *O → *OH → * + H2O. However, for VCr, VMn, VFe, VCu, and CrMn DACs, the high ΔG values (>0.8 eV) of *OH → * + H2O induce the poisoning of active sites, despite the low ΔG values for the hydrogenation step of *NO, and *NH2, and the other steps (Fig. S5†). These results well reveal that our screening criterion is effective and straightforward for NORR activity towards NH3 and the Cu2 DAC possesses the highest electrocatalytic activity with the lowest ΔG of 0.30 eV for the PDS (UL = −0.30 V).
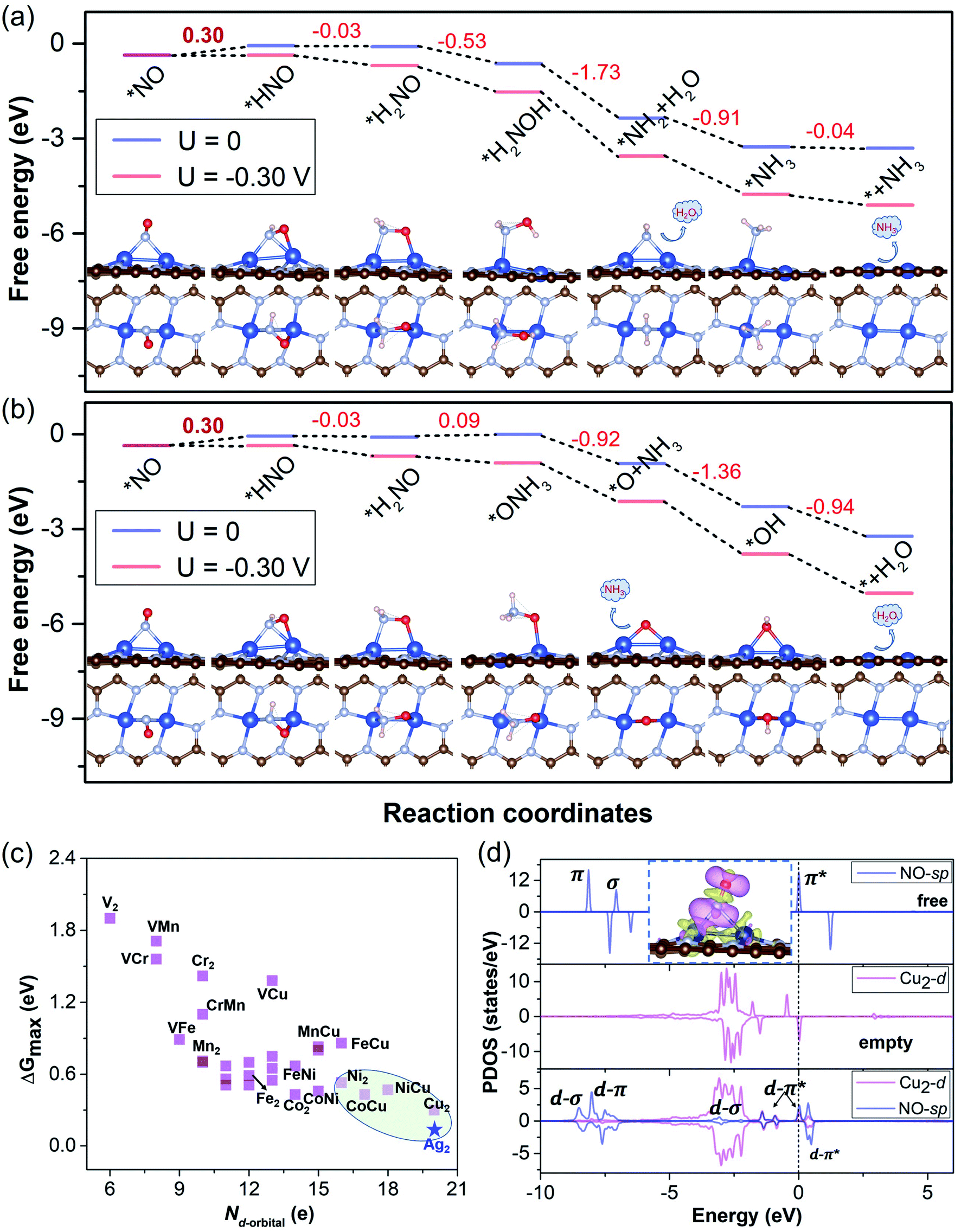 | ||
| Fig. 5 (a and b) The free energy diagrams for the NORR towards NH3 over the Cu2 DAC via the two most relevant mechanisms. The corresponding free energy changes and structures of the key steps along the reaction pathways are presented and the ΔGmax values of the PDS are in bold. (c) The ΔGmax of the NORR versus the total number of d-orbital valence electrons (Nd-orbital) of free metal-dimer atoms (data from 28 M1M2@NG DAC candidates (M1/M2 = V, Cr, Mn, Fe, Co, Ni, and Cu)). (d) The PDOS with orbital interaction for the Cu2 DAC, including the NO-sp orbitals for free NO molecules (blue), metal-d orbitals for Cu2@NG without NO adsorption (purple), and metal-d orbitals and NO-sp orbitals for Cu2@NG with NO adsorption, respectively. The Fermi level was set to 0. The inset shows the CDD (0.002 e Å−3) for *NO over the Cu2 DAC. The purple and yellow maps in the CDD denote electron accumulation and depletion, respectively. | ||
To gain insight into the high electrocatalytic activity of the Cu2 DAC for the NORR, we revealed the intrinsic nature of the descriptor based on the d-orbital features of metal DACs, which may play an important role in the chemical and electrocatalytic activity. Based on the charge analysis of the scaling relationship with adsorption properties and free energy changes (for the details and results, see Note S4 with Fig. S14 in the ESI†) from 28 M1M2@NG DAC systems, here we found that the points for Cu-based