
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNa-β-Al2O3 stabilized Fe2O3 oxygen carriers for chemical looping water splitting: correlating structure with redox stability†
Nur Sena
Yüzbasi
 a,
Andac
Armutlulu
a,
Thomas
Huthwelker
b,
Paula M.
Abdala
*a and
Christoph R.
Müller
*a
a,
Andac
Armutlulu
a,
Thomas
Huthwelker
b,
Paula M.
Abdala
*a and
Christoph R.
Müller
*a
aLaboratory of Energy Science and Engineering, ETH Zurich, Leonhardstrasse 27, 8092, Zurich, Switzerland. E-mail: abdalap@ethz.ch; muelchri@ethz.ch
bSwiss Light Source, Paul Scherrer Institut, 5232, Villigen PSI, Switzerland
First published on 12th April 2022
Abstract
Chemical looping is an emerging technology to produce high purity hydrogen from fossil fuels or biomass with the simultaneous capture of the CO2 produced at the distributed scale. This process requires the availability of stable Fe2O3-based oxygen carriers. Fe2O3–Al2O3 based oxygen carriers exhibit a decay in the H2 yield with cycle number, due to the formation of FeAl2O4 that possesses a very low capacity for water splitting at typical operating conditions of conventional chemical looping schemes (700–1000 °C). In this study, the addition of sodium (via a sodium salt) in the synthesis of Fe2O3–Al2O3 oxygen carriers was assessed as a means to counteract the cyclic deactivation of the oxygen carrier. Detailed insight into the oxygen carrier's structure was gained by combined X-ray powder diffraction (XRD), X-ray absorption spectroscopy (XAS) at the Al, Na and Fe K-edges and scanning transmission electron microscopy/energy-dispersive X-ray spectroscopy (STEM/EDX) analyses. The addition of sodium prevented the formation of FeAl2O4 and stabilized the oxygen carrier via the formation of a layered structure, Na-β-Al2O3 phase. The material, i.e. Na-β-Al2O3 stabilized Fe2O3, showed a stable H2 yield of ca. 13.3 mmol g−1 over 15 cycles.
Introduction
Hydrogen is an emerging energy carrier with potential applications in the transport, industry and power sectors as a fuel that yields water vapour as the only combustion product. If produced sustainably, H2 has the potential to become a near-zero emissions energy carrier, hence reducing energy-related CO2 emissions.1–3 Currently, the majority of H2 is produced from natural gas via steam methane reforming (SMR) without carbon dioxide capture, which is an energy intensive process and emits a significant amount of CO2.3,4 In addition, further energy-intensive purification steps are required to obtain H2 of sufficiently high purity allowing its use in polymer electrolyte membrane fuel cells (PEMFC).5,6 Hence, for H2 to become an energy carrier in a sustainable framework, it must be produced in an efficient and sustainable manner, i.e. from renewable sources or with CO2 capture.7,8A chemical looping (CL) scheme based on the cyclic redox reactions of iron oxide/iron offers the possibility to produce high purity H2 with inherent CO2 capture (Fig. 1a).7–14 This process could be operated using different reducing gases such as CO, synthesis gas (produced by biomass gasification) or natural gas.15,16 For instance, CO is used to reduce Fe2O3 to lower oxidation states (ideally to metallic Fe which gives the highest H2 yield), following reactions (1)–(3). During Fe2O3 reduction, a pure stream of CO2 is obtained readily through the condensation of water vapour. The oxidation of metallic Fe with steam produces H2 (water splitting reactions (4) and (5)) of high purity.17 To close the cycle, Fe3O4 is oxidized to Fe2O3 in air (reaction (6)).
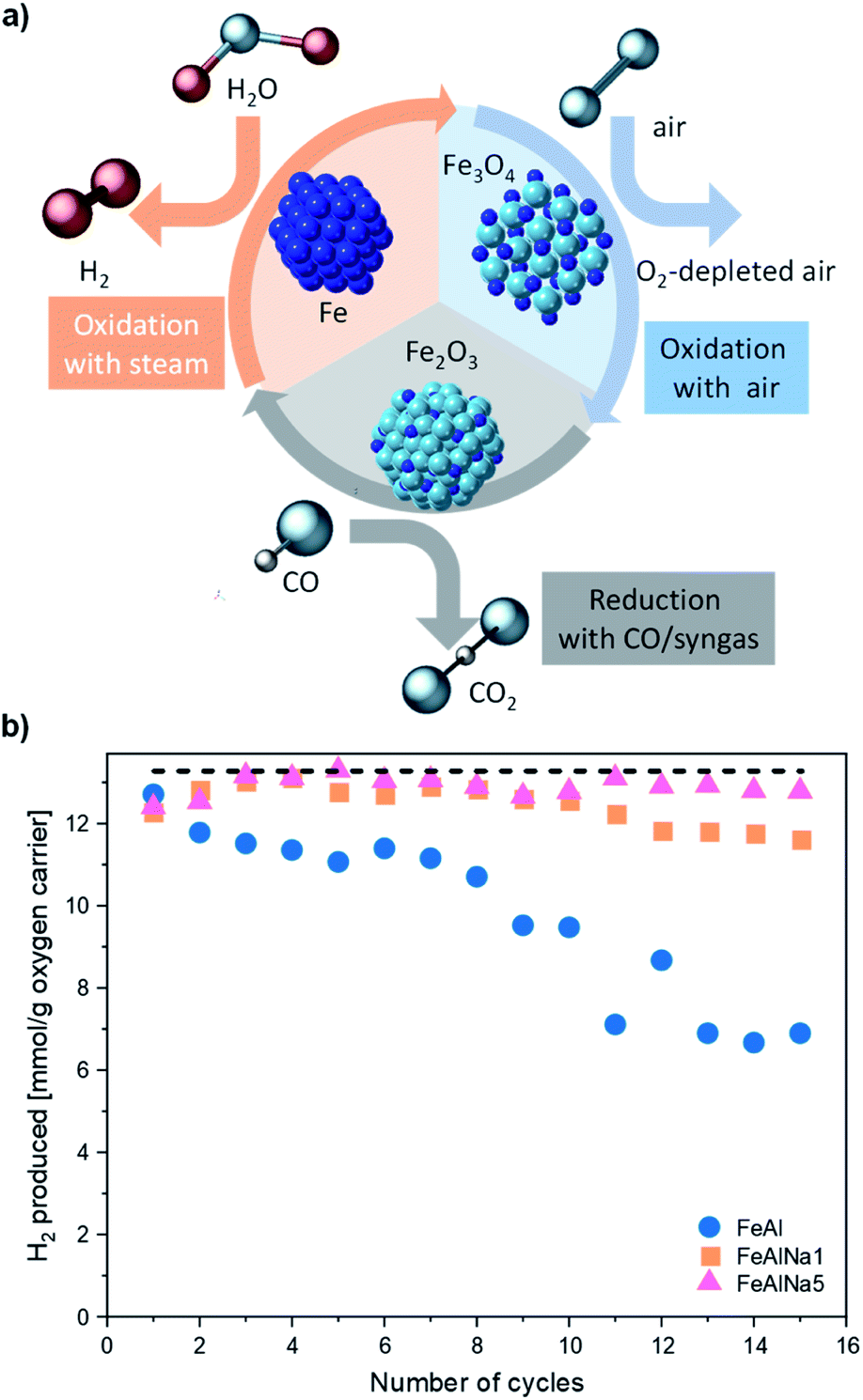 | ||
| Fig. 1 (a) Schematic of the Fe2O3–Fe based chemical looping process for the production of H2 using a synthesis gas (H2 and CO) as the fuel. (b) H2 yield as a function of cycle number for FeAl, FeAlNa1 and FeAlNa5. The redox experiments were performed at 800 °C in a packed-bed reactor using 10 vol% CO in N2 for reduction and 23 vol% H2O in N2 followed by 5 vol% O2 in N2 for re-oxidation. The dashed horizontal line represents the theoretically expected H2 yield (13.3 mmol g−1) for an oxygen carrier that contains 80 wt% Fe2O3. | ||
Reduction of iron oxide in CO
| 3Fe2O3 + CO ↔ 2Fe3O4 + CO2 | (1) |
| Fe3O4 + CO ↔ 3FeO + CO2 | (2) |
| FeO + CO ↔ Fe + CO2 | (3) |
Oxidation in steam
| Fe + H2O ↔ FeO + H2 | (4) |
| 3FeO + 4H2O ↔ Fe3O4 + 4H2 | (5) |
Oxidation in air
| 2Fe3O4 + ½O2 → 3Fe2O3 | (6) |
