
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Atomistic and electronic structure of metal clusters supported on transition metal carbides: implications for catalysis†
Hector
Prats
 * and
Michail
Stamatakis
*
* and
Michail
Stamatakis
*
Department of Chemical Engineering, University College London, Roberts Building, Torrington Place, London WC1E 7JE, UK. E-mail: m.stamatakis@ucl.ac.uk; h.garcia@ucl.ac.uk
First published on 20th December 2021
Abstract
Novel research avenues have been explored over the last decade on the use of transition metal carbides (TMCs) as catalytically active supports for metal nanoclusters, which display high catalytic activity despite the poor reactivity (or even inertness) of the bulk metal. It has been postulated that TMCs polarise the electron density of adsorbed metal particles in such a way that their catalytic activity ends up being superior to those dispersed on more traditional metal oxide supports. Herein, we investigate the structural and electronic properties of small clusters of precious metals (Rh, Pd, Pt and Au) and more affordable metals (Co, Ni and Cu) supported on TMCs with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry (TiC, ZrC, HfC, VC, NbC, TaC, MoC and WC) by means of periodic density functional theory calculations. Our high-throughput screening studies indicate that it is possible not only to have strongly bonded and stably dispersed metal nanoparticles on TMC surfaces, but also to manipulate their charge by carefully selecting elements with desired electronegativity for the host TMC and the metal cluster. By doing so, it is possible to tune the amount of charge density on the cluster hollow sites, which can facilitate the bonding of certain molecules. Moreover, we identify Pt, Pd and Rh clusters supported on hexagonal TMC (001) facets as the candidates with the highest potential catalytic activity—as estimated by the significant polarisation of the cluster electron density—and stability—as quantified by the strongly negative values of adsorption energy per atom and formation energy.
:1 stoichiometry (TiC, ZrC, HfC, VC, NbC, TaC, MoC and WC) by means of periodic density functional theory calculations. Our high-throughput screening studies indicate that it is possible not only to have strongly bonded and stably dispersed metal nanoparticles on TMC surfaces, but also to manipulate their charge by carefully selecting elements with desired electronegativity for the host TMC and the metal cluster. By doing so, it is possible to tune the amount of charge density on the cluster hollow sites, which can facilitate the bonding of certain molecules. Moreover, we identify Pt, Pd and Rh clusters supported on hexagonal TMC (001) facets as the candidates with the highest potential catalytic activity—as estimated by the significant polarisation of the cluster electron density—and stability—as quantified by the strongly negative values of adsorption energy per atom and formation energy.
1. Introduction
Transition metal carbides (TMCs) have been attracting an increasing amount of interest in the last few decades in the field of heterogeneous catalysis due to the following features: (i) they are very resistant and refractory, with melting points in the vicinity of 3000 °C,1 (ii) they exhibit high resistance to carbon deposition or sulphur poisoning,2 two of the main weaknesses of commercial catalysts, and (iii) their economic cost is much lower than that of Pt-group metals. In addition, some TMCs display catalytic activities like those of noble and Pt-group metals for a considerable number of reactions, such as hydrogen oxidation,3 SO2 decomposition4 or the water–gas shift reaction.5Apart from the use of TMCs per se as active materials in catalyst formulations, novel research avenues have been explored, over the last decade, on the use of TMCs as catalytically active supports.6–8 This line of research originated from the theoretical discovery that TiC can modify the electronic structure of supported Au particles, thereby drastically increasing their catalytic activity through strong metal–support interactions (SMSIs) between Au and TiC.9 Subsequent experimental studies showed that small Au nanoparticles supported on TiC can catalyse CO oxidation and desulfurisation processes at low temperatures, with higher activities and selectivities than those obtained when using oxide supports.10 In fact, many studies have shown that manipulating metal–support interactions can be used to tune activity and selectivity in oxide-supported metal particles,11 and this also holds for TMC-supported catalysts. For instance, Posada-Pérez et al. showed that small Au nanoparticles dispersed on MoC are highly active for the low-temperature water–gas shift reaction12 as a consequence of beneficial metal–support interactions, and subsequent kinetic Monte Carlo simulations based on density functional theory (DFT) calculations provided strong evidence for a cooperative effect,13 where MoC is responsible for the adsorption and dissociation of water molecules, while Au nanoparticles contribute to the formation of COOH, a key reaction intermediate. In another joint computational and experimental study, Prats et al. showed that small Ni clusters supported on TiC can chemisorb methane, which is subsequently activated at room temperature.14 The importance of this study lies in the fact that activation of methane at low temperature is a major challenge due to the high stability of the C–H bond (i.e., 4.5 eV dissociation energy in vacuum) and the absence of low-energy empty orbitals,15,16 which make C–H bond cleavage the most probable rate-determining step.17 Moreover, Rodriguez et al. showed that small Au, Cu and Ni particles in contact with TiC display a very high activity for CO2 hydrogenation,8 which can be orders of magnitude higher than those of Au(100), Cu(100), or Ni(100). As shown recently by Lozano-Reis et al., the enhanced activity is attributed to the polarisation that the TMC surface inflicts on the supported particle electronic density,18 which greatly reduces the dissociation energy barriers for CO2 and H2.19
In this work, we undertake a comprehensive theoretical study focusing on small clusters Mn (n = 3, 4) of precious metals (M = Rh, Pd, Pt and Au) and more affordable metals (M = Co, Ni and Cu) supported on TMCs with 1:1 stoichiometry (TiC, ZrC, HfC, VC, NbC, TaC, MoC and WC). Our study employs periodic DFT calculations within a high-throughput screening framework to obtain the structural and electronic properties of these materials. The results obtained from this study yield valuable insight about the catalytic properties of these materials and are expected to motivate and guide further theoretical and experimental studies on metal cluster-supported TMC as catalysts for reactions of practical importance. The rest of this manuscript is organised as follows. The ‘Computational models and methods’ section describes the bulk and slab models adopted and provides the details of the DFT calculations and the relevant formulae used to estimate the adsorption and formation energies, as well as the charge density difference. Subsequently, the ‘Atomistic structure and binding strength’ section begins with a description of the size and configurations studied for the supported clusters, followed by an analysis of the most stable configurations and their adsorption and formation energies. Next, the ‘Electronic properties’ section presents an analysis of the charge transfer, electron density polarization, density of states and magnetic moment of the metal clusters. Then, the ‘Effect of carbon vacancies’ section evaluates the effect of the surface C vacancies on the TMC interaction with the cluster. Finally, in the ‘Conclusions’ section, a brief summary of the main results is provided along with a discussion on the potential catalytic properties of these materials.
2. Computational models and methods
This section introduces all the cubic and hexagonal TMC facets considered for the present screening, the main details of the DFT calculations, the bulk and slab models employed, as well as the energetic properties that have been calculated.2.1. Selected TMC facets
For all carbides under consideration except MoC, the most stable phase corresponds to a face-centred cubic (fcc) crystal packing belonging to space group Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m (i.e., TiC, ZrC, HfC, VC, NbC and TaC). The lowest energy surfaces for all these carbides have been shown to be the (001) faces.20 For MoC, fcc and hexagonal closed packed (hcp) phases can be synthesised by employing different carburising agents,21 which belong to space groups Fmm and P