DACs with more d electrons are concentrated in the region of higher electrocatalytic activity, though those for V and Cr-based DACs with fewer d electrons are relatively scattered in the region of lower electrocatalytic activity (Fig. 5c). This suggests that Cu-based DACs have more d electrons and thus higher electrocatalytic activity while V and Cr-based DACs with fewer d electrons induce their higher chemical activity (lower electrocatalytic activity), as observed elsewhere.5,33 Thus, within reasonable limits, the Nd-orbital can be a simple and approximate descriptor to evaluate the electrocatalytic activity of current DAC systems for the NORR, the determination of which does not need DFT calculations.
The classical Sabatier principle in catalysis determines that the adsorption energy of the reactants should be moderate. We also analyzed the appropriate affinity between *NO and the Cu2 DAC using the charge density difference (CDD) and partial density of states (PDOS), as shown in Fig. 5d. The adsorption and activation of NO over metal catalysts depend on the “acceptance–donation” mechanism,20 which denotes that the metal–NO bond formation stems from the electron acceptance of empty metal-d orbitals from the NO-σ orbitals, and the elongation/activation of the N–O bond derives from the electron donation of metal-d orbitals to NO-π* orbitals. This ‘‘acceptance–donation’’ mechanism can be confirmed from the CDD of *NO over the Cu2 DAC, where electron redistribution occurs upon NO adsorption with electron accumulation (purple region) and depletion (yellow region). Thus, the cooperation and reorganization of unoccupied and occupied d-orbitals of Cu-dimer activate the *NO. We can find the electronic state hybridization between Cu2-d (purple) and NO-sp (blue) orbitals from the PDOS upon NO adsorption on the Cu2 DAC. The d–π* orbital hybridization near the Fermi level indicates the activation of *NO, which induces low ΔG for the hydrogenation of *NO to from *HNO.