Owing to its favourable thermodynamics iron oxide is one of the most attractive oxygen carriers for the chemical-looping based production of hydrogen.7–11,13,17–27 However, pure Fe2O3 shows a rapid decay in its H2 yield with number of redox cycles owing to sintering.17 Sintering leads to an appreciable decrease in the reduction rate of iron oxide, which results in an incomplete reduction and in turn a low H2 yield in the subsequent re-oxidation step.7,27,28 To mitigate deactivation by sintering, strategies based on the addition of a high Tammann temperature stabilizing oxide, e.g. Al2O3,7,10,22,26,29 SiO2,7,9 TiO2,30 CeO2,14 MgAl2O4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 21,31 and ZrO2.13,23,27 have been proposed. Indeed, these oxides have shown to increase the stability of iron based oxygen carriers over multiple cycles by reducing FeOx/Fe particle sintering.23 Among the investigated oxides, Al2O3 is particularly interesting owing to its relatively low cost, high Tammann temperature of ∼885 °C and high mechanical resistance.32–34 However, Al2O3 can react with iron oxide forming the spinel phase FeAl2O4 (hercynite) which leads to a decay in the H2 yield due to the limited capacity of FeAl2O4 to split water at the typical operating conditions of conventional CL (700–1000 °C).10,22,26,35 Hydrogen production using iron aluminate-based materials has been proposed for solar driven thermochemical cycles that cycles between spinel iron based solid solutions through an oxygen vacancy mediated mechanism; however, this is limited to high steam-to-hydrogen ratios and the H2 capacity does not exceed 0.5 mmol H2 per g FeAl2O4.36–38
21,31 and ZrO2.13,23,27 have been proposed. Indeed, these oxides have shown to increase the stability of iron based oxygen carriers over multiple cycles by reducing FeOx/Fe particle sintering.23 Among the investigated oxides, Al2O3 is particularly interesting owing to its relatively low cost, high Tammann temperature of ∼885 °C and high mechanical resistance.32–34 However, Al2O3 can react with iron oxide forming the spinel phase FeAl2O4 (hercynite) which leads to a decay in the H2 yield due to the limited capacity of FeAl2O4 to split water at the typical operating conditions of conventional CL (700–1000 °C).10,22,26,35 Hydrogen production using iron aluminate-based materials has been proposed for solar driven thermochemical cycles that cycles between spinel iron based solid solutions through an oxygen vacancy mediated mechanism; however, this is limited to high steam-to-hydrogen ratios and the H2 capacity does not exceed 0.5 mmol H2 per g FeAl2O4.36–38
Approaches to prevent the formation of FeAl2O4 during redox operation have included the addition of alkali or alkali earth metal oxides to the oxygen carrier, yet the mechanisms behind the mitigation of FeAl2O4 formation remains elusive.23,34,39 In particular, the effect of the addition of sodium on the redox performance of Fe2O3–Al2O3 based oxygen carriers is controversially discussed and the underlying mechanisms at the atomic scale that would explain the improved macroscopic redox performance of such oxygen carriers are currently not well understood. For instance, a previous study proposed that formulations based on Fe2O3, Al2O3, Na2O and/or MgO could potentially hinder the formation of FeAl2O4 by forming NaAlO2 or MgAl2O4 phases that are thermodynamically more stable.31 Indeed, X-ray powder diffraction (XRD) analysis confirmed the presence of NaAlO2 and MgAl2O4 phases in the two studied oxygen carriers with compositions Na:Al:Fe = 0.25:0.25:0.50 and Na:Mg:Fe:Al = 0.20:0.15:0.28:0.37 (molar ratios). However, it was found that only MgAl2O4 effectively hindered the formation of inactive FeAl2O4 while NaAlO2 only partially prevented its formation. This was explained by the gradual replacement of Al3+ by Fe3+ in NaAlO2 (NaAl1−yFeyO2) during redox cycling leading to the formation of free Al2O3 that subsequently reacts with FeOx to form FeAl2O4. A further study found that the addition of sodium in concentrations below 5 wt% stabilizes the reactivity of Fe2O3, yet the structure of the oxygen carriers was not studied in detail.20,40 Typically, X-ray powder diffraction (XRD) has been applied to identify the phases present in the oxygen carrier, however, this analysis fails at describing the local structure of the materials and it is insensitive to amorphous phases and phases in low concertation. Thus, the fine structural features of Na-modified Fe2O3–Al2O3 oxygen carriers remain elusive.
