
Cobalt single-atom-decorated nickel thiophosphate nanosheets for overall water splitting†
Jian
Zhang
 *ab,
Niannian
Zhou
a,
Ming
Du
b,
Yonghua
Li
b,
Yan
Cui
c,
Xing'ao
Li
*ab,
Xinbao
Zhu
d and
Wei
Huang
be
*ab,
Niannian
Zhou
a,
Ming
Du
b,
Yonghua
Li
b,
Yan
Cui
c,
Xing'ao
Li
*ab,
Xinbao
Zhu
d and
Wei
Huang
be
aNew Energy Technology Engineering Lab of Jiangsu Province, College of Science, Nanjing University of Posts & Telecommunications (NUPT), Nanjing 210023, P. R. China. E-mail: iamjzhang@njupt.edu.cn; lxahbmy@126.com
bState Key Laboratory of Organic Electronics and Information Displays, Institute of Advanced Materials (IAM), Nanjing University of Posts and Telecommunications (NUPT), Nanjing 210023, Jiangsu, P. R. China
cKey Laboratory of Broadband Wireless Communication and Sensor Network Technology, Ministry of Education, Nanjing University of Posts and Telecommunications, Nanjing 210003, P. R. China
dCollege of Chemical Engineering, Nanjing Forestry University, Nanjing 210037, P. R. China
eInstitute of Flexible Electronics (IFE), Northwestern Polytechnical University (NPU), Xi'an 710072, P. R. China
First published on 22nd November 2021
Abstract
Formidable challenges in the development of efficient single-atom catalysts including accessible fabrication processes, unambiguous interactions with matrixes, and accelerated kinetics of catalytic mechanisms, pose an obstacle in maximizing the catalytic performance. Herein, it is proposed, for the first time, to bond single-atom Co on the surface of single-layered nickel thiophosphate (SA Co NiPS3) forming a uniform unsaturated coordination mode of isolated Co. Theoretical studies indicate that the SA Co NiPS3 electrode possesses a higher intrinsic activity than that of traditional Co alternative-doped NiPS3via the enhancement of conductivity and the reduction of energy barriers for bifunctional hydrogen/oxygen evolution reactions (HERs/OERs). Notably, a water splitting electrolyzer assembled using SA Co NiPS3 nanosheets as both the cathode and the anode achieves 50 mA cm−2 at 1.60 V, which outperforms the Pt/C//RuO2 benchmark (1.74 V@50 mA cm−2). Besides, this electrocatalyst exhibits an outstanding durability up to 6 days with negligible decay. This work shines fresh light on igniting 2D metal thiophosphate electrocatalytic activity that even surpasses noble-metal-based electrocatalysts via the single-atom engineering.
1. Introduction
Hydrogen, as a good substitute for traditional carbon-based fuels in future energy consumption, has gained increasing research interest, mainly due to its high energy density, zero-emission, and renewability.1,2 Water splitting for hydrogen evolution via electrocatalysis is one of the most effective methods because of high energy conversion efficiency, cost-effectiveness, and environmental green nature.3–9 Noble-metal-based materials such as Pt and RuO2 have proved to be the most active electrocatalysts for hydrogen evolution reactions (HERs) and oxygen evolution reactions (OERs), respectively; however, scarcity and high cost of noble metals hamper their large-scale implementation.10,11 Exploration of catalysts with competitive properties even surpassing noble-metal-based materials on catalytic activity and stability is a critical challenge for wide use of water splitting.Recently, various noble-metal-free materials have been explored as alternative overall water splitting electrocatalysts including alloys,12 sulfides,13–15 carbides,16 phosphides,17 hydroxides,18 and (oxy)hydroxides,19 among many others. Noteworthy, lamellar metal thiophosphates (MPS3),20 which have an analogous sandwich configuration to the typical metal disulfides such as MoS2 and WS2, can also be employed as highly attractive electrocatalysts due to their tunable electronic structure and compositional diversity. Inspired by the modification of MoS2, large-sized and mono-layered NiPS3 were prepared via an electrochemically exfoliating process, delivering a robust overall water splitting performance.21 Besides, atom-scale modulation of electronic configuration via in situ substitution doping of elements in the MPS3 lattice has been demonstrated as an effective approach to motivate the intrinsic catalytic activity. For example, when half of Fe atoms in FePS3 have been substituted by Ni atoms forming NiFePS3, the electrocatalytic performance was significantly enhanced on HER, OER and overall water splitting for the unique lattice distortion, which decreased the kinetic energy barriers of water splitting.22 Unfortunately, a large gap in their electrocatalytic water splitting activity still exists compared to the noble-metal-based benchmark due to the difficulty in maximizing the intrinsic activity of their active sites.23
In parallel, single atoms anchored on the surface of supports can realize a theoretical efficiency of 100% of atomic utilization for catalysis. This single-atom (SA) catalyst strategy has been implemented on various 2D materials to activate the inert basal planes drastically in recent years.24 According to the results of computational and experimental studies, the electron density distribution of the 2D metal disulfide matrix can be flexibly adjusted via electronic interaction between isolated metal atoms and the matrix, thereby optimizing the intermediate adsorption and desorption possibilities of single-atom decorative metal disulfides.25,26 Qi and coworkers presented the utilization of Co SAs as a fuse to trigger the electrocatalytic activity of ultrathin MoS2; Co SAs covalently bound to the distorted 1T MoS2 catalyst exhibited extraordinary HER activities.27 In another study, Liu's group obtained similar results: Co SAs promoting mono-layered MoS2 displayed an even much higher HER activity than that of the state-of-the-art Pt catalyst.28 Additionally, Co SAs can act as promoters for other 2D materials such as TaS2,29 Mo2C30 and N-doped graphene.31 Although Co partial substitution of interlayered metals in MPS3 has been proved to be an efficient method to enhance the electrocatalytic performance, the noble-metal alternative target is still out of reach because of the restricted electronic modulation causing by the coverage of the outer P–S layer.32 Nevertheless, to the best of our knowledge, fully inspiring the intrinsic activity of ultrathin NiPS3via SA Co anchoring has not been reported yet, and more importantly, this origin underlying the electrocatalytic activity influenced by the SA Co merging is blank and it can be explored reasonably by the intricate interactions of electronic states between SA Co and NiPS3, following the metal disulfides transcendentally.
In this work, for the first time, we successfully synthesized SA Co anchored on mono-layered NiPS3 (SA Co NiPS3) by a gentle cyclic voltammetry leaching process over Co nanodisks/NiPS3 precursors (Co@NiPS3). This isolated Co atoms have a uniform unsaturated coordination mode through three Co–S covalent bonds, which was confirmed by aberration-corrected scanning transmission electron microscopy (STEM), X-ray photoelectron spectroscopy (XPS) and X-ray absorption fine structure (XAFS) spectroscopy. As expected, electrochemical evaluates over SA Co NiPS3 electrodes in 1.0 M KOH exhibit superior HER and OER activities with very low overpotentials of 1.19 and 1.46 V, respectively at a current density of 50 mA cm−2. More significantly, when tested as overall water splitting electrocatalysts, a SA Co NiPS3-based two-electrode setup delivers a current density of 50 mA cm−2 at only 1.60 V and good stability with only ∼3.4% decay over 6 days of uninterrupted measurement, which obviously surpass commercial Pt/C//RuO2 electrodes. These interesting results open up an avenue in designing SA-based MPS3 catalysts with remarkable water splitting performance.