On the basis of pivotal NORR performance, it is necessary to explore multifunctional electrocatalysts that can also promote the electroreduction of NO3− and NO2− species to realize the NOxRR towards NH3 by protonation. To conduct the electrocatalytic NO3RR and NO2RR on catalysts, NO3− and NO2− species are expected to be adsorbed over the Cu2 DAC, in view of the NO3− affinity of Cu surfaces.65 We found that the Cu2 DAC possesses moderate affinity for NO3− and NO2− species (Fig. 6a), which stems from the electron donation of the Cu2 DAC to adsorbates with 0.78 and 0.66 electrons, respectively. Through free energy correction for the most stable *NO3 and *NO2 over the Cu2 DAC, we obtained the ΔG (*NO3) and ΔG (*NO2) as −0.73 and −0.90 eV, smaller than that for *H (0.79 eV). From the CDD results, it was found that π* antibonding orbitals of NO3− and NO2− species accept electrons from the Cu2 DAC, yielding the elongation of the N–O bond and activation of NO3− and NO2− species. Therefore, *NO3 is promoted to accept the hydrogenation of O atoms to in turn generate *NO2 and *NO. And *NO2 is by means of two steps of hydrogenation to generate the *NO, then following by NORR to form the final product NH3. The corresponding ΔGmax (UL) for the NO3RR to NH3 (Fig. 6b) and the NO2RR to NH3 (Fig. 6c) is 0.36 (−0.36) and 0.32 eV (−0.32 V), comparable with 0.30 eV (−0.30 V) of the NORR to NH3, revealing high NOxRR activity with multifunctional electrocatalytic ability of the Cu2 DAC.
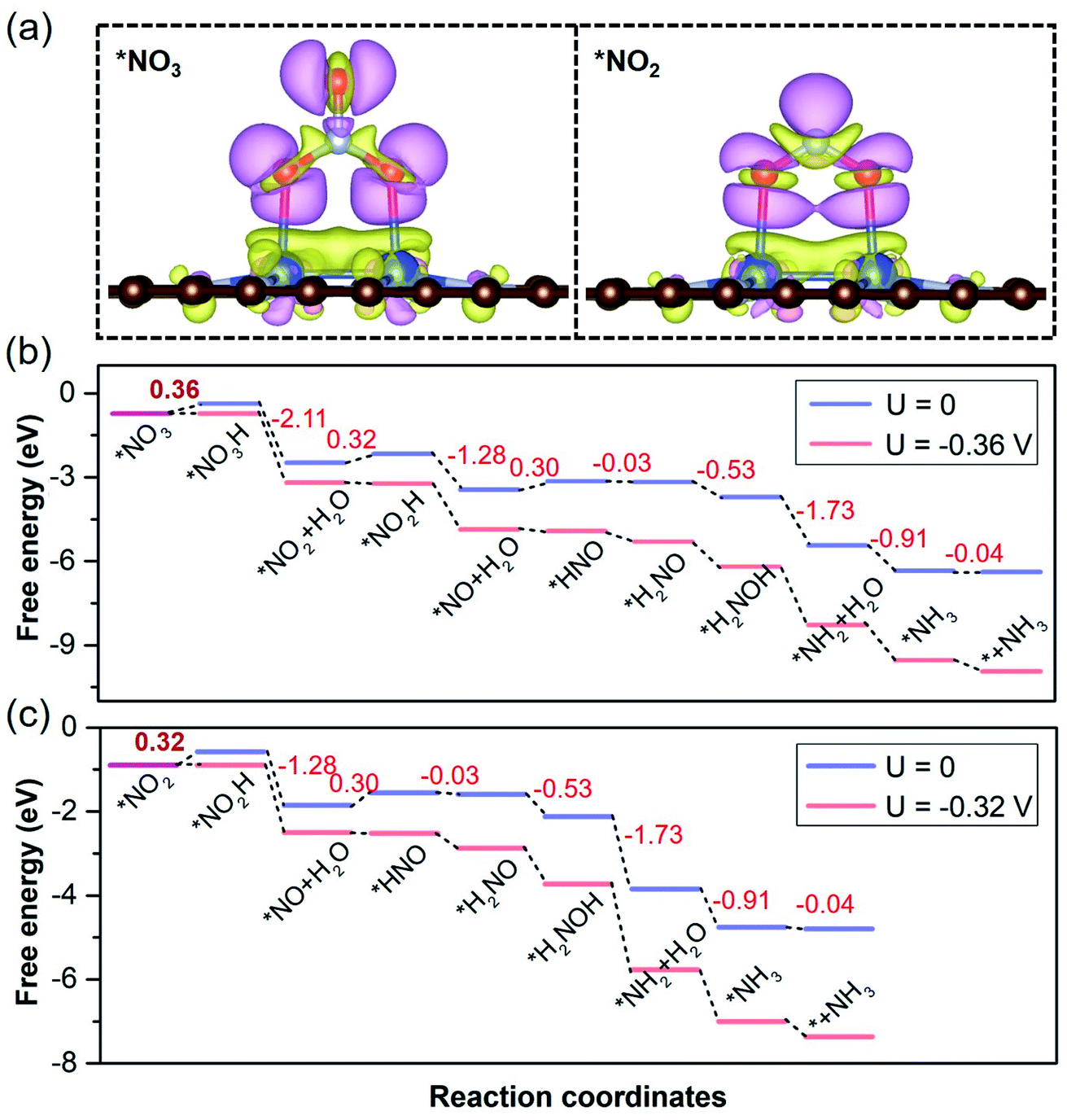 | ||
| Fig. 6 (a) The most stable configurations with the CDD (0.002 e Å−3) for *NO3 and *NO2 over the Cu2 DAC. The purple and yellow maps in the CDD denote electron accumulation and depletion, respectively. (b) The free energy diagrams for the NO3RR towards NH3 over the Cu2 DAC. (c) The free energy diagrams for the NO2RR towards NH3 over the Cu2 DAC. In (b) and (c), the corresponding free energy changes of the key steps along the reaction pathway are presented and the ΔGmax values of PDS are in bold. | ||
We also performed the screening strategy on 4d (Rh, Pd, and Ag) and 5d (Ir, Pt and Au) late transition-metal based homonuclear DACs to search for suitable electrocatalysts for the NOxRR, because these metal atoms have more d-orbital electrons with potentially high electrocatalytic activity. Through firstly the screening processes for NORR electrocatalysts, the DAC of Ag2@NG was identified as the best one due to its small ΔG of the potential-determining step (0.14 eV) and high NH3 selectivity (Table S1†). The free energy diagrams for the NORR towards NH3 over the Ag2 DAC via the most relevant mechanism are calculated (Fig. S6a†) and the corresponding structures of key steps along the reaction pathways are presented in Fig. S7.