m (i.e., TiC, ZrC, HfC, VC, NbC and TaC). The lowest energy surfaces for all these carbides have been shown to be the (001) faces.20 For MoC, fcc and hexagonal closed packed (hcp) phases can be synthesised by employing different carburising agents,21 which belong to space groups Fmm and P![[6 with combining macron]](https://www.rsc.org/images/entities/char_0036_0304.gif) m2, respectively. Concerning the MoC cubic phase, the lowest energy surface corresponds to the (001) face.22 In the case of the MoC hexagonal phase, the Mo- and C-terminated (001) faces have been theoretically predicted to be the lowest energy ones, with similar stability.22 For WC, the most stable phase has the hcp crystal structure,23 but a fcc phase can also be synthesised from a reaction between WCl4 and CaC2.24 For the WC hexagonal phase, the lowest energy surface is the W-terminated (001) face, while for the WC cubic phase it is again the (001) face.20 In this work, we considered all the above-mentioned surfaces, as illustrated in Fig. 1. The calculated surface energies are reported in Section S1 in the ESI.†
m2, respectively. Concerning the MoC cubic phase, the lowest energy surface corresponds to the (001) face.22 In the case of the MoC hexagonal phase, the Mo- and C-terminated (001) faces have been theoretically predicted to be the lowest energy ones, with similar stability.22 For WC, the most stable phase has the hcp crystal structure,23 but a fcc phase can also be synthesised from a reaction between WCl4 and CaC2.24 For the WC hexagonal phase, the lowest energy surface is the W-terminated (001) face, while for the WC cubic phase it is again the (001) face.20 In this work, we considered all the above-mentioned surfaces, as illustrated in Fig. 1. The calculated surface energies are reported in Section S1 in the ESI.†
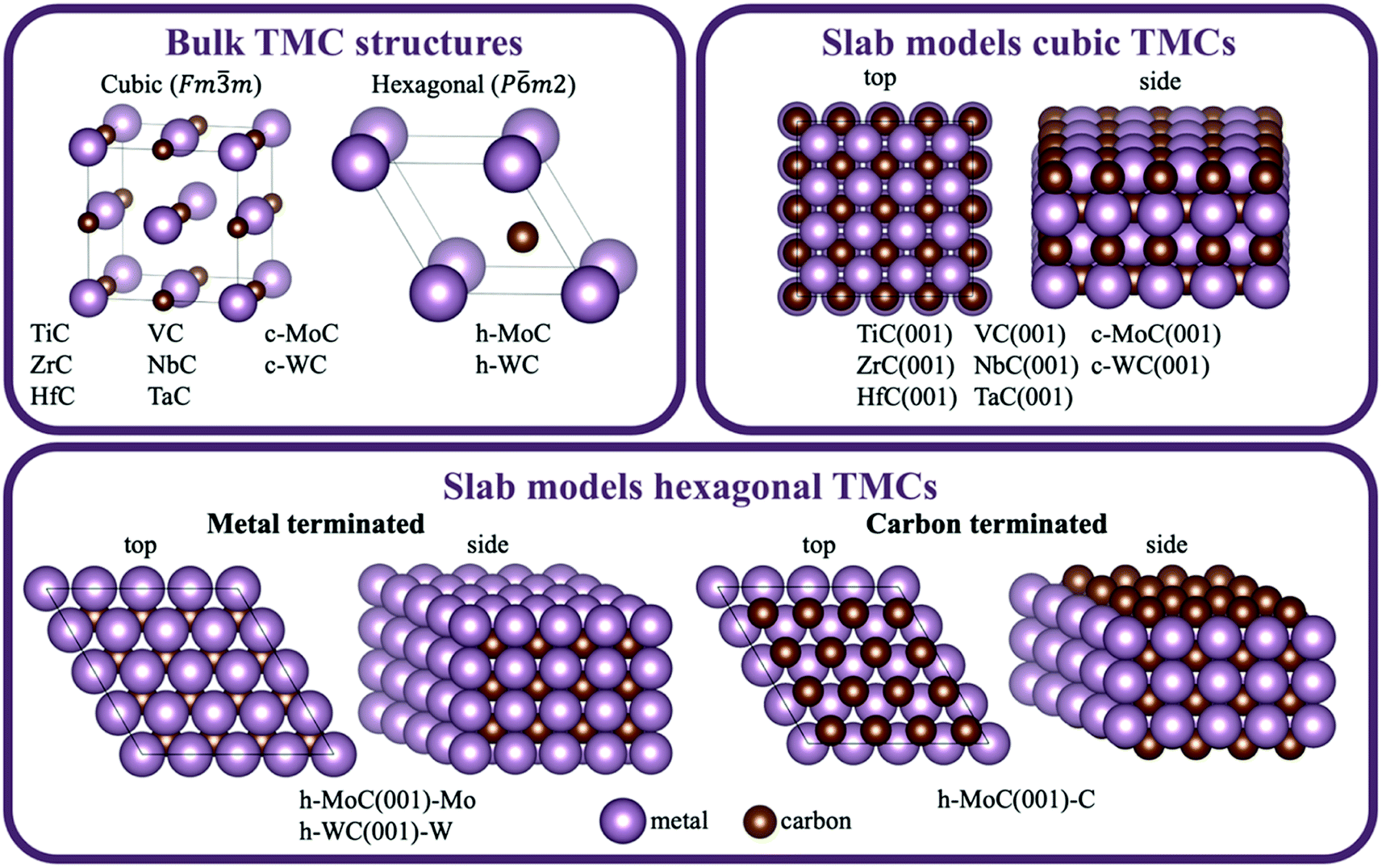 | ||
| Fig. 1 Bulk and slab models used in the present work. The studied systems belonging to each bulk or slab model are indicated below the drawing. | ||
Note that, in the literature, the cubic and hexagonal phases of MoC are denoted δ-MoC and α-MoC, respectively.22 For WC, the cubic phase has been designated as γ-WC, β-WC, α-WC or simply WC25, while the hexagonal phase is denoted as δ-WC25 or also simply WC.23 To ease the notation and avoid confusion, in this work, the cubic (c-) and hexagonal (h-) phases are referred to as c-MoC and h-MoC for MoC, and c-WC and h-WC for WC, respectively.
2.2. DFT calculations for bulk and slab models
For the materials of interest to this study, periodic spin polarised DFT calculations were performed using the Vienna Ab initio Simulation Package (VASP), version 5.4.1.26 These calculations employed the PBE exchange correlation functional,27 which has been demonstrated to provide most accurate results among GGA functionals in describing the atomic and electronic structure of TMCs.28 Moreover, dispersion —van der Waals— interactions were included through the D3 method as proposed by Grimme and coworkers.29 A plane-wave basis set was used with a kinetic energy cutoffs of 520 and 415 eV for bulk and surface calculations, respectively. Core electrons were treated with the projector-augmented wave (PAW) method.30,31 All bulk structures were obtained from the Materials Project open dataset.32 For bulk geometry relaxation, electronic and force convergence tolerances of 10−6 eV and 10−3 eV Å−1, respectively, were imposed, and a dense Γ-centred k-point grid of 80/a × 80/b × 80/c was used, with non-integer values rounded up to the nearest integer. Noticeably, all calculated lattice parameters are in good agreement with available experimental data, with the largest deviation being below 2% for both TMCs and TMs, as shown in Sections S2 and S3 in the ESI,† respectively.The slab models for the surfaces were constructed from the optimised bulk structures. For slab geometry relaxation, electronic and force convergence tolerances of 10−5 eV and 10−2 eV Å−1, respectively, were imposed, and a Γ-centred k-point grid of 60/a × 60/b × 1 was used, with non-integer values rounded up to the nearest integer. In all the slabs, the bottom half of the slab in the vertical z-direction was constrained at the bulk positions, while the top half of the slab, the metal clusters and the adsorbed species were fully relaxed. Periodic slab images were separated in the z-direction (perpendicular to the surface) by 14 Å of vacuum, and a dipole correction was applied. Convergence tests with respect to k-point grid and vacuum length are found in Sections S4 and S5 in the ESI,† respectively. All crystal structure manipulations and data analysis were carried out using the Python Materials Genomics package (pymatgen)33 and the Atomic Simulation Environment (ASE).34
2.3. Calculated properties
Several properties have been investigated. The stability of the metal clusters on TMC surfaces has been evaluated by calculating the adsorption energy per atom, Eatads, and the formation energy of the cluster, Ef, defined as: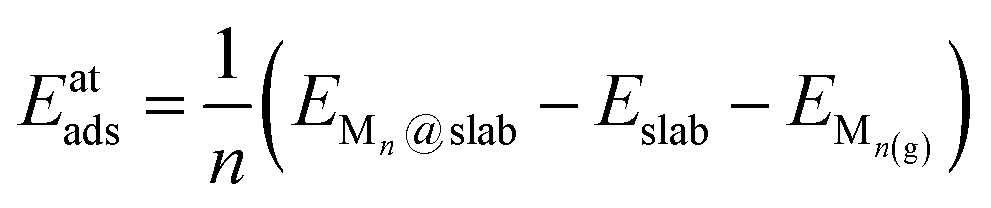 | (1) |
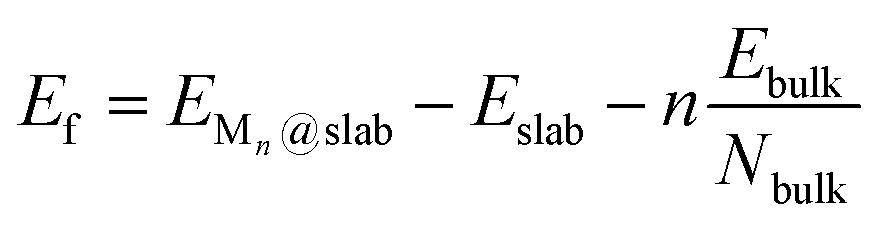 | (2) |
Similarly, the adsorption energies of the clusters adsorbed on C vacancy sites, Eatads-vac, were calculated as
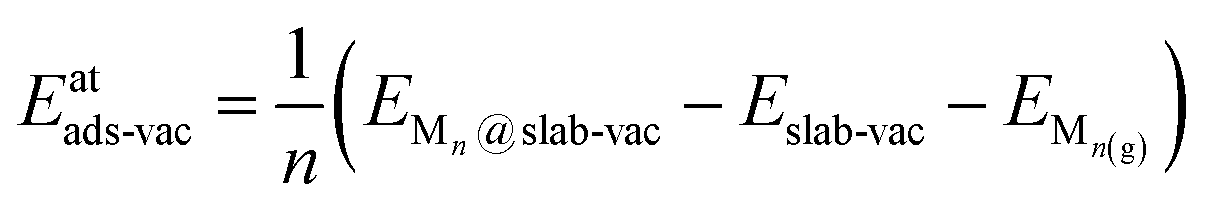 | (3) |
| ΔEatads-vac = Eatads-vac − Eatads | (4) |
By this definition, ΔEatads-vac < 0 indicates that the adsorption energy is more negative in the presence of surface C vacancies (i.e., stronger binding).
To further understand the metal–support interactions, the net atomic charges on supported metal clusters and surface carbon atoms (Qat) were computed through a Bader analysis of the electron density37 using the VTST Tools by G. Henkelman et al.38 Moreover, charge density difference (CDD) plots were used to evaluate the polarisation of the electronic density of the supported clusters by means of the VESTA software.39 The CDD is defined as:
| Δρ = ρMn@slab − ρslab − ρMn | (5) |
2.4. Data sharing
A dataset containing all relevant VASP input files (INCAR, POSCAR and KPOINTS) and output files (vasprun.xml, CONTCAR, CHGCAR, ACF.dat, ACF.dat and BFC.dat) has been made available in the NOMAD repository42 (https://dx.doi.org/10.17172/NOMAD/2021.09.25-1). Moreover, a comma-separated values (CSV) file with the calculated values of all properties has been included in the ESI.3. Atomistic structure and binding strength
We now investigate the most stable configurations for supported metal clusters and their interaction with the TMC support. Several trends regarding the atomic structure of these systems and the binding strength are also presented and discussed.3.1. Metal cluster sizes and configurations
Previous experimental studies on Au particles supported on TiC(001) have shown that the catalytic activity towards O2 (ref. 43) or SO2 (ref. 44) dissociation improves substantially when the size of these particles is very small (<0.6 nm). In general, these particles consist of one single Au layer (i.e., are two-dimensional),9 which would indicate particles such as planar Au4, and theoretical calculations have highlighted the high degree of charge polarisation in these small particles.9 The same trend has been observed experimentally for CO2 hydrogenation on Au/TiC, Cu/TiC, and Ni/TiC catalysts towards CO, methanol, and methane, where the highest activity was found for small two-dimensional particles of the admetals in close contact with TiC(001).8 Recently, Ni particles dispersed on TiC(001) have been shown to activate CH4 at room temperature in a joint computational and experimental study,14 where the energy barriers where shown to increase with particle size (the barriers reported were 0.18, 0.25 and 0.38 eV for Ni4, Ni9, and Ni13, respectively). Moreover, it has been shown by DFT calculations that very small Ni particles (i.e., Ni4) dispersed on TiC interact more strongly with the carbide than bigger Nin (n = 9, 13, 16) particles, leading to a more polarised electron density18 and lower energy barriers not only for CH4 activation but also for CO2 and H2 dissociation.19 Consequently, we investigated the adsorption of three- and four-atom clusters on hexagonal and cubic TMCs, respectively, as these are the ones most likely responsible for the catalytic activity. These particular sizes were selected as representative models, since they feature compact, high symmetry structures that maximise the atomic coordination and are thus highly likely to be energetically stable.Due to the large number of systems involved in the present screening, the effect of cluster size or coverage on the atomic and electronic properties of such systems has not been studied. For a discussion on how the cluster size can affect these properties, the reader is referred to the work by Lozano-Reis et al.18 Moreover, it is worth pointing out that another interesting approach to tune the catalytic activity of TMCs is by transition metal doping. Interestingly, López et al.45,46 used DFT calculations to show that doping TiC(001) with other elements can modify its interaction with CO2. However, there are no experimental studies on these systems yet, and the number of elements that can be used as dopants is limited.