As discussed above, a difficulty in improving our understanding of the effect of the addition of sodium to Fe2O3–Al2O3 on its redox behaviour is the lack of knowledge of the atomic-scale structure of these materials. Thus, this work aims at shedding light on the structure–performance relationships of sodium modified Fe2O3–Al2O3 oxygen carriers for H2-production by addressing the following questions: (i) how is the atomic scale structure of Fe2O3–Al2O3 based oxygen carriers modified by the addition of Na? (ii) what is the Na, Al and Fe environment in the oxygen carrier? and (iii) how does the redox performance relate to the oxygen carriers' structure? To address these questions we performed a detailed structural and redox analysis of sodium modified Fe2O3–Al2O3 and a sodium free benchmark utilizing XRD and X-ray absorption spectroscopy (XAS) data at the Fe, Al and Na K-edges.
Experimental
Synthesis of the oxygen carriers
Fe2O3:Al2O3 and Na-modified Fe2O3:Al2O3 oxygen carriers were synthesized using a sol–gel method22 with the following compositions: (i) Fe:Al molar ratio: 0.66:0.34 (75 wt% Fe2O3, 25 wt% Al2O3), referred to as FeAl; (ii) Na:Fe:Al = 0.02:0.66:0.32 (76 wt% Fe2O3, 23 wt% Al2O3, 1 wt% Na2O), referred to as FeAlNa1; and (iii) Na:Fe:Al 0.11:0.64:0.25 (76 wt% Fe2O3, 19% wt% Al2O3 and 5 wt% Na2O), referred to as FeAlNa5, (Table S1†). In a typical synthesis, aluminum isopropoxide (Al(OCH(CH3)2)3, purity ≥ 98 wt%) was mixed with water and the mixture was hydrolysed for two hours at 75 °C under constant stirring. Nitric acid was used to peptize the slurry. The required amount of the iron and sodium precursors, iron nitrate (Fe(NO3)3·9H2O, purity ≥ 98 wt%) and sodium nitrate (NaNO3, purity ≥ 99 wt%) respectively, were mixed with water to obtain a 1 M solution that was added subsequently to the slurry and refluxed for 12 h at 90 °C. The molar ratio of Al3+:H2O:H+ was fixed to 0.5:50:0.07. The resulting gel was dried at 100 °C overnight to remove the solvents. A xerogel was obtained after calcination at 900 °C for 2 hours. The calcined materials were crushed and sieved to a particle size range of 300–425 μm for further characterization.
Reference materials for structural characterization
The following reference materials for XAS analysis were synthesized. Na-β-Al2O3, with a molar ratio of Na:Al = 1:11 (NaAl11O17, P63/mmc space group39) was synthesized by mixing stoichiometric amounts of γ-Al2O3 and Na2CO3 followed by calcination at 1350 °C for 6 hours. Na-γ-Al2O3 (sodium dispersed at the surface of γ-Al2O3) with a molar ratio of Na:Al = 1:15 was synthesized by wet impregnation of a solution of NaNO3 (purity > 99 wt%) onto γ-Al2O3. The material was dried at 100 °C in an oven overnight, followed by calcination at 900 °C for 2 h. In addition, commercially available α-Al2O3 (Alfa Aesar, aluminum oxide, α-phase, 99.9% metal basis) and γ-Al2O3 (Alfa Aesar, aluminum oxide, γ-phase, 99.98% metal basis) were utilized as reference materials. The XRD pattern of the references Na-β-Al2O3, Na-γ-Al2O3, α-Al2O3 and γ-Al2O3 are provided in Fig. S1.† All of the materials were exposed to ambient conditions before characterization. Thus, ambient humidity likely hydrated the surface of all studied oxygen carriers and references, and possibly also the interlayers in the case of Na-β-Al2O3.
Characterization of the oxygen carriers
The crystalline phases of the calcined and cycled oxygen carriers were studied by XRD, using a PANalytical Empyrean X-ray powder diffractometer, equipped with a X'Celerator Scientific ultra-fast line detector and Bragg–Brentano HD incident beam optics using Cu Kα radiation (45 kV and 40 mA). A secondary monochromator was employed to suppress unwanted fluorescence originating from iron. Patterns were collected in the range of 2θ = 5–90°, with a step size of 0.016° s−1 and a total acquisition time of 4 h. Rietveld refinements were performed using FullProf software.41The local structure of the materials was characterized by XAS at the Fe, Na and Al K-edges. The Na K-edge and Al K-edge XAS measurements were carried out at the Phoenix II, elliptical undulator beamline at the Swiss Light Source (SLS) at the Paul Scherrer Institute (PSI), Switzerland. In a typical experiment a small quantity of material was pressed onto an indium foil and fixed to a copper plate.42 XAS measurements were performed in fluorescence mode. The X-ray fluorescence signal was detected using a 1-element Si-drift diode detector SDD (Ketek, Germany). Total electron yield was also collected in the case of Al K-edge measurements. In samples with a high content of absorbing atoms, i.e. non-diluted and not thin enough samples, the fluorescence detected XAS signal was corrected due to self-absorption. The current of the incoming beam (I0) was measured using the total electron yield signal from a 0.5 μm thin polyester foil that was coated with a 50 nm thick layer of nickel. The beam passed through this foil approximately 1 m upstream of the sample in a vacuum chamber held at ∼10−6 mbar. Self-absorption correction in the Al K-edge spectra was applied. The Fe K-edge XAS spectra were collected at the BM31 beamline at the ESRF, Grenoble, France. The samples were ground, mixed with cellulose to optimize for X-ray absorption and pelletized. The data were collected in transmission mode using a Si(111) double crystal monochromator. Post-processing, self-absorption correction and analysis of the XAS data were performed using the Demeter 0.9.20 software package.43
High-resolution field emission scanning electron microscopy (Zeiss ULTRA 55 plus) was employed to visualize the surface morphology of the oxygen carriers before and after cyclic redox tests. Furthermore, elemental mapping of the synthesized materials was achieved via a Leo Gemini 1530 SEM equipped with an energy dispersive X-ray spectrometer (EDX). A FEI Talos F200X operated at 200 kV was used in both transmission electron microscopy (TEM) and scanning TEM (STEM) modes, with a probe size of approximately 0.8 nm. The instrument is equipped with SuperX EDX comprising four SDD detectors. The STEM/EDX analyses were complemented with atomic number sensitive, high angle annular dark field (HAADF) STEM.
The surface area as well as the pore size distribution of the calcined oxygen carriers were determined using a Quantochrome NOVA 4000e N2 adsorption analyser. The samples were degassed at 300 °C for two hours prior to the acquisition of the N2 isotherms. The Brunauer–Emmett–Teller (BET)44 and Barrett–Joyner–Halenda (BJH)45 models were used to calculate, respectively, the surface area and the pore size distribution of the materials.