† Next, we obtained ΔG (*NO3) and ΔG (*NO2) as −0.73 and −0.74 eV, also smaller than that for *H (0.86 eV). The protonation of NO3− and NO2− species to NH3 was studied (Fig. S6b and c†) and the corresponding ΔGmax (UL) for the NO2RR and NO3RR to NH3 is 0.14 (−0.14) and 0.23 eV (−0.23 V), comparable with its electrocatalytic activity for the NORR to NH3. Moreover, the result of deduced Ag2@NG is added in Fig. 5c, and is located near the region of high electrocatalytic activity. This confirms that the Ag2 DAC (Ag and Cu metal elements belong to the same group with more d-orbital electrons) also possesses high NOxRR activity with multifunctional electrocatalytic ability and again shows that our proposed screening strategy is feasible and efficient.
4. Conclusions
In summary, to investigate the multifunctional electrocatalytic ability of DACs for the NOxRR towards NH3, we firstly investigated the electrocatalytic activity of 28 DACs for the NORR by using the proposed strategy based on a first-principles screening method, which consists of the steps of “stability of catalyst”, “NO adsorption”, “NORR activity”, and “NH3 selectivity”. Specifically, the “NORR activity” criterion includes two steps of key hydrogenation processes, i.e., (1) *NO → *NOH/*HNO and (2) *NH2 → *NH3 and *OH → * + H2O, instead of considering the free energy changes for entire reaction steps. Through screening, our results reveal that the Cu2@NG DAC is an excellent NORR electrocatalyst towards NH3 with a rather low limiting potential of −0.30 V and great NH3 selectivity, while its high electrocatalytic activity is also reflected in the NO2RR (−0.32 V) and NO3RR (−0.36 V) to identify the multifunctional electrocatalytic ability of Cu2@NG. We also find a simple descriptor of the total number of metal-dimer d-orbital valence electrons for electrocatalytic activity. The binding affinity of NO is deeply explained by the physical mechanism including the “acceptance–donation” mechanism and orbital interaction between the active metal of the catalyst and NO. The proposed strategy helps us to find another NOxRR multifunctional electrocatalyst from 4d and 5d late transition-metal based DACs (Ag2@NG). Our work effectively optimizes the screening strategy for the exploring of NOxRR multifunctional electrocatalysts and gives an in-depth insight into the atomic mechanisms of NOx electroreduction.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work is supported by the National Natural Science Foundation of China (Grant No. 12074099) and the Program for Science & Technology Innovation Talents in Universities of Henan Province (Grant No. 20HASTIT028).References
- C. Ling, X. Niu, Q. Li, A. Du and J. Wang, J. Am. Chem. Soc., 2018, 140, 14161–14168 CrossRef CAS PubMed.
