
Damage restoration in rigid materials via a keloid-inspired growth process†
Yuanlai
Fang
a,
Juan
Xue
a,
Hong
Wang
a,
Li
Yang
a,
Shihua
Dong
*a and
Jiaxi
Cui
 *ab
*ab
aInstitute of Fundamental and Frontier Sciences, University of Electronic Science and Technology of China, Chengdu, Sichuan 611731, China. E-mail: Jiaxi.cui@uestc.edu.cn; tomkudom@gmail.com
bYangtze Delta Region Institute (Huzhou), University of Electronic Science and Technology of China, Huzhou 313001, P. R. China
First published on 22nd November 2021
Abstract
Living organisms can self-heal wounds in their rigid and strong bodies via the growth of keloids. In contrast, it is still challenging for current self-healing strategies to efficiently self-heal catastrophic damage in rigid materials without compromising intrinsic mechanical properties. Inspired by the formation of keloids, we here report a novel growth-induced self-healing strategy for restoring heavy damage in rigid polymer materials. This strategy is based on the combination of traditional extrinsic and intrinsic self-healing mechanisms and involves in situ polymerization to form new matrices in damage regions as well as chain exchange on the interface between original and newborn polymers to integrate the matrices. We demonstrate this strategy using rigid crosslinked copolymers made from 2-hydroxyethyl acrylate and 2-hydroxyethyl methacrylate (P-HEA-co-HEMA). The copolymers show a high Young's modulus up to 1.0 GPa but can still self-heal with a high efficiency of 86.1% even when the damage region almost destroys all the mechanical properties. Since both in situ polymerization and chain exchange reactions are common in different polymer material systems, we thus envision the great potential of our growth-based strategy in designing high-performance self-healing materials.
Self-healing constitutes a powerful strategy to prolong the lifespan of polymeric materials.1–3 Two kinds of self-healing methods, i.e., extrinsic and intrinsic self-healings, have been developed.4–6 The latter mainly relies on exchange reactions among various dynamic or reversible interactions.7–11 These reactions are usually motivated by exterior stimulations, such as heat, light, pH, etc.12–14 For efficient self-healing, the surfaces of the cracks should be in tight contact and the polymer chains should be mobile for the occurrence of exchange reactions to recover the overall properties, as illustrated in Fig. 1a.4,15–17 On the other hand, extrinsic self-healing polymers normally load liquid healing agents in microcapsules or hollow fibers.18 Upon damage, the free-flowing self-healing agents will be released to fill the cracks and undergo polymerization to repair damages when the propagation