2. Results and discussion
First, density functional theory (DFT) calculations were implemented to predict the effect of the two different configurations of Co atoms on the electronic properties and electrocatalytic hydrogen evolution activity of NiPS3. The original NiPS3 monolayer exhibits a sandwich structure comprising octahedral NiS6 clusters surrounded by three inverted trigonal bipyramidal P2S6 clusters, as shown in Fig. 1a. Based on the theoretical optimized results among different structures regarding single Co atom-decorated NiPS3, there are Co atoms substituting Ni atoms (CoNiPS3) and Co atoms anchored on the surface of NiPS3 (SA Co NiPS3). According to the calculated results, CoNiPS3 is the more preferential configuration compared to SA Co NiPS3 because of the milder formation energy (−2.7833 and −6.9561 eV for CoNiPS3 and SA Co NiPS3, respectively), which can explain the fact that it is blank for the SA metal decorating on NiPS3 until now. As shown in Fig. 1b, all the total density of states (TDOS) calculated for NiPS3, CoNiPS3, and SA Co NiPS3 are discontinuous near the Fermi levels (Ef), suggesting the semiconductor features of NiPS3 kept well after Co merging with different coordination modes. In particular, after decorating Co atoms on/into NiPS3 cells, CoNiPS3 and SA Co CoPS3 show smaller energy bands of 1.45 eV and 1.38 than that of NiPS3 (1.53 eV), which implies that more charge carriers are involved in the catalytic reaction, thus significantly boosting the HER performance.33 Then, DFT calculations were further carried out to evaluate the effects of saturated and unsaturated Co atoms on the catalytic activity of mono-layered NiPS3via monitoring the hydrogen adsorption free energy (ΔGH*) of three different catalysts. It is generally accepted that an almost zero ΔGH* means a good HER electrocatalyst, which possesses the approachable H adsorption and satisfactory H2 desorption.34 As displayed in Fig. 1c, the ΔGH* value obviously decreases from 0.397 eV (NiPS3) to 0.168 eV (CoNiPS3) when one Ni atom was in situ substituted by one Co atom, indicating the lower energy barriers for H2 formation over CoNiPS3, in line with previous reports on MPS3.22,35 Surprisingly, ΔGH* of SA Co NiPS3 displays a much closer value to the thermoneutral value of only 0.078 eV, which benefits the compromise of reaction barriers between adsorption and desorption processes, resulting in a further enhanced HER performance. A similar tendency was also discovered on the theoretical value of H2O molecule adsorption energy, as displayed in Fig. 1d. The SA Co NiPS3 catalyst displays the strongest binding strength with the initial H2O (0.234 eV) transcending that of NiPS3 (0.132 eV) and CoNiPS3 (0.192 eV), guaranteeing a smooth activation of H2O for the efficient HER, and the theoretical conclusion matching well with the experimental results. The above-mentioned theoretical calculation results motivate us to implement the assembly of the Co SA-anchored single-layered NiPS3 with uniform unsaturated coordination mode as electrocatalysts for water splitting applications.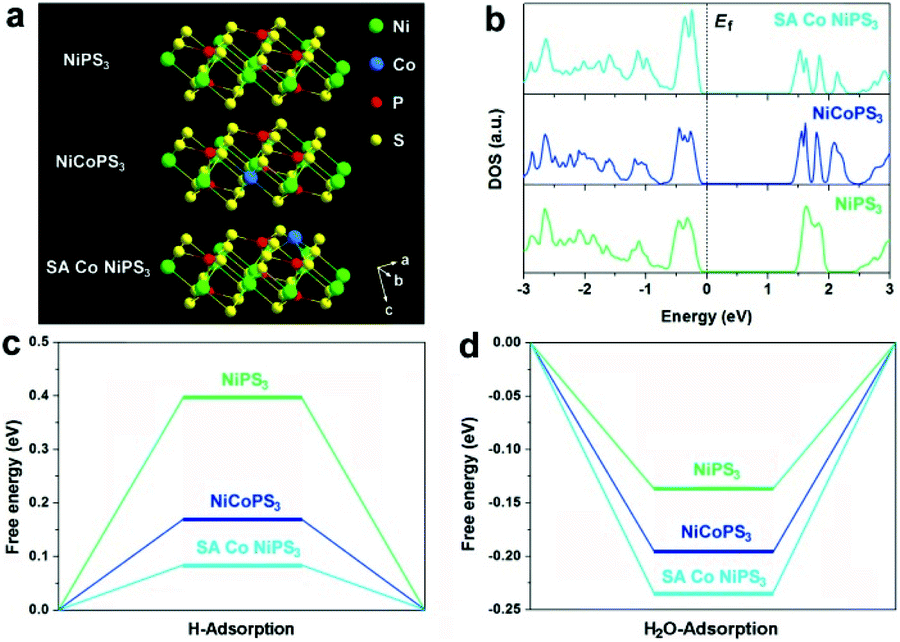 | ||
| Fig. 1 (a) Optimized structure of NiPS3, NiCoPS3, and SA Co NiPS3. (b) Calculated TDOS for NiPS3, NiCoPS3, and SA Co NiPS3. (c) DFT calculated free-energy diagram for the HER of NiPS3, NiCoPS3, and SA Co NiPS3. (d) H2O molecular absorption energy diagram for NiPS3, NiCoPS3, and SA Co NiPS3. | ||
Fig. 2a displays the schematic of the preparation of the SA Co NiPS3 catalysts via an ultrasonic assembly followed by a leaching method, which ensures the successful introduction of single Co atoms on the surface other than substitution doping of Ni. Uniform Co nanodisks (Fig. S1, ESI†), as a smart intermediation, were first assembled on the basal plane of mono-layered NiPS3 (Fig. S2, ESI†) to achieve Co@NiPS3 heterojunction via sonication-induced Co–S bonding, which was removed next by an electrochemical CV leaching process,36 finally, leaving single Co atoms anchored on NiPS3 (SA Co NiPS3) plane. Phonons at ultrasonic power may provide sufficient energy to overcome the barrier for the formation of 2D/2D heterojunctions (Co@NiPS3) via Co–S bond formation.37 TEM image, Raman spectra (Fig. S3, ESI†), and XRD patterns (Fig. S4, ESI†) present that the Co nanodisks are well contacted with NiPS3 after sonication. From Fig. 1b, the TEM image of SA Co NiPS3 clearly shows a smooth lamellar morphology with a diameter of 50–100 nm, and the nearly transparent feature indicates the ultrathin thickness of the catalyst. The regular bright spots with a hexagonal symmetry from the SAED pattern (inset of Fig. 2b) indicate a single-crystalline structure exposed with the (001) facets along their primary surfaces. Meanwhile, the good structure stability of SA Co NiPS3 can also be demonstrated via morphology comparison (Fig. S2b and 1b†), which even suffers from sonication as well as cyclic voltammetry leaching processes (Fig. S5, ESI†). To clarify the accurate distribution of the Co atoms, aberration-corrected HAADF-STEM characterization is executed as shown in Fig. 1c. First, one set of lattice fringes with distances of about 0.583 and 1.011 nm were marked, which matched well with that of the standard NiPS3 crystal.38 Second, the isolated bright spots on the hexagonal crystal base were highlighted by purple circles, which are assigned to Co atoms comfortably according to the atomic weight. Besides, the corresponding line profiles following the two orientations (marked by the blue rectangle) were extracted from the STEM image and monitored in Fig. 1d, further testifying the homogeneous distribution of Co single atoms on the basal plane of NiPS3, and the location of the Co atom overlaps with that of the P atom.