- X.-F. Li, Q.-K. Li, J. Cheng, L. Liu, Q. Yan, Y. Wu, X.-H. Zhang, Z.-Y. Wang, Q. Qiu and Y. Luo, J. Am. Chem. Soc., 2016, 138, 8706–8709 CrossRef CAS PubMed.
- T. Wang, Q. Liu, T. Li, S. Lu, G. Chen, X. Shi, A. M. Asiri, Y. Luo, D. Ma and X. Sun, J. Mater. Chem. A, 2021, 9, 884–888 RSC.
- C. Choi, S. Back, N.-Y. Kim, J. Lim, Y.-H. Kim and Y. Jung, ACS Catal., 2018, 8, 7517–7525 CrossRef CAS.
- D. Ma, Y. Wang, L. Liu and Y. Jia, Phys. Chem. Chem. Phys., 2021, 23, 4018–4029 RSC.
- C. Liu, Q. Li, C. Wu, J. Zhang, Y. Jin, D. R. MacFarlane and C. Sun, J. Am. Chem. Soc., 2019, 141, 2884–2888 CrossRef CAS PubMed.
- J. Long, S. Chen, Y. Zhang, C. Guo, X. Fu, D. Deng and J. Xiao, Angew. Chem., 2020, 132, 9798–9805 CrossRef.
- Y. Li, S. Xiao, X. Li, C. Chang, M. Xie, J. Xu and Z. Yang, Mater. Today Phys., 2021, 19, 100431 CrossRef CAS.
- Y. Zeng, C. Priest, G. Wang and G. Wu, Small Methods, 2020, 4, 2000672 CrossRef CAS.
- J. Sun, D. Alam, R. Daiyan, H. Masood, T. Zhang, R. Zhou, P. J. Cullen, E. C. Lovell, A. Jalili and R. Amal, Energy Environ. Sci., 2021, 14, 865–872 RSC.
- L. Li, C. Tang, X. Cui, Y. Zheng, X. Wang, H. Xu, S. Zhang, T. Shao, K. Davey and S. Qiao, Angew. Chem., 2021, 133, 2–9 CrossRef.
- P. Gao, Z. Xue, S. Zhang, D. Xu, G. Zhai, Q. Li, J. Chen and X. Li, Angew. Chem., 2021, 133, 20879–20884 CrossRef.
- R. Daiyan, T. Tran-Phu, P. Kumar, K. Iputera, Z. Tong, J. Leverett, M. H. A. Khan, A. Asghar Esmailpour, A. Jalili, M. Lim, A. Tricoli, R.-S. Liu, X. Lu, E. Lovell and R. Amal, Energy Environ. Sci., 2021, 14, 3588–3598 RSC.
- H. Wan, A. Bagger and J. Rossmeisl, Angew. Chem., Int. Ed., 2021, 60, 21966–21972 CrossRef CAS PubMed.
- M. Duca and M. T. M. Koper, Energy Environ. Sci., 2012, 5, 9726–9742 RSC.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- Z. Wang, J. Zhao, J. Wang, C. R. Cabrera and Z. Chen, J. Mater. Chem. A, 2018, 6, 7547–7556 RSC.
- A. Wu, J. Yang, B. Xu, X.-Y. Wu, Y. Wang, X. Lv, Y. Ma, A. Xu, J. Zheng, Q. Tan, Y. Peng, Z. Qi, H. Qi, J. Li, Y. Wang, J. Harding, X. Tu, A. Wang, J. Yan and X. Li, Appl. Catal., B, 2021, 299, 120667 CrossRef CAS.
- Q. Wu, B. Huang, Y. Dai, Y. Ma and T. Heine, ArXiv Prepr. arXiv211001947, 2021.
- H. Niu, Z. Zhang, X. Wang, X. Wan, C. Kuai and Y. Guo, Small, 2021, 17, 2102396 CrossRef CAS PubMed.
- L. Lv, Y. Shen, J. Liu, X. Meng, X. Gao, M. Zhou, Y. Zhang, D. Gong, Y. Zheng and Z. Zhou, J. Phys. Chem. Lett., 2021, 12, 11143–11150 CrossRef CAS PubMed.
