
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInducing atomically dispersed Cl–FeN4 sites for ORRs in the SiO2-mediated synthesis of highly mesoporous N-enriched C-networks†
Xiong
Zhang
a,
Lai
Truong-Phuoc
a,
Xuemei
Liao
*ab,
Vasiliki
Papaefthimiou
a,
Matteo
Pugliesi
c,
Giulia
Tuci
c,
Giuliano
Giambastiani
 *ac,
Sergey
Pronkin
*a and
Cuong
Pham-Huu
*a
*ac,
Sergey
Pronkin
*a and
Cuong
Pham-Huu
*a
aInstitute of Chemistry and Processes for Energy, Environment and Health (ICPEES), ECPM, UMR 7515 CNRS-University of Strasbourg, 25 Rue Becquerel, 67087 Strasbourg Cedex 02, France. E-mail: giambastiani@unistra.fr; sergey.pronkin@unistra.fr; cuong.pham-huu@unistra.fr
bSchool of Food and Biological Engineering, Xihua University, 610039, Chengdu, China. E-mail: xmliao@mail.xhu.edu.cn
cInstitute of Chemistry of Organometallic Compounds, ICCOM-CNR, Consorzio INSTM, Via Madonna del Piano, 10 – 50019, Sesto F. no, Florence, Italy. E-mail: giuliano.giambastiani@iccom.cnr.it
First published on 22nd November 2021
Abstract
Atomically dispersed iron sites within N-enriched C-networks are promising low-cost catalytic materials for electrochemical applications. At odds with their often-outstanding performance in challenging electrocatalytic processes (i.e. oxygen reduction reaction, ORR) their fabrication strategy frequently relies on trial-and-error approaches. Moreover, the complex chemical nature of these hybrids is often dictated by the use of highly aggressive etching/doping thermo-chemical treatments. Therefore, the development of simplified chemical protocols based on cheap and abundant raw materials ensuring highly reproducible synthetic paths with the prevalent generation of discrete single-atom sites in a definite coordination environment remains a challenging issue to be properly addressed. In this contribution, the synthesis of hierarchically porous and N-enriched C-networks prevalently containing Cl–FeN4 sites is proposed. The outlined procedure takes advantage of citrate ions as carriers for N-sites and a sacrificial C-source for the synthesis of N/C matrices. At the same time, the chelating character of citrate polyions fosters the complexation of transition metals for their ultimate atomic dispersion in C/N matrices. The procedure is finally adapted to the use of common inorganic hard templates and porogens for the control of the material morphology. Avoiding any thermo-chemical etching/doping phase, the as-prepared catalytic material has shown remarkably high ORR performance in an alkaline environment. With a half-wave potential (E1/2) of 0.88 V, a kinetic current density up to 109.6 A g−1 (normalized to the catalyst loading at 0.8 V vs. RHE) and outstanding stability, it largely outperforms commercial Pt/C catalysts and certainly ranks among the most performing ORR Fe-single-atom-catalysts (Fe-SACs) reported so far.
1. Introduction
The kinetically sluggish oxygen reduction reaction (ORR) is certainly among the electrochemical processes that gained steadily growing interest during the last few years from the catalysis and material chemistry community. It represents the central core of many energy-related transformations, and its tight dependence on the use of noble metal-based catalysts has seriously hampered any large-scale implementation and commercialization of energy storage and conversion devices such as fuel cells and metal–air batteries.1,2 The rate of the ORR at a cathodic compartment of an electrochemical cell can be several orders lower than that of the semi-reaction occurring at the anodic counterpart (i.e. hydrogen oxidation reaction – HOR) and their ultimate performance depends on the use of Pt or platinum group metal (PGM) alloys as electrocatalysts.3,4 The limited natural abundance of Pt and PGMs, their relatively high costs and moderate tolerance to alcohol (i.e. MeOH in alcohol cross-over phenomena) or gas impurities (i.e. CO) have extensively stimulated fundamental research towards alternative, costless and definitively more sustainable catalytic systems for the ORR process to occur.5–10 Earth-abundant, transition metal (TM)-based electrocatalysts represent an ideal solution to the replacement of costly and rarer PGM-based systems, provided that comparable or even higher performance in the process can be ensured. Of course, any transition towards a new catalysts' series needs that some other fundamental issues such as catalyst stability and durability can be fully addressed under varying operational conditions along with a reduction of energy wastes (generation of undesired by-products) while ensuring high process efficiency.Supported single-metal atom catalysts (SACs) have recently emerged as promising alternatives to classical TM-based heterogeneous systems within a series of challenging catalytic transformations, including several of those conventionally carried out by PGM-based catalysts.11–13 Carbon-based SACs are made of atomically dispersed active metal centers within inert carbonaceous supports featured by variable morphologies and eventually doped with light hetero-elements (i.e. N, O, and S). Their stability and catalytic performance are largely controlled by the coordination environment of metal ions embedded in the neighboring hetero-doped carbonaceous network. Such an atomistic approach to the design and synthesis of heterogeneous catalysts has unveiled unique opportunities to process intensification and their selectivity improvement other than offering valuable solutions to the drastic reduction of the active metal loadings.14–18
At odds with the deep interest aroused by SACs, as witnessed by the exponential growth of manuscripts and review articles published in the last few years, their fabrication strategy has often relied on trial-and-error approaches, where the complex chemical nature of these hybrids was frequently dictated by high-temperature etching/doping thermal phases if not by the thermal decomposition of costly and time-consuming precursors. Seminal examples of highly catalytically active materials in the form of atomically dispersed metal ions within complex carbonaceous networks were obtained by the carbonization of sophisticated coordination compounds such as metal–organic frameworks (MOFs),19–21 metal–organic polymers22,23 and a mixture of coordinating polymers with N-containing molecules.24,25 On a similar ground, severe chemically conditions (i.e. pyrolysis under NH3, HCN or CH3CN atmosphere) for the etching/doping of preformed C-networks or strong-acid leaching treatments for the etching/removal of unstable phases26,27 have often been used to obtain highly performing SACs or for improving their electrochemical performance.27–35
In spite of these seminal achievements, milder chemical solutions employing costless reagents remain challenging issues to be addressed for the large-scale synthesis and implementation of these materials. Both this problem poses serious concerns to any practical exploitation of SAC technology. This is even more relevant if considering the limits of such hard synthetic strategies where both density and identity of the SA metal active phases are scarcely controlled and the catalysts produced rarely contain only discrete single-atom sites in a definite coordination environment.
We have recently described an effective and general strategy for the production of highly metal loaded and atomically dispersed Fe-based composites as performing electrocatalysts for the oxygen reduction reaction, starting from cheap and food-grade raw components in combination with key thermo-chemical etching/doping phases (a NH3 atmosphere).27 The employed methodology took advantage from a proprietary synthetic procedure originally applied to the preparation of N-enriched mesoporous carbon-based 1D–3D networks36–38 or organic–inorganic composites39–41 to be employed as metal-free catalysts for a wide series of industrially relevant transformations. Accordingly, Fe-SAs were prepared with D-glucose (C6H12O6) as the main C-source, ammonium carbonate [(NH4)2CO3] as the N-source and citric acid (C6H8O7) as a sacrificial carrier for Fe3+ and NH4+ ions. The presence of a potentially tridentate chelating agent (i.e. citrate ions) was regarded as an “open gate” for water-soluble metal ions to be accommodated within a C–N network. To make a step-forward and prepare highly mesoporous and N-doped C-networks containing discrete FeNx sites, we propose herein a deeply simplified, costless and general synthetic procedure that does not include any thermo-chemical etching/doping material treatment (NH3, CH3CN or HCN), adapted to the use of common inorganic hard templates (i.e. SiO2 nanospheres) and porogens (i.e. ZnCl2) for the control of the ultimate material morphology. The effect of macro- and meso-porous networks on the ORR performance of Fe–N–C SACs in electrocatalysis has already been a seminal precedent in the literature. While mesopores were claimed to be key channels for the reaction flow transit and for the electrolyte wetting of the whole catalyst surface area, macropores largely reduced pressure drop phenomena and facilitated the reagent accessibility to the active sites.42
To this aim, citric acid was employed as a metal ion chelating agent, N-carrier and main sacrificial C-source for the production of the final composite. To provide with a higher extent of mesoporosity, we easily combined our methodology with the use of a hard template in the form of SiO2 nanospheres and ZnCl2 salts. While the former was thought to promote the generation of hierarchically porous structures,43,44 the latter was employed to prevent the undesired generation of iron carbide NPs while fostering the generation of micropores.44
As a result, the optimized catalyst exhibited a homogeneous and relatively dense distribution of iron single-site moieties whose nature was unambiguously established on the basis of PXRD, HAADF-STEM and XAS analyses. The latter suggested the dominating presence of FeN4 moieties within a distorted square-pyramidal coordination geometry containing an axial chlorine atom. The catalyst prepared from a cheap reagent mixture and without undergoing any costly and risky thermo-chemical treatment (i.e. annealing under a reactive atmosphere, NH3, CH3CN or HCN) showed easy and complete reproducibility other than outstanding activity, stability and alcohol tolerance when employed in ORRs in an alkaline environment. It certainly ranks among the most performing state-of-the-art ORR Fe-SAC catalysts, and the role of the hard silica template along with that of ZnCl2 salt has been thoroughly discussed along the contribution.