Redox performance
Cyclic redox experiments were performed in a packed bed reactor, as described in detail in a previous study.23 The operating temperature was 800 °C to ensure a sufficiently high reduction rate allowing for a full reduction of Fe2O3 to Fe in the set time. A typical redox cycle consists of the following steps: (i) reduction in CO (10 vol% CO in N2) for 15 minutes (1.5 L min−1), (ii) purging with N2 (1.5 L min−1) for 1 minute, (iii) oxidation of the reduced oxygen carrier with steam (23 vol% H2O in N2) for 7 minutes (1.94 L min−1), (iv) purging with N2 (1.5 L min−1) for 1 minute, and (v) oxidation with 5 vol% O2 in N2 (2 L min−1) for 5 minutes.Results and discussion
First, we discuss the cyclic redox behaviour of the synthesized oxygen carriers to determine the effect of the addition of sodium on the redox performance of the materials. Subsequently, we probe in detail the structure of the oxygen carriers in the calcined stated and after exposure to 15 redox cycles to establish structure–performance correlations.Cyclic redox performance
The cyclic redox performance of the oxygen carriers was assessed over 15 redox cycles in a packed bed reactor at 800 °C. In each cycle, the oxygen carriers were reduced in CO (10 vol% CO in N2) and oxidized first in H2O (23 vol% H2O in N2) and in a second step in O2 (5 vol% O2 in N2). The concentration profiles of the off gases for the different oxygen carriers during redox cycling are given in Fig. S2.† For each cycle, the H2 yield during steam oxidation was calculated by integrating the molar flowrate of H2 produced (obtained from off-gas concentration profiles) with respect to time by using eqn (7):21 | (7) |
The H2 yield expressed as mmol H2 per g of oxygen carrier as a function of cycle number is given in Fig. 1. The H2 yield of FeAl decreased rapidly over 15 cycles from 12.7 mmol H2 per g oxygen carrier in the first cycle, to 11.1 mmol H2 per g oxygen carrier in the fifth cycle and to 6 mmol H2 per g oxygen carrier in the fifteenth cycle. The addition of sodium enhanced the redox stability of FeAlNa1 and increased the H2 yield compared to FeAl; however, from the eighth redox cycle onwards also for this material the H2 yield started to decrease. A more pronounce stabilization effect was observed in FeAlNa5. The H2 yield of FeAlNa5 was stable over 15 redox cycles and was close to the theoretically expected value of 13.3 mmol g−1 oxygen carrier. These results show that the introduction of sodium has a large impact on the cyclic behaviour of Al2O3 stabilized, Fe2O3-based oxygen carriers.
The effect of sodium on the reduction kinetics of iron oxide can be evaluated using the gas concentration profiles obtained in the first cycle. The concentration profiles of the off gases given in Fig. S3a,† show that FeAlNa1 and FeAlNa5 have very comparable reduction rates. The fractional conversion of the oxygen carriers tested are only marginally different in the first reduction step, which indicates that the presence of sodium does not affect appreciably the reduction kinetics of Fe2O3. The reduction characteristics of the oxygen carriers were assessed further by CO and H2-TPR experiments (Fig. S4†). In the case of steam oxidation, in the first cycle the conversion of FeAl was faster than the conversion of FeAlNa1 and FeAlNA5 indicating that the addition of sodium does not have a positive impact on oxidation kinetics (Fig. S5†). Over cycling, the water splitting kinetics decreased for the deactivating oxygen carriers (Fig. S6†). However, the overall oxidation duration of FeAlNa5 is fairly unaffected by the cycle number.
To obtain insight into the origin of the improved redox stability of FeAlNa1 and FeAlNa5 compared to FeAl, the morphological and structural characteristics of the calcined and cycled materials (15 cycles) were probed by XRD, XAS and STEM/EDX.
Structure of the calcined and cycled oxygen carriers by XRD and XAS
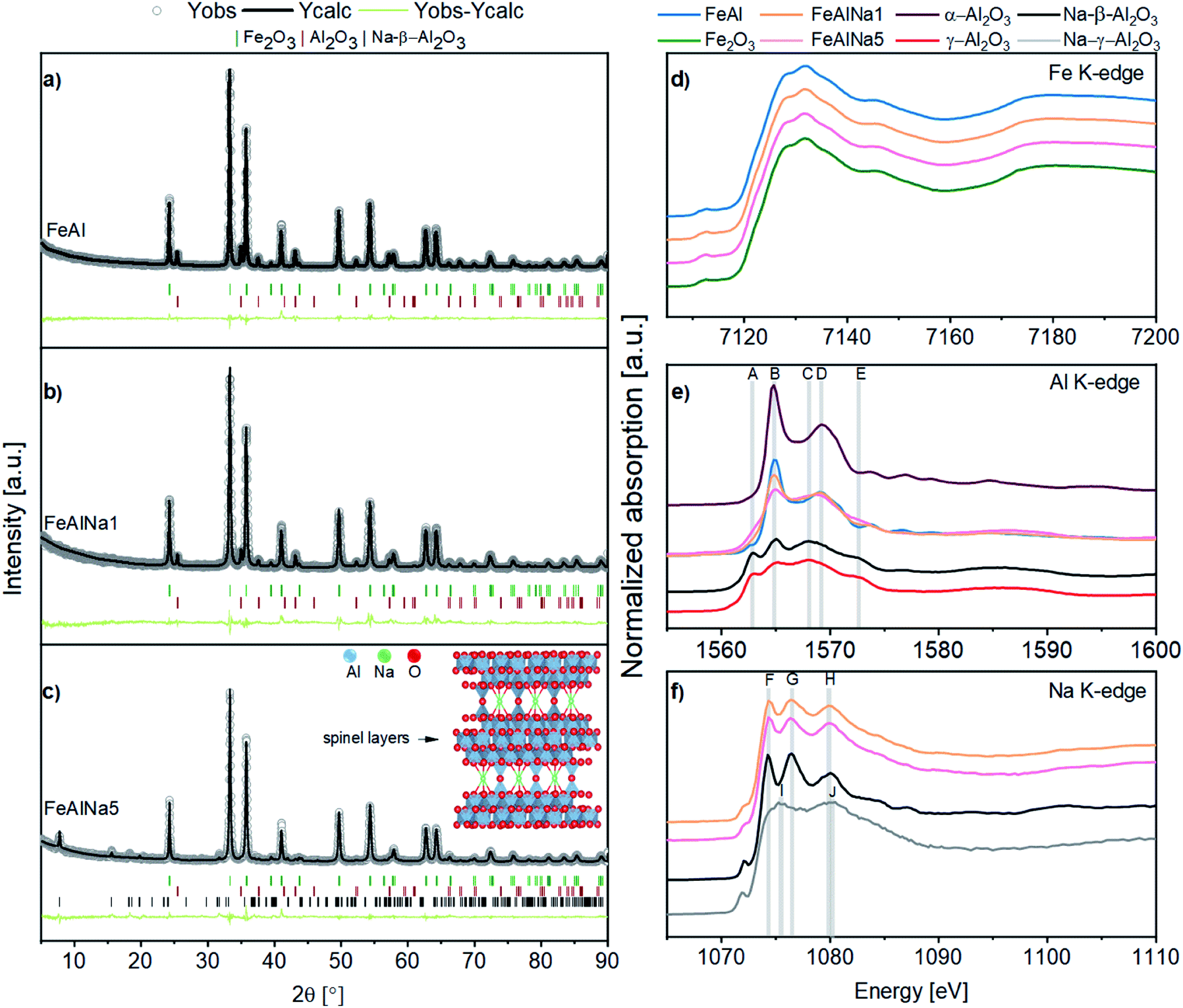 | ||
| Fig. 2 (a)–(c) XRD patterns of the calcined oxygen carriers and the corresponding Rietveld refinements. The inset in (c) sketches the crystal structure of Na-β-Al2O3 containing layers of γ-alumina that are connected by bridging oxygen and sodium ions. XANES spectra at the (d) Fe K-edge, (e) Al K-edge, (f) Na K-edge of the calcined oxygen carriers and reference materials. | ||
First, we analysed the local environment around iron. Fe K-edge XANES (X-ray absorption near edge structure) spectra of the calcined oxygen carriers are provided in Fig. 2d. The spectra of all of the oxygen carriers contain the same features as the α-Fe2O3 reference. The EXAFS (extended X-ray absorption fine structure) functions and the corresponding Fourier transforms (FT) are given in Fig. S7† and further confirm that the local environment around Fe corresponds to the local Fe environment in α-Fe2O3 in all three calcined materials. Hence, the XAS data confirm that the local environment of Fe is not affected by the presence of sodium. Importantly, it also shows that there is no formation of a solid solution or mixed oxide between Fe2O3 and Al2O3 upon calcination at 900 °C.