of cracks breaks the wall of the capsules or hollow fibers.18–20 Newborn materials can fill the damage and glue the cracking interface, whereas the inadequate adhesion between the matrices and the newborn polymers may become weak points, which deteriorate the self-healing efficiencies, as depicted in Fig. 1b.18,21,22 In general, it is extremely challenging for rigid polymers to achieve excellent self-healing properties based on traditional self-healing mechanisms, because the poor chain mobility limits the exchange reactions or polymer diffusion to enhance adhesion.23,24 Hence, more comprehensive and useful self-healing mechanisms are anticipated to balance the drawbacks of traditional intrinsic and extrinsic self-healing to realize appealing self-healing of rigid polymers.
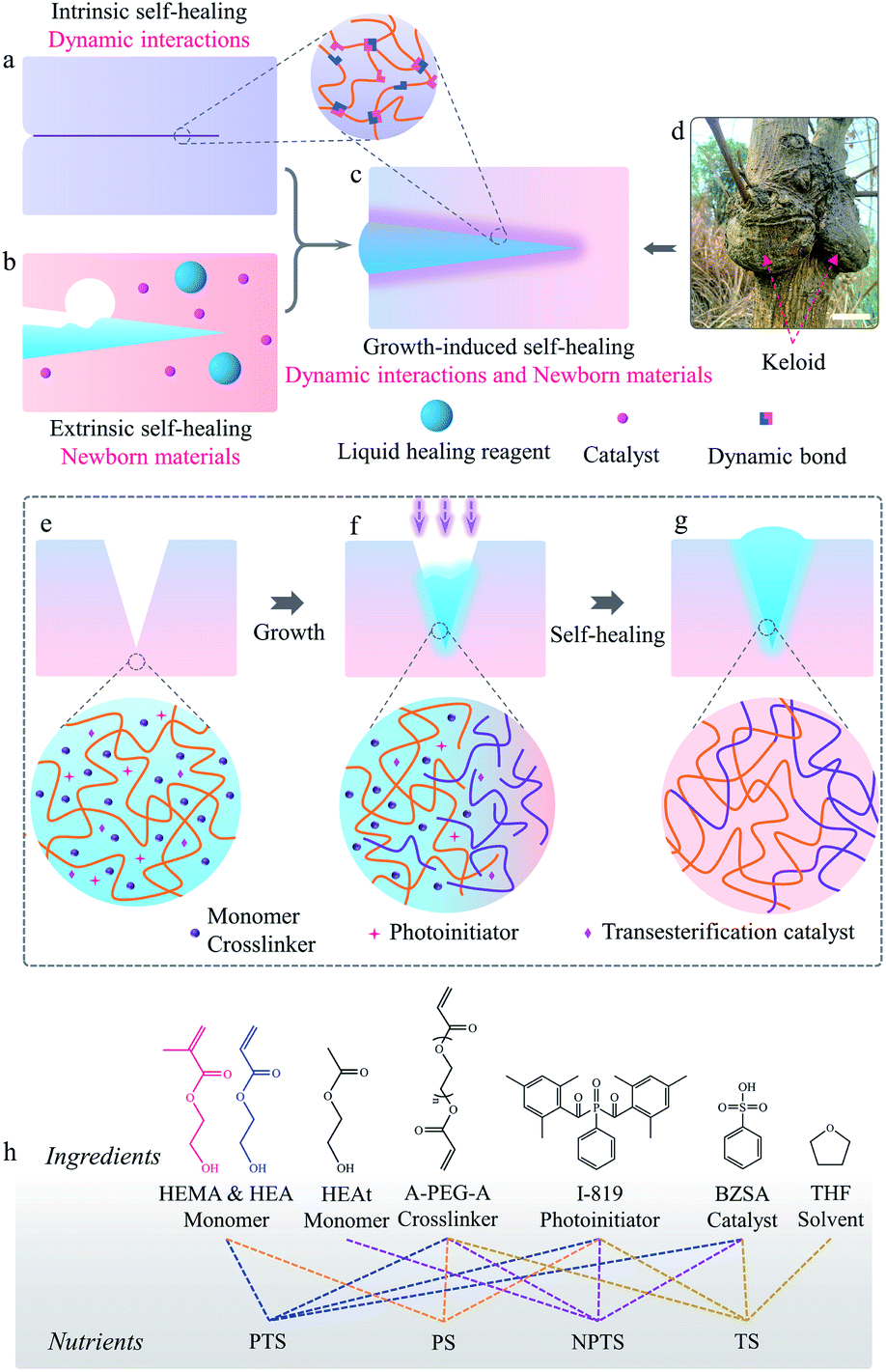 | ||
| Fig. 1 Keloid-inspired growth-induced self-healing strategy. Self-healing mechanism diagram of (a) intrinsic self-healing, (b) extrinsic self-healing, and (c) growth-induced self-healing. (d) Keloid from the self-healing wounds of a tree (scale bar, 10 cm). The procedure of the growth-induced self-healing mechanism: (e) cracked and swollen P-HEA-co-HEMA, (f) growing P-HEA-co-HEMA, and (g) keloid residual and repaired P-HEA-co-HEMA. (h) Chemical structures of the different ingredients within the nutrients, and compositions for different nutrients. In the nutrients, “P” means polymerizable, “NP” means non-polymerizable, “T” means transesterification, and “S” means self-healing. | ||
Living organisms show excellent self-healing ability, which can be observed in tree barks and animal skins.25–27 When injuries occur anywhere on them, abundant nutrients are transported to the injured regions to accelerate cell proliferation.28 New tissues thus grow out from the wound surroundings to generate keloid-like structures, without the formation of any interfaces between newborn and original tissues (Fig. 1d).25,29 Inspired by this natural phenomenon, we propose a new strategy for self-healing high Young's modulus polymers by combining the merits of both extrinsic and intrinsic self-healing methods in a self-growth process (Fig. 1c and d).19,30–34 In this new strategy, polymerizable nutrients entrapped in polymer matrices permeated into the cracked matrices (Fig. 1e). Light is applied to induce the polymerization of nutrients to form newborn polymers grown from the matrices (Fig. 1f). When a chain exchange mechanism (such as transesterification) is applied into the systems for the reconfiguration of original and newborn polymers, interface-free matrices are obtained. Keloid-like structures could even form to strengthen the damage regions to prevent further fractures there (Fig. 1g).