For each combination of supported metal and carbide, six different configurations were studied, as shown in Fig. 2. The present study involves a set of 7 different metal clusters on 11 possible surfaces (i.e., 8 from cubic TMCs and 3 from hexagonal TMCs), resulting in 336 (7 × 8 × 6) and 126 (7 × 3 × 6) DFT geometry optimisations for the cubic and hexagonal TMC surfaces, respectively. Each configuration is labelled by first indicating the adsorption site of the cluster atoms (i.e., tC and hC for top carbon and hollow carbon sites, respectively, tM for top metal sites, br for bridge sites, and hX for a hollow site with no atoms in the perpendicular z-direction), and then the nature of the atom in the middle of the cluster (i.e., C for carbon, M for metal, and X for none). For the four-atom clusters, there are two configurations in which the cluster presents a tetrahedral structure. In these cases, the second part of the label is th. As an example, tC–M refers to a configuration in which the atoms of the cluster are adsorbed on top carbon sites and there is a metal atom located in the centre of the cluster.
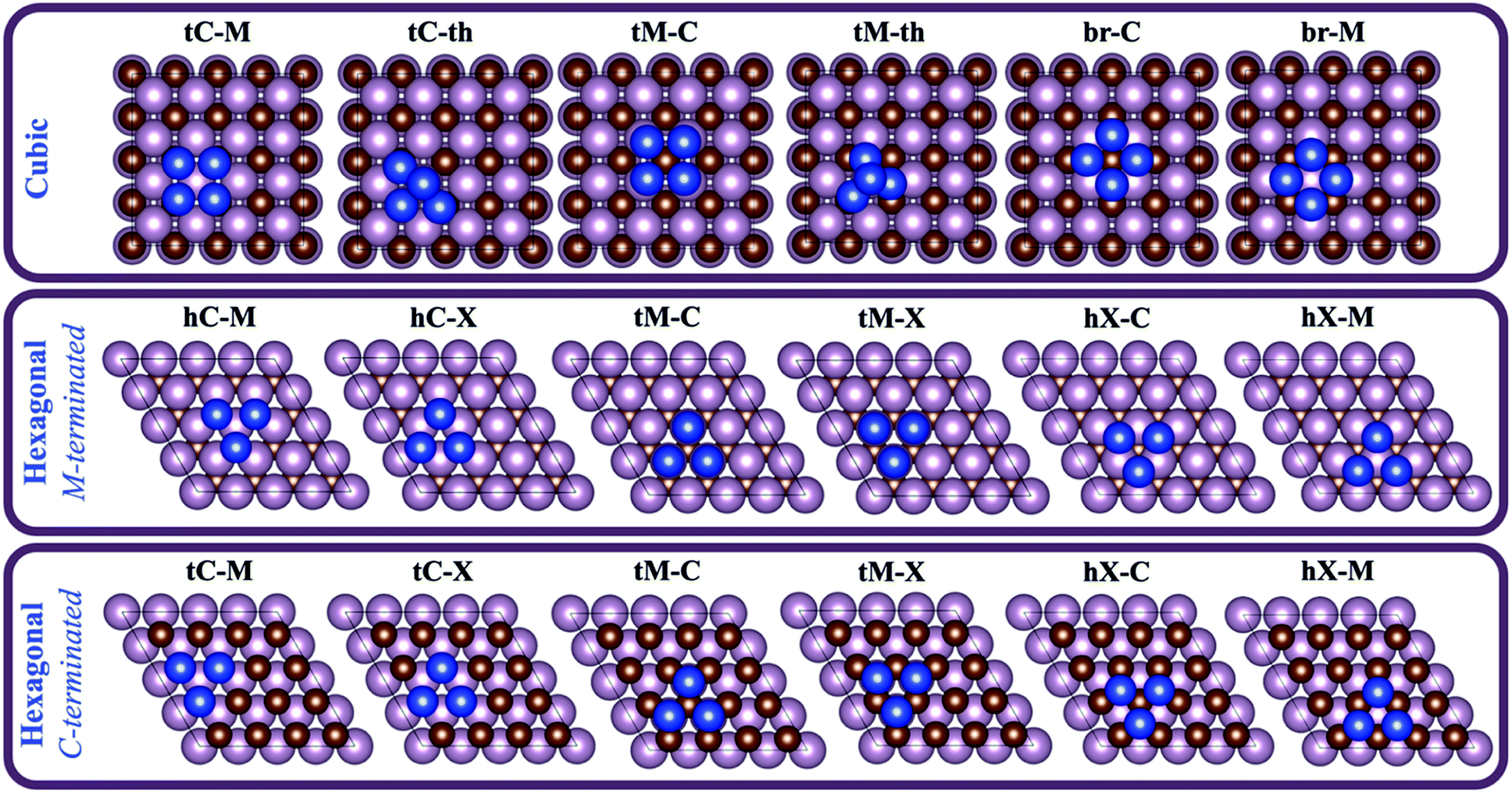 | ||
| Fig. 2 Top views of the different configurations considered in the present study for cubic (top panel) and hexagonal (mid and bottom panels) carbides. Magenta and brown spheres represent metal and carbon atoms from the carbide, respectively, and blue spheres represent cluster metal atoms. | ||
3.2. Most stable configurations
The relative energies between the different configurations are plotted in Fig. S3 in the ESI.† For clusters adsorbed on cubic TMC surfaces, the most stable configuration is one of the following: tC-M, br-C or tC-th. Out of these, tC-M is the most well-represented, with 31 out of the 56 systems considered having this as the most stable configuration, while br-C is the most stable configuration in 17 out of the 56 systems, and finally tC-th is the most stable configuration for the remaining 8 systems. These results indicate that the cluster atoms prefer to interact with the surface C atoms rather than with the surface metal atoms. In fact, the least stable configuration in almost all cases is tM-C, followed by br-M, in which the cluster is less coordinated to C atoms. There is also a broad preference for the formation of 2D clusters over 3D, except for Au clusters, where the tC-th tetrahedral configuration is normally lower in energy than the flat ones. On hexagonal M-terminated surfaces, the most stable configurations are hC-M (in 8 out of 14 systems) and hC-X (in 6 out of 14 systems), with very similar stability. These are the configurations in which the cluster is closer to C atoms, again suggesting that the interaction with C atoms is stronger, which is also evidenced by the fact that tM-C and tM-X configurations are the highest in energy. On the C-terminated surfaces, the lowest energy configurations are hX-M (5 out of 7), hX-M and tM-X (1 out of 7 each). This type of surface exposes a high density of low-coordinated C atoms, resulting in similar energies for all tM and hX configurations, while tC configurations are much higher in energy since the cluster is only coordinated to 3C atoms instead of 6–7 (Fig. 2). Therefore, we conclude that for both cubic and hexagonal TMCs, supported metal clusters bind to surface C rather than surface metal atoms. It is worth mentioning that the preference of supported clusters to coordinate with surface C atoms was already reported for small Cu and Au particles on TiC(001) from X-ray photoemission spectroscopy (XPS) spectra and DFT calculations,9,47 where a positive shift in the binding energy of the C 1s core level was observed upon particle deposition, while only small changes in peak position were observed in the Ti 2p core level peaks. In fact, the higher melting points of TMCs compared to the corresponding metals or metal alloys indicates that C–metal bonds are indeed stronger than metal–metal bonds.48In general, the adsorption of the metal cluster only produces a small displacement of the nearest C atom(s) in the vertical direction, which ranges between −0.2 Å (down) and +0.2 Å (up), as shown in Fig. S4 in the ESI.† However, for Co, Ni and Rh clusters on cubic TMC supports having a br-C configuration, there is a large displacement of the central C atom which can rise to +2.3 Å, placing itself in the centre of the 4-atom cluster and largely stabilizing it. As discussed above, the reason for this displacement is the strong interaction between the cluster and the surface C atoms. For further information, see the discussion in Section S7 in the ESI.†
3.3. Adsorption and formation energies