2. Experimental section
2.1 Materials and chemicals
Citric acid [C6H8O7 anhydrous, >99.5%, MW: 192.12 g mol−1] and ammonium carbonate [(NH4)2CO3, MW: 96.09 g mol−1] were provided by Fisher Chemicals and ACROS Organics, respectively. Zinc chloride (ZnCl2, anhydrous, >98%; MW: 136.286 g mol−1) and silica (SiO2, fumed nanospheres, ∅ mean size: 7 nm) were purchased by Sigma-Aldrich. Iron(III) chloride hexahydrate (FeCl3·6H2O; >98%, MW: 270.296 g mol−1) and Nafion® (5 wt% in isopropanol/water) were obtained from Alfa Aesar and Fluka, respectively. Unless otherwise stated, all reagents and solvents were used as provided by commercial suppliers without any further purification/treatment.2.2 Material synthesis
In a typical procedure, 2 g of citric acid (10.4 mmol) and 5 g of (NH4)2CO3 (52 mmol) were dissolved in 25 mL ultrapure water (Veolia Ultra Analytique, 18.2 MΩ cm, TOC < 2 ppb). Afterwards, the solution was treated under vigorous stirring with 1.2 g of silica and a uniform gel suspension was formed. FeCl3·6H2O (0.024 g, 0.087 mmol) and ZnCl2 (0.2435 g, 1.75 mmol) [Fe![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Zn = 1:20 mol mol−1 and roughly 1:10 w/w] were then dissolved in 3 mL of distilled water and the resulting solution was added dropwise to the above-mentioned colloidal suspension. After stirring for 30 minutes, the mixture was transferred to an oven and dried at 110 °C overnight. The resulting solid was then crushed into a fine powder and calcined at 900 °C for 2 h in an Ar atmosphere.
:Zn = 1:20 mol mol−1 and roughly 1:10 w/w] were then dissolved in 3 mL of distilled water and the resulting solution was added dropwise to the above-mentioned colloidal suspension. After stirring for 30 minutes, the mixture was transferred to an oven and dried at 110 °C overnight. The resulting solid was then crushed into a fine powder and calcined at 900 °C for 2 h in an Ar atmosphere.
The as-obtained solid was suspended in 120 mL of a 2 M NaOH solution and maintained under stirring at 80 °C for 8 h to remove the silica template. The solid recovered by successive water-washing/centrifugation/drying steps underwent a second calcination step at 900 °C for 2 h in an Ar atmosphere. The collected material was stored at room temperature in an ambient atmosphere and named hereafter as FeZn/CNP (1). For comparison, two additional samples were prepared following the above-described procedure except for the use of ZnCl2 or SiO2 in the mixture. These samples were indicated as Fe/CNP (2) (without ZnCl2 in the mixture) and FeZn/CN (3) (without SiO2 template), respectively.
2.3 Characterization techniques
Scanning electron microscopy (SEM) was carried out using a Zeiss 2600F with a resolution of 5 nm. Samples were deposited onto a double-face graphite tape in order to avoid charging effects during the measurements. The loading amount of Fe was determined by inductively coupled plasma-atomic emission spectroscopy (ICP-AES, Optima 2000 PerkinElmer Inductively Coupled Plasma (ICP) Dual Vision instrument). Transmission electron microscopy (TEM) was carried out using a JEOL 2100F working at 200 kV accelerating voltage, equipped with a probe corrector for spherical aberrations and a point-to-point resolution of 0.2 nm. X-ray photoelectron spectroscopy (XPS) measurements were performed using an ultrahigh vacuum spectrometer equipped with a RESOLVE 120 MCD5 hemispherical electron analyzer. The Al Kα line (Al Kα, hv = 1486.6 eV) of a dual anode X-ray source was used as the incident radiation. The constant pass energy mode with pass energies of 100 and 20 eV was used to record survey and high-resolution spectra respectively. X-ray diffractogram (XRD) patterns were recorded with Cu Kα radiation (40 mA, 45 kV) in the 10–80° 2θ range, using a step size and step time of 0.05° and 80 s, respectively. The Raman spectra were recorded using a LabRAM ARAMIS Horiba Raman spectrometer equipped with a Peltier cooled CCD detector. A laser line (532 nm/100 mW (YAG) with a Laser Quantum MPC600 PSU) was used to excite the sample. X-ray absorption spectroscopy (XAS) measurements were carried out in the fluorescence mode at the BL14W1 station of the Shanghai Synchrotron Radiation Facility (SSRF, 3.5 GeV, 250 mA in maximum, Si (311) double crystals). IFEFFIT software packages were used to process the data. The extended X-ray absorption fine structure (EXAFS) contribution was separated from different coordination shells using a Hanning window (dk = 1.0 Å−1).2.4 Electrochemical measurements
Each catalytic material was prepared in the form of a Nafion-based ink to be drop-casted/evaporated on a glassy carbon support of a RD or RRD electrode. In a typical procedure, 5 mg of the catalyst (powder) was dispersed in a solution containing 750 μL ultrapure water, 250 μL isopropanol and 50 μL Nafion (5 wt% in isopropanol/water). The resulting mixture was then sonicated for 30 min to get a homogeneous black ink. Afterwards, 16 μL of the ink was drop-casted onto the glassy carbon surface of the electrode (RDE Metrohm, 5 mm in diameter or RRDE PINE AFE6R2GCPT, a glassy carbon disk with a diameter of 5 mm, a Pt ring with an inner diameter of 6.5 mm and an outer diameter of 7.5 mm), and left to dry under ambient conditions. By this way, a dried electrode (working electrode, WE) charged with 377 μg cm−2 of catalyst was employed for electrochemical measurements in a three-electrode cell, using a Ag/AgCl electrode and a graphite rod as the reference electrode (RE) and the counter electrode (CE), respectively. The amount of 377 μg cm−2 was fixed for all catalytic materials (1–3) under study, irrespective to their different Fe-loadings determined by ICP-AES analysis. The catalyst loading was calibrated in accordance with the highest number of exchanged electrons (nE) measured in ORRs with the model catalyst 1 (FeZn/CNP, vide infra). Two compartments separated by a porous glass membrane were used for housing the WE and CE, respectively. All potentials were then calibrated to the reversible hydrogen electrode (RHE) according to the following equation: ERHE = EAg/AgCl + 0.197 + 0.0592pH. Electrochemical measurements were finally performed using a Bio-Logic SP-300 potentiostat and the EC-Lab 11.32 software. Linear sweep voltammetry (LSV) was conducted in a 0.1 M O2-saturated KOH electrolyte at a scan rate of 10 mV s−1 and a rotating rate of 1600 rpm. For comparison, a standard Pt/C electrode (20 μg cm−2 of Pt on Vulcan XC-72, E-TEK, Inc. USA) was used as the benchmark catalytic system in the electrochemical tests. The accelerated durability tests (ADT) were measured by cycling the catalysts between 0.6 and 1.0 V at a scan rate of 50 mV s−1 in an O2 atmosphere. The chronoamperometry response was collected by polarizing the catalyst at a rotating speed of 1600 rpm at 0.65 V (vs. RHE). Methanol poisoning experiments were performed in a 0.1 M O2-saturated KOH by the addition of 1% (v/v) of MeOH. With RDE, the number of electrons involved in the oxygen reduction reaction (ORR) was determined by the Koutecky–Levich equation (eqn (1)) as follows: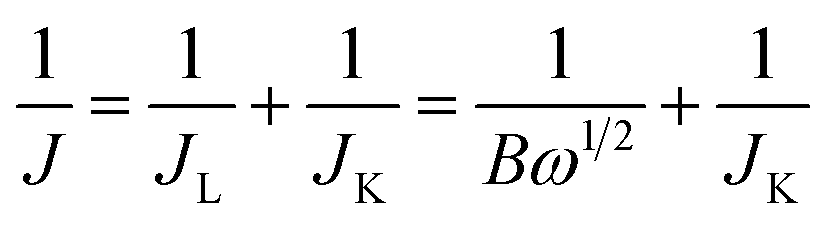 | (1) |
| B = 0.62nFCODO2/3υ−1/6 |
485 C mol−1), CO the bulk concentration of O2 (1.2 × 10−6 mol cm−3), DO the diffusion coefficient of O2 in 0.1 M KOH (1.9 × 10−5 cm2 s−1), and υ the kinetic viscosity of the electrolyte (0.01 cm2 s−1).