Copolymer systems made from 2-hydroxyethyl acrylate (HEA) and 2-hydroxyethyl methacrylate (HEMA) were used to demonstrate our idea. HEA is a monomer that can form a soft flexible polymer while HEMA is a monomer that composes a rigid polymer chain.35 Materials, denoted as P-HEA-co-HEMAs, were prepared via photopolymerization of HEA and HEMA in the presence of poly(ethylene glycol)diacrylate (A-PEG-A, crosslinker), as illustrated in Schemes S1 and S2.† The modulus of the materials can be tuned using the crosslinking density and the molar ratio of HEA to HEMA. A P-HEA-co-HEMA matrix with 60 mol% (40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :60) HEMA monomer and 2 mol% of A-PEG-A crosslinker, showing a Young's modulus of 1.0 GPa, a tensile strength of 18.4 MPa, and an elongation of 198.2% at break (Fig. S1 and S2, Tables S1 and S2†), was chosen to investigate the growth-induced self-healing performances. Samples with high HEMA fractions (e.g., 80 mol%) would have brittle matrices (Fig. S3†). P-HEA-co-HEMA cannot be melted at high temperatures (e.g., 130 °C) or dissolved by good solvents (DMSO), as portrayed in Fig. S4.†
:60) HEMA monomer and 2 mol% of A-PEG-A crosslinker, showing a Young's modulus of 1.0 GPa, a tensile strength of 18.4 MPa, and an elongation of 198.2% at break (Fig. S1 and S2, Tables S1 and S2†), was chosen to investigate the growth-induced self-healing performances. Samples with high HEMA fractions (e.g., 80 mol%) would have brittle matrices (Fig. S3†). P-HEA-co-HEMA cannot be melted at high temperatures (e.g., 130 °C) or dissolved by good solvents (DMSO), as portrayed in Fig. S4.†
The growth-induced self-healing process started from feeding a “nutrient” into the systems. Fig. 1h shows the components used for preparing different nutrient solutions. Nutrient PTS (Polymerization and Transesterification Self-healing) is constituted by HEA, HEMA, A-PEG-A, I-819 (photoinitiator), and benzenesulfonic acid (BZSA, transesterification catalyst) with the same HEA/HEMA ratio for the preparation of P-HEA-co-HEMA matrices. Nutrients are stable under light-free conditions (testing time: 90 days, Fig. S5†). To explore the role of polymerization and transesterification in the self-healing process, several control nutrients, including PS (Polymerization Self-healing), NPTS (Non-Polymerization and Transesterification Self-healing), and TS (Transesterification Self-healing), were also prepared. The PS nutrient contained polymerizable HEA and HEMA monomers but no BZSA catalyst and thus only polymerization was expected. Since growth from PS would not induce chain exchange reactions to prevent the formation of an interface between newborn and original matrices, it was expected to show a similar effect to the extrinsic self-healing strategy.36 NPTS was employed to verify the importance of polymerization for self-healing. Transesterification is a dynamic reaction in response to a temperature stimulus, and is usually considered to design a lot of intrinsic self-healing polymers.10,37 Thus, TS represented the intrinsic self-healing method.
The nutrient solutions could be fed into the systems by immersing the samples into the nutrient solutions or dropping nutrient solutions on the surfaces of the samples to allow swelling (Fig. S6 and S7†). P-HEA-co-HEMAs show a saturated swelling ratio of 1.85 to PTS nutrients. Note that the swelling ratio could be well controlled. For example, a P-HEA-co-HEMA thin film swelled with 5 wt% PTS could be prepared, and shows similar mechanical properties (Young's modulus: 0.8 GPa, tensile strength: 15.1 MPa, yielding stress: 12.1 MPa, and elongation at break: 157.6%, Fig. S8†) to the original sample due to the low swelling ratio. Such swollen P-HEA-co-HEMA shows good fatigue resistance to withstand tensile deformations (1000 cycles as depicted in Fig. S9†). These absorbed nutrients could be integrated into the matrices by UV-irradiation to induce polymerization of the monomer and crosslinker in the nutrients. The grown samples did not lose their weight after washing treatments. In contrast, without light-induced polymerization, the nutrients absorbed in the samples could be removed by washing with EtOH. Polymerization of acrylate-based compounds is exothermic, which would elevate the temperature of the irradiated regions to trigger transesterification with the assistance of the BZSA catalyst for preventing the formation of any possible interfaces and mechanic tension (Fig. S10†).31,38 To confirm this mechanism, the temperature of the irradiated region was recorded and it was detected that the temperature of the mask exposed area increased from 20.7 °C to 82.1 °C during photopolymerization (Fig. S11†). In comparison, the temperature mildly increased by 1.9 °C for the sample swelled with the NPTS nutrient (Fig. S12†).
Growth-induced self-healing processes were first studied in a qualitative method. Fig. 2a and d show the schematic diagram of the cracked and self-healed P-HEA-co-HEMAs, respectively. When the rigid P-HEA-co-HEMA was knifed with a surgical blade, a macroscopical crack was generated as shown in Fig. 2b. The crack could be completely repaired via a growth-induced self-healing process, and a keloid formed as a result of the polymerization of the PTS nutrients surrounding the crack (Fig. 2c).