- H. Niu, Z. Zhang, X. Wang, X. Wan, C. Shao and Y. Guo, Adv. Funct. Mater., 2021, 31, 2008533 CrossRef CAS.
- Y. Li, Q. Zhang, C. Li, H.-N. Fan, W.-B. Luo, H.-K. Liu and S.-X. Dou, J. Mater. Chem. A, 2019, 7, 22242–22247 RSC.
- W. Ren, X. Tan, W. Yang, C. Jia, S. Xu, K. Wang, S. C. Smith and C. Zhao, Angew. Chem., Int. Ed., 2019, 58, 6972–6976 CrossRef CAS PubMed.
- Y. Zhao, K. R. Yang, Z. Wang, X. Yan, S. Cao, Y. Ye, Q. Dong, X. Zhang, J. E. Thorne, L. Jin, K. L. Materna, A. Trimpalis, H. Bai, S. C. Fakra, X. Zhong, P. Wang, X. Pan, J. Guo, M. Flytzani-Stephanopoulos, G. W. Brudvig, V. S. Batista and D. Wang, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 2902 CrossRef CAS PubMed.
- Z. He, K. He, A. W. Robertson, A. I. Kirkland, D. Kim, J. Ihm, E. Yoon, G.-D. Lee and J. H. Warner, Nano Lett., 2014, 14, 3766–3772 CrossRef CAS PubMed.
- C. Wu, X. Zhang, Z. Xia, M. Shu, H. Li, X. Xu, R. Si, A. I. Rykov, J. Wang, S. Yu, S. Wang and G. Sun, J. Mater. Chem. A, 2019, 7, 14001–14010 RSC.
- T. He, A. R. P. Santiago, Y. Kong, M. A. Ahsan, R. Luque, A. Du and H. Pan, Small, 2021, 2106091 Search PubMed.
- Y. Cheng, S. He, J.-P. Veder, R. De Marco, S. Yang and S. Ping Jiang, ChemElectroChem, 2019, 6, 3478–3487 CrossRef CAS.
- X. Lv, W. Wei, B. Huang, Y. Dai and T. Frauenheim, Nano Lett., 2021, 21, 1871–1878 CrossRef CAS PubMed.
- L. Zhang, G. Fan, W. Xu, M. Yu, L. Wang, Z. Yan and F. Cheng, Chem. Commun., 2020, 56, 11957–11960 RSC.
- H. Li, Z. Zhao, Q. Cai, L. Yin and J. Zhao, J. Mater. Chem. A, 2020, 8, 4533–4543 RSC.
- X. Guo, J. Gu, S. Lin, S. Zhang, Z. Chen and S. Huang, J. Am. Chem. Soc., 2020, 142, 5709–5721 CrossRef CAS PubMed.
- D. Ma, Z. Zeng, L. Liu, X. Huang and Y. Jia, J. Phys. Chem. C, 2019, 123, 19066–19076 CrossRef CAS.
- Z. W. Chen, J.-M. Yan and Q. Jiang, Small Methods, 2019, 3, 1800291 CrossRef.
- Z.-Y. Wu, M. Karamad, X. Yong, Q. Huang, D. A. Cullen, P. Zhu, C. Xia, Q. Xiao, M. Shakouri, F.-Y. Chen, J. Y. Kim, Y. Xia, K. Heck, Y. Hu, M. S. Wong, Q. Li, I. Gates, S. Siahrostami and H. Wang, Nat. Commun., 2021, 12, 2870 CrossRef CAS PubMed.
- G.-F. Chen, Y. Yuan, H. Jiang, S.-Y. Ren, L.-X. Ding, L. Ma, T. Wu, J. Lu and H. Wang, Nat. Energy, 2020, 5, 605–613 CrossRef CAS.
- P. Li, Z. Jin, Z. Fang and G. Yu, Energy Environ. Sci., 2021, 14, 3522–3531 RSC.
- W. Ye, S. Chen, Y. Lin, L. Yang, S. Chen, X. Zheng, Z. Qi, C. Wang, R. Long, M. Chen, J. Zhu, P. Gao, L. Song, J. Jiang and Y. Xiong, Chem, 2019, 5, 2865–2878 CAS.