Fig. 3A shows the average Eatads and Ef values by metal and by support for the most stable configurations of each cluster–support pair (see Fig. S3 in the ESI† for the values for the values of all possible configurations). In general, clusters bind more strongly to hexagonal TMCs than to cubic ones, with average adsorption energies per atom of ∼−2 eV on the cubic carbides and up to −4 eV on the hexagonal ones (top right panel in Fig. 3A). The main reason behind this trend is discussed in the following. Whilst it is true that the (001) surface is the most stable face in cubic TMCs,20 the less stable (111) surface is generally more reactive.49–51 As summarised by Johansson,52 the electronic structure of cubic (111) TMC surfaces is characterised by the existence of more surface states near the Fermi level than in the case of (001) surfaces, for which the density of states around the Fermi level is relatively small. This results in a higher chemical reactivity of the (111) surfaces. For instance, it has been shown that H2 and CO adsorb dissociatively on ZrC(111) but don't adsorb on ZrC(001) at room temperature.53 Moreover, as shown by Quesne et al.,20 the d-band centres in the (111) surfaces are higher than in the (001) surfaces. This result indicates that we could expect the (111) surfaces to be more active, and therefore to interact strongly with the supported clusters. Interestingly, a close inspection of the atomic structure of cubic TMC(111) surfaces and hexagonal TMC(001) surfaces shows that they present identical local coordination in the first double layer of C and metal atoms. However, unlike the cubic (111) surfaces which are difficult to synthesise, hexagonal (001) surfaces for h-MoC and h-WC are easily generated. This structural similarity is reflected in the stronger binding of metal clusters to hexagonal TMCs compared to cubic TMCs reported in Fig. 3A (top panels).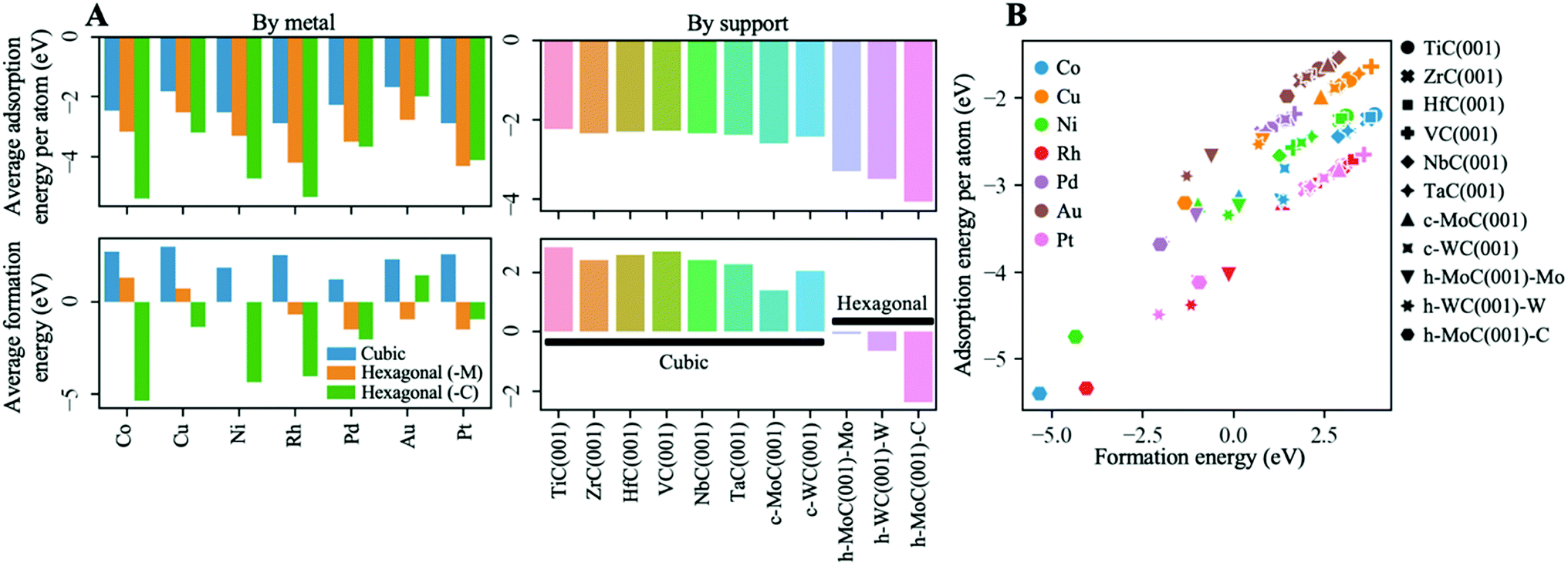 | ||
| Fig. 3 (A) Averages of the adsorption energy per atom, Eatads (top), and formation energy of the cluster, Ef (bottom). Eatads and Ef are defined in eqn (1) and (2). Averages by metal and by support are plotted in the left and right panels, respectively. In the left panels, the averages have been separated in three categories: cubic (blue), hexagonal M-terminated (orange) and hexagonal C-terminated (green) TMC supports. Note that the latter does not correspond to an average, as there is only one data point (i.e., h-MoC(001)-C). (B) Scatter plot of the adsorption energy per atom against the cluster formation energy. Only the most stable configurations for each cluster–support pair are considered in all plots. | ||
In the case of cubic TMCs, the cluster–TMC interactions are of similar magnitude for all structures. However, for the hexagonal TMCs, the C-termination interacts more strongly with the clusters than the M-termination. This result can be explained by the preference of the cluster to interact with surface C atoms rather than metal atoms, as discussed in the previous section.
An inspection of the adsorption energy by metals (top left panel in Fig. 3A) shows that, in general, the strongest binding is found for Rh and Pt clusters, while the lowest is found for Cu and Au clusters. This trend can be explained by the fact that Cu and Au are coinage metals, and therefore have filled d states that are lower in energy. According to the d-band model by Hammer and Nørskov,54 this leads to a higher occupation of antibonding states when interacting with the d bands of carbides, and therefore weaker binding.
Despite the strong binding of the clusters, the formation energies on the cubic TMC supports have positive values (bottom right plot in Fig. 3A), with averages by metal Ēf ranging from 1.2 eV for Pd to 3.0 eV for Cu (bottom left plot in Fig. 3A). This means that the formation of big metallic particles on the cubic TMCs is preferred, from a purely energetic point of view, as the intrametallic bonds within the cluster atoms are stronger than the cluster–TMC bonds. Even so, the strong interaction between the cluster and the support implies that the diffusion barriers of these clusters should be quite high, so, in general, these systems should be quite resistant to aggregation/sintering, as has been observed experimentally on several occasions.7,8,12,14,55,56 As shown in Fig. S3C,† the only cluster with Ef < 0 on a cubic TMC is Ni@c-MoC(001), which arises from the abovementioned special configuration in which a surface C atom rises, placing itself in the centre of the Ni4 cluster and largely stabilising it (see Section S7 in the ESI† for further details). On the other hand, the formation energies on the hexagonal TMCs are negative, with average values up to −2 eV for the C-terminated h-MoC(001) surface (bottom right panel in Fig. 3A), confirming the superior stability of the small metal clusters on the hexagonal TMC supports. An inspection of the formation energy by metals on the hexagonal TMC supports (bottom left panel in Fig. 3A) shows that, on average, the strongest resistance to aggregation/sintering corresponds to Pd and Pt clusters on the M-terminated TMC supports (i.e., Ēf = −1.5 eV), and to Co on the C-terminated h-MoC(001) surface (i.e., Ef = −5.3 eV).
A scatter plot of the adsorption energy per atom against the cluster formation energy is shown in Fig. 3B. The most stable systems, from a purely energetic point of view, are Co, Ni and Rh clusters supported on the C-terminated h-MoC(001) surface, which are located in the left-bottom corner of the plot. The opposite corner of the plot contains most Cu and Au clusters on cubic TMCs, which feature the highest formation energies and less negative adsorption energies. Despite that, small Cu and Au particles in contact with TiC(001) were shown experimentally to be good catalysts for CO2 activation and the catalytic synthesis of methanol.7 Therefore, we expect that all the small metal clusters supported on TMCs considered in this work will remain anchored at a given site, without tending to aggregate into larger clusters and, more importantly, without any tendency to escape from the TMC surface.