 | ||
| Fig. 2 Morphology of the growth-induced self-healing. (a) Schematic diagram of the cracked sample; photos of the cracked (b) and the self-healed (c) P-HEA-co-HEMAs (scale bar, 0.5 cm); (d) schematic diagram of the healed sample; the optical micrographs of the initial crack (e) and the healed cracks obtained from the PTS (f), NPTS (g), or TS (h), respectively. The pictures were taken from the side and top views of the sample, and the exterior and interior morphologies were recorded for the repaired cracks (scale bar, 50 μm). | ||
The crack could be detected using an optical microscope from the top and side views, as portrayed in Fig. 2e. When the PTS nutrient was chosen for growth-induced self-healing, the crack would be filled with the newborn P-HEA-co-HEMAs (Fig. 2f). From the side and interior views, the matrix and the newborn polymers were integrated uniformly due to transesterification, and no obvious interface was observed. In comparison, the crack treated via the same process with other nutrients did not exhibit any self-healing properties. For example, in the case of using NPTS or TS as the nutrient, the cracks remained (Fig. 2g and h). In these cases, no polymerization occurred to generate new polymer matrices to fill the damage. Although the condition of heating and BZSA could trigger dynamic transesterifications in the case of using the TS nutrient, the rigid P-HEA-co-HEMAs limited the intimate contact of the crack surfaces to achieve transesterification-founded intrinsic self-healing.10 Control experiments using an optical microscope proved that the polymerization of the monomer and crosslinker in the nutrients was essential for growth-induced self-healing.
To evaluate our strategy in recovering the mechanical properties of rigid polymers, virginal P-HEA-co-HEMA specimens were knifed with surgical blades, and the samples were almost separated into two parts (Fig. S13†). The mechanical performance of the cracked P-HEA-co-HEMAs degraded (Fig. 3a and Table 1). The healed P-HEA-co-HEMAs obtained from the PTS nutrients recovered the mechanical properties compared with virginal ones (Fig. 3a and Table 1). Fig. 3c and d show the pictures of the virginal and repaired P-HEA-co-HEMAs (PTS) before and during stretching for tension measurements, respectively. It could be detected that the self-healed keloid region was strengthened, which showed slow diminution compared with the neat substrates under tension. The Young's modulus, tensile strength, yielding stress, and elongation at break of the repaired P-HEA-co-HEMAs (PTS) are 0.9 GPa, 18.1 MPa, 16.1 MPa, and 175.9%, respectively, in comparison with the values of 1.0 GPa, 18.4 MPa, 17.2 MPa, and 198.2% for the virginal P-HEA-co-HEMAs. The self-healing efficiency is 86.1% (Table 1). The virginal and the repaired P-HEA-co-HEMAs could sustain successive loading/unloading deformation over 1000 times (Fig. 3e, f, and S14†), suggesting excellent fatigue resistance for both virginal and healed samples.