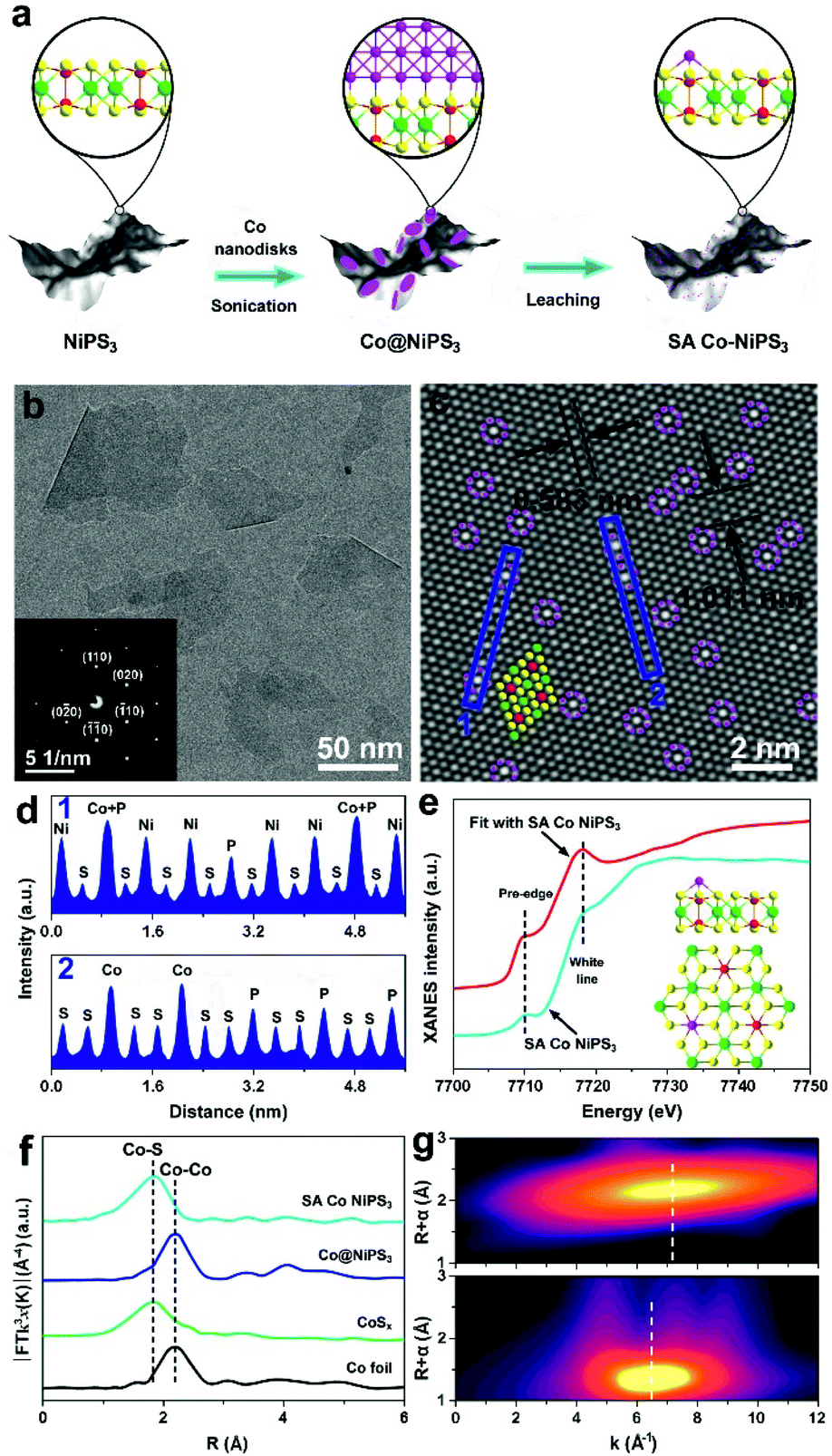 | ||
| Fig. 2 (a) Schematic of the synthesis process of the SA Co NiPS3. (b) TEM images of SA Co NiPS3; the inset shows the corresponding SAED pattern. (c) Aberration-corrected HAADF-STEM image of SA Co NiPS3 together with the line profiles (d) extracted from the areas marked with blue rectangles of (c). (e) Co K-edge XANES of SA Co NiPS3 and fitted curve. The inset shows the atomic structure of SA Co NiPS3. (f) FT-EXAFS spectra of various Co-based catalysts at the Co K-edge. (g) Corresponding wavelet transform analysis for the k2-weighted Co K-edge EXAFS of Co foil (top) and SA Co NiPS3 (bottom). | ||
In previous reports about metal-based semiconductor catalysts, the metal atoms have been usually verified as the catalyst active center. For the SA metal catalyst system, the local coordination environment and the electronic structure of the SA metal also play a crucial role in the catalytic process.39 To investigate the electronic properties and coordination features of Co species in SC Co NiPS3, EXAFS and XAFS techniques were conducted on a series of Co-based samples. The XANES spectrum of the Co K-edge for SA Co NiPS3 with Co foil, CoSx and Co@NiPS3 as contrasts (Fig. S6†) indicates that the Co SAs possess higher valence states than that of CoSx and Co@NiPS3 according to the profile of SA Co NiPS3 (between those of the Co foil and CoSx or Co@NiPS3),40 caused by the free electrons associated with the Co SAs, which were partially depleted to the bonding S atoms. It is generally accepted that the pre-edge characteristic is due to the 1s → 3d orbital forbidden transition, which would be excluded by dipole selection rules for a symmetry site.41 Taking the SA Co NiPS3 as a model (the inset of Fig. 2e: Co atom was located upon the P atom through the coordination of three adjacent S atoms), we also simulated the XANES spectrum,42 as displayed in Fig. 2e. Clearly, the same pre-edge and white line energy values were marked at 7710.3 and 7717.9 eV, respectively, further demonstrating our expectation beginning: the single Co atom is right at the P atom atop site, consistent with the above-mentioned HAADF-STEM results (Fig. 2c). For EXAFS spectra, the best-fit results of Co@NiPS3 and SA Co NiPS3 are summarized in Fig. S7 and Table S1,† respectively. From Fig. 2f, undoubtedly, a single strong signal recorded at ∼1.78 Å in the R-space of the EXAFS spectrum suggests the exclusive existence of the Co–S bond in SA Co NiPS3.43 To more deeply uncover the dispersion of Co atoms on the basal planes of mono-layered NiPS3, wavelet transform of the Co K-edge EXAFS oscillations on the Co foil and SA Co NiPS3 are displayed in Fig. 2g. For the Co foil, only one wavelet transform focus at 7.2 Å−1 was found, which corresponds with the previous reports on Co–Co active centers.44 The single signal is concentrated at 6.5 Å−1 over the SA Co NiPS3 catalyst, in line with the Co–S bond features.27 This visual analysis of wavelet transform EXAFS results again confirmed the fact that Co species were individually dispersed as single atoms, consistent with the HAADF-STEM images.