For rotation ring-disk electrode (RRDE) tests, the working electrode (PINE AFE6R2GCPT, a glassy carbon disk with a diameter of 5 mm, a Pt ring with an inner diameter of 6.5 mm and an outer diameter of 7.5 mm) was prepared with the same catalyst loading while the ring potential was constantly kept at 1.2 V vs. RHE. The number of electron transfer and HO2− yield were evaluated using the following equations (eqn (2) and (3)):
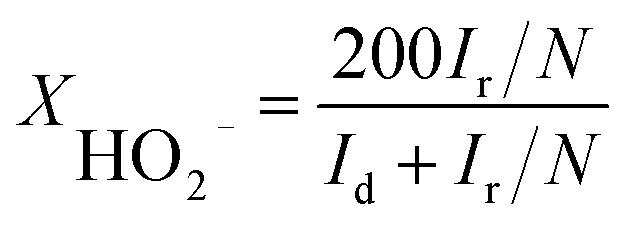 | (2) |
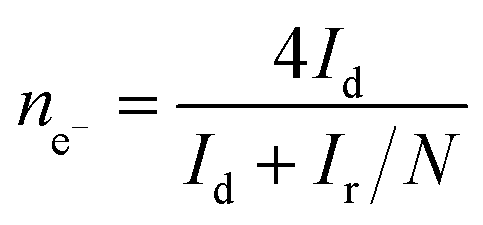 | (3) |
3. Results and discussion
3.1 Synthesis of FeZn/CNP catalyst (1) and their ZnCl2-free (Fe/CNP, 2) or SiO2-free (FeZn/CN, 3) counterparts
The synthetic procedure of new SACs takes advantage from the chelating action of the citrate polyion and its ability to generate relatively stable and water-soluble complexes with a high variety of transition metals.45 In our synthetic scheme, citric acid plays a three-fold role: it represents the unique sacrificial C-source for the preparation of N-enriched C-based networks; it is the molecular carrier for harvesting NH3 released from the (NH4)2CO3 decomposition under the form of ammonium citrate [HO–C3H4(COO)3Hx(NH4)3−x] (x = 0–3); it allows the complexation of transition metal ions (i.e. Fe3+ and Zn2+) and their convey within the N-doped C-material (Scheme 1). SiO2 is an already well-known hard template for the pyrolytic generation of mesoporous C-networks.44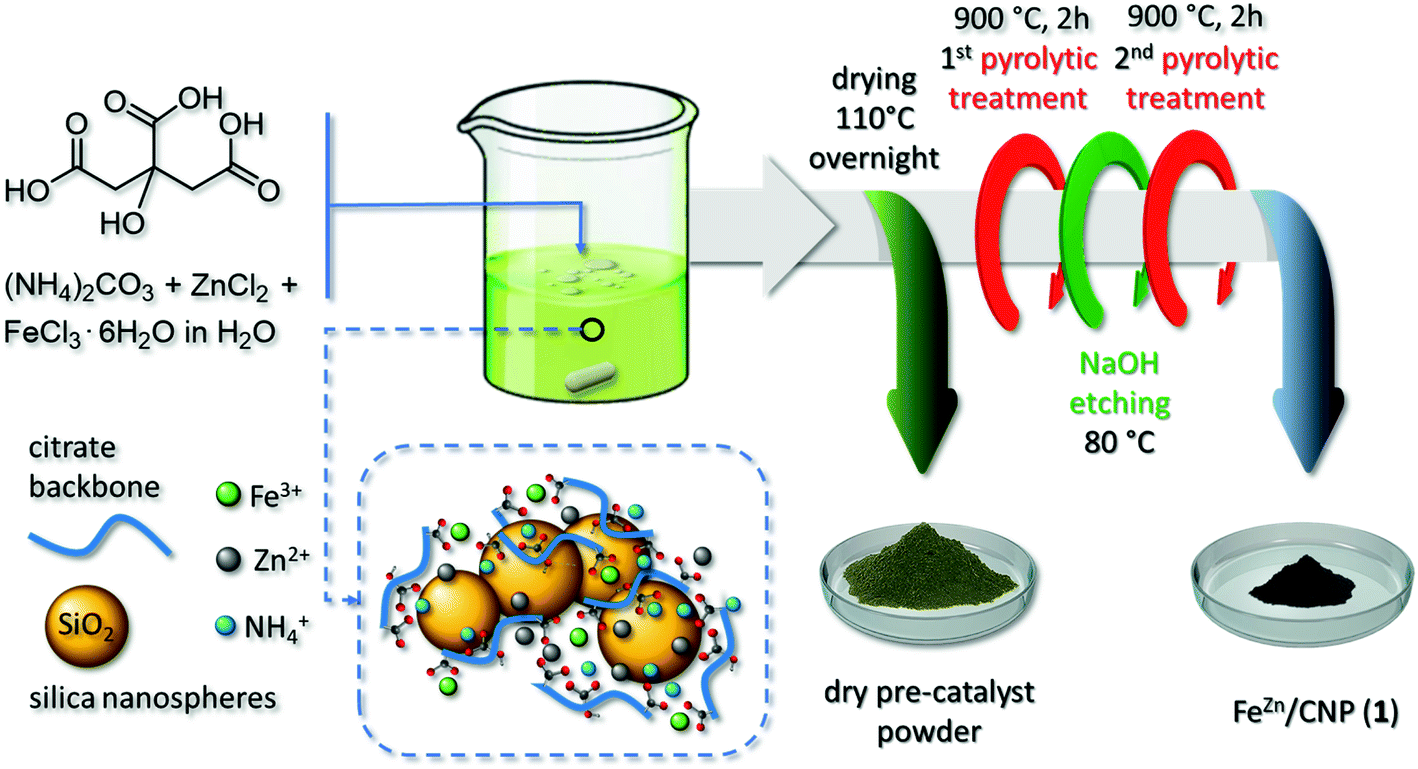 | ||
| Scheme 1 Synthetic procedure for the preparation of the atomically dispersed FeZn/CNP (1). | ||
The reaction of citric acid and (NH4)2CO3 was accompanied by a vigorous CO2 effervescence during which NH4+ ions were trapped in the form of tri-, di- or mono-basic ammonium citrates.36,37 Afterwards, Zn2+ and Fe3+ salts were added under stirring as a unique water solution. The use of a zinc salt in the synthesis of Fe-SACs is equally well known to prevent the generation of iron carbide (Fe3C) species during material pyrolysis and to act as a porogen by favoring the generation of micropores.44
The pH value of the resulting colloidal suspension was ≈8.5, hence largely over that of the isoelectric point of a SiO2 aqueous suspension (pH ≈ 2.5). It implied a sticky interaction between the negatively charged surfaces of silica nanospheres and the ammonium-rich citrate frameworks. The suspension was then evaporated to dryness (110 °C overnight), to afford a yellow-green powdery sample (Scheme 1). The latter underwent a first pyrolysis treatment (900 °C, Ar atmosphere) during which a black powder of the desired pre-catalyst was formed. Its subsequent treatment with a 2 M NaOH solution at 80 °C was employed to etch and remove the silica template46 before submitting the sample to a second pyrolysis step (see the Experimental section for details). During the thermal phases, Zn was vaporized leaving behind micropore channels while Fe atoms remained trapped in the C/N matrix as stable FeNx moieties (vide infra). The as-prepared mesoporous catalyst was the result of a long optimization procedure where variable citric acid/(NH4)2CO3 ratios and Fe3+/Zn2+ ratios were used. A complete account of the properties of materials obtained using various reagent ratios will be discussed elsewhere; the reagent composition described herein was selected as the best compromise between chemical composition, Fe-loading and morphological properties of SACs. The catalyst was arbitrarily named FeZn/CNP (1), where Zn apex indicates the combined use of a Zn salt while CNP stands for a C- and N-containing porous network, and it was thoroughly characterized. To address the role of the Zn salt and that of SiO2 nanospheres in the ultimate properties of 1, two new Fe-containing materials were prepared following the above-described synthetic procedure except for the use of ZnCl2 (Fe/CNP, 2) or the silica template (FeZn/CN, 3).
3.2 Catalyst characterization
All as-prepared materials (1–3) were initially analyzed with respect to their specific surface area (SSA) and pore-size distribution by N2 physisorption analysis at the temperature of liquid nitrogen (T = 77 K). A comparative analysis of these data was used to assess the role of SiO2 as the hard template and ZnCl2 as the pore forming agent in the process. As Fig. 1A and Table 1 show, samples 1 and 2 display largely superimposable (type IV) isotherm profiles featured by moderate H3 hysteresis loop, typical of mesoporous structures with slit-shaped pores.47 Both samples obtained from the silica nanosphere as the hard template showed a relatively high SSA (from 828 to 926 m2 g−1) along with a largely prevalent mesoporous nature.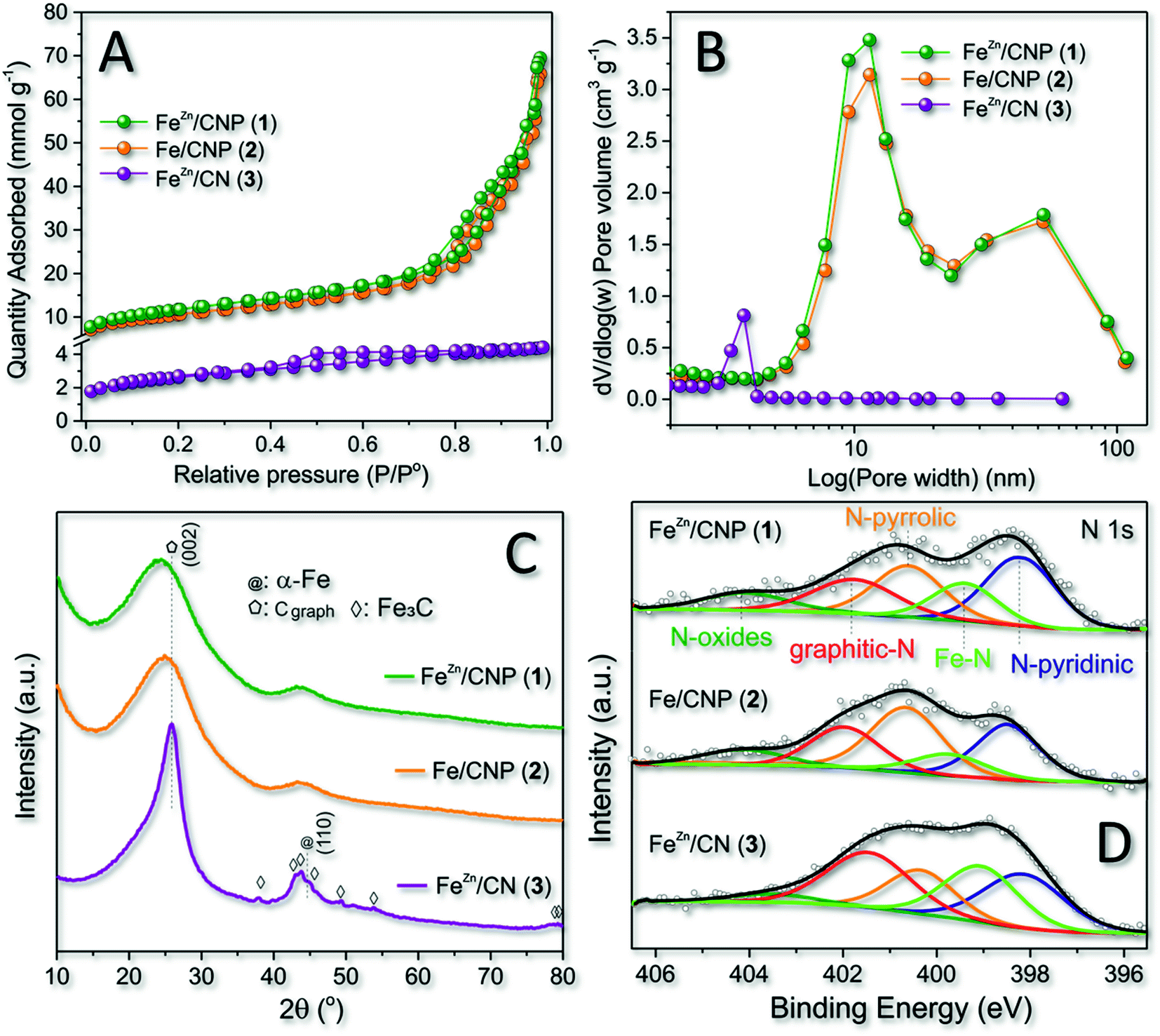 | ||
| Fig. 1 (A) N2 adsorption–desorption isotherm linear plots of FeZn/CNP (1) (green curve), Fe/CNP (2) (orange curve) and FeZn/CN (3) (violet curve) recorded at 77 K along with (B) the respective pore-size distributions (BJH method). (C) Wide-angle XRD profiles of 1–3. (D) XPS spectra of the high-resolution N 1s core region of 1–3 with relative curve fitting. | ||
| Entry | Sample | SSAa (m2 g−1) | Total pore volumeb (cm3 g−1) | Micropore volumec (cm3 g−1) | Average pore sized (nm) | I D/IGe | Fef wt% | XPS data | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N speciesh (at%) | |||||||||||||
| Ng at% | Pyridinic | Pyrrolic | Graphitic | Oxidized | Fe–N | ||||||||
| a Brunauer–Emmett–Teller (BET) specific surface area (SSA) measured at T = 77 K. b Total pore volume determined using the adsorption branch of N2 isotherm at P/P0 = 0.98. c Determined by t-plot method. d Determined by BJH desorption average pore width (4V/A). e From Raman spectra fitted with Lorentzian peaks (Fig. S1). f Determined by ICP analysis on the properly digested samples. g Determined by XPS analysis. h Determined by deconvolution peaks of the high resolution XPS N 1s core region. | |||||||||||||
| 1 | FeZn/CNP (1) | 925 | 2.40 | 0.09 | 13.3 | 4.04 | 0.40 | 6.35 | 30.8 | 23.3 | 18.6 | 9.5 | 17.8 |
| 2 | Fe/CNP (2) | 828 | 2.28 | 0.09 | 14.0 | 4.17 | 0.43 | 6.84 | 28.5 | 29.7 | 21.0 | 9.6 | 11.2 |
| 3 | FeZn/CN (3) | 204 | 0.15 | 0.02 | 3.3 | 2.36 | 1.85 | 3.1 | 25.2 | 17.9 | 27.9 | 4.4 | 24.6 |