4. Electronic properties
In this section, the metal–support interactions are further investigated by computing the net atomic charges on supported metal clusters and surface carbon atoms through a Bader analysis of the electron density, as well as the CDD plots. These investigations are also motivated by the theoretical discovery that TiC can modify the electronic structure of supported Au9 and Ni18 particles, even though the charge transfer is small (i.e., QatAu < 0.15 and QatNi ≈ 0e), which drastically increases their catalytic activity through strong meta–support interactions. Thus, it is interesting to check if the net charges of TMC–supported metal clusters are always small and to find which systems exhibit a more polarised electron density. Several trends regarding charge transfer and electron density polarisation are presented and discussed.4.1. Charge transfer
Fig. 4A shows a heatmap of the net atomic charges for all metal clusters in all different TMC supports in the lowest energy configuration. Note that these atomic charges have been calculated as the total net charge of the cluster divided by the number of atoms in the cluster. In general, charge transfer is small, as for 60% of the systems studied |Qat| < 0.25 and only 10% of them exhibit |Qat| ≥ 0.50e. Moreover, it occurs in both directions, as in two thirds of the systems studied the clusters pull electron density from the carbide support, ending up negatively charged, while in the other third the charge transfer occurs from the metal to the support. Interestingly, the charge transfer resulting from cluster adsorption is much higher -in absolute value- in hexagonal surfaces than in cubic ones. Moreover, for hexagonal carbides, supported clusters are oxidised in the C-termination and reduced in M-termination. This trend can be rationalised based on the electronegativity of the cluster metal atoms (Fig. 5A) and the partially ionic character of TMCs. In C-terminated hexagonal surfaces, the more electronegative surface C atoms pull electron density from the supported cluster, oxidizing it. Similarly, in M-terminated hexagonal surfaces, the less electronegative surface metal atoms yield electron density to the supported cluster, reducing it. These cases correspond to the two extreme situations in which the difference in electronegativity between the surface atoms and the supported cluster atoms is highest, leading to higher charge transfer. However, in cubic surfaces, half of the surface atoms correspond to C and the other half to metals, leading to a lower electronegativity difference between surface atoms and cluster atoms and, therefore, lower or negligible charge transfer.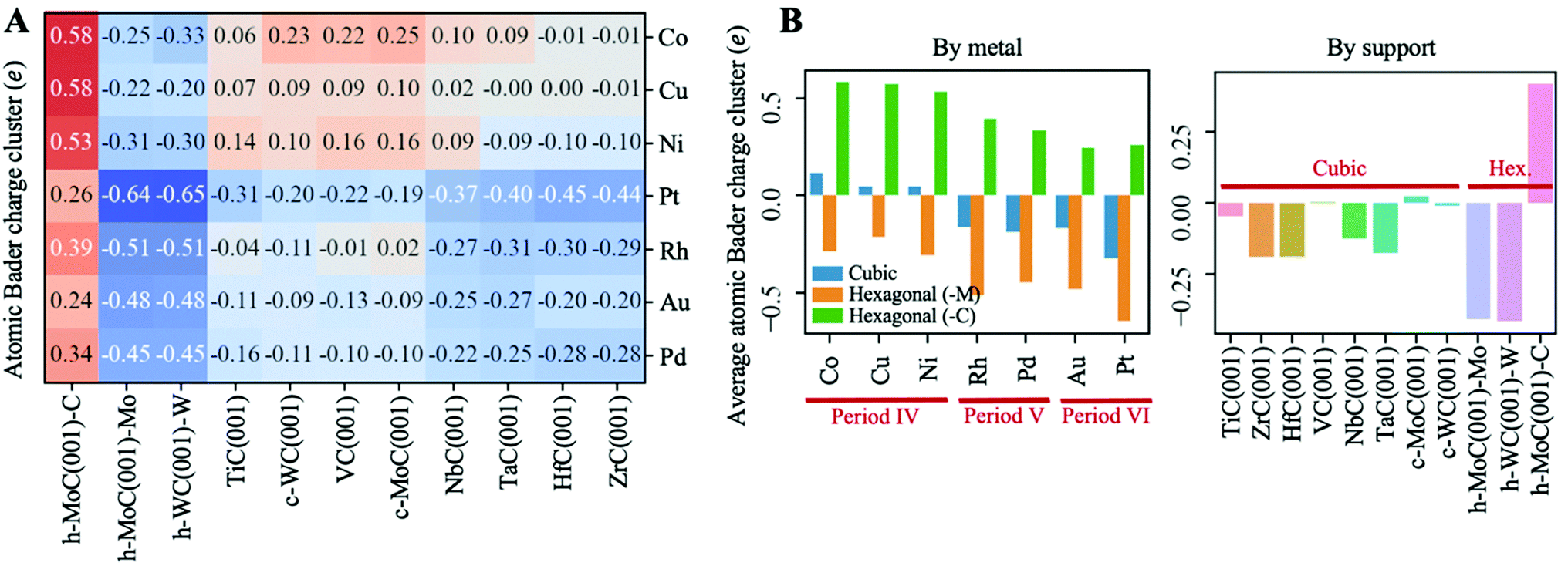 | ||
| Fig. 4 (A) Heatmap of net atomic Bader charges for each cluster–TMC pair. (B) Average net atomic Bader charges by metal (left panel) and by support (right panel). All Bader charges are calculated for the lowest energy configurations. | ||
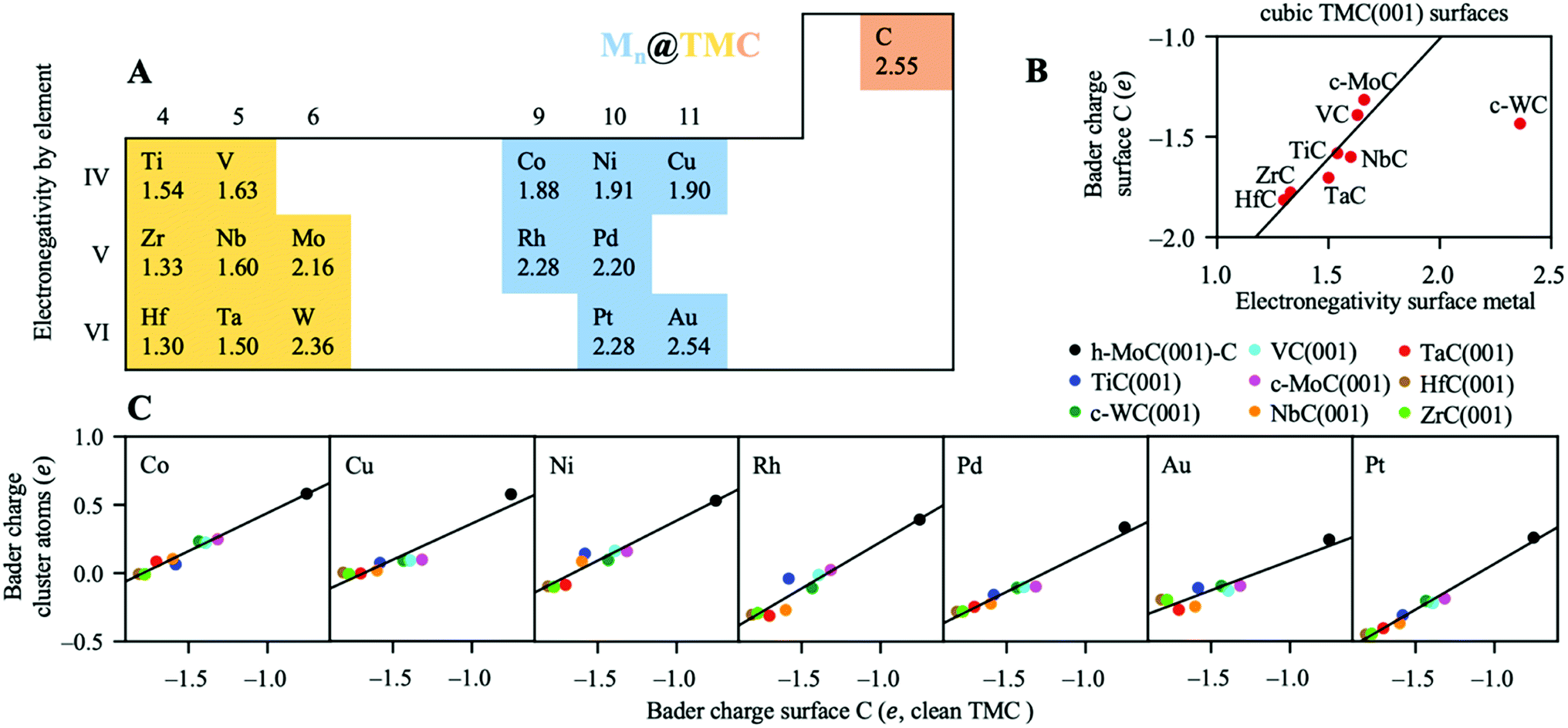 | ||
| Fig. 5 (A) Electronegativity of all elements involved in the present study. (B) Net atomic Bader charges of surface C atoms in the clean TMCs as a function of the electronegativity of the TMC metal atoms. (C) Net atomic Bader charges of the supported clusters plotted against net atomic Bader charges of surface C atoms in the clean TMCs. All Bader charges are calculated for the lowest energy configurations. | ||
Fig. 4B shows the average atomic Bader charges by metal and support. As shown in the left panel, supported clusters become more reduced (or less oxidised) when going down a group in the periodic table. This trend can also be explained in terms of electronegativity; since it increases when going down a group, the cluster atoms become more prone to grasping electron density from the TMC support and getting oxidised. Note that charged clusters are thought to have a stronger resistance against aggregation/sintering, since the coulombic repulsion between two neighbouring charged clusters would prevent recombination thereof towards a bigger particle.
Noticeably, chemical bonding between atoms in TMCs involves three main contributions:57 a metallic one arising from the rearrangement of the metal–metal bonds; a covalent one due to the formation of typical chemical bonds between metal and C atoms, and an ionic one arising from the metal-to-C charge transfer. In general, the most ionic TMCs are those in which the electronegativity difference between C and the metal atoms is largest. This conclusion is also supported by the shifts of the C 2s levels towards lower binding energies measured by XPS58 and near edge X-ray absorption fine structure spectroscopy (NEXAFS)59 techniques. Experimental data obtained by these techniques suggest that, within the carbide structure, C atoms are less negatively charged when bonded to metal atoms towards the middle (as opposed towards the left) of the d-block in the periodic table. For instance, Zr and Hf are the least electronegative atoms from the present set and, when they form carbides, these metals are the ones that oxidise the most and where the C atoms have the most negative charge. This is illustrated in Fig. 5B, which shows that there is a correlation between the charge of surface C atoms and the electronegativity of the surface metal atoms in the cubic TMC surfaces. Table S4† reports the calculated Bader charge values of surface C atoms on all cubic TMC(001) surfaces. The reason why c-WC(001) does not follow this trend might be the very similar electronegativity between W and C atoms (Fig. 5A) and the fact that WC is more stable in its hexagonal h-WC phase.
Finally, Fig. 5C shows that the net atomic charges of the supported cluster atoms are correlated with the net charges of surface C for the clean TMCs. The higher the electron density of the C atoms before adsorption, the higher the density the metal cluster can pull (or the lower density it can yield) once the cluster and the TMC interact. Thus, those TMCs with a higher ionic character (e.g. ZrC or HfC) are more prone to yield electron density to metallic clusters. Note that hexagonal M-terminated surfaces were not included in the plots, since they don't have surface C. In summary, we show that it is possible to manipulate the charge state (partially oxidised or partially reduced) of small metal particles dispersed on TMCs by choosing TMC metal and cluster metal atoms with custom electronegativities.