AFM was employed to accurately monitor the thickness of the as-prepared ultrathin catalysts. The AFM picture of NiPS3 (Fig. 3a) and the corresponding height profile (Fig. 3b) clearly displayed that the average thickness of exfoliated NiPS3 is ∼0.95 nm, which agrees well with the thickness of mono-layered NiPS3.45 Suffering from the ultrasonic and leaching processes, mono-layered NiPS3 well maintained the thickness, as shown in Fig. 3c and d, which exhibited superior structure stability simultaneously. HAADF-STEM (Fig. 3e) and corresponding STEM-EDX element mapping images (Fig. 3f–i) of the SA Co NiPS3 catalyst present that Ni, Co, S and P were all uniformly distributed over the 2D basal surface. Additionally, the EDX mapping spectrum reveals that the atomic ratio of Ni![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :P:S is close to 1:1:2.89, which conforms to the theoretical value and the atomic ratio of Ni:Co is about 1:0.053, matching well with the result of cursory EDS (1:0.062) and accurate ICP-AES (1:0.057), as shown Fig. S8 and Table S2† respectively. For these mono-layer-based catalyst samples, XPS surface analysis was more accurate for uncovering the electronic properties and bonding configurations compared to other nanostructures. For blank single-layered NiPS3 flakes (the top in Fig. 3j), six obvious peaks at 881.8, 875.6, 871.7, 864.1, 858.6, and 854.0 eV that deconvoluted from the high-resolution Ni 2p XPS curve could be assigned to the spin–orbit doublets of 2p1/2 and 2p3/2 and the corresponding satellites from Ni2+ species, respectively.46 After Co nanodisk merging, the six primary XPS signals from the Co@NiPS3 heterojunction kept well except the overall shifts to higher binding energies (∼0.3 eV) slightly, suggesting the decrease in the electron density around Ni atoms.47 Further shifts toward higher binding energies (∼0.4 eV) can be found for the SA Co NiPS3 sample in comparison with Co@NiPS3, demonstrating the stronger electron delocalization from Ni atoms, which was influenced by the introduced Co SAs. This unidirectional electron migration behavior may guarantee enough electron requirements in a highly active HER process. Furthermore, Fig. 3k summarizes the comparison on Co 2p spectra features of Co nanodisks, Co@NiPS3, and SA Co NiPS3. After 30 h of continuous ultrasonic treatment, a distinct Co–S signal appeared at ∼776.9 eV, indicating that the strong interaction in Co@NiPS3 was successfully established between Co nanodisks and NiPS3 nanosheets. It is not difficult to find that the Co–Co bond disappears thoroughly and the Co–S bond heightens to a primary peak from the Co 2p curve of SA Co NiPS3 after continuous 200th electrochemical leaching, which coincides with the results of XAFS and EDX.
:P:S is close to 1:1:2.89, which conforms to the theoretical value and the atomic ratio of Ni:Co is about 1:0.053, matching well with the result of cursory EDS (1:0.062) and accurate ICP-AES (1:0.057), as shown Fig. S8 and Table S2† respectively. For these mono-layer-based catalyst samples, XPS surface analysis was more accurate for uncovering the electronic properties and bonding configurations compared to other nanostructures. For blank single-layered NiPS3 flakes (the top in Fig. 3j), six obvious peaks at 881.8, 875.6, 871.7, 864.1, 858.6, and 854.0 eV that deconvoluted from the high-resolution Ni 2p XPS curve could be assigned to the spin–orbit doublets of 2p1/2 and 2p3/2 and the corresponding satellites from Ni2+ species, respectively.46 After Co nanodisk merging, the six primary XPS signals from the Co@NiPS3 heterojunction kept well except the overall shifts to higher binding energies (∼0.3 eV) slightly, suggesting the decrease in the electron density around Ni atoms.47 Further shifts toward higher binding energies (∼0.4 eV) can be found for the SA Co NiPS3 sample in comparison with Co@NiPS3, demonstrating the stronger electron delocalization from Ni atoms, which was influenced by the introduced Co SAs. This unidirectional electron migration behavior may guarantee enough electron requirements in a highly active HER process. Furthermore, Fig. 3k summarizes the comparison on Co 2p spectra features of Co nanodisks, Co@NiPS3, and SA Co NiPS3. After 30 h of continuous ultrasonic treatment, a distinct Co–S signal appeared at ∼776.9 eV, indicating that the strong interaction in Co@NiPS3 was successfully established between Co nanodisks and NiPS3 nanosheets. It is not difficult to find that the Co–Co bond disappears thoroughly and the Co–S bond heightens to a primary peak from the Co 2p curve of SA Co NiPS3 after continuous 200th electrochemical leaching, which coincides with the results of XAFS and EDX.
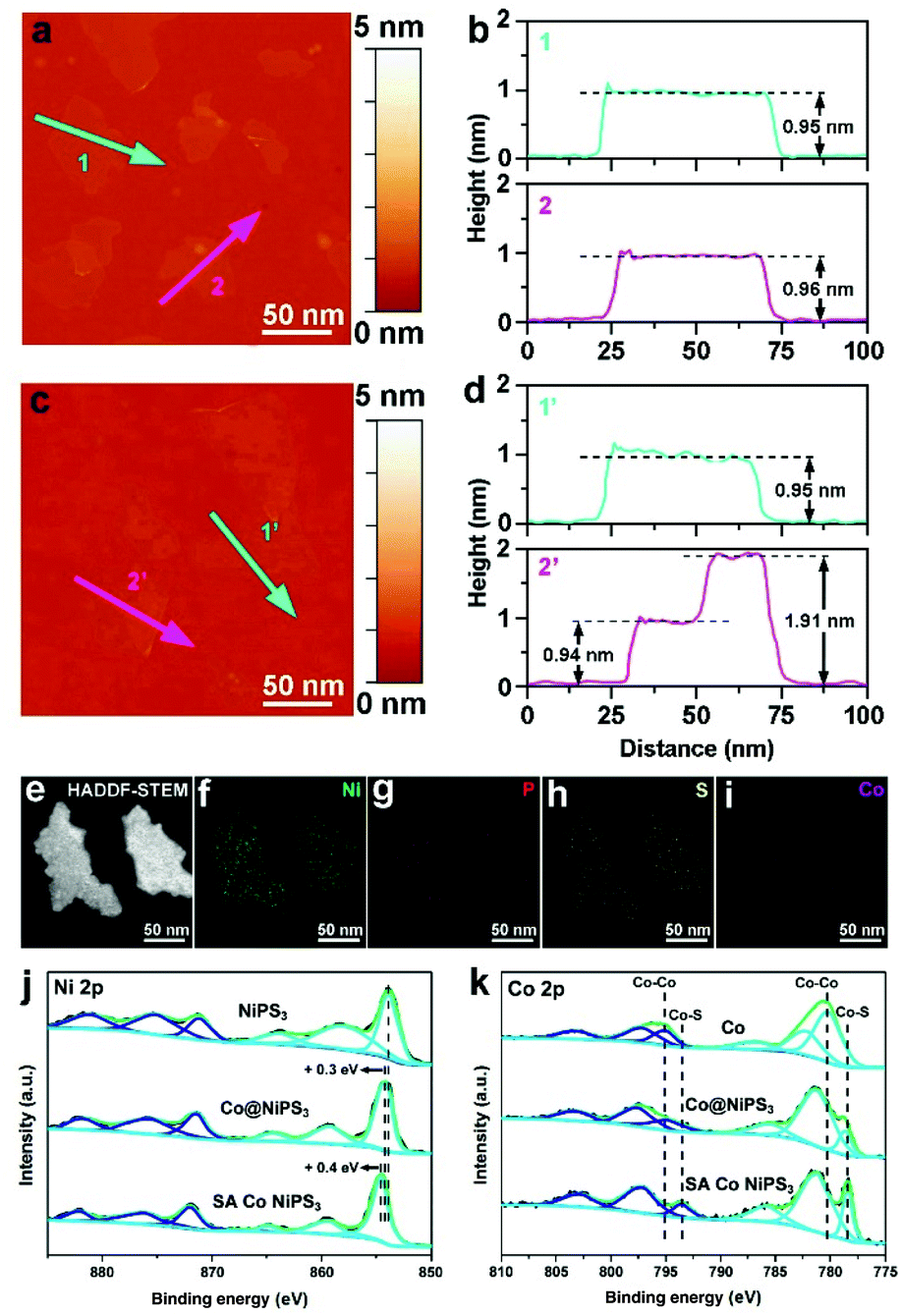 | ||
| Fig. 3 AFM images of NiPS3 (a) and SA Co NiPS3 (b). The corresponding height profiles of NiPS3 (c) and SA Co NiPS3 (d). (e) HAADF-STEM image and corresponding STEM-EDX element mappings on SA Co NiPS3: Ni (f), P (g), S (h), and Co (i). High-resolution XPS spectra for Ni 2p (j) of NiPS3, Co@NiPS3, and SA Co NiPS3. Co 2p (k) of Co nanodisks, Co@NiPS3, and SA Co NiPS3. | ||