It should be noticed that mesopores were certainly responsible for the slightly higher SSA value measured on sample 1. While the role of the SiO2 template on the SSA and pore-size distribution of samples 1 and 2 was evident and definitively clear by comparing these material features with those of sample 3 (Table 1, entries 1, 2 vs. 3 and Fig. 1B), the role of Zn-salt as a porogen for the generation of micropores appeared under our condition less important if not negligible.44 Indeed, the micropore component in 1 (with Zn) was even less relevant than that in 2 (without Zn; Table 1, entry 1 vs. 2). However, where the SiO2 template did not drive the final material porosity (i.e. sample 3), a carbon sample featured by small mesopores (close to the micropore region) was formed (Fig. 1B).
Wide-angle XRD profiles of the three samples at comparison (Fig. 1C) added further hints to the comprehension of the role played by SiO2 and ZnCl2 on the composition of 1–3. At a first glance, the absence of the mesoporous template in 3 contributed to more extensive material graphitization. Indeed, XRD of 3 presented a relatively strong peak around 26°, corresponding to the (002) facets of graphite. At the same time, peaks located at 37.8, 43.9, 45.0, 46.0, 49.2, and 54.5° were unambiguously indexed to Fe3C species.48 At odds with 3, samples 1 and 2 presented very similar XRD patterns with two broad and distinctive peaks at ≈24 and ≈44°, typically indexed as [0 0 2] and [1 0 0] diffractions of mesoporous carbon samples.49 Notably, neither 1 nor 2 seemed to be contaminated by the presence of Fe3C as well as no peaks associated with other Fe metal phases were observed. Our experimental evidence resized the role of ZnCl2 as both the porogen and the co-reagent for preventing the generation of Fe3C species during the material thermal phases, while the use of SiO2 seemed to hold a key role on the control of both material properties. Indeed, samples 1 and 2 prepared in the presence of SiO2 used as the hard template possessed similar morphologies (SSA and pore-size distribution) and iron phase distributions, apparently under the form of atomically dispersed nuclei (vide infra). However, the Fe3C phase was detected in 3 despite the use of ZnCl2 in the mixture. Raman spectra of 1–3 (Fig. S1†) were additionally recorded for the sake of completeness to provide further details on the structural defect degree of differently synthesized mesoporous carbon networks. As Fig. S1† shows, all sample profiles were fitted by four Lorentzian peaks50 with two dominating components at 1596 cm−1 and 1352 cm−1, ascribed to G (graphitic) and D (disordered) bands of carbonaceous samples, respectively.51 The ratio of integrated intensity of the D and G bands (ID/IG) is classically claimed as a descriptive tool of the density of structural defects in C-based materials: the lower the ID/IG ratio, the higher the material graphitization degree. It can be inferred that samples 1 and 2 presented high disordered structures (see the ID/IG ratio in Table 1), whereas the material produced without SiO2 (3) showed a higher graphitization extent. This latter evidence is in good accordance with the XRD results (Fig. 1C), and it was also taken as an indirect proof of the essentially atomic dispersion of iron ions in 1 and 2. Indeed, where larger iron aggregates were formed (3), they could initiate metal-catalyzed graphitization paths during the high-temperature pyrolysis steps.52–54
XPS survey spectra (Fig. S2† and Table 1) of 1–3 were acquired to quantitatively provide the N at% content in each sample. Deconvolution of the high-resolution profile of 1–3 at the N 1s core region was then used to establish the nature of different N-configurations available (Fig. 1D and Table 1) as well as to highlight the presence of distinctive metal–N coordination modes. Five components were assigned in the 398.2–404.2 eV range with those at 398.4 ± 0.2 (N-pyridinic) and 400.7 ± 0.1 eV (N-pyrrolic) being the most intense for samples 1–2. As Fig. 1D shows, deconvolution pointed out an additional component at 399.4 ± 0.1 eV that was attributed to iron-coordinated N sites (Fe–Nx) in good accordance with seminal literature precedents.43,54–56 As far as the iron loading in samples 1–2 was concerned, any attempt to get a semi-quantitative estimation based on XPS analysis failed due to the presumably very low metal charge. ICP-AES measurements made this possible and the measured iron content was 0.40, 0.43 and 1.85 wt% for samples 1, 2 and 3, respectively (Table 1).
Overall, 1 remains the sample combining the higher SSA with a supposed atomic distribution of the catalytically active sites. Moreover, the use of ZnCl2 in the mixture was found to appreciably increase (≈40% with respect to sample 2) the relative percentage of Fe–N active sites density in the final composite (Fig. 1D and Table 1).57 For these reasons, 1 was selected as the catalyst for all main characterizations and catalytic trials discussed hereafter.
The morphology of 1 was examined by scanning electron microscopy (SEM, Fig. 2A and S3†) and transmission electron microscopy (TEM, Fig. 2B, D and S3†). As SEM images showed, the sample presented a homogeneous 3D hierarchical flower-like morphology made of highly intertwined and thin graphite-like sheets. TEM analysis (Fig. 2B and D) recorded at different magnifications revealed the presence of abundant pores with an average diameter between 8 and 12 nm, consistent with the average size of the nanosphere silica template. No evidence for the presence of Fe-clusters was given on the whole sample scanned area. However, high-angle annular dark-field scanning TEM (HAADF-STEM) images (Fig. 2E and F) revealed the presence of bright spots (highlighted in Fig. 2F) assigned to heavier Fe atoms in the C/N matrix. At the same time, EDX elemental mapping recorded for 1 (Fig. 2C(a–d) and S4†) showed the homogeneous distribution of C (red), N (yellow), O (light blue) and Fe (pink) elements in the sample.
 | ||
| Fig. 2 (A) SEM images of 1. (B and D) TEM images of 1 at different magnifications. (E and F) HAADF-STEM images of 1 at different magnifications. C(a–d) EDX elemental mapping for sample 1. Color codes: C (red), N (yellow), O (light blue) and Fe (pink). (F) Highlights the bright spots assigned to heavier Fe atoms within green circles. | ||
The SEM images of samples 2 and 3 are also given as the ESI† material for completeness. As Fig. S5† shows, the morphology of 2 was not so different from that of 1 in accordance with the role exerted on both samples from the silica template as well as the similar SSA and pore-size distribution values (Fig. 1A and B). As far as the morphology of 3 was concerned, the absence of the SiO2 nanosphere as the sacrificial template gave rise to substantially flat flakes of the C/N composite featured by moderate mesoporosity, SSA and pore-size distribution (Fig. 1A and B).