4.2. Electron density polarisation
Despite no significant charge transfer in most of the studied systems —especially for clusters supported on cubic carbides— the TMC support can induce a significant polarisation of the cluster electron density. This effect has, for instance, previously been shown for small Au clusters in contact with TiC(001) by measuring the binding energy of the C 1s core level through XPS.9 Thus, we evaluated the degree of charge polarisation on the full set of materials by performing a CDD analysis. The full set of CDD plots for all systems investigated are compiled in Fig. S5–S11 in the ESI,† while Fig. 6 shows some representative plots for Cu, Co, Pd and Pt clusters on selected TMCs.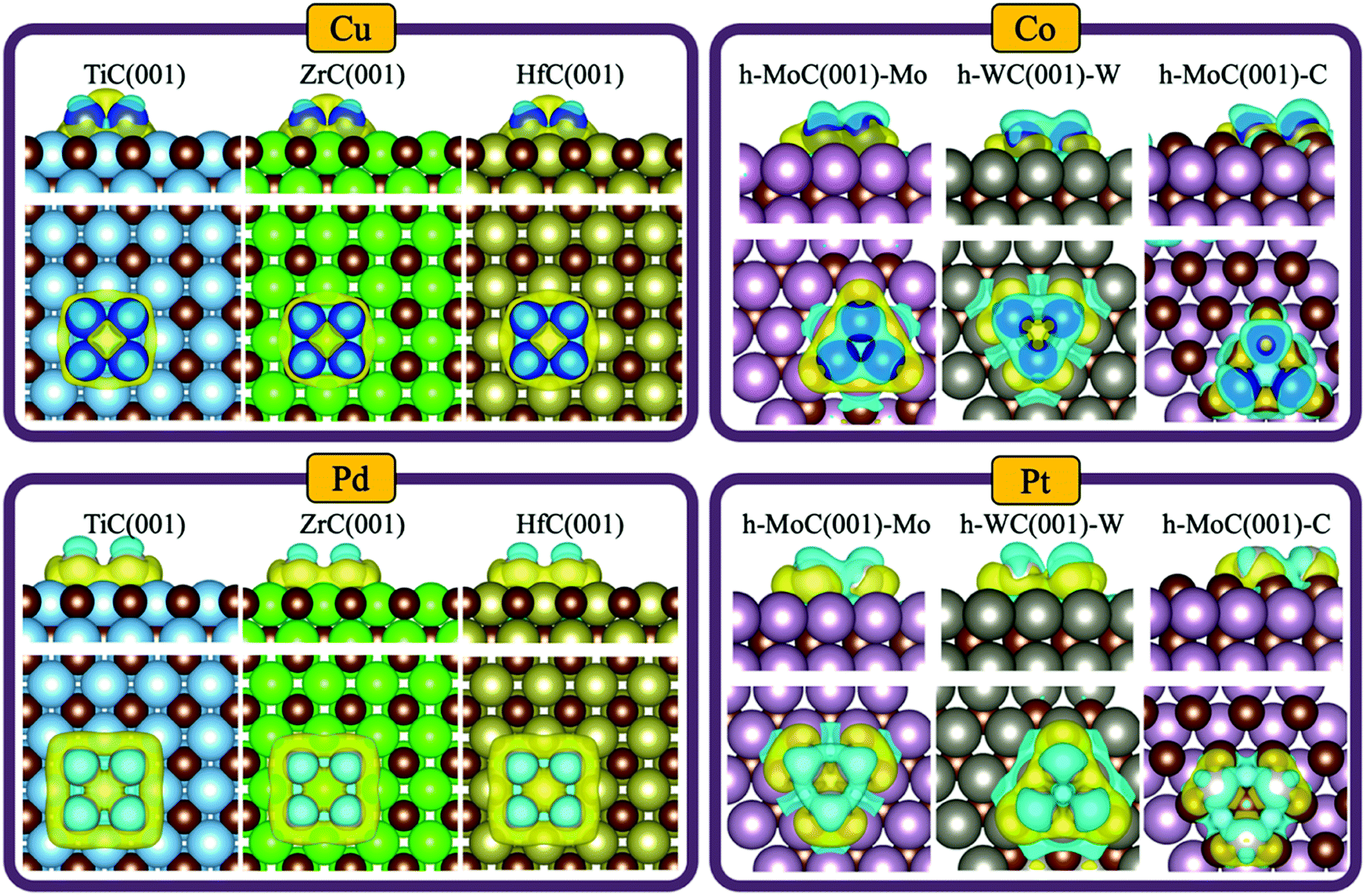 | ||
| Fig. 6 Charge density difference plots for Cu, Co, Pd and Pt clusters supported on selected TMC facets. The isosurface level is taken as 0.0015e bohr−3. Yellow regions denote accumulation of charge density, while blue regions denote charge density depletion. | ||
Interestingly, there is an accumulation of charge density on the interface between the cluster and the support and a charge depletion on top of the cluster atoms in all systems except for the tetrahedral clusters, where the topmost atom is typically partially oxidised (e.g., see Pt3@c-MoC in Fig. S9 in the ESI†). This polarisation should facilitate bonding of electron-acceptor molecules (e.g., O2, CO2, SO2, etc…) to the interface, and bonding of electron-donor molecules (e.g., H2, CO, NH3, etc…) on top of the cluster atoms. In general, the most polarised clusters are those of Pt, Pd and Rh, while the least polarised ones are those of Co and Cu. This is observed in the left-side plots in Fig. 6, which show that the charge polarisation around Cu clusters is much less pronounced than for Pd clusters. This trend can be explained, in part, due to the higher polarizability of larger atoms such as Pt, Pd and Rh, which have more loosely held electrons and more diffuse orbitals, contrary to smaller atoms with tightly bound electrons, such as Co and Cu. Note that there is a clear polarisation of the electronic density even if the net charge transfer is negligible, as shown, for instance, for the case of Cu4@HfC(001) (top left panel in Fig. 6), where there is no net charge transfer (Fig. 4A). If we compare the systems with the highest metal→TMC and TMC→metal charge transfer, which are Cu3@h-MoC(001)-C and Pt3@h-WC(001)-W, respectively (Fig. 4A), we can see that the only significant difference is the polarisation of the hollow site at the centre of the cluster. While Cu3@h-MoC(001)-C, with an atomic Bader charge of +0.58e, shows depletion of charge density on this site, Pt3@h-WC(001)-W, with an atomic Bader charge of −0.65e, shows a significant accumulation of charge density on the centre of the cluster.
By choosing the appropriate metal and support, one can obtain a catalyst with the ideal charge polarisation to interact with the reactants of a particular reaction. For instance, the CDD plots for Pt in Fig. S9† suggest that Pt clusters supported on cubic TMCs will strongly interact with molecules such as CO2 due to the significant accumulation of charge density on the cluster centre, which makes them more promising for CO2 hydrogenation reactions, where the relatively stable CO2 molecule must be activated. On the other hand, Pt clusters supported on hexagonal TMCs will favour the adsorption and activation of molecules such as NH3 due to the strong depletion of charge density on the cluster centre, which makes them more suitable for ammonia decomposition.
4.3. Density of states and magnetisation
The strong chemical interaction between the supported metal clusters and the TMC support is also evidenced by the density of states (DOS) plots and the final magnetic moment of the supported clusters. Due to the large number of systems involved in this screening study, a complete overview and discussions on the DOS of such materials is out of the scope of the present work. However, we have calculated their DOS on four representative systems, which involve Ni clusters supported on two cubic TMC and two hexagonal TMC surfaces (one M-terminated and the other C-terminated), exhibiting Bader charges ranging from −0.31 to 0.53e. A comparison between the total and partial DOS for the Ni/TMC and clean TMC systems in Fig. S12–S15 in the ESI† reveal that the region near the Fermi level for the clean carbides is flatter than when the Ni cluster is adsorbed, because of Ni3d populated states around this region. The partial DOS plots reveal mixing of the Ni-d states with the C-p and metal-d states of the TMC support around the Fermi level, thus showing a strong chemical interaction between the cluster and the surface. For the interested reader, a complete discussion on the DOS for the clean TMCs is found in the work by Quesne et al.20To complete the study, we focus now on the effect that the TMC support has on the magnetic properties of the supported clusters. The interest here relies in the fact that any catalytic reaction involving radical species can be affected by a change in the spin alignment and magnetic properties of the catalytic centre. Table S5 in the ESI† shows that 5 out of 7 three-atom TM clusters and 6 out of 7 four-atom TM clusters in the gas-phase present a magnetic moment different from zero, with average magnetic moments per atom ranging from 0.33 to 2.5 μB. However, the strong chemical interaction with the TMC quenches the magnetic moment of the metal clusters upon adsorption. This result is illustrated by the histograms in Fig. S16 in the ESI,† which show that only in 8 out of the 77 systems included in this screening (i.e., considering only the most stable configuration for each cluster) the supported cluster has a non-zero magnetic moment in its adsorbed state.
5. Effect of carbon vacancies
In this section, we evaluate the effect of surface C vacancies on the TMC interaction with the supported metal clusters. An interesting feature of TMCs is that they are almost never stoichiometric in practice, and often exhibit significant deviations from the ideal 1:1 metal–C ratio.1,60 For instance, the C-vacancy concentration in TiC can be as large as 50% while still maintaining the fcc crystal structure.1 In the case of vanadium carbides, the sub-stoichiometric V8C7 phase is slightly more stable than the stoichiometric VC phase,61 and can be modelled as a VC supercell with ordered C vacancies, one for every 8 V atoms on each layer.62 In a recent study, Rasander et al.63 studied the C vacancy formation in fcc and hcp TMCs by means of DFT calculations. They showed that, for fcc TMCs, C vacancies are more likely to form as the number of valence electrons in the system increases. Also, their formation depends on the growth conditions: for metal-rich conditions, fcc will always favour C vacancy formation, while for C-rich conditions it is energetically favourable to form C vacancies only in ScC, VC, NbC, CrC, c-MoC and c-WC. The main reasons for the deviation of the ideal stoichiometry in these TMCs are: (i) the high temperature used in their synthesis, which leads to an increased configurational entropy favouring a high vacancy concentration, along with high vacancy–migration barriers, which prevent the system from recovering towards a stoichiometric structure, when cooled to low temperatures;64 (ii) the slow diffusion rates for C penetration into the metal lattices during the synthesis;65 and (iii) the formation of electronic states below the Fermi level which stabilise the vacancies.66
To evaluate the effect of surface C vacancies on the binding strength of supported metal clusters, we performed additional DFT geometry optimisations on a subset of systems by removing the closest surface C atom to the cluster and re-optimising it. Specifically, this subset includes, for each metal–support combination, the most stable configuration, as well as all configurations that are up to 2 eV higher in energy than that (see Fig. S3A in the ESI† for relative energies between different configurations). Thus, there was a total of 304 new DFT optimisations that were performed. The histogram in Fig. 7A shows that, in most cases, surface C vacancies increase the binding strength of these clusters, making them more stable (i.e., the adsorption energies per atom become 0.2 eV more negative on average). The clusters with the most negative shifts in adsorption energy are those of Pt and Au, which are the most electronegative ones (Fig. 5A).