To understand the effect of SAs Co in NiPS3 on their HER activity, the electrocatalytic activity towards the HER over the three NiPS3-based catalysts and Pt/C benchmark were evaluated by a standard three-electrode system in an alkaline electrolyte (1.0 M KOH) (see the Experimental section for details, ESI†). Fig. 4a displays the polarization curves recorded by linear sweep voltammetry (LSV) tests. Clearly, the HER performance from the pristine NiPS3 monolayer was comparable to that of ultrathin MPS3 catalysts recently reported.22,48–50 In comparison, the Co@NiPS3 electrode only requires an overpotential of 153.5 mV (vs. RHE) to generate a current density of 10 mA cm−2, much lower than that of the blank NiPS3 monolayers. This boosting can be ascribed to the electronic modulation of the metal atoms, implied by the results of XPS. Impressively, after anchoring Co SAs, the HER activity of SA Co NiPS3 significantly enhances closing to the commercial Pt/C benchmark (only 20 mV gap at 10 mA cm−2), which is among the best values in layered electrocatalysts (Table S3†). Based on the Butler–Volmer equation and its derivations, the Tafel plots of these four electrodes were fitted to obtain the Tafel slopes for confirming the rate-determining steps of the catalysts (Fig. 4b). As shown in Fig. 4b, the pure NiPS3 and Co@NiPS3 heterojunctions output the Tafel slopes of 77.2 and 102.1 mV dec−1, respectively, which can be classified as the Volmer rate-determining step, a step of the primary discharge of protons.51,52 In sharp contrast, the SA Co NiPS3 electrode exhibits very low Tafel slopes of 38.8 mV dec−1, close to that of Pt/C (36.0 mV dec−1) at which the Heyrovsky reaction pathway is the rate-determining step. The lower Tafel slope from SA Co NiPS3 than the original NiPS3 monolayer as well as the Co@NiPS3 heterojunction indicates much better charge transfer over the SA Co NiPS3 flake, agreeing well with the electrochemical impedance spectroscopy tests that display the lowest reaction resistance of the SA Co NiPS3 electrocatalyst (Fig. S9†). The potentiostatic test (Fig. S10†) presents no significant change in the current density (only 5.2% decline) under a potential of −0.10 V (vs. RHE) for 6 days, suggesting an outstanding catalytic stability from the SA Co NiPS3 catalyst. Besides, the robust stability is further verified by the comparison of polarization curves before and after 10000 cycles tests (Fig. S11†).
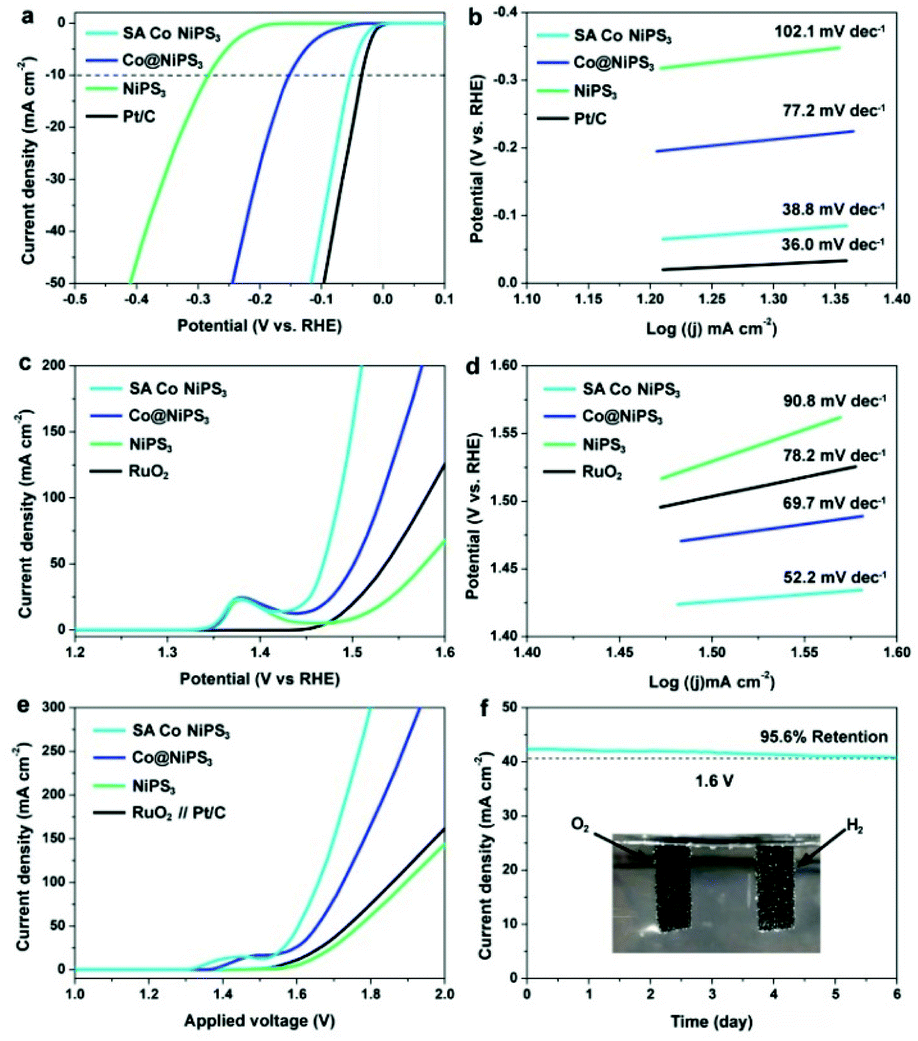 | ||
| Fig. 4 (a) HER polarization curves, (b) corresponding Tafel plots, (c) OER polarization curves, (d) corresponding Tafel plots, and (e) polarization curves for water splitting of NiPS3, Co@NiPS3, and SA Co NiPS3. Pt/C, RuO2, and RuO2//Pt/C were employed as the reference standard for the HER, OER, and overall water splitting, respectively. (f) The chronoamperometric curves of SA Co NiPS3 for electrocatalytic water splitting at 1.6 V applied potential. The inset in (f) is the optical image of water electrolysis using SA Co NiPS3 as both the cathode and the anode. All of the obtained potentials were directly calibrated into a reversible hydrogen electrode (RHE) and performed with iR correction (80%). | ||
The electrocatalytic ability of the SA Co NiPS3 NRs for OER application was also detected by LSV in 1.0 M KOH electrolyte. Fig. 4c records the OER activity of the three as-prepared electrodes above and RuO2 as the reference. An obvious peak located at ∼1.38 V (vs. RHE) can be clearly observed in the LSV curves of the three NiPS3-based anodes due to the Ni2+/Ni3+ redox reaction.53 Focusing on the profile of polarization curves, beyond expectation, SA Co NiPS3 and Co@NiPS3 display an extraordinary OER activity with an onset potential of 146.7 mV (vs. RHE) and an overpotential of 151.8 mV (vs. RHE) at a current density of 50 mA cm−2, surpassing the commercial RuO2 benchmark. An exploration of the electrocatalytic OER kinetics over the three NiPS3-based samples with RuO2 was further probed based on the Tafel plot method, as shown in Fig. 4d. As expected, the Tafel slope of the SA Co NiPS3 electrode (52.2 mV dec−1) is far below that of Co@NiPS3 (69.7 mV dec−1), NiPS3 (90.8 mV dec−1), and RuO2 benchmark (78.2 mV dec−1). Overall, the SA Co NiPS3 sample presents impressive OER catalytic activity, even superior to most of the reported 2D electrocatalysts in an alkaline environment (Table S4†). Meanwhile, the chronopotentiometry test suggests superior cyclic stability of the SA Co NiPS3 catalyst with the 96.3% retention of voltage (Fig. S12†). Considering the results of the durability measurement before and after 10000 cycles (Fig. S13†), together with the contrast of XPS characterization before and after HER/OER cycles35 (Fig. S14–S17†), we have sufficient evidence to confirm the astounding activities and stabilities over the as-synthesised bi-functional SA-based electrocatalysts. As a common cognition in heterocatalysis, specific surface area and porosity features of the catalyst are crucial factors that significantly impact catalytic performance. N2 adsorption–desorption isotherms were carried out to estimate the specific surface area and porosity of the three catalysts (Fig. S18 and S19†). Notably, the NiPS3 (39.5 m2 g−1) and SA Co NiPS3 (41.7 m2 g−1) nanoflakes have analogous surface