Fig. 2e displays the Al K-edge XANES spectra of the oxygen carriers and reference materials. To interpret these XANES data we will first discuss the XANES data of the reference materials α-Al2O3, γ-Al2O3 and Na-β-Al2O3. The crystal structure of α-Al2O3 is described by a hexagonal unit cell with Al in an octahedral coordination. The corresponding XANES spectrum (Fig. 2e) exhibits a doublet peak in the white-line region, with a sharp intense peak at 1565 eV (labelled as B), and a broader feature at 1568 eV (labelled as D), typical for Al in an octahedral coordination and in agreement with previously data.49,50 The reference γ-Al2O3 has a defect spinel type structure, in which Al atoms occupy both octahedral and tetrahedral sites.51 In the XANES spectrum of γ-Al2O3, features A, B, C and E are observed at 1563, 1565, 1568 and 1573 eV, respectively. In γ-Al2O3 feature B is considerably less intense than in α-Al2O3 and feature C has been attributed to tetrahedrally coordinated Al.52,53 Na-β-Al2O3 is a layered structure containing γ-Al2O3 layers that are linked by sodium and oxygen atoms (inset in Fig. 2c).54,55 The fact that the Al K-edge XANES spectrum of Na-β-Al2O3 exhibits similar features as γ-Al2O3 confirms the presence of γ-Al2O3 layers in Na-β-Al2O3.54
Next, we investigate the Al environment in the three oxygen carries. The Al K-edge XANES spectrum of FeAl shows features B and D, indicating that Al3+ is in an octahedral coordination similar to α-Al2O3,49 in line with XRD. With increasing sodium content, the intensity of feature B decreases, which implies that the fraction of α-Al2O3 in the material decreases with increasing sodium content. In agreement with our previous XRD analysis is the fact that the Al K-edge XANES spectrum of FeAlNa1 is dominated by features due to α-Al2O3. On the other hand, in FeAlNa5 features A, C and E are prominent which is indicative of the presence of spinel-type γ-Al2O3 or Na-β-Al2O3 (according to XRD). Features A, C and E are also present in FeAlNa1, but with a considerably lower intensity. Hence, our XANES results indicate that FeAlNa1 and FeAlNa5 contain a mixture of α-Al2O3 and a spinel-type Al2O3 phase. While α-Al2O3 is the main Al-based phase in FeAlNa1, Na-β-Al2O3 (or γ-Al2O3) is the main Al-containing phase in FeAlNa5. Na-β-Al2O3 and γ-Al2O3 share the same Al K-edge features, yet combining the information of XRD and Na-Kedge XAS (vide infra), we assign the observed features to Na-β-Al2O3 rather than to γ-Al2O3. Lastly, we assess the Na environment in the oxygen carriers and reference materials. The Na K-edge XANES spectrum of Na-β-Al2O3 exhibits three main peaks labelled as F, G and H, located at 1074, 1076 and 1080 eV, respectively (Fig. 2f). To the best of our knowledge, this is the first experimentally collected Na K-edge spectrum of Na-β-Al2O3. The spectrum of Na-γ-Al2O3 (i.e. the reference material prepared by wet impregnation of a solution of NaNO3 onto γ-Al2O3) shows features different to Na-β-Al2O3 and are labelled I (1075 eV) and J (1080 eV) (Fig. S8†). These Na K-edge XANES features of Na-γ-Al2O3 are assigned to Na ions dispersed on the surface of γ-Al2O3 (in a solvated state).55 Thus, using Na K-edge XANES we can distinguish between two different Na environments: (i) a Na-β-Al2O3 like environment, i.e. Na that is located between layers of γ-Al2O3 layers (ordered) and (ii) a Na-γ-Al2O3 like environment in which Na is located on the surface of γ-Al2O3 (surface Na+). The Na K-edge XANES spectra of FeAlNa1 and FeAlNa5 exhibit features F, G and H at, respectively 1074.4 eV (A), 1076.4 eV (B) and 1080.2 eV (C), suggesting a similar Na local environment in these two calcined oxygen carriers as in Na-β-Al2O3 (Fig. 2f). The presence of Na-β-Al2O3 in FeAlNa5 aligns with XRD results. The absence of diffraction peaks due to Na-β-Al2O3 in FeAlNa1 is possibly linked to its small quantity in FeAlNa1. However, we noticed that the features F, G and H in the Na K-edge XANES spectra of FeAlNa1 and FeAlNa5 are less intense compared to the reference Na-β-Al2O3. This is possibly due to the presence of two different Na environments in the calcined oxygen carriers. This observation led us to propose that the Na K-edge XANES spectra of the oxygen carriers can be fitted by a linear combination of Na in an environment of Na-β-Al2O3 and surface Na+ in the form of Na-γ-Al2O3. Indeed, linear combination fitting yielded Na to be ca. 60% in a Na-β-Al2O3 environment and 40% in a Na-γ-Al2O3 type environment in both FeAlNa1 and FeAlNa5 (Fig. S9†).