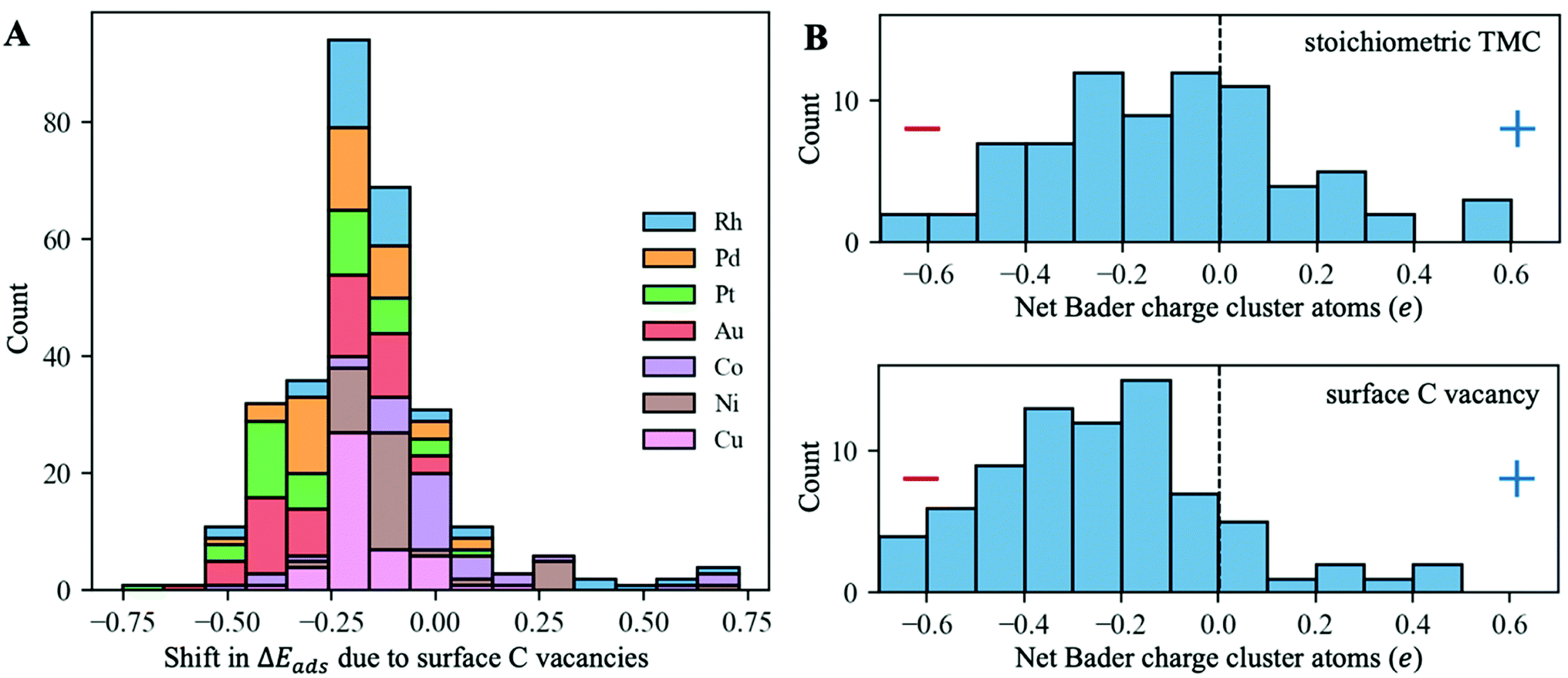 | ||
| Fig. 7 (A) Histogram of ΔEatads-vac, the shifts in the atomic adsorption energies of the metal clusters due to the presence of surface C vacancies (negative values indicate stronger binding with C vacancies). All configurations up to 2 eV higher in energy than the most stable one for each cluster–support pair were considered. (B) Distribution of net atomic Bader charges for the supported clusters on the stoichiometric TMC (top) and in the presence of surface C vacancies (bottom). Only the most stable configurations were considered for these plots. | ||
This increase in the binding strength can be explained by a higher charge transfer from the TMC support to the metal cluster in the presence of surface C vacancies. The histograms in Fig. 7B show the distribution of net Bader atomic charges of the supported cluster atoms on the stoichiometric TMC (top panel) and when they are adsorbed on a C vacancy site (bottom panel). In the first case, there is a fairly even distribution between positively and negatively charged clusters, with many of them having a net Bader charge close to zero, as was discussed in the previous section. However, when a very electronegative surface C atom is removed, the cluster metal atoms can pull a higher amount of electron density from the support and most of them end up with a negative Bader charge.
6. Conclusions
We have performed a high-throughput screening study of the structural and electronic properties of small metal clusters of Co, Ni, Cu, Rh, Pd, Pt and Au supported on TiC, ZrC, HfC, VC, NbC, TaC, MoC and WC by means of periodic DFT calculations, which captured several general trends in the anchoring of these clusters and on their chemical properties. Metal clusters bind to surface C atoms in a 2D configuration, exhibiting adsorption energies per atom that range from −2 to −3 eV on the cubic TMC (001) facets, and from −3 to −5 eV on the more reactive hexagonal TMC (001) facets. The strongest binding, on average, is found for Rh and Pt clusters, while Cu and Au are the ones that bind less strongly. The formation of bigger metallic particles is preferred on the cubic TMC surfaces from a purely energetic point of view. However, due to the strong metal–support interactions, the diffusion barriers of these clusters should be quite high, making them resistant to aggregation/sintering, especially those supported on the hexagonal TMC (001) facets.The charge transfer between the cluster and the support can occur in both directions and, in general, is small on cubic TMCs and higher on hexagonal TMCs. The degree and direction of charge transfer can be explained by comparing the electronegativity difference between the less electronegative host metal atoms, the more electronegative cluster atoms, and the highly electronegative C atoms. Thus, the average atomic charges of the supported clusters are −0.4e on M-terminated hexagonal TMCs, where the surface metal atoms yield electron density to the cluster, and +0.4e on C-terminated hexagonal TMCs, where surface C atoms grasp electron density from both the TMC metal atoms and the cluster. On cubic TMCs, the degree of charge transfer depends also on the ionicity of the support, which decreases along the series. In this way, the most negative Bader charges are found for Period VI clusters (i.e., Au and Pt) supported on TMCs with a higher ionic character (i.e., ZrC and HfC). The presence of surface C vacancies leads to a stronger support-to-cluster charge transfer, which increases the binding strength of the clusters and therefore their stability.
Despite no significant charge transfer in most of the studied systems, the TMC support always induces a significant polarisation of the cluster electron density, resulting in an accumulation of charge density at the interface between the cluster and the support and a charge depletion on top of the cluster atoms. The most pronounced polarisation is observed for the larger Pt, Pd and Rh clusters, which have more loosely held electrons and more diffuse orbitals. Based on the charge polarisation induced by the TMC substrate, one can expect significant differences between the chemical properties of supported small metal clusters and the corresponding extended metal surfaces. Similar arguments have been invoked in the past to explain the enhancement in the catalytic activity of Au, Cu and Ni particles deposited on TiC(001) with respect to Au(100), Cu(100) and Ni(100) for the reverse water–gas shift reaction (CO2 + H2 → CO + H2O),8 as well as the high catalytic activity of Au/TiC56 and Au/c-MoC12 for the water–gas shift reaction (CO + H2O → CO2 + H2) at low-temperature or Ni/TiC for the room-temperature CH4 activation.14 TiC and c-MoC supports are not unique in their ability to polarise small metal clusters; as our computational study demonstrates, this polarisation of the electron density is a general feature of such systems, without Au, Cu or Ni clusters supported on TiC or c-MoC standing out compared to the rest. Therefore, it is expected that the carbide-supported metal clusters investigated herein will exhibit strong interactions with the aforementioned reactant molecules (e.g. CO2, CO or CH4), and that several of these materials would be promising catalysts for the pertinent chemistries.
Furthermore, the fact that all planar clusters present accumulation of charge density on the interface and charge depletion on top of the cluster atoms suggests that the activated complexes encountered in these chemistries would have similar geometries, thereby giving rise to thermochemical linear scaling among these materials. Thus, the ability to manipulate the amount of electron density in the hollow site of the planar clusters by choosing the appropriate host and cluster metal atoms can be used to facilitate the activation of certain molecules on these sites. For instance, when Cu4 clusters are supported on TaC(001), the hollow site exhibits an accumulation of charge density, favouring the adsorption of electron-acceptor molecules (O2, CO2, SO2…), while when they are supported on c-MoC(001), they exhibit a depletion of charge density, thus facilitating the bonding of electron-donor molecules (H2, CO, NH3…).
Overall, the present results suggest that the deposition of small metal particles on TMCs can lead to stable catalysts with unique catalytic properties. We identify Pt, Pd and Rh clusters supported on hexagonal TMC (001) facets as the candidates with the highest potential catalytic activity and stability, as estimated from the degree of polarisation of the electron density and the values for the adsorption energy and formation energy. We hope that the trends identified in this study will provide a solid theoretical background from which potential catalytic activity and stability can be estimated and understood, paving the road for further studies on the interaction and catalytic conversion of chemical species with TMC-supported metallic nanoclusters.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie Grant Agreement No. 891756. We gratefully acknowledge the use of the UCL High Performance Computing Facility Kathleen@UCL in the completion of the simulations of this work. We are grateful to the UK Materials and Molecular Modelling Hub for computational resources, which is partially funded by EPSRC (EP/P020194/1 and EP/T022213/1). We also thank Dr Francesc Illas and Dr Francesc Viñes for useful discussions.References
- L. E. Toth, Transition Metal Carbides and Nitrides, Academic Press, New York, NY, 1971 Search PubMed.
- S. T. Oyama, The Chemistry of Transition Metal Carbides and Nitrides, Blackie Academic and Professional, Glasgow, 1996 Search PubMed.
- R. B. Levy and M. Boudart, Science, 1973, 181, 547–549 CrossRef CAS.