areas and pore size distributions because of the good structural stability from the advanced preparation strategies. After cycle tests, the BET data maintained well, which also proved the stable performance of SA Co NiPS3. From Fig. S20 in the ESI,† the electrochemically active surface area (ECSA) assessed by Cdl displays that SA Co NiPS3 (31.8 mF cm−2) output much larger ECSA than that of Co@NiPS3 (17.3 mF cm−2) and NiPS3 (7.8 mF cm−2), implying that the enhanced electrocatalytic activity was triggered from the Co SAs anchoring. Motivated by the high catalytic activity from the obtained NiPS3-based catalysts toward both the HER and the OER in a KOH (1 M) solution, we further employed these NiPS3-based electrodes as both the anode and the cathode for overall water splitting in the same electrolysis media. As recorded in Fig. 4e, a current density of 50 mA cm−2 can be obtained at a cell voltage as low as 1.603 V from the polarization curve of SA Co NiPS3, beyond that of the other two NiPS3-based samples (1.658 and 1.769 V for Co@NiPS3 and NiPS3, respectively). Impressively, the catalytic activity from the SA Co NiPS3 electrode toward overall water splitting was much higher than that of Pt/C coupled with the IrO2/C electrode system (1.731 V@50 mA cm−2). In addition, 6 days chronoamperometric (Fig. 4f) and 10000 cycles testing (Fig. S21†) over the optimal electrocatalyst (SA Co NiPS3) exhibit only a slight deactivation, proving the excellent stability of the SA Co NiPS3 in an overall water splitting application. Besides, the outstanding stability of SA Co NiPS3 was further confirmed by the comparison of the nondestructive characterization before and after water splitting tests, including XRD, aberration-corrected HAADF-STEM, EDS (Fig. S22†), and Raman spectroscopy (Fig. S23†). This performance from SA Co NiPS3 toward overall water splitting was comparable to that of the other 2D-based electrocatalysts reported presently (Table S5†).
To gain insights into the effect of a single Co atom on the electrocatalysis activity of NiPS3, DFT calculations were performed on SA Co NiPS3 with Co in situ replaced NiPS3 as the reference. For the HER, the advantage of hydrogen evolution over SA Co NiPS3 was verified through the obtained lower ΔGH* value from the SA Co NiPS3 (Fig. 1c). In the four electron mechanism of the OER process as presented in Fig. 5a and b, a single bond between oxygen intermediates and the surface of catalyst is the only attended mode for simplification.54 All the possible catalytic active sites influenced by the Co atom were fixed as the adjacent S, P, and Co atoms themselves. Bonding with the three different sites, the Gibbs free energies of the four steps over NiCoPS3 and SA Co NiPS3 are shown in Fig. 5c and d: low adsorbed energy difference between ΔGO* and ΔGOH* is desirable; the largest difference between two connected energy levels is identified as the rate determining step.55 For the three different sites on NiCoPS3, the rate determining step in each case is inconsistent: the second step for connecting with S–Co and P–S–Co (OH* → O*); the third step for connecting with Co (O* → OOH*), with the calculated determining potentials of 1.73, 1.01, and 0.49 V respectively. In view of the much lower determining potential, the substituted Co atom site in Co@NiPS3 was identified for optimal catalytic activities, which should be due to the partial charge transfer effect of Ni atoms (Fig. 3j). Changing the Co coordination mode into an unsaturated state, the SA Co NiPS3 sample demonstrated a similar rate determining step to Co@NiPS3. However, the connection with Co path delivers an ultralow determining potential of 0.19 V, the lowest theoretical potential barrier value in all possible attended mode,56,57 which agrees well with our experimental results.
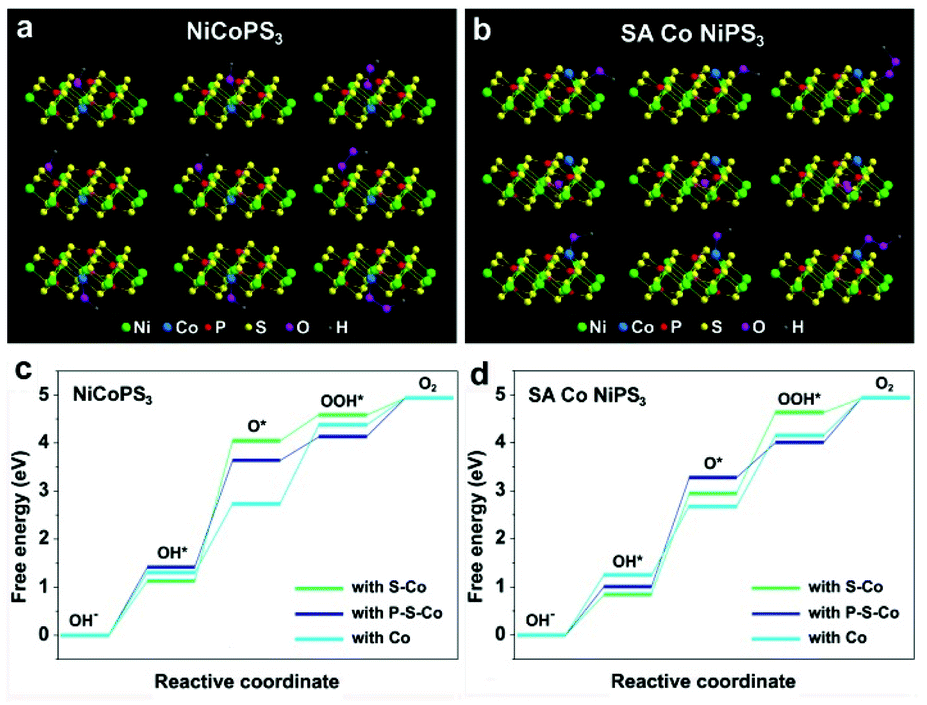 | ||
| Fig. 5 DFT calculations of the NiCoPS3 and SA CoNiPS3 for OER: (a) three different connect sites around the alternative Co atom and (b) the Gibbs free energy diagram for the four steps of OERs on NiCoPS3. (c) Three different connect sites around the anchoring Co atom and (d) the Gibbs free energy diagram for the four steps of OER on SA Co NiPS3 in alkaline media. | ||
The promising catalytic performance of the designed SA-based NiPS3 electrocatalyst could be ascribed to the following factors: (i) stable monolayered configuration provides abundant reaction sites and large ECSA, increasing electrochemical performance; (ii) the synergistic effect of bimetallic composition results in higher intrinsic activities, better conductivity, and lower energy barriers, which benefit for the electrocatalytic activity; (iii) the almost zero determining potentials from the SA Co site favor the OER activity, which acts as a crucial factor for the overall water splitting process; (iv) unsaturated SA Co design anchoring on the basal plane of NiPS3 single layers causes monodirectional electron migration from nanoflakes to enthetic Co, triggering the activity of water splitting thoroughly. Overall, these combined factors synergistically contribute to the efficient and stable electrocatalytic water splitting performance of the SA Co NiPS3 nanoflakes.