The cycled materials (labelled with _cyc) exhibit dominant Bragg reflections due to fcc-Fe (Fig. 3a). However, the presence of magnetite (Fe3O4), wuestite-FeO (only in FeAl) and hercynite (spinel FeAl2O4) in FeAl_cyc and FeAlNa1_cyc points to an incomplete reduction. Importantly, the XRD pattern of FeAlNa5_cyc does not show peaks due to FeAl2O4, but peaks due to Na-β-Al2O3 (Fig. 3a), indicating that Na-β-Al2O3 is stable under redox conditions. The XANES spectra and FT-EXAFS functions of the three cycled oxygen carriers are shown in Fig. 3 and S10,† respectively. From the increase in the intensity of the white line it is clear that the degree of reduction followed the order FeAl_cyc < FeAlNa1_cyc < FeAlNa1_cyc. The Fe K-edge XANES spectra (Fig. 3b) of the oxygen carriers were fitted by a linear combination of an Fe foil, FeO and FeAl2O4 and yielded 48, 79 and 94 wt% metallic Fe0 in, respectively, FeAl_cyc, FeAlNa1_cyc and FeAlNa5_cyc. These results confirm that in FeAlNa5 the formation of FeAl2O4 during reduction is hindered and iron oxide can reduced almost completely over 15 redox cycles. To some extent, there is also a stabilization effect in FeAlNa1, yet the formation of FeAl2O4 could not avoided fully. This is line with the drop in the hydrogen yield of FeAlNa1 with cycle number.
 | ||
| Fig. 3 (a) XRD patterns of the cycled oxygen carriers (15 redox cycles, reduction step) together with simulated diffraction patterns of the reference materials. XANES spectra at the (b) Fe K-edge (the arrows show the direction of change), (c) Al K-edge (the arrows point to distinct features in FeAlNa_cyc and FeAlNa_cyc); and (d) Na K-edge of the cycled materials. | ||
Turning to the analysis of the Al K-edge XANES data of the cycled oxygen carriers, we did not observe any signatures due to α-Al2O3 in any of the cycled oxygen carriers, instead features characteristic of spinel type phases are present. The Al K-edge XANES spectrum of FeAlNa5_cyc (Fig. 3c) shows the same characteristic features as the Na-β-Al2O3 reference (Fig. 2e). This indicates that during redox cycling the small fraction of α-Al2O3 that was initially present in calcined FeAlNa5 transforms fully to Na-β-Al2O3. Comparing the spectrum of FeAlNa5_cyc with the spectra of FeAl_cyc and FeAlNa1_cyc revealed a more intense feature at ca. 1569.6 eV for FeAl_cyc and FeAlNa1_cyc (marked with an arrow in Fig. 3c). This feature can be attributed to the substitution of Al sites by Fe in a spinel structure in analogy to the spinel MgAl2O452 which is isostructural to FeAl2O4. Hence, consolidating with our XRD and Fe K-edge XAS results, the feature at 1569.6 eV in the Al K-edge XANES spectra of FeAl_cyc and FeAlNa1_cyc is attributed to the formation of FeAl2O4.
The Na K-edge XANES spectra of the cycled materials are shown in Fig. 3d. We observe that in FeAlNa5_cyc the local environment of Na in the form of Na-β-Al2O3 is preserved during redox cycling. This is evidenced by the presence of the three features F, G and H in FeAlNa5_cyc. Linear combination fitting of the spectrum of FeAlNa5_cyc yields 71% of Na in a Na-β-Al2O3 framework and 29% of Na in the form of surface Na (Na-γ-Al2O3 type). Hence, the fraction of Na in a Na-β-Al2O3-type environment in FeAlNa5 increases with redox cycling, in line with Al K-edge XANES analysis. On the other hand, the Na K-edge XANES spectrum of FeAlNa1_cyc exhibits, compared to FeAlNa5, a different change in the Na environmental with redox cycling. The three sharp features (labelled F, G and H) in calcined FeAlNa1 are not detected in FeAlNa1_cyc (Fig. S11†). Instead a broader doublet (features I and J) at 1075 and 1080.5 eV appears after redox cycling, indicative that FeAlNa1_cyc closely resembles the spectrum of the Na-γ-Al2O3 reference. This change in the local environment of Na upon redox cycling, indicates that in FeAlNa1 Na is initially present in a Na-β-Al2O3 like environment (in the layered structure), but migrates and disperses on the material's surface during redox cycling.
Morphology and elemental mapping of the calcined and cycled carriers
The morphology of the freshly calcined oxygen carriers, as probed by electron microscopy is affected only marginally by the addition of sodium (Fig. S12†). The calcined oxygen carriers FeAl, FeAlNa1 and FeAlNa5 were composed of particles with an average grain size of ∼100 ± 20 nm (based on the analysis of 50 particles). The particle size of Fe2O3, calculated using STEM-EDX maps of the calcined oxygen carries (Fig. S13†), was in the range of 80–120 nm for the oxygen carries tested (119 ± 33 nm, 135 ± 36 nm and 87 ± 40 nm for FeAl, FeAlNa1 and FeAlN5, respectively). The weight composition of the materials a determined by SEM/EDX (Table S1†) are in good agreement with the nominal compositions of the oxygen carriers. The elemental mapping of the oxygen carriers revealed that the sol–gel synthesis led to a high dispersion of Fe, Na and Al, indicating that the different phases were highly dispersed in the materials (Fig. S14†). The calcined oxygen carriers FeAl, FeAlNa1 and FeAlNa5 have similar BET surface areas and BJH pore volumes (Table S1†).HAADF-STEM images and the corresponding EDX maps of Fe, Al and Na are given in Fig. 4. In the calcined materials FeAl, FeAlNa1, and FeAlNa5, discrete Fe2O3- and Al2O3-rich particles are formed, and Na is distributed evenly in FeAlNa1. In FeAlNa5, the EDX signals of Al and Na overlap, which is in line with the outcome of the XRD and XAS analysis (Fig. 2) pointing to the formation of a Na-β-Al2O3 phase. Similarly, the EDX maps of FeAl_cyc and FeAlNa1_cyc show overlapping signals of Fe and Al, which are in agreement with the formation of the spinel FeAl2O4 as determined by XRD and XAS. No such overlap is observed in FeAlNa5_cyc, which features discrete Al- and Fe-rich particles. Furthermore, the overlapping signals of Na and Al in FeAlNa5_cyc provide further evidence for the formation of Na-β-Al2O3 as determined by the XRD and XAS analyses.