- J. A. Rodriguez, P. Liu, J. Dvorak, J. Gomes, Y. Takahashi and K. Nakamura, Surf. Sci., 2003, 543, L675–L682 CrossRef CAS.
- F. Viñes, J. A. Rodriguez, P. Liu and F. Illas, J. Catal., 2008, 260, 103–112 CrossRef.
- J. A. Rodriguez, P. Liu, D. J. Stacchiola, S. D. Senanayake, M. G. White and J. G. Chen, ACS Catal., 2015, 5, 6696–6706 CrossRef CAS.
- A. B. Vidal, L. Feria, J. Evans, Y. Takahashi, P. Liu, K. Nakamura, F. Illas and J. A. Rodriguez, J. Phys. Chem. Lett., 2012, 3, 2275–2280 CrossRef CAS.
- J. A. Rodriguez, J. Evans, L. Feria, A. B. Vidal, P. Liu, K. Nakamura and F. Illas, J. Catal., 2013, 307, 162–169 CrossRef CAS.
- J. A. Rodriguez, F. Viñes, F. Illas, P. Liu, Y. Takahashi and K. Nakamura, J. Chem. Phys., 2007, 127, 211102 CrossRef CAS.
- J. A. Rodriguez and F. Illas, Phys. Chem. Chem. Phys., 2012, 14, 427–438 RSC.
- T. W. van Deelen, C. Hernández Mejía and K. P. de Jong, Nat. Catal., 2019, 2, 955–970 CrossRef CAS.
- S. Posada-Pérez, R. A. Gutiérrez, Z. Zuo, P. J. Ramírez, F. Viñes, P. Liu, F. Illas and J. A. Rodriguez, Catal. Sci. Technol., 2017, 7, 5332–5342 RSC.
- H. Prats, S. Posada-Pérez, J. A. Rodriguez, R. Sayós and F. Illas, ACS Catal., 2019, 9, 9117–9126 CrossRef CAS.
- H. Prats, R. A. Gutiérrez, J. J. Piñero, F. Viñes, S. T. Bromley, P. J. Ramírez, J. A. Rodriguez and F. Illas, J. Am. Chem. Soc., 2019, 141, 5303–5313 CrossRef CAS.
- P. Tang, Q. Zhu, Z. Wu and D. Ma, Energy Environ. Sci., 2014, 7, 2580–2591 RSC.
- R. Horn and R. Schlögl, Catal. Lett., 2015, 145, 23–39 CrossRef CAS.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS.
- P. Lozano-Reis, R. Sayós, J. A. Rodriguez and F. Illas, Phys. Chem. Chem. Phys., 2020, 22, 26145–26154 RSC.
- P. Lozano-Reis, H. Prats, R. Sayós, J. A. Rodriguez and F. Illas, J. Phys. Chem. C, 2021, 125, 12019–12027 CrossRef CAS.
- M. G. Quesne, A. Roldan, N. H. de Leeuw and C. R. A. Catlow, Phys. Chem. Chem. Phys., 2018, 20, 6905–6916 RSC.
- T. Xiao, A. P. E. York, K. S. Coleman, J. B. Claridge, J. Sloan, J. Charnock and M. L. H. Green, J. Mater. Chem., 2001, 11, 3094–3098 RSC.
- J. R. dos S. Politi, F. Viñes, J. A. Rodriguez and F. Illas, Phys. Chem. Chem. Phys., 2013, 15, 12617–12625 RSC.
- D. J. Siegel, L. G. M. Hector Jr and J. B. Adams, Surf. Sci., 2002, 498, 321–336 CrossRef CAS.
- A. M. Nartowski, I. P. Parkin, M. MacKenzie, A. J. Craven and I. MacLeod, J. Mater. Chem., 1999, 9, 1275–1281 RSC.
- A. S. Kurlov and A. I. Gusev, Inorg. Mater., 2006, 42, 156–163 CrossRef.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- P. Janthon, S. M. Kozlov, F. Viñes, J. Limtrakul and F. Illas, J. Chem. Theory Comput., 2013, 9, 1631–1640 CrossRef CAS.
- S. Grimme, J. Antony and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner, G. Ceder and K. A. Persson, APL Mater., 2013, 1, 011002 CrossRef.
- S. P. Ong, W. D. Richards, A. Jain, G. Hautier, M. Kocher, S. Cholia, D. Gunter, V. Chevrier, K. A. Persson and G. Ceder, Comput. Mater. Sci., 2013, 68, 314–319 CrossRef CAS.
- A. Hjorth Larsen, J. Jørgen Mortensen, J. Blomqvist, I. E. Castelli, R. Christensen, M. Dułak, J. Friis, M. N. Groves, B. Hammer, C. Hargus, E. D. Hermes, P. C. Jennings, P. B. Jensen, J. Kermode, J. R. Kitchin, E. L. Kolsbjerg, J. Kubal, K. Kaasbjerg, S. Lysgaard, J. B. Maronsson, T. Maxson, T. Olsen, L. Pastewka, A. Peterson, C. Rostgaard, J. Schiøtz, O. Schütt, M. Strange, K. S. Thygesen, T. Vegge, L. Vilhelmsen, M. Walter, Z. Zeng and K. W. Jacobsenet, J. Phys.: Condens. Matter, 2017, 29, 273002 CrossRef.
- X. Liu, C. Z. Wang, M. Hupalo, W. C. Lu, M. C. Tringides, Y. X. Yao and K. X. Ho, Phys. Chem. Chem. Phys., 2012, 14, 9157–9166 RSC.
- H. Prats, G. Alonso, R. Sayós and P. Gamallo, Microporous Mesoporous Mater., 2020, 308, 110462 CrossRef CAS.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- W. Tang, E. Sanville and G. Henkelman, J. Phys.: Condens. Matter, 2009, 21, 084204 CrossRef CAS.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- P. E. Blöchl, O. Jepsen and O. K. Andersen, Phys. Rev., 1994, 49, 16223 Search PubMed.
- V. Wang, N. Xu, J. C. Liu, G. Tang and W. T. Geng, Comput. Phys. Commun., 2021, 267, 108033 CrossRef CAS.
- C. Draxl and M. Scheffler, MRS Bull., 2018, 43, 676–682 CrossRef.
- J. A. Rodriguez, L. Feria, T. Jirsak, Y. Takahashi, K. Nakamura and F. Illas, J. Am. Chem. Soc., 2010, 132, 3177–3186 CrossRef CAS.
- J. A. Rodriguez, P. Liu, F. Viñes, F. Illas, Y. Takahashi and K. Nakamura, Angew. Chem., 2008, 120, 6787–6791 CrossRef.
- M. López, L. Broderick, J. J. Carey, F. Viñes and F. Illas, Phys. Chem. Chem. Phys., 2018, 20, 22179–22186 RSC.
- M. López, F. Viñes, M. Nolan and F. Illas, J. Phys. Chem. C, 2020, 124, 15969–15976 CrossRef.
- L. Feria, J. A. Rodriguez, T. Jirsak and F. Illas, J. Catal., 2011, 279, 352–360 CrossRef CAS.
- O. Cedillos-Barraza, D. Manara, K. Boboridis, T. Watkins, S. Grasso, D. D. Hayaseelan, R. J. M. Konings, M. J. Reece and W. E. Lee, Sci. Rep., 2016, 6, 37962 CrossRef CAS.
- H. H. Hwu and J. G. Chen, Chem. Rev., 2005, 105, 185–212 CrossRef CAS.
- T. Noda, M. Yamazaki, K. Ozawa, K. Edamoto and S. Otani, Surf. Sci., 2000, 450, 27–33 CrossRef CAS.
- A. Vojvodic, C. Ruberto and B. I. Lundqvist, J. Phys.: Condens. Matter, 2010, 22, 375504 CrossRef CAS.
- L. I. Johansson, Surf. Sci. Rep., 1995, 21, 177 CrossRef CAS.
- T. Nakane, T. Noda, K. Ozawa and K. Edamoto, Surf. Sci., 1999, 433, 180 CrossRef.
- B. Hammer and J. K. Nørskov, Adv. Catal., 2000, 45, 71–129 CAS.
- S. Posada-Pérez, P. J. Ramírez, J. Evans, F. Viñes, P. Liu, F. Illas and J. A. Rodriguez, J. Am. Chem. Soc., 2016, 138, 8269–8278 CrossRef.
- J. A. Rodriguez, P. J. Ramírez, G. G. Asara, F. Viñes, J. Evans, P. Liu, J. M. Ricart and F. Illas, Angew. Chem., Int. Ed., 2014, 53, 11270–11274 CrossRef CAS.
- J. G. Chen, Chem. Rev., 1996, 96, 1497 Search PubMed.
- L. I. Johansson, Surf. Sci. Rep., 1995, 21, 177 CrossRef CAS.
- J. C. Chen, J. Eng. Jr and S. P. Kelty, Catal. Today, 1998, 43, 147 CrossRef CAS.
- K. Schwarts, Crit. Rev. Solid State Mater. Sci., 1987, 13, 211–257 CrossRef.
- X. Chong, Y. Jiang, R. Zhou and J. Feng, Sci. Rep., 2016, 6, 34007 CrossRef CAS.
- A. Pajares, H. Prats, A. Romero, F. Viñes, P. Ramírez de la Piscina, R. Sayós, N. Homs and F. Illas, Appl. Catal., B, 2020, 267, 118719 CrossRef CAS.
- M. Rasander and A. Delin, arXiv, preprint, August 21, 2018, available from: https://arxiv.org/abs/1808.07114.
- V. P. Zhukov, N. I. Medvedeva and V. A. Gubanov, Phys. Status Solidi B, 1989, 151, 408 CrossRef.
- B. M. Klein, D. A. Papaconstantopoulos and L. L. Boyer, Phys. Rev. B: Condens. Matter Mater. Phys., 1980, 22, 1946 CrossRef CAS.
- L. M. Huisman, A. E. Carlsson, C. D. Gelatt Jr and H. Ehrenreich, Phys. Rev. B: Condens. Matter Mater. Phys., 1980, 22, 991 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta08468b |
| This journal is © The Royal Society of Chemistry 2022 |