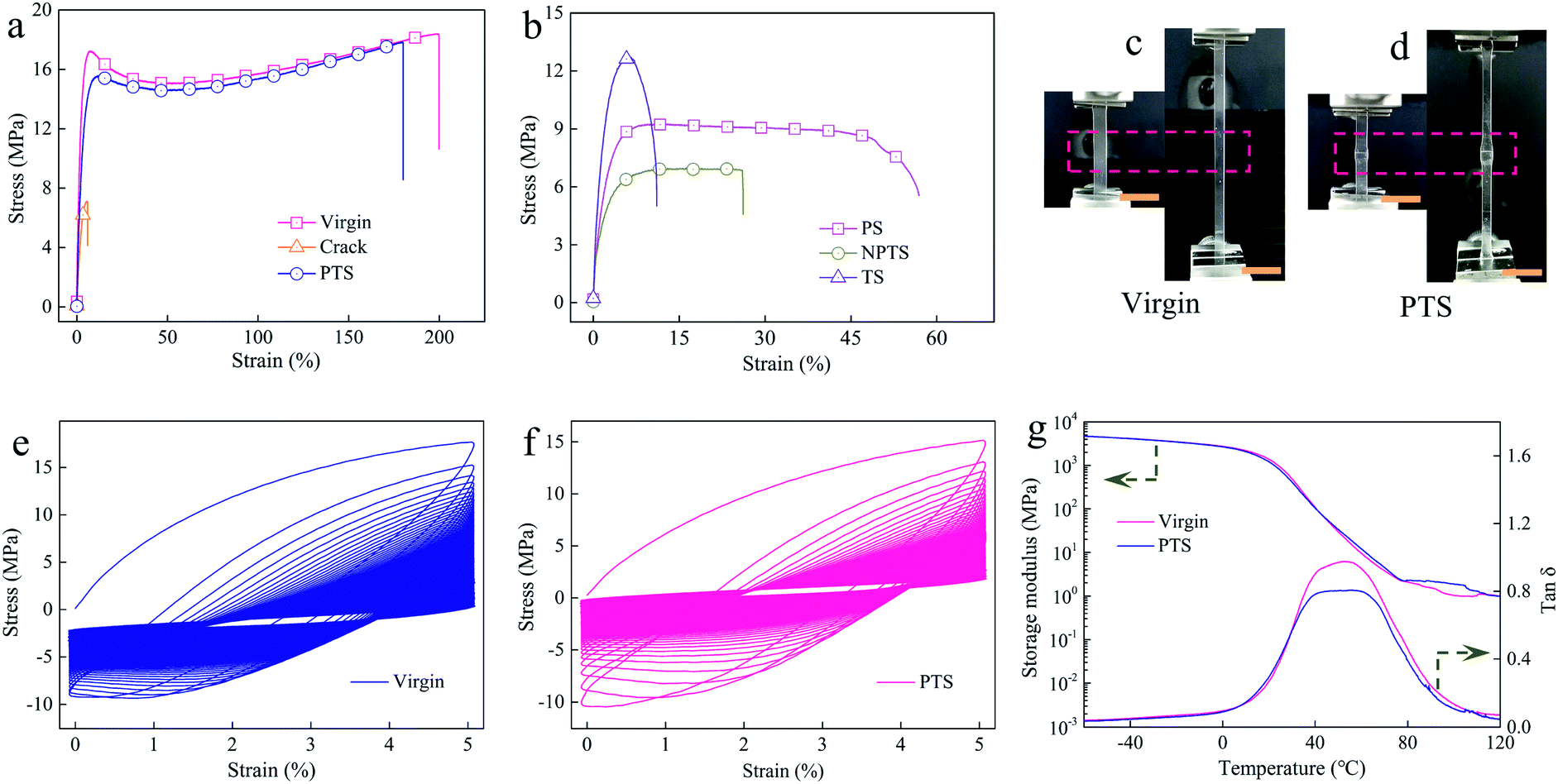 | ||
| Fig. 3 Mechanical properties of the samples. (a) Tensile stress–strain curves for the virginal, cracked, and growth-induced self-healed P-HEA-co-HEMAs (PTS). (b) Tensile stress–strain curves of the healed P-HEA-co-HEMA samples obtained from the control PS, NPTS and TS nutrients. Photos before and during the tensile test for a virginal P-HEA-co-HEMA (c) and a healed P-HEA-co-HEMA obtained from PTS (d). Scale bar in (c) and (d): 1 cm. Stress–strain curves for successive loading/unloading tensile deformation for 1000 cycles of (e) the virginal and (f) the repaired (PTS) P-HEA-co-HEMAs, respectively. (g) Storage modulus and tanδ from the virginal P-HEA-co-HEMAs and healed P-HEA-co-HEMAs obtained from PTS. | ||
| Sample | Young's modulus (GPa) | Tensile strength (MPa) | Yield stress (MPa) | Elongation at break (%) | Toughness (MJ m−3) | Self-healing efficiencies (%) |
|---|---|---|---|---|---|---|
| Virgin | 1.0 ± 0.1 | 18.4 ± 2.1 | 17.2 ± 3.1 | 198.2 ± 13.8 | 32.31 | — |
| Crack | 0.4 ± 0.1 | 7.0 ± 2.3 | — | 5.8 ± 1.9 | 0.30 | — |
| PTS | 0.9 ± 0.1 | 18.1 ± 1.0 | 16.1 ± 1.4 | 175.9 ± 16.5 | 27.81 | 86.1 |
| PS | 0.6 ± 0.2 | 10.1 ± 2.1 | — | 57 ± 18.0 | 4.85 | 15.0 |
| NPTS | 0.7 ± 0.1 | 7.0 ± 0.3 | — | 26.0 ± 12.1 | 1.68 | 5.2 |
| TS | 0.9 ± 0.3 | 12.7 ± 4.6 | — | 10.2 ± 3.8 | 1.13 | 3.5 |
The size of the keloid could be well controlled by regulating the growth, as compared in Fig. S15.† The small keloid-based self-healed P-HEA-co-HEMAs exhibited a good self-healing efficiency of 80.8% yet. Our strategy could also be applied to soft systems. For example, when an elastomer made from HEA was repaired by its PTS nutrient, an excellent self-healing efficiency of 106.2% was achieved (Fig. S16 and Table S3†). Dynamic mechanical analysis (DMA) was further employed to evaluate the self-healing effect. From the tanδ curves, it was found that the glass transformation temperature (Tg) is around 50 °C for both virginal and repaired ones (Fig. 3g). This Tg indicated that the polymer segments were in glass states at room temperature. Under this condition, self-healing is normally difficult.34,35
In contrast, all control samples did not show prominent self-healing ability. In the case of using PS as the nutrient for growth, new cracks would form in the interface regions between the healed matrices and the original substrates (Fig. S17†). The formation of cracks was attributed to the generation of intrinsic mechanical tension during photopolymerization since no chain exchange reactions occurred to integrate the virginal and new polymer chains. As a result, the PS nutrient-based repaired P-HEA-co-HEMAs only display a poor self-healing efficiency of 15.0% (Fig. 3b and Table 1). On the other hand, when NPTS was used for the treatment, the samples did not exhibit any self-healing behavior because no polymerization and transesterification were