 | ||
| Fig. 4 HAADF images and elemental maps (STEM-EDX) of the calcined oxygen carriers and after cycling experiments (15 cycles, reduction step). | ||
Structure–performance relationships
Combing the results of the cyclic redox tests, with our XRD, XAS and HAADF-STEM-EDX analyses we can propose the following structure–performance correlations. The reaction of α-Al2O3 with Fe/FeOx during redox cycling leads to the formation of (inactive) FeAl2O4 that in turn correlates with a rapid decay in the H2 yield in FeAl. The high redox stability of FeAlNa5 correlates with the formation of Na-β-Al2O3 and the absence of α-Al2O3 in this oxygen carrier. The formation of Na-β-Al2O3 in FeAlNa5 hinders effectively the formation of FeAl2O4 during repeated redox cycles, leading in turn to a material with a stable hydrogen yield. The Na-β-Al2O3 phase is stable at reductive and oxidative environments and is highly dispersed within the Fe/FeOx matrix. The oxygen carrier with a lower Na content, i.e. FeAlNa1, shows a dominating presence of α-Al2O3 and a small fraction of Na-β-Al2O3 initially freshly calcined state. Such a phase composition can only partially stabilize the oxygen carrier. Indeed, in FeAlNa1, Na is initially present in two environments i.e. as Na-β-Al2O3 and dispersed as surface Na. During the redox cycling of FeAlNa1, all of the α-Al2O3 reacts with FeOx forming FeAl2O4, while sodium is found in a non-crystalline form (dispersed at the surface of the particle). A schematic representation of the phase evolution during redox cycling is given in Fig. S15.† Therefore, our study points to the critical role of the layered Na-β-Al2O3 phase in stabilizing the hydrogen yield of Al2O3-stabilized, Fe2O3 based oxygen carriers.Conclusions
In this work, we probe the effect of the addition of sodium to Fe2O3–Al2O3-based oxygen carriers, on their redox stability, phase composition and phase dynamics over redox cycles. Application of XRD, XAS at the Al, Na and Fe K-edges and STEM/EDX to the freshly calcined and cycled materials allowed us to draw the following conclusions:(i) α-Al2O3 reacts with Fe/FeOx during redox cycling leading to the formation of FeAl2O4; the formation of which correlates directly with the rapid decrease in the H2 yield of an oxygen carrier.
(ii) Depending on the loading of sodium in the oxygen carrier, sodium can be found in a crystalline Na-β-Al2O3 phase or at the surfaces of the particles.
(iii) Na in the environmental of the layered structure Na-β-Al2O3 effectively stabilizes the redox performance of an oxygen carrier, while surface sodium cannot prevent fully material deactivation.
This work, demonstrates how probing of the local and average structure, such as multi edge XAS analysis in combination with XRD and electron microscopy allows for the formulation of structure–performance correlations. In addition, this work showcases the application of XAS in the tender X-ray region (Na and Al K-edges) that have not been investigated extensively particularly in the field of chemical looping and can, therefore, be of interest to the broader functional materials community. Our study brings fundamental understanding of the role of Na-β-Al2O3 for the stabilization of Fe2O3-based oxygen carriers. Nonetheless, further studies that explore different reducing gases (such as CH4), operation temperatures and long-term stability (>100 cycles) are required.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the Swiss National Science Foundation (406640_136707) and the Swiss Office of Energy (BFE, SI/500652) for financial support. This project has received partial funding from the European Union's Horizon 2020 research and innovation program (grant agreement No. 800419). This publication was created as part of NCCR Catalysis, a National Centre of Competence in Research funded by the Swiss National Science Foundation. We also thank Scientific Centre for Optical and Electron Microscopy (ScopeM) for providing training and access to scanning electron microscopes. The authors are grateful to the Swiss Light Source (SLS) at the Paul Scherrer Institute (Villigen, Switzerland), for providing beamtime at the PHOENIX beamline, and to the European Synchrotron Facility (ESRF) and the Swiss-Norwegian beamline (SNBL at ESRF) for providing beamtime for XAS measurements. We thank Dr Felix Donat for the insightful discussions on thermodynamic analysis.Notes and references
- BP, Energy Outlook 2017 Edition, 2017 Search PubMed.
- IEA, Technology Roadmap: Hydrogen and Fuel Cells, 2015 Search PubMed.
- J. A. Turner, Science, 2004, 305, 972–974 CrossRef CAS PubMed.
- M. Zerta, P. R. Schmidt, C. Stiller and H. Landinger, Int. J. Hydrogen Energy, 2008, 33, 3021–3025 CrossRef CAS.
- T. V. Choudhary and D. Goodman, Catal. Today, 2002, 77, 65–78 CrossRef CAS.
- G. Postole and A. Auroux, Int. J. Hydrogen Energy, 2011, 36, 6817–6825 CrossRef CAS.
- C. Bohn, J. Cleeton, C. Müller, S. Chuang, S. Scott and J. Dennis, Energy Fuels, 2010, 24, 4025–4033 CrossRef CAS.
- A. Thursfield, A. Murugan, R. Franca and I. S. Metcalfe, Energy Environ. Sci., 2012, 5, 7421–7459 RSC.
- Q. Zafar, T. Mattisson and B. Gevert, Ind. Eng. Chem. Res., 2005, 44, 3485–3496 CrossRef CAS.
- P. R. Kidambi, J. P. E. Cleeton, S. A. Scott, J. S. Dennis and C. D. Bohn, Energy Fuels, 2012, 26, 603–617 CrossRef CAS.
- J. R. Scheffe, M. D. Allendorf, E. N. Coker, B. W. Jacobs, A. H. McDaniel and A. W. Weimer, Chem. Mater., 2011, 23, 2030–2038 CrossRef CAS.
- R. D. Solunke and G. Veser, Ind. Eng. Chem. Res., 2010, 49, 11037–11044 CrossRef CAS.
- N. S. Yüzbasi, P. M. Abdala, Q. Imtiaz, S. M. Kim, A. M. Kierzkowska, A. Armutlulu, W. van Beek and C. R. Müller, Phys. Chem. Chem. Phys., 2018, 20, 12736–12745 RSC.
- L.-S. Fan and F. Li, Ind. Eng. Chem. Res., 2010, 49, 10200–10211 CrossRef CAS.
- G. Voitic and V. Hacker, RSC Adv., 2016, 6, 98267–98296 RSC.
- M. Rydén and M. Arjmand, Int. J. Hydrogen Energy, 2012, 37, 4843–4854 CrossRef.