- J. Wang, Z. Huang, W. Liu, C. Chang, H. Tang, Z. Li, W. Chen, C. Jia, T. Yao, S. Wei, Y. Wu and Y. Li, J. Am. Chem. Soc., 2017, 139, 17281–17284 CrossRef CAS PubMed.
- J. Wang, W. Liu, G. Luo, Z. Li, C. Zhao, H. Zhang, M. Zhu, Q. Xu, X. Wang, C. Zhao, Y. Qu, Z. Yang, T. Yao, Y. Li, Y. Lin, Y. Wu and Y. Li, Energy Environ. Sci., 2018, 11, 3375–3379 RSC.
- X. Han, X. Ling, D. Yu, D. Xie, L. Li, S. Peng, C. Zhong, N. Zhao, Y. Deng and W. Hu, Adv. Mater., 2019, 31, 1905622 CrossRef CAS PubMed.
- T. Deng, C. Cen, H. Shen, S. Wang, J. Guo, S. Cai and M. Deng, J. Phys. Chem. Lett., 2020, 11, 6320–6329 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Nørskov, Energy Environ. Sci., 2010, 3, 1311–1315 RSC.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- J. Rossmeisl, A. Logadottir and J. K. Nørskov, Chem. Phys., 2005, 319, 178–184 CrossRef CAS.
- http://webbook.nist.gov/chemistry/ .
- V. Wang, N. Xu, J.-C. Liu, G. Tang and W.-T. Geng, Comput. Phys. Commun., 2021, 267, 108033 CrossRef CAS.
- Y. Wang, X. Qin and M. Shao, J. Catal., 2021, 400, 62–70 CrossRef CAS.
- G. Brancato, N. Rega and V. Barone, J. Chem. Phys., 2008, 128, 144501 CrossRef PubMed.
- K. Mathew, R. Sundararaman, K. Letchworth-Weaver, T. A. Arias and R. G. Hennig, J. Chem. Phys., 2014, 140, 084106 CrossRef PubMed.
- G. J. Martyna, M. L. Klein and M. Tuckerman, J. Chem. Phys., 1992, 97, 2635–2643 CrossRef.
- L. Zhang, J. Liang, Y. Wang, T. Mou, Y. Lin, L. Yue, T. Li, Q. Liu, Y. Luo, N. Li, B. Tang, Y. Liu, S. Gao, A. A. Alshehri, X. Guo, D. Ma and X. Sun, Angew. Chem., 2021, 133, 1–7 CrossRef.
- X. Peng, Y. Mi, H. Bao, Y. Liu, D. Qi, Y. Qiu, L. Zhuo, S. Zhao, J. Sun, X. Tang, J. Luo and X. Liu, Nano Energy, 2020, 78, 105321 CrossRef CAS.
- D. Kim, D. Shin, J. Heo, H. Lim, J.-A. Lim, H. M. Jeong, B.-S. Kim, I. Heo, I. Oh, B. Lee, M. Sharma, H. Lim, H. Kim and Y. Kwon, ACS Energy Lett., 2020, 5, 3647–3656 CrossRef CAS.
- S. L. Foster, S. I. P. Bakovic, R. D. Duda, S. Maheshwari, R. D. Milton, S. D. Minteer, M. J. Janik, J. N. Renner and L. F. Greenlee, Nat. Catal., 2018, 1, 490–500 CrossRef.
- Y. Xiao and C. Shen, Small, 2021, 2100776 CrossRef CAS PubMed.
- Q. Wu, H. Wang, S. Shen, B. Huang, Y. Dai and Y. Ma, J. Mater. Chem. A, 2021, 9, 5434–5441 RSC.
- J. Zhao and Z. Chen, J. Am. Chem. Soc., 2017, 139, 12480–12487 CrossRef CAS PubMed.
- S. Chatterjee, C. Griego, J. L. Hart, Y. Li, M. L. Taheri, J. Keith and J. D. Snyder, ACS Catal., 2019, 9, 5290–5301 CrossRef CAS.
- T. Hu, C. Wang, M. Wang, C. M. Li and C. Guo, ACS Catal., 2021, 14417–14427 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2ta00192f |
| This journal is © The Royal Society of Chemistry 2022 |