The material characterization acquired for 1 was consistent with previous analyses (XPS and XRD) and thus with an atomistic distribution of the iron particles in the N-doped carbonaceous network. XPS also suggested the existence of iron species within an N-coordination sphere (Fe–Nx). To gain additional insights into the nature and coordination environment of iron atoms in 1, we also performed XAS spectroscopy. Fig. 3A refers to the X-ray absorption near edge structure spectra (XANES) at the Fe K edge of 1 along with that of three common Fe0, Fe2+ and Fe3+ species (i.e. metallic Fe foil, FeO and Fe2O3) in comparison. It has been demonstrated that the position of pre-edge (1s → 3d transition) and edge (1s → 4p transition) resonances were sensitive to the iron oxidation state, whereas the intensity of the pre-edge peak was related to the coordination symmetry of FeNx species. In particular, the higher the intensity of 1s → 3d transition, the lower the symmetry around the Fe sites.58 As far as the iron oxidation state was concerned, it should be noticed that the absorption edge transition (1s → 4p) of 1 was largely superimposable to that of the Fe2O3 reference sample, thus indicating a prevalent +3 valence for the isolated Fe atoms.59 This was in accordance with previous reports where the pyrolysis of Fe-containing organic precursors led to the generation of iron sites in their higher valence state.60,61
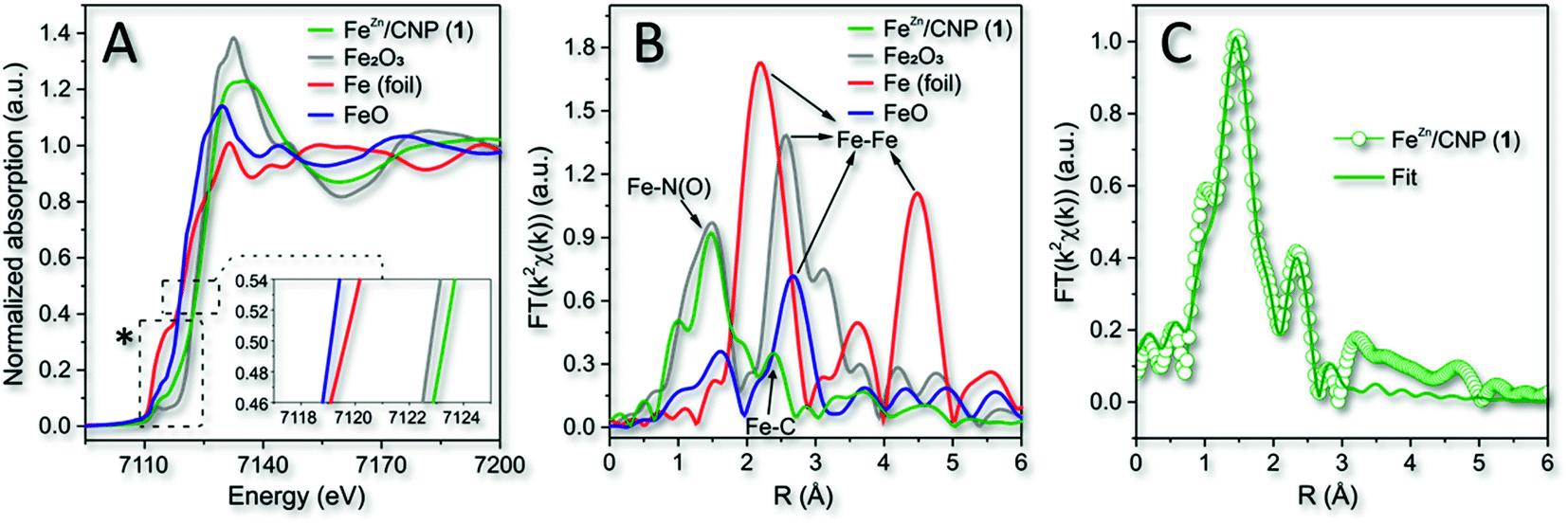 | ||
| Fig. 3 (A) Normalized XANES spectra at the Fe K-edge of 1, metallic Fe foil, FeO and Fe2O3. Inset refers to the enlargement of the main edges. *See Fig. S6A† for details on pre-edge peaks. (B) k2-weighted Fourier transform of EXAFS data. (C) EXAFS fitting curve of 1 in R space. No phase-shift correction was applied to the Fourier transforms. | ||
The phase-uncorrected Fourier transform extended X-ray absorption fine structure (FT-EXAFS, Fig. 3B) of the samples in comparison exhibited for 1 a prominent peak at 1.47 Å, assigned to Fe–N(O) in the first coordination shell, while another peak centered at 2.39 Å was ascribed for the same sample to the Fe–C distance of the second neighbor shell.62 Fe–Fe backscattering signals at 2.20 Å in the Fe foil and 2.58 Å, in FeO and Fe2O3 samples, respectively, were definitively absent in 1.60 This evidence was consistent with previous conclusions based on XRD and HAADF-STEM analysis.
Fe-phthalocyanine (FePc) is traditionally benchmarked as a FeN4 structure in a typical square-planar configuration with a high D4h symmetry.63 However, the latter shows a typical pre-edge peak at 7118 eV (due to a 1s → 4pz shakedown transition),63,64 definitively missing in 1 (Fig. S6A†). Pre-edge peak of 1 felt at 7114 eV instead, hence revealing a slight deviation from the highly symmetric D4h group.65 The curve-fitting of FT-EXAFS (Fig. 3C and Table S1†) on 1 contributed to add further hints to the iron coordination number and its chemical environment. It unveiled an average Fe-coordinated number of ≈3.4, thus suggesting a dominating Fe–N4 configuration in the system. Interestingly, an additional contribution to the metal coordination sphere was due to the axial coordination of a chlorine atom (Fe–Cl; Fig. 3C, S6B and Table S1†). Therefore, a distorted square-pyramidal coordination geometry of the type Cl–FeN4 similar to that occurring in the model iron(III) meso-tetraphenyl porphyrin chloride (FeTPPCl) was inferred for 1. Notably, both XANES and FT-EXAFS spectra of the model molecular complex FeTPPCl59,64,66 showed close similarities to the profiles recorded for 1 (Fig. 3A and B). All fitting parameters listed in Table S1† suggested for 1 the presence of Fe atoms prevalently coordinated to four N atoms at 1.97 Å and to one axial chlorine atom at 2.32 Å.
The atomically dispersed nature of 1 together with the Cl–FeN4-like structure of its active sites embedded in extensive and highly mesoporous carbonaceous networks and their well-known activity in challenging reduction processes,43,56,63–68 has prompted us to evaluate its performance in the model electrocatalytic oxygen reduction reaction (ORR).
3.3 Electrochemical performance of 1–3 as ORR catalysts in an alkaline environment
Cyclic voltammetry (CV) carried out in an Ar and O2 atmosphere was initially used to assess the ability of 1 to efficiently reduce dioxygen in an alkaline environment. Finally, 2 and 3 were scrutinized under identical electrochemical conditions (0.1 M KOH as the electrolyte at a scan rate of 50 mV s−1) for the sake of comparison. All CV spectra were finally plotted together in Fig. S7† for the sake of completeness. As Fig. S7† shows, all samples presented a distinct reduction peak only when CV was operated under O2-saturated conditions. Most importantly, 1 and 2 exhibited more pronounced ORR cathode peaks with 1 revealing a positive overpotential shift with respect to 2. Such a shift suggested the ability of 1 to start the ORR process at less reduction potentials. Afterwards, RRDE voltammograms of the three samples were registered under O2-saturated 0.1 M KOH electrolyte solution and compared with the benchmark Pt/C electrode (20 μg cm−2 of Pt on Vulcan XC-72, E-TEK, Inc, USA). For 1–3, the amount of the deposited catalyst was fixed to 377 μg cm−2 irrespective of the different Fe-loadings measured for each sample by ICP-AES analysis. The optimal loading was assumed for the model catalyst 1 (FeZn/CNP) in accordance with the highest number of exchanged electrons (nE) measured in a range of catalyst weights (see also Table S2†).Polarization curves are displayed in Fig. 4A, while the onset potential (Eon), half-wave potential (E1/2) and kinetic current density values at 0.8 V (JK @ 0.8 V) are detailed in Table 2. Noteworthy, 1 outperformed all catalytic materials from this series including the Pt/C reference electrode with whom it shared the Eon value only. E1/2 and JK were undoubtedly superior in 1. The latter also showed an overpotential of about 10 mV in Eon with respect to its counterpart 2 prepared without ZnCl2 and up to 60 mV with respect to 3 obtained without the use of the SiO2 as the hard template.