3. Conclusion
In conclusion, enlightened by the single-atom metals boosting the electrocatalytic property of layered MoS2, we have designed and assembled single-atom Co decorating on the basal plane of NiPS3 monolayers via a top-down assembly/leaching strategy. The as-prepared SA Co NiPS3 catalyst exhibits impressive catalytic activities with ultralow overpotentials of 1.21 and 1.47 V at 50 mA cm−2 for HERs and OERs, respectively. More importantly, this catalytic performance has great potential to achieve overall water splitting via delivering a very low cell voltage of 1.60 V at 50 mA cm−2 with excellent durability, outperforming the noble metallic Pt/C//RuO2 couple, which holds promise for applications in large-scale water splitting electrolyzers. Additionally, theoretical investigations demonstrate that Co SA anchored on the basal plane of NiPS3 monolayers is a crucial active site for HERs with ultrafast hydrogen adsorption/desorption kinetics; meanwhile, for OERs with the lowest determining potentials, both originates from strong hybridization between the single Co atoms and water. Our work about the SA Co-decorated NiPS3 highlights the area of modifying the monolayer metal thiophosphate catalysts on the atomic scale, which could significantly make a footprint in advanced technological development for large-scale industrial-grade applications.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We acknowledge National Natural Science Foundation of China (52072182, U1732126, and 51872145), Natural Science Foundation of Jiangsu Province (BK20211278), China Postdoctoral Science Foundation (No. 2019M650120 and 2020M671554), and National Synergetic Innovation Center for Advanced Materials (SICAM).Notes and references
- H. Huang, M. Yan, C. Yang, H. He, Q. Jiang, L. Yang, Z. Lu, Z. Sun, X. Xu, Y. Bando and Y. Yamauchi, Adv. Mater., 2019, 31, 1903415 CrossRef CAS PubMed.
- W. Chen, G. Huang, H. Song and J. Zhang, J. Mater. Chem. A, 2020, 8, 20963–20969 RSC.
- T. Ouyang, X. Wang, X. Mai, A. Chen, Z. Tang and Z. Liu, Angew. Chem., Int. Ed., 2020, 59, 11948–11957 CrossRef CAS PubMed.
- J. Wang, W. Liu, X. Li, T. Ouyang and Z. Liu, Chem. Commun., 2020, 56, 1489–1492 RSC.
- J. Wei, M. Cao, X. Guo, S. Ye and Z. Liu, Small Struct., 2021, 2, 2100047 CrossRef.
- J. Chen, Q. Long, K. Xiao, T. Ouyang, N. Li, S. Ye and Z. Liu, Sci. Bull., 2021, 66, 1063–1072 CrossRef CAS.
- K. Xiao, J. Wei, W. Han and Z. Liu, J. Power Sources, 2021, 487, 229408 CrossRef CAS.
- J.-H. Qin, P. Xu, Y.-D. Huang, L.-Y. Xiao, W. Lu, X.-G. Yang, L. Ma and S.-Q. Zang, Chem. Commun., 2021, 57, 8468–8471 RSC.
- J. H. Qin, H. Zhang, P. Sun, Y. D. Huang, Q. Shen, X. G. Yang and L. F. Ma, Dalton Trans., 2020, 49, 17772–17778 RSC.
- J. Li, H. Huang, X. Cao, H. Wu, K. Pan, Q. Zhang, N. Wu and X. Liu, Chem. Eng. J., 2021, 416, 127677 CrossRef CAS.
- J. Li, X. Liu and J. Zhang, ChemSusChem, 2020, 13, 2996–3004 CrossRef CAS.
- C. Zhang, X. Liang, R. Xu, C. Dai, B. Wu, G. Yu, B. Chen, X. Wang and N. Liu, Adv. Funct. Mater., 2021, 31, 2008298 CrossRef CAS.
- N. Yao, P. Li, Z. Zhou, R. Meng, G. Cheng and W. Luo, Small, 2019, 15, 1901993 CrossRef.
- Y. Yang, H. Yao, Z. Yu, S. Islam, H. He, M. Yuan, Y. Yue, K. Xu, W. Hao, G. Sun, H. Li, S. Ma, P. Zapol and M. Kanatzidis, J. Am. Chem. Soc., 2019, 141, 10417–10430 CrossRef CAS PubMed.
- J. Zhang, L. Wang, X. Liu, X. Li and W. huang, J. Mater. Chem. A, 2015, 3, 535–541 RSC.
- Z. Lv, W. Ma, M. Wang, J. Dang, K. Jian, D. Liu and D. Huang, Adv. Funct. Mater., 2021, 31, 2102576 CrossRef CAS.
- T. Zhao, X. Shen, Y. Wang, R. Hocking, Y. Li, C. Rong, K. Dastafkan, Z. Su and C. Zhao, Adv. Funct. Mater., 2021, 31, 2100614 CrossRef CAS.
- Z. Chen, M. Ju, M. Sun, L. Jin, R. Cai, Z. Wang, L. Dong, L. Peng, X. Long, B. Huang and S. Yang, Angew. Chem., Int. Ed., 2021, 60, 9699–9705 CrossRef CAS PubMed.
- J. Zhang, R. Cui, C. Gao, L. Bian, Y. Pu, X. Zhu, X. Li and W. Huang, Small, 2019, 17, 1904688 CrossRef PubMed.
- J. Song, S. Qiu, F. Hu, Y. Ding, S. Han, L. Li, H. Chen, X. Han, C. Sun and S. Peng, Adv. Funct. Mater., 2021, 31, 2100618 CrossRef CAS.
- X. Li, Y. Fang, J. Wang, B. Wei, K. Qi, H. Hoh, Q. Hao, T. Sun, Z. Wang, Z. Yin, Y. Zhang, J. Lu, Q. Bao and C. Su, Small, 2019, 17, 1902427 CrossRef PubMed.
- Q. Liang, L. Zhong, C. Du, Y. Zheng, Y. Luo, J. Xu, S. Li and Q. Yan, Adv. Funct. Mater., 2018, 28, 1805075 CrossRef.
- H. Li, C. Tsai, L. Ai, L. Cai, A. Contryman, A. Fragapane, J. Zhao, H. Han, H. Manoharan and F. Abild-Pedersen, Nat. Mater., 2015, 15, 48–53 CrossRef PubMed.
- Y. Wang, H. Su, Y. He, L. Li, S. Zhu, H. Shen, P. Xie, X. Fu, G. Zhou, C. Feng, D. Zhao, F. Xiao, X. Zhu, Y. Zeng, M. Shao, S. Chen, G. Wu, J. Zeng and C. Wang, Chem. Rev., 2020, 120, 12217–12314 CrossRef CAS PubMed.
- N. Xuan, J. Chen, J. Shi, Y. Yue, P. Zhuang, K. Ba, Y. Sun, J. Shen, Y. Liu, B. Ge and Z. Sun, Chem. Mater., 2019, 31(2), 429–435 CrossRef CAS.
- S. Ji, Y. Chen, X. Wang, Z. Zhang, D. Wang and Y. Li, Chem. Rev., 2020, 120, 11900–11955 CrossRef CAS PubMed.
- K. Qi, X. Cui, L. Gu, S. Yu, X. Fan, M. Luo, S. Xu, N. Li, L. Zheng, Q. Zhang, J. Ma, Y. Gong, F. Lv, K. Wang, H. Huang, W. Zhang, S. Guo, W. Zheng and P. Liu, Nat. Commun., 2019, 10, 5231 CrossRef PubMed.