- C. D. Bohn, C. R. Müller, J. P. Cleeton, A. N. Hayhurst, J. F. Davidson, S. A. Scott and J. S. Dennis, Ind. Eng. Chem. Res., 2008, 47, 7623–7630 CrossRef CAS.
- J. P. E. Cleeton, C. D. Bohn, C. R. Müller, J. S. Dennis and S. A. Scott, Int. J. Hydrogen Energy, 2009, 34, 1–12 CrossRef CAS.
- D. Hosseini, F. Donat, S. M. Kim, L. Bernard, A. M. Kierzkowska and C. R. Müller, ACS Appl. Energy Mater., 2018, 1, 1294–1303 CrossRef CAS.
- W.-C. Huang, Y.-L. Kuo, P.-C. Su, Y.-H. Tseng, H.-Y. Lee and Y. Ku, Chem. Eng. J., 2018, 334, 2079–2087 CrossRef CAS.
- Q. Imtiaz, N. S. Yüzbasi, P. M. Abdala, A. M. Kierzkowska, W. van Beek, M. Broda and C. R. Müller, J. Mater. Chem. A, 2016, 4, 113–123 RSC.
- A. Kierzkowska, C. Bohn, S. Scott, J. Cleeton, J. Dennis and C. Muller, Ind. Eng. Chem. Res., 2010, 49, 5383–5391 CrossRef CAS.
- W. Liu, J. S. Dennis and S. A. Scott, Ind. Eng. Chem. Res., 2012, 51, 16597–16609 CrossRef CAS.
- C. Müller, C. Bohn, Q. Song, S. Scott and J. Dennis, Chem. Eng. J., 2011, 166, 1052–1060 CrossRef.
- J. B. Yang, N. S. Cai and Z. S. Li, Energy Fuels, 2008, 22, 2570–2579 CrossRef CAS.
- N. S. Yüzbasi, A. Kierzkowska and C. Müller, Energy Procedia, 2017, 114, 436–445 CrossRef.
- N. S. Yüzbasi, A. M. Kierzkowska, Q. Imtiaz, P. M. Abdala, A. Kurlov, J. L. M. Rupp and C. R. Müller, J. Phys. Chem. C, 2016, 120, 18977–18985 CrossRef.
- Z. Ma, S. Zhang and R. Xiao, Energy Convers. Manage., 2019, 188, 429–437 CrossRef CAS.
- M. Ishida, K. Takeshita, K. Suzuki and T. Ohba, Energy Fuels, 2005, 19, 2514–2518 CrossRef CAS.
- C. Chung, L. Qin, V. Shah and L.-S. Fan, Energy Environ. Sci., 2017, 10, 2318–2323 RSC.
- W. Liu, M. Ismail, M. T. Dunstan, W. Hu, Z. Zhang, P. S. Fennell, S. A. Scott and J. S. Dennis, RSC Adv., 2015, 5, 1759–1771 RSC.
- M. Arjmand, A.-M. Azad, H. Leion, T. Mattisson and A. Lyngfelt, Ind. Eng. Chem. Res., 2012, 51, 13924–13934 CrossRef CAS.
- J. Adanez, A. Abad, F. Garcia-Labiano, P. Gayan and L. F. de Diego, Prog. Energy Combust. Sci., 2012, 38, 215–282 CrossRef CAS.
- Q. Imtiaz, P. M. Abdala, A. M. Kierzkowska, W. Van Beek, S. Schweiger, J. L. Rupp and C. R. Müller, Phys. Chem. Chem. Phys., 2016, 18, 12278–12288 RSC.
- A. Turnock and H. Eugster, J. Petrol., 1962, 3, 533–565 CrossRef CAS.
- K. J. Warren, J. T. Tran and A. W. Weimer, Energy Environ. Sci., 2022, 15, 806–821 RSC.
- C. L. Muhich, B. W. Evanko, K. C. Weston, P. Lichty, X. Liang, J. Martinek, C. B. Musgrave and A. W. Weimer, Science, 2013, 341, 540–542 CrossRef CAS PubMed.
- C. L. Muhich, B. D. Ehrhart, V. A. Witte, S. L. Miller, E. N. Coker, C. B. Musgrave and A. W. Weimer, Energy Environ. Sci., 2015, 8, 3687–3699 RSC.
- Q. Song, W. Liu, C. D. Bohn, R. N. Harper, E. Sivaniah, S. A. Scott and J. S. Dennis, Energy Environ. Sci., 2013, 6, 288–298 RSC.
- L. Liu and M. R. Zachariah, Energy Fuels, 2013, 27, 4977–4983 CrossRef CAS.
- J. Rodriguez-Carvajal, Phys. B, 1993, 192, 55 CrossRef CAS.
- M. Galib, M. Baer, L. Skinner, C. Mundy, T. Huthwelker, G. Schenter, C. Benmore, N. Govind and J. L. Fulton, J. Chem. Phys., 2017, 146, 084504 CrossRef CAS PubMed.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- E. P. Barrett, L. G. Joyner and P. P. Halenda, J. Am. Chem. Soc., 1951, 73, 373–380 CrossRef CAS.
- N. Zhu, F. Guo, S. Yan, L. Chen and A. Li, Acta Chim. Sin., 1992, 50, 527–532 CAS.
- W. A. England, A. J. Jacobson and B. C. Tofield, Solid State Ionics, 1982, 6, 21–27 CrossRef CAS.
- C. Peters, M. Bettman, J. t. Moore and M. Glick, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1971, 27, 1826–1834 CrossRef CAS.
- A. Omegna, R. Prins and J. A. van Bokhoven, J. Phys. Chem. B, 2005, 109, 9280–9283 CrossRef CAS PubMed.
- D. R. Neuville, L. Cormier, A.-M. Flank, V. Briois and D. Massiot, Chem. Geol., 2004, 213, 153–163 CrossRef CAS.
- R. Prins, J. Catal., 2020, 392, 336–346 CrossRef CAS.
- D. R. Neuville, D. De Ligny, L. Cormier, G. S. Henderson, J. Roux, A.-M. Flank and P. Lagarde, Geochim. Cosmochim. Acta, 2009, 73, 3410–3422 CrossRef CAS.
- Y. Kato, K.-i. Shimizu, N. Matsushita, T. Yoshida, H. Yoshida, A. Satsuma and T. Hattori, Phys. Chem. Chem. Phys., 2001, 3, 1925–1929 RSC.
- A. Marcelli, A. Mottana and G. Cibin, J. Appl. Crystallogr., 2000, 33, 234–242 CrossRef CAS.
- M. Digne, P. Raybaud, P. Sautet, D. Guillaume and H. Toulhoat, Phys. Chem. Chem. Phys., 2007, 9, 2577–2582 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d1ta10507h |
| This journal is © The Royal Society of Chemistry 2022 |