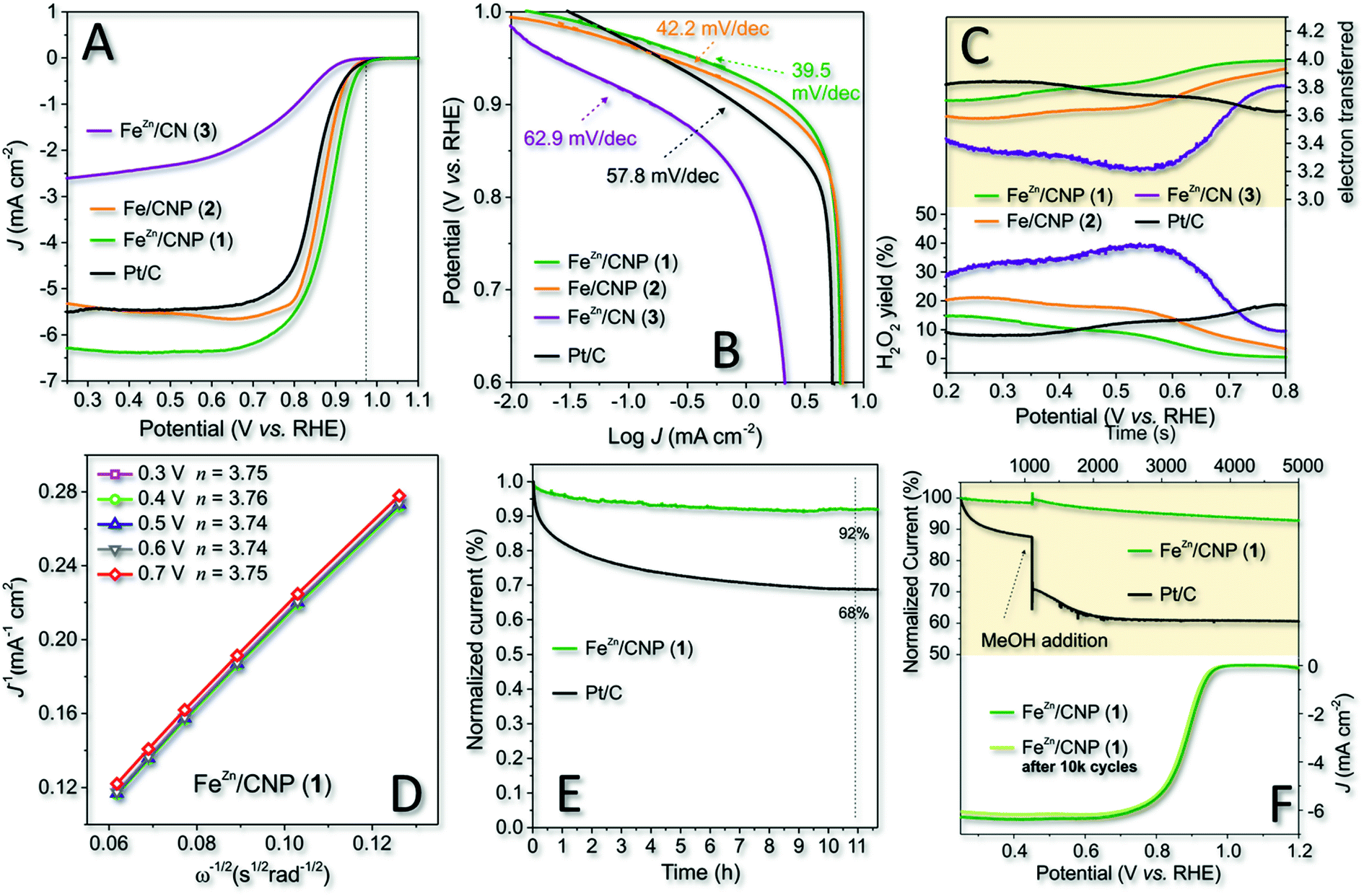 | ||
| Fig. 4 (A) RRDE polarization curves of 1–3 along with that of the benchmark Pt/C (20 μg cm−2 of Pt on Vulcan®), recorded in O2-saturated 0.1 M KOH electrolyte solution at a scan rate of 10 mV s−1 and a rotating electrode speed of 1600 rpm; (B) Tafel plots of 1–3 and Pt/C (20 μg cm−2 of Pt on Vulcan®); (C) electron transfer number and H2O2 yield plots of 1–3 and Pt/C as measured at a scan rate of 10 mV s−1 in an O2-saturated 0.1 M KOH solution (rpm = 1600). (D) K–L plots of 1 as obtained from the respective LSVs in the 0.3–0.7 V range; (E) chronoamperometric (CA) responses of 1 and Pt/C (20 μg cm−2 of Pt on Vulcan®) at comparison and at applied potential of 0.65 V (rpm = 1600); (F) methanol poisoning (upper side) and durability tests (bottom side) for 1. | ||
| Entry | Catalyst | E on (mV) | E 1/2 (V) | J L (mA cm−2) | Tafel slope (mV dec−1) | J K (mA cm−2) |
|---|---|---|---|---|---|---|
| a Onset potential values vs. RHE. b Half-wave potential values reported vs. RHE. c Limited diffusion current density. d Kinetic current density calculated from K–L equation at 0.80 V vs. RHE. | ||||||
| 1 | FeZn/CNP (1) | 970 | 0.88 | 6.35 | 39.5 | 41.31 |
| 3 | Fe/CNP (2) | 960 | 0.86 | 5.56 | 42.2 | 19.05 |
| 2 | FeZn/CN (3) | 910 | 0.77 | 2.45 | 62.9 | 1.8 |
| 4 | Pt/C | 970 | 0.85 | 5.44 | 57.8 | 25.98 |
At a first glance, the superior performance of 1 was attributed to the higher material specific surface area and porosity. Anyhow, its lower iron content with respect to catalysts 2 and 3 was also thought as a distinctive feature of this more performing system in ORRs. In spite of the resized role of ZnCl2 as the porogen in the mixture of 1 under our synthetic conditions, it certainly contributed to the control of the ultimate catalyst properties and performance in electrocatalysis by preventing the generation of Fe3C or iron clusters in general and favoring an increase of the SAFe active site density (see Table 1 and 1vs.2, XPS deconvoluted result). As a proof of concept, the comparative analysis of 1 and 2 pointed out the superior performance of the former in spite of its lower iron content.
The calculated kinetic current density values were as high as 41.31 mA cm−2 and 109.6 A g−1 as normalized values to the electrode geometrical area and catalyst loading, respectively. Both values were the highest measured for all investigated samples from this series. Overall, 1, besides ranking among the most performing state-of-the-art Fe-SACs ORR catalysts (Table S3†), represents an effective, cheaper, and definitively more sustainable alternative to commercial Pt-based systems for the process.
To gain additional mechanistic insights into the excellent catalytic activity of 1 in the ORR in an alkaline environment, we studied and compared the process kinetics of 1–3 and Pt/C from the respective Tafel plots (Fig. 4B). The results indicated that 1 exhibited a Tafel slope of 39.5 mV dec−1 whereas 2, 3 and commercial Pt/C showed slope values of 42.2, 62.9 and 57.8 mV dec−1, respectively. The Tafel slope variation reflects the rate-determining step (RDS) of the process and the lower the slope value, the higher the catalyst ORR kinetic. Accordingly, 1 confirmed its superior kinetic in the ORR compared to the other electrocatalysts under study. Literature precedents led us to think that the enhanced reaction kinetics of 1 was also associated with the distinctive nature of Cl–FeN4 single sites and the presence of an axial Cl atom.56,69 Li Y., Wang D. and co-workers have recently demonstrated how tuning the electronic structure of active iron centers by the near-range interaction with a coordinated chlorine atom significantly improved the catalyst ORR performance in an alkaline environment.43
From a mechanism viewpoint, a Tafel slope of 39.5 mV dec−1 suggested that the order of reaction was −1 in OH−, with a FeIII–OH/FeII–OH2 redox transition and a RDS associated with the generation of a superoxide FeII–O2− species.70
The ring current values measured at the Pt-ring of RRDE for 1–3 and Pt/C as electrocatalysts are summarized in Fig. 4C (bottom side), and they are expressed as the percentage of H2O2 produced by each catalyst as a function of the applied potential. Accordingly, the average number of electrons transferred (nE) for each electrocatalyst in the whole range of potential values was inferred (Fig. 4C; upper side). Noteworthy, the yield of H2O2 (<10%) produced with 1 at higher potential values (0.45–0.8 V) was even lower than that measured on the benchmark Pt/C catalyst. Moreover, 1 exhibited a prevalent 4e− reduction process within the whole range of potential values. Such a behavior at the higher potential values was consistent with a dominant inner-sphere electron transfer (ISET) mechanism at work. However, the relatively higher production of H2O2 by the Pt/C catalyst at higher potentials suggested the outer-sphere electron transfer (OSET) as an operative mechanism to a larger extent on the benchmark catalyst.71 As expected, 2 and 3 offered the worse electrochemical performance with a percentage comprised between 20% and 40% of H2O2 produced, respectively. Accordingly, nE was also constantly and appreciably lower than that measured for 1 in the whole scanned potential range.
Focusing on 1 as the most performing catalyst from this series, we confirmed the average number of electrons transferred per O2 molecule (nE) as evaluated in the diffusion and kinetically limited region (0.3 to 0.7 V vs. RHE) by means of the Koutecky–Levich (K–L) equation.72 Linear sweep voltammograms (LSVs) recorded for this catalyst at variable RRDE spin rates (600–2500 rpm) are outlined in Fig. S8,† while K–L plots obtained at different potential values (from 0.3 to 0.7 V) are shown in Fig. 4D. All curves presented excellent linearity, hence implying a first-order reaction toward dissolved O2. Accordingly, the calculated nE value per O2 molecule was about 3.75, hence approaching with 1 the ideal four-electron reduction process. LSVs, K–L plots and average number of exchanged electrons (nE) per O2 molecule have also been acquired/calculated for catalysts 2 and 3 under identical electrochemical conditions and reported in Fig. S8† for the sake of completeness.