- Y. Luo, S. Zhang, H. Pan, S. Xiao, Z. Guo, L. Tang, U. Khan, B. Ding, M. Li, Z. Cai, Y. Zhao, W. Lv, Q. Feng, X. Zou, J. Lin, H. Cheng and B. Liu, ACS Nano, 2020, 14, 767–776 CrossRef CAS PubMed.
- Z. Li, Z. Wang, S. Xi, X. Zhao, T. Sun, J. Li, W. Yu, H. Xu, T. Herng, X. Hai, P. Lyu, M. Zhao, S. Pennycook, J. Ding, H. Xiao and J. Lu, ACS Nano, 2021, 15, 7105–7113 CrossRef CAS PubMed.
- D. Kuznetsov, Z. Chen, P. Kumar, A. Tsoukalou, A. Kierzkowska, P. Abdala, O. Safonova, A. Fedorov and C. Müller, J. Am. Chem. Soc., 2019, 141, 17809–17816 CrossRef CAS PubMed.
- H. Fei, J. Dong, M. Arellano-Jiménez, G. Ye, N. Kim, E. Samuel, Z. Peng, Z. Zhu, F. Qin, J. Bao, M. Yacaman, P. Ajayan, D. Chen and J. James, Nat. Commun., 2015, 6, 8668 CrossRef CAS PubMed.
- H. Huang, F. Li, Q. Xue, Y. Zhang, S. Yin and Y. Chen, Small, 2019, 15, 1903500 CrossRef CAS PubMed.
- B. Qiu, L. Cai, Y. Wang, Z. Lin, Y. Zuo, M. Wang and Y. Chai, Adv. Funct. Mater., 2018, 28, 1706008 CrossRef.
- J. Greeley, T. Jaramillo, J. Bonde, I. Chorkendorff and J. Norskov, Nat. Mater., 2006, 5, 909–913 CrossRef CAS PubMed.
- B. Song, K. Li, Y. Yin, T. Wu, L. Dang, M. Cabán-Acevedo, J. Han, T. Gao, X. Wang, Z. Zhang, J. Schmidt, P. Xu and S. Jin, ACS Catal., 2017, 7, 8549–8557 CrossRef CAS.
- L. Fan, P. Liu, X. Yan, L. Gu, Z. Yang, S. Qiu and X. Yao, Nat. Commun., 2016, 7, 10667 CrossRef CAS PubMed.
- X. Shi, S. Posyasev, M. Huttula, V. Pankratov, J. Hoszowska, J. Dousse, F. Zeeshan, Y. Niu, A. Zakharov, T. Li, O. Miroshnichenko, M. Zhang, X. Wang, Z. Huang, S. Saukko, D. Gonzalez, S. Dijken, M. Altalo and W. Cao, Small, 2018, 14, 1704526 CrossRef PubMed.
- D. Goossens, D. James, J. Dong, R. Whitfield, L. Noren and R. Withers, J. Phys.: Condens. Matter, 2011, 23, 065401 CrossRef CAS PubMed.
- J. Fu, J. Dong, R. Si, K. Sun, J. Zhang, M. Li, N. Yu, B. Zhang, M. Humphrey, Q. Fu and J. Huang, ACS Catal., 2021, 11, 1952–1961 CrossRef CAS.
- Z. Chen, C. Liu, J. Liu, J. Li, S. Xi, X. Chi, H. Xu, I. Park, X. Peng, X. Li, W. Yu, X. Liu, L. Zhong, K. Leng, W. Huang, M. Koh and K. Loh, Adv. Mater., 2019, 31, 1906437 Search PubMed.
- J. Zheng, S. Wu, L. Lu, C. Huang, P. Ho, A. Kirkland, T. Sudmeier, R. Arrigo, D. Gianoliod and S. Tsang, Chem. Sci., 2021, 12, 688–695 RSC.
- W. Liu, L. Zhang, W. Yan, X. Liu, X. Yang, S. Miao, W. Wang, A. Wang and T. Zhang, Chem. Sci., 2016, 7, 5758–5764 RSC.
- H. Kasai, K. Tolborg, M. Sist, J. Zhang, V. Hathwar, M. Filso, S. Cenedese, K. Sugimoto, J. Overgaard and B. Iversen, Nat. Mater., 2018, 17, 249–252 CrossRef CAS PubMed.
- W. Liu, W. Hu, L. Yang and J. Liu, Nano Energy, 2020, 73, 104750 CrossRef CAS.
- J. Wang, X. Li, B. Wei, R. Sun, W. Yu, H. Hoh, H. Xu, J. Li, X. Ge, Z. Chen, C. Su and Z. Wang, Adv. Funct. Mater., 2020, 30, 1908708 CrossRef CAS.
- Q. Liang, Y. Zheng, C. Du, Y. Luo, J. Zhang, B. Li, Y. Zong and Q. Yan, Small Methods, 2017, 1, 1700304 CrossRef.
- S. Gage, C. Ngo, V. Molinari, M. Causa, R. Richards, F. Gentile, S. Pylypenko and D. Esposito, J. Phys. Chem. C, 2018, 122, 339–348 CrossRef CAS.
- J. Zhang, R. Cui, X. a. Li, X. Liu and W. Huang, J. Mater. Chem. A, 2017, 5, 23536–23542 RSC.
- R. Gusmeao, Z. Sofer and M. Pumera, Adv. Funct. Mater., 2019, 29, 1805975 CrossRef.
- Q. Liang, L. Zhong, C. Du, Y. Luo, J. Zhao, Y. Zheng, J. Xu, J. Ma, C. Liu, S. Li and Q. Yan, ACS Nano, 2019, 13, 7975–7984 CrossRef CAS PubMed.
- Y. Cui, Y. Xue, R. Zhang, J. Zhang, X. Li and X. Zhu, J. Mater. Chem. A, 2019, 7, 21911–21917 RSC.
- Y. Luo, X. Li, X. Cai, X. Zou, F. Kang, H. Cheng and B. Liu, ACS Nano, 2018, 12, 4565–4573 CrossRef CAS PubMed.
- S. Hao, L. Chen, C. Yu, B. Yang, Z. Li, Y. Hou, L. Lei and X. Zhang, ACS Energy Lett., 2019, 4, 952–959 CrossRef CAS.
- B. Zhang, C. Zhu, Z. Wu, E. Stavitski, Y. Lui, T. Kim, H. Liu, L. Huang, X. Luan, L. Zhou, K. Jiang, W. Huang, S. Hu, H. Wang and J. Francisco, Nano Lett., 2020, 20, 136–144 CrossRef CAS PubMed.
- H. Xu, T. Liu, S. Bai, L. Li, Y. Zhu, J. Wang, S. Yang, Y. Li, Q. Shao and X. Huang, Nano Lett., 2020, 20, 5482–5489 CrossRef CAS PubMed.
- D. Hu, X. Wang, X. Chen, Y. Wang, A. Hong, J. Zhong, X. Bu, P. Feng and T. Wu, J. Mater. Chem. A, 2020, 8, 11255–11260 RSC.
- F. Diongi, Z. Zeng, I. Sinev, T. Merzdorf, S. Deshpande, M. Lopez, S. Kunze, I. Zegkinoglou, H. Sarodnik, D. Fan, A. Bergmann, J. Drnec, J. de Araujo, M. Gliech, D. Teschner, J. Zhu, W. Li, J. Greeley, B. Roldan Cuenya and P. Strasser, Nat. Commun., 2020, 11, 2522 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta08599a |
| This journal is © The Royal Society of Chemistry 2022 |