involved. Besides, we also mimicked the traditional intrinsic self-healing process using the TS nutrient. The samples were annealed at 80 °C to trigger transesterification. However, a low self-healing efficiency of 3.5% was obtained (Fig. 3b and Table 1). It was because, in the heavy damage regions, the surfaces could not efficiently contact each other for allowing chain exchange, due to the absence of newborn matrices. These control experiments indicated that both polymerization and transesterification from the nutrients were fundamental for growth-induced self-healing.39
Since the “keloids” in the healed samples mainly consisted of nutrients, it was possible to modulate the mechanical properties of the healed regions by the components in the nutrients. To show this flexibility, a soft PTS nutrient (100% soft HEA monomer) and a hard PTS nutrient (100% hard HEMA monomer) were selected to investigate their influence on self-healing performance. Fig. 4a shows the tensile stress–strain curves of the repaired P-HEA-co-HEMA specimens made from normal, soft, and hard PTSs, respectively. The samples obtained from normal and hard PTSs exhibit nearly the same self-healing performance (self-healing efficiencies: 86.1% and 81.1%, respectively, Table S4†). These healed samples might break at new points rather than in the healed region, when fractured (inset of Fig. 4a and b). The fracture positions could be controlled using the soft PTS nutrient to create the keloid structures. Since the grown keloids were significantly weaker than the bulk substrates, the keloids were preferentially destroyed rather than matrices under tension (inset of Fig. 4a and b). The hard and soft keloids resulted from the steric effect from the methyl group of the PHEMA main chains, whereas the PHEA showed a more flexible conformation transition (Fig. 4c). Such fracture position-controllable properties might be useful for security announce. The healed P-HEA-co-HEMAs obtained from normal PTS were quite strong and could sustain a dumbbell, whose weight is 12025 times over that of the specimens (Fig. 4d). Noteworthily, the repaired P-HEA-co-HEMAs perfectly maintain their original shapes due to their rigid nature (Fig. 4e) when compared with the relaxed specimens (Fig. 4f).
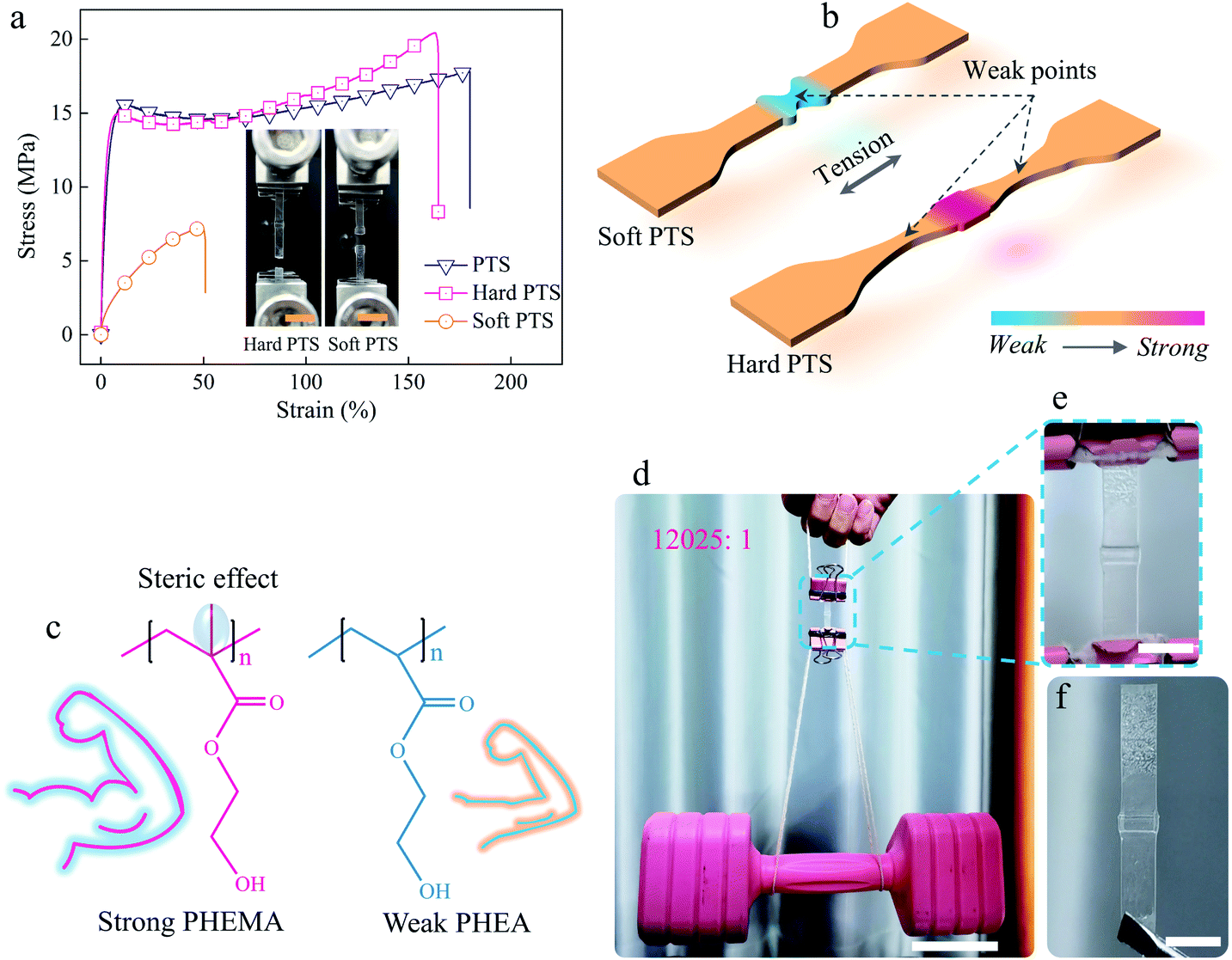 | ||
| Fig. 4 Influence of the PTS nutrient components. (a) Tensile stress–strain curves of the growth-induced self-healed P-HEA-co-HEMA samples obtained from normal, soft, and hard PTS nutrients, respectively. The insets show the fractured samples of the healed P-HEA-co-HEMAs based on hard and soft PTS, respectively (scale bar, 1 cm). (b) The diagram of the repaired samples during the tensile tests for the soft and hard PTS nutrients, respectively. (c) The chemical structures of rigid PHEMA and flexible PHEA. (d) A growth-induced self-healed P-HEA-co-HEMA (normal PTS) could sustain a dumbbell without deformation (scale bar, 5 cm). (e) and (f) Partial enlargement of the repaired P-HEA-co-HEMA in loaded and relaxed states, respectively (scale bar, 0.5 cm). | ||