Finally, 1 and Pt/C electrodes were subjected to stress conditions with the aim of investigating their stability under long-term electrochemical ORR runs. In a first experiment, the chronoamperometry (CA) response of both electrocatalysts was evaluated in an O2-saturated atmosphere at 0.65 V for 11 h (Fig. 4E). In addition, an accelerated durability test (ADT) was performed separately on 1 by long-term cycling the catalyst in a 0.1 KOH solution, in an O2-saturated atmosphere at 50 mV s−1 as the scan rate, between 0.6 and 1.0 V (vs. RHE) (Fig. 4F, bottom side). As far as CA was concerned, after 11 h continuous operational tests, 1 retained about 92% of its initial current density (J). Remarkably, the CA response of commercial Pt/C after the same time was up to 26% lower than that measured on 1, thus highlighting the superior stability of the latter. Similarly, the ADT test after 10000 cycles showed only a negligible shift of E1/2 in 1 towards negative values (≈5 mV), suggesting again the outstanding catalyst stability under operative conditions (Fig. 4F, bottom side).
As an additional trial, the tolerance of 1 towards alcohol poisoning was investigated. Methanol tolerance is a fundamental feature for the successful exploitation of electrocatalysts at the cathode of direct methanol fuel cells (DMFC).73 Catalyst poisoning in Pt-based electrocatalysts by alcohols (crossover) is a well-known phenomenon since a long time ago.74 Accordingly, electrochemical solutions used for the ORR with 1 and Pt/C were separately treated with a given amount of methanol (1% v/v; Fig. 4F, upper side). As Fig. 4F shows, the alcohol addition caused a sudden and irreversible decrease in the current density on Pt/C, whereas 1 maintained its electrochemical performance almost unchanged.
All these electrochemical trials taken together highlighted the remarkable catalytic activity, durability and tolerance of 1, which therefore holds great potential for the cost-effective development of highly efficient PGM-free electrocatalysts to be applied in the ORR.
4. Conclusion
To summarize, we described a simple, cheap and highly reproducible synthetic methodology for the production of hierarchically porous N-enriched C-networks containing atomically dispersed iron sites, prevalently (if not exclusively) in the form of Cl–FeN4 nuclei with the metal ion housed within a distorted square-pyramidal coordination sphere. The synthetic protocol takes advantage of the chelating action of the citrate polyion towards a variety of transition metals for their atomic convey within the final composite. At the same time, this simple organic building block holds the key role of sacrificial C-source and NH4+ ion carrier in the generation of N-rich C-networks. A further sacrificial template (fused SiO2 nanospheres) and a classical porogen (ZnCl2) have been finally used to control the morphology and SAFe active-sites density of the composite, respectively.The presence of a relatively high N-loading in the as-prepared material, obtained through the simple use of an inorganic leavening agent [i.e. (NH4)2CO3] without incurring into more risky and costly thermo-chemical treatments for the sample doping/etching, adds further relevance to the described protocol.
Nonetheless, the outstanding electrochemical performance of FeZn/CNP (1), its stability and durability as a catalyst for the oxygen-reduction reaction (ORR) in an alkaline environment have pointed out the great potential of a simple and cost-effective procedure for the development of highly efficient PGM-free electrocatalysts.
Looking beyond the nature and composition of the catalyst synthesized and employed in the present work, the proposed methodology remains of general application for a relatively wide series of transition metal ions that can undergo complexation/coordination by the citrate (carrier) polyion.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
X. Z. would like to thank the China Scholarship Council (CSC) for the PhD grant during his stay at ICPEES. G. G. and C. P.-H. thank the TRAINER project (Catalysts for Transition to Renewable Energy Future) of the “Make our Planet Great Again” program (Ref. ANR-17-MPGA-0017) for support. The Italian teams would also like to thank the Italian MIUR through the PRIN 2017 Project Multi-e (20179337R7) “Multielectron transfer for the conversion of small molecules: an enabling technology for the chemical use of renewable energy” for financial support to this work. X. L. thank the National Natural Science Foundation of China (No. 21706216) and the Sichuan Science and Technology Program (2020YFG0162) for support. S. P. finally thank the Institut Carnot MICA (COM-Gra 2020) for support.References
- M. K. Debe, Nature, 2012, 486, 43–51 CrossRef CAS PubMed.
- Q. Li, R. Cao, J. Cho and G. Wu, Adv. Energy Mater., 2014, 4, 1301415 CrossRef.
- S. Sui, X. Wang, X. Zhou, Y. Su, S. Riffat and C.-j. Liu, J. Mater. Chem. A, 2017, 5, 1808–1825 RSC.
- J. Zhang, Y. Yuan, L. Gao, G. Zeng, M. Li and H. Huang, Adv. Mater., 2021, 33, 2006494 CrossRef CAS PubMed.
- M. D. Bhatt and J. Y. Lee, Energy Fuels, 2020, 34, 6634–6695 CrossRef.
- X. Huang, T. Shen, S. Sun and Y. Hou, ACS Appl. Mater. Interfaces, 2021, 13, 6989–7003 CrossRef CAS.
- X. Wang, Z. Li, Y. Qu, T. Yuan, W. Wang, Y. Wu and Y. Li, Chem, 2019, 5, 1486–1511 CAS.
- M. Shao, Q. Chang, J.-P. Dodelet and R. Chenitz, Chem. Rev., 2016, 116, 3594–3657 CrossRef CAS PubMed.
- C. W. Bezerra, L. Zhang, K. Lee, H. Liu, A. L. Marques, E. P. Marques, H. Wang and J. Zhang, Electrochim. Acta, 2008, 53, 4937–4951 CrossRef CAS.
- F. Jaouen, E. Proietti, M. Lefèvre, R. Chenitz, J.-P. Dodelet, G. Wu, H. T. Chung, C. M. Johnston and P. Zelenay, Energy Environ. Sci., 2011, 4, 114–130 RSC.
- G. Wu, K. L. More, C. M. Johnston and P. Zelenay, Science, 2011, 332, 443–447 CrossRef CAS PubMed.
- M. Lefèvre, E. Proietti, F. Jaouen and J.-P. Dodelet, Science, 2009, 324, 71–74 CrossRef.
- H. T. Chung, D. A. Cullen, D. Higgins, B. T. Sneed, E. F. Holby, K. L. More and P. Zelenay, Science, 2017, 357, 479–484 CrossRef CAS.
- B. Singh, M. B. Gawande, A. D. Kute, R. S. Varma, P. Fornasiero, P. McNeice, R. V. Jagadeesh, M. Beller and R. Zboril, Chem. Rev., 2021, 121, 13620–13697 CrossRef CAS.
- X.-F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS PubMed.
- S. K. Kaiser, Z. Chen, D. F. Akl, S. Mitchell and J. Pérez-Ramírez, Chem. Rev., 2020, 120, 11703–11809 CrossRef CAS.
- J. Li, M. F. Stephanopoulos and Y. Xia, Chem. Rev., 2020, 120, 11699–11702 CrossRef CAS.
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem., 2018, 2, 65–81 CrossRef CAS.
- Y. Chen, S. Ji, Y. Wang, J. Dong, W. Chen, Z. Li, R. Shen, L. Zheng, Z. Zhuang and D. Wang, Angew. Chem., Int. Ed., 2017, 56, 6937–6941 CrossRef CAS.
- Q.-L. Zhu, W. Xia, L.-R. Zheng, R. Zou, Z. Liu and Q. Xu, ACS Energy Lett., 2017, 2, 504–511 CrossRef CAS.
- X. Chen, D.-D. Ma, B. Chen, K. Zhang, R. Zou, X.-T. Wu and Q.-L. Zhu, Appl. Catal., B, 2020, 267, 118720 CrossRef CAS.
- Z. Miao, X. Wang, M. C. Tsai, Q. Jin, J. Liang, F. Ma, T. Wang, S. Zheng, B. J. Hwang and Y. Huang, Adv. Energy Mater., 2018, 8, 1801226 CrossRef.
- F. L. Meng, Z. L. Wang, H. X. Zhong, J. Wang, J. M. Yan and X. B. Zhang, Adv. Mater., 2016, 28, 7948–7955 CrossRef CAS.
- L. T. Song, Z. Y. Wu, F. Zhou, H. W. Liang, Z. Y. Yu and S. H. Yu, Small, 2016, 12, 6398–6406 CrossRef CAS PubMed.
- S. Zhang, H. Zhang, Q. Liu and S. Chen, J. Mater. Chem. A, 2013, 1, 3302–3308 RSC.
- X. Cui, L. Gao, S. Lei, S. Liang, J. Zhang, C. D. Sewell, W. Xue, Q. Liu, Z. Lin and Y. Yang, Adv. Funct. Mater., 2021, 31, 2009197 CrossRef CAS.
- X. Zhang, L. Truong-Phuoc, X. Liao, G. Tuci, E. Fonda, V. Papaefthymiou, S. Zafeiratos, G. Giambastiani, S. Pronkin and C. Pham-Huu, ACS Catal., 2021, 11, 8915–8928 CrossRef CAS.
- G. Lalande, D. Guay, J. Dodelet and G. Denes, J. Electrochem. Soc., 1998, 145, 2411 CrossRef.
- H. Wang, R. Cote, G. Faubert, D. Guay and J. Dodelet, J. Phys. Chem. B, 1999, 103, 2042–2049 CrossRef CAS.
- L. Bai, Z. Duan, X. Wen and J. Guan, J. Catal., 2019, 378, 353–362 CrossRef CAS.
- X. Cui, S. Yang, X. Yan, J. Leng, S. Shuang, P. M. Ajayan and Z. Zhang, Adv. Funct. Mater., 2016, 26, 5708–5717 CrossRef CAS.
- J. Xiao, Y. Xu, Y. Xia, J. Xi and S. Wang, Nano Energy, 2016, 24, 121–129 CrossRef CAS.
- U. I. Kramm, I. Herrmann-Geppert, P. Bogdanoff and S. Fiechter, J. Phys. Chem. C, 2011, 115, 23417–23427 CrossRef CAS.
- H. Perez, V. Jorda, J. Vigneron, M. Frégnaux, A. Etcheberry, A. Quinsac, Y. Leconte and O. Sublemontier, C, 2019, 5, 26 CAS.
- B. Stöhr, H. P. Boehm and R. Schlögl, Carbon, 1991, 29, 707–720 CrossRef.
- H. Ba, Y. Liu, L. Truong-Phuoc, C. Duong-Viet, X. Mu, W. H. Doh, T. Tran-Thanh, W. Baaziz, L. Nguyen-Dinh and J.-M. Nhut, Chem. Commun., 2015, 51, 14393–14396 RSC.
- H. Ba, Y. Liu, L. Truong-Phuoc, C. Duong-Viet, J.-M. Nhut, D. L. Nguyen, O. Ersen, G. Tuci, G. Giambastiani and C. Pham-Huu, ACS Catal., 2016, 6, 1408–1419 CrossRef CAS.
- C. Pham-Huu, G. Giambastiani, Y. Liu, H. Ba, L. Nguyen-Dinh and J.-M. Nhut, Method for preparing highly nitrogen-doped mesoporous carbon composites, EP 3047905 A1, 2016.
- H. Ba, J. Luo, Y. Liu, C. Duong-Viet, G. Tuci, G. Giambastiani, J.-M. Nhut, L. Nguyen-Dinh, O. Ersen, D. S. Su and C. Pham-Huu, Appl. Catal., B, 2017, 200, 343–350 CrossRef CAS.
- C. Duong-Viet, J.-M. Nhut, T. Truong-Huu, G. Tuci, L. Nguyen-Dinh, Y. Liu, C. Pham, G. Giambastiani and C. Pham-Huu, Catal. Sci. Technol., 2020, 10, 5487–5500 RSC.
- C. Duong-Viet, J.-M. Nhut, T. Truong-Huu, G. Tuci, L. Nguyen-Dinh, C. Pham, G. Giambastiani and C. Pham-Huu, Catalysts, 2021, 11, 226 CrossRef CAS.
- S. H. Lee, J. Kim, D. Y. Chung, J. M. Yoo, H. S. Lee, M. J. Kim, B. S. Mun, S. G. Kwon, Y.-E. Sung and T. Hyeon, J. Am. Chem. Soc., 2019, 141, 2035–2045 CrossRef CAS PubMed.
- Y. Han, Y. Wang, R. Xu, W. Chen, L. Zheng, A. Han, Y. Zhu, J. Zhang, H. Zhang, J. Luo, C. Chen, Q. Peng, D. Wang and Y. Li, Energy Environ. Sci., 2018, 11, 2348–2352 RSC.
- G. Chen, P. Liu, Z. Liao, F. Sun, Y. He, H. Zhong, T. Zhang, E. Zschech, M. Chen and G. Wu, Adv. Mater., 2020, 32, 1907399 CrossRef CAS PubMed.
- T. A. Hudson, K. J. Berry, B. Moubaraki, K. S. Murray and R. Robson, Inorg. Chem., 2006, 45, 3549–3556 CrossRef CAS.
- R. Ma, G. Lin, Q. Ju, W. Tang, G. Chen, Z. Chen, Q. Liu, M. Yang, Y. Lu and J. Wang, Appl. Catal., B, 2020, 265, 118593 CrossRef CAS.
- K. S. Sing, D. Everett, R. Haul, L. Moscou, R. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603–619 CAS.
- G. Ren, X. Lu, Y. Li, Y. Zhu, L. Dai and L. Jiang, ACS Appl. Mater. Interfaces, 2016, 8, 4118–4125 CrossRef CAS PubMed.
- Y. Wang, X. Bai, F. Wang, H. Qin, C. Yin, S. Kang, X. Li, Y. Zuo and L. Cui, Sci. Rep., 2016, 6, 26673 CrossRef CAS.
- H. J. Seong and A. L. Boehman, Energy Fuels, 2013, 27, 1613–1624 CrossRef CAS.
- F. Tuinstra and J. L. Koenig, J. Chem. Phys., 1970, 53, 1126–1130 CrossRef CAS.
- Z. Zhang, J. Sun, F. Wang and L. Dai, Angew. Chem., Int. Ed., 2018, 57, 9038–9043 CrossRef CAS PubMed.
- D. Zhai, H. Du, B. Li, Y. Zhu and F. Kang, Carbon, 2011, 49, 725–729 CrossRef CAS.
- X. Ao, W. Zhang, Z. Li, J.-G. Li, L. Soule, X. Huang, W.-H. Chiang, H. M. Chen, C. Wang and M. Liu, ACS Nano, 2019, 13, 11853–11862 CrossRef CAS.
- Q. Li, H. Liu, L.-C. Zhang, H. Chen, H. Zhu, Y. Wu, M. Xu and S.-J. Bao, J. Power Sources, 2020, 449, 227497 CrossRef CAS.
- Z. Li, R. Wu, S. Xiao, Y. Yang, L. Lai, J. S. Chen and Y. Chen, Chem. Eng. J., 2022, 430, 132882 CrossRef CAS.
- J.-C. Li, Z.-Q. Yang, D.-M. Tang, L. Zhang, P.-X. Hou, S.-Y. Zhao, C. Liu, M. Cheng, G.-X. Li and F. Zhang, NPG Asia Mater., 2018, 10, e461 CrossRef CAS.
- C. Genovese, M. E. Schuster, E. K. Gibson, D. Gianolio, V. Posligua, R. Grau-Crespo, G. Cibin, P. P. Wells, D. Garai and V. Solokha, Nat. Commun., 2018, 9, 1–12 CrossRef CAS.
- J. Gu, C.-S. Hsu, L. Bai, H. M. Chen and X. Hu, Science, 2019, 364, 1091–1094 CrossRef CAS PubMed.
- T. N. Huan, N. Ranjbar, G. Rousse, M. Sougrati, A. Zitolo, V. Mougel, F. Jaouen and M. Fontecave, ACS Catal., 2017, 7, 1520–1525 CrossRef CAS.
- W. Liu, L. Zhang, X. Liu, X. Liu, X. Yang, S. Miao, W. Wang, A. Wang and T. Zhang, J. Am. Chem. Soc., 2017, 139, 10790–10798 CrossRef CAS PubMed.
- W.-J. Jiang, L. Gu, L. Li, Y. Zhang, X. Zhang, L.-J. Zhang, J.-Q. Wang, J.-S. Hu, Z. Wei and L.-J. Wan, J. Am. Chem. Soc., 2016, 138, 3570–3578 CrossRef CAS PubMed.
- A. Zitolo, V. Goellner, V. Armel, M.-T. Sougrati, T. Mineva, L. Stievano, E. Fonda and F. Jaouen, Nat. Mater., 2015, 14, 937–942 CrossRef CAS PubMed.
- Q. Jia, N. Ramaswamy, H. Hafiz, U. Tylus, K. Strickland, G. Wu, B. Barbiellini, A. Bansil, E. F. Holby, P. Zelenay and S. Mukerjee, ACS Nano, 2015, 9, 12496–12505 CrossRef CAS PubMed.
- N. Ramaswamy, U. Tylus, Q. Jia and S. Mukerjee, J. Am. Chem. Soc., 2013, 135, 15443–15449 CrossRef CAS PubMed.
- H. Li, Z. Zhang, M. Dou and F. Wang, Chem.–Eur. J., 2018, 24, 8848–8856 CrossRef CAS PubMed.
- D. J. Wasylenko, C. Rodríguez, M. L. Pegis and J. M. Mayer, J. Am. Chem. Soc., 2014, 136, 12544–12547 CrossRef CAS PubMed.
- Y. Feng and N. Alonso-Vante, Phys. Status Solidi B, 2008, 245, 1792–1806 CrossRef CAS.
- Z. Cao, S. B. Zacate, X. Sun, J. Liu, E. M. Hale, W. P. Carson, S. B. Tyndall, J. Xu, X. Liu, X. Liu, C. Song, J.-H. Luo, M.-J. Cheng, X. Wen and W. Liu, Angew. Chem., Int. Ed., 2018, 57, 12675–12679 CrossRef CAS PubMed.
- J. H. Zagal and M. T. Koper, Angew. Chem., Int. Ed., 2016, 55, 14510–14521 CrossRef CAS PubMed.
- N. Ramaswamy and S. Mukerjee, Adv. Phys. Chem., 2012, 2012, 491604 Search PubMed.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, John Wiley & Sons, Inc., New York, 2nd edn, 2001 Search PubMed.
- C. Y. Du, T. S. Zhao and W. W. Yang, Electrochim. Acta, 2007, 52, 5266–5271 CrossRef CAS.
- Y. M. Tan, C. F. Xu, G. X. Chen, N. F. Zheng and Q. J. Xie, Energy Environ. Sci., 2012, 5, 6923–6927 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta09519f |
| This journal is © The Royal Society of Chemistry 2022 |