Conclusions
In summary, we have proposed a new self-healing strategy to restore heavy damages in rigid polymers by combining the merits of extrinsic and intrinsic self-healing methods. Our strategy is inspired by the growth of keloids in plants and involves the growth of new matrices from fed nutrients and chain exchange occurring between newborn and original matrices to prevent the formation of any interfaces. By using P-HEA-co-HEMA as an example, we have demonstrated that the self-growth strategy allows for efficient restoration (86.1%) of heavy damages in rigid matrices with Young's moduli of up to 1.0 GPa. Moreover, the keloid-like structures could be selected to be hard or soft for controlling fracture positions. Since in situ polymerization and chain exchange reactions are quite common in many polymer systems, this strategy could be extended to plenty of rigid materials for their mechanical property recovery and modulation.Author contributions
Conceptualization: Yuanlai Fang and Jiaxi Cui; investigation: Yuanlai Fang, Juan Xue, and Hong Wang; formal analysis: Li Yang, Shihua Dong, and Jiaxi Cui; supervision: Shihua Dong and Jiaxi Cui; writing – original draft: Yuanlai Fang; writing – review & editing: Yuanlai Fang, Shihua Dong, and Jiaxi Cui.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (51973023 and 52003035), Sichuan Science and Technology Program (2021JDRC0014 and 2021JDRC0106), and China Postdoctoral Science Foundation (No. 2021M700678).References
- C. C. Hornat and M. W. Urban, Prog. Polym. Sci., 2020, 102, 101208 CrossRef CAS.
- W. Zou, J. Dong, Y. Luo, Q. Zhao and T. Xie, Adv. Mater., 2017, 29, 1606100 CrossRef PubMed.
- C. Kim, H. Ejima and N. Yoshie, J. Mater. Chem. A, 2018, 6, 19643–19652 RSC.
- S.-M. Kim, H. Jeon, S.-H. Shin, S.-A. Park, J. Jegal, S. Y. Hwang, D. X. Oh and J. Park, Adv. Mater., 2017, 30, 1705145 CrossRef PubMed.
- M. J. Barthel, T. Rudolph, A. Teichler, R. M. Paulus, J. Vitz, S. Hoeppener, M. D. Hager, F. H. Schacher and U. S. Schubert, Adv. Funct. Mater., 2013, 23, 4921–4932 CrossRef CAS.
- G. B. Lyon, A. Baranek and C. N. Bowman, Adv. Funct. Mater., 2016, 26, 1477–1485 CrossRef CAS.
- Y. Fang, X. Du, Y. Jiang, Z. Du, P. Pan, X. Cheng and H. Wang, ACS Sustainable Chem. Eng., 2018, 6, 14490–14500 CrossRef CAS.
- J. Cui and A. d. Campo, Chem. Commun., 2012, 48, 9302–9304 RSC.
- V. T. Tran, M. T. I. Mredha, J. Y. Na, J.-K. Seon, J. Cui and I. Jeon, Chem. Eng. J., 2020, 394, 124941 CrossRef CAS.
- M. Capelot, D. Montarnal, F. Tournilhac and L. Leibler, J. Am. Chem. Soc., 2012, 134, 7664–7667 CrossRef CAS PubMed.
- T. L. Sun, T. Kurokawa, S. Kuroda, A. B. Ihsan, T. Akasaki, K. Sato, M. A. Haque, T. Nakajima and J. P. Gong, Nat. Mater., 2013, 12, 932–937 CrossRef CAS PubMed.
- Y. Fang, J. Li, X. Du, Z. Du, X. Cheng and H. Wang, Polymer, 2018, 158, 166–175 CrossRef CAS.
- Y. Fang, X. Du, S. Yang, H. Wang, X. Cheng and Z. Du, Polym. Chem., 2019, 10, 4142–4153 RSC.
- Y. Zhang, H. Ying, K. R. Hart, Y. Wu, A. J. Hsu, A. M. Coppola, T. A. Kim, K. Yang, N. R. Sottos, S. R. White and J. Cheng, Adv. Mater., 2016, 28, 7646–7651 CrossRef CAS PubMed.
- J. Canadell, H. Goossens and B. Klumperman, Macromolecules, 2011, 44, 2536–2541 CrossRef CAS.
- M. Röttger, T. Domenech, R. van der Weegen, A. Breuillac, R. Nicolaÿ and L. Leibler, Science, 2017, 356, 62 CrossRef PubMed.
- C. Bao, Y.-J. Jiang, H. Zhang, X. Lu and J. Sun, Adv. Funct. Mater., 2018, 28, 1800560 CrossRef.
- S. R. White, N. R. Sottos, P. H. Geubelle, J. S. Moore, M. R. Kessler, S. R. Sriram, E. N. Brown and S. Viswanathan, Nature, 2001, 409, 794–797 CrossRef CAS PubMed.
- S. R. White, J. S. Moore, N. R. Sottos, B. P. Krull, W. A. Santa Cruz and R. C. R. Gergely, Science, 2014, 344, 620 CrossRef CAS PubMed.
- S. H. Cho, H. M. Andersson, S. R. White, N. R. Sottos and P. V. Braun, Adv. Mater., 2006, 18, 997–1000 CrossRef CAS.
- M. W. Keller, S. R. White and N. R. Sottos, Adv. Funct. Mater., 2007, 17, 2399–2404 CrossRef CAS.
- H. Jin, C. L. Mangun, D. S. Stradley, J. S. Moore, N. R. Sottos and S. R. White, Polymer, 2012, 53, 581–587 CrossRef CAS.
- Y. Yanagisawa, Y. Nan, K. Okuro and T. Aida, Science, 2018, 359, 72 CrossRef CAS PubMed.
- S.-M. Kim, H. Jeon, S.-H. Shin, S.-A. Park, J. Jegal, S. Y. Hwang, D. X. Oh and J. Park, Adv. Mater., 2018, 30, 1705145 CrossRef PubMed.
- P. Cheng, J. Zhang, J. Huang, Q. Miao, C. Xu and K. Pu, Chem. Sci., 2018, 9, 6340–6347 RSC.
- H. Cochard and S. Delzon, Ann. For. Sci., 2013, 70, 659–661 CrossRef.
- T.-L. Tuan and L. S. Nichter, Mol. Med. Today, 1998, 4, 19–24 CrossRef CAS PubMed.
- S. Ahn, C. O. Chantre, H. A. M. Ardoña, G. M. Gonzalez, P. H. Campbell and K. K. Parker, Biomaterials, 2020, 255, 120149 CrossRef CAS PubMed.
- Q. Miao, D. C. Yeo, C. Wiraja, J. Zhang, X. Ning, C. Xu and K. Pu, Angew. Chem., Int. Ed., 2018, 57, 1256–1260 CrossRef CAS PubMed.
- Y. Li, Z. Zhang, W. J. Mo, M. Rong, M. Zhang and D. Liu, Polymer, 2020, 192, 122299 CrossRef CAS.
- L. Xue, X. Xiong, B. P. Krishnan, F. Puza, S. Wang, Y. Zheng and J. Cui, Nat. Commun., 2020, 11, 963 CrossRef CAS PubMed.
- T. Matsuda, R. Kawakami, R. Namba, T. Nakajima and J. P. Gong, Science, 2019, 363, 504 CrossRef CAS PubMed.
- M. B. Gordon, J. M. French, N. J. Wagner and C. J. Kloxin, Adv. Mater., 2015, 27, 8007–8010 CrossRef CAS PubMed.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965 CrossRef CAS PubMed.
- R. C. R. Gergely, W. A. Santa Cruz, B. P. Krull, E. L. Pruitt, J. Wang, N. R. Sottos and S. R. White, Adv. Funct. Mater., 2018, 28, 1704197 CrossRef.
- D. Y. Zhu, M. Z. Rong and M. Q. Zhang, Prog. Polym. Sci., 2015, 49–50, 175–220 CrossRef CAS.
- L. Cao, J. Fan, J. Huang and Y. Chen, J. Mater. Chem. A, 2019, 7, 4922–4933 RSC.
- S. Wang, L. Yang, H. Wang, L. Xue, J. Chen and J. Cui, ACS Appl. Polym. Mater., 2019, 1, 3227–3232 CrossRef CAS.
- S. Wang, H. Wang, P. Zhang, L. Xue, J. Chen and J. Cui, Mater. Horiz., 2021, 8, 1481–1487 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta08713d |
| This journal is © The Royal Society of Chemistry 2022 |