
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and characterisation of diketopyrrolopyrrole-based hydrogels†
Valentina
Gauci
 a,
Annela
Seddon
b and
Dave J.
Adams
*a
a,
Annela
Seddon
b and
Dave J.
Adams
*a
aSchool of Chemistry, University of Glasgow, Glasgow, G12 8QQ, UK. E-mail: dave.adams@glasgow.ac.uk
bSchool of Physics, HH Wills Physics Laboratory, University of Bristol, Tyndall Avenue, Bristol, BS8 1TL, UK
First published on 25th April 2022
Abstract
Diketopyrrolopyrrole (DPP) based materials can be easily tuned by functionalising with groups that extend the conjugation and thus alter the electronic properties. When attaching thiophenes to give dithiophene-diketopyrrolopyrroles (DTDPPs), a donor–acceptor–donor system is created that is suitable for charge-transfer applications. This core also promotes π-stacking and hydrophobic interactions. Here, we describe a number of DTDPPs functionalised with amino acids that undergo pH-trigerred gelation. We show that the optical properties of our DTDPPs are affected by whether the amino acids have aromatic or aliphatic side chains. We also describe the effect of solvent polarity. We have successfully produced hydrogels via a pH trigger with examples containing phenylalanine (F), valine (V), leucine (L) and alanine (A) amino acids. Viscosity and small angle X-ray scattering measurements show the presence of micellar structures in solution in water at pH 10.5, with gelation starting at a pH less than 7 due to the formation of a fibrous network.
Introduction
The diketopyrrolopyrrole (DPP) chromophore comprises two fused lactam rings, which can have additional aromatic moieties attached (such as benzene, furan or thiophene) that provide extended conjugation within the structure (Fig. 1a).1 Adding unsaturated groups that increase the rigidity of the structure allows for planarity and thus better π–π stacking of the aromatic core (Fig. 1b).1–4 The free –NH group on the DPP core reduces the solubility of the pigment due to hydrogen-bonding.5 Thus, it is common to alkylate this site to increase solubility of the pigment. Attaching other functional groups can be used to vary the intermolecular interactions between molecular planes to result in solubility changes and variation in the self-assembled structures.6–8 | ||
| Fig. 1 (a) Molecular structure of the synthesised DTDPP-R-OH used here; (b) a cartoon of the solution-to-gel process for DTDPP-based low molecular weight gelators. | ||
The DPP fused rings can perform as electron acceptors or donors. The lactam component is electron-withdrawing and hence gives the DPP unit high electron-affinity properties.9 When symmetrically attaching electron-donating units (such as thiophene, Fig. 1a), the DPP is thus the electron acceptor and this creates a donor–acceptor–donor (D–A–D) type architecture.8,10,11 These D–A–D π-systems are interesting due to the intramolecular charge transfer (ICT) that is present, which gives interesting optical and electronic properties.12
Gels are soft materials that include a large volume of solvent entrapped within a three-dimensional network made of macromolecules. In polymer gels, these macromolecules are polymer chains, and in supramolecular gels, the network is formed by assembled aggregates. The sol-to-gel process for a supramolecular gel (shown schematically in Fig. 1b) is typically not spontaneous and occurs in the presence of a trigger (e.g., a pH change, solvent change, enzymatic action, or salt addition).13–15
Forming a network of self-assembled DPPs has the potential to bring about many advantages including chemical stability, robustness of the material, as well as forming a highly ordered network that can enhance properties such as charge transport.1,8,16 Self-assembled DPPs including polymers,17 films18 and crystals19 used in dyes20,21 and electronic applications1,8,17–19 can be found in literature. However, despite this interest, there are a limited number of examples of self-assembled DPP gels.
The DPP gelator designed by Thool et al. includes groups that promote self-assembly by means of H-bonding, van der Waals interactions, and π–π stacking, and this produces an organogel prepared via a temperature trigger.2 The supramolecular architectures present were favourable for energy transfer and hence it was used in organic solar cells with high efficiency values. Nyayachavadi et al. designed a cross-linked organogel by means of photopolymerization of diacetylene units.22 Their study investigated how different amide groups on the gelator molecule influences the occurrence of cross-linking, self-assembly and gelation. Draper et al. reported the first DPP hydrogel that was formed using pH trigger (with glucono-δ-lactone; GdL), where they symmetrically linked phenylalanine to the core.23 The study compared the xerogel to the monomer in solution and showed that the former assembled via H-aggregates. They focused on conductivity measurements of the materials produced where it is shown that applying shear to realign worm-like micelles (obtained at high pH) will increase the conductivity. Biswas et al. produced DPP-based hydrogels linked with different amino acids, via an enzymatic trigger and showed the difference in aggregation depending on the different amino acids.24 The hydrogel was used for the photocatalytic oxidation of aliphatic and aromatic substrates.24
Here, we report the synthesis of a small library of dithiophene-DPP (DTDPP) amphiphiles that are symmetrically functionalised with amino acids and form supramolecular hydrogels via pH triggered gelation. This library greatly expands the number of DPP-based supramolecular gels. The thiophene moiety attached to the DPP core allows for extended conjugation that promotes π–π intermolecular interactions and hydrophobic interactions. The free –NH group on the DPP core has been substituted with a chain that can easily undergo peptide coupling, and the hydrophobic amino acids linked here promote H-bonding as well as additional interactions depending on their side group. This work explores both aromatic and aliphatic amino acids. The molecular design is based on the concept that several components of the molecule can bring about different intermolecular interactions for self-assembly (Fig. 1a). The terminal carboxylic acid group is the pH switch that is used to trigger gelation. To initially dissolve the gelator in water, a solution at a high pH (pH = 10.5) is prepared where the carboxylic acid will be deprotonated. Upon addition of glucono-δ-lactone (GdL) and slow hydrolysis to gluconic acid,25,26 the pH of the solution gradually lowers, re-protonating the carboxylic acid and reducing solubility, triggering self-assembly and formation of a hydrogel.
Results and discussion
The first step of the synthetic procedure (Scheme 1) was a base-catalysed condensation reaction with dimethyl succinate and 2-thiophenecarbonitrile that produced the DTDPP core at a yield of 58%. This DTDPP product was then reacted via an SN2 reaction to give DTDPP-OtBu. Compared to our previous work,23 we used t-butyl bromoacetate instead of the chloro-counterpart. Despite the yield being low, the greater nucleophilicity allowed for shorter reaction times and the omission of the KI catalyst. Acetone was found to be the optimal solvent on comparing DMF,27 NMP,28 and acetone in terms of yield and ease of workup. The t-butyl group on DTDPP-OtBu was subsequently removed via a deprotection reaction to isolate DTDPP-OH. The peptide coupling reaction was carried out with isobutylchloroformate (IBCF) and N-methylmorpholine (NMM). The carboxylic acid group on the amino acid was protected with a t-butyl group resulting in DTDPP-R-OtBu (R = –CH2Ph for F/phenylalanine, –CH(CH3)2 for V/valine, –CH2CH(CH3)2 for L/leucine, –CH3 for A/alanine, Fig. 1). Deprotection then yielded the final product of DTDPP-R-OH (Fig. 1). Both deprotection reactions that give DTDPP-OH and DTDPP-R-OH were carried out with trifluoroacetic acid (TFA) and are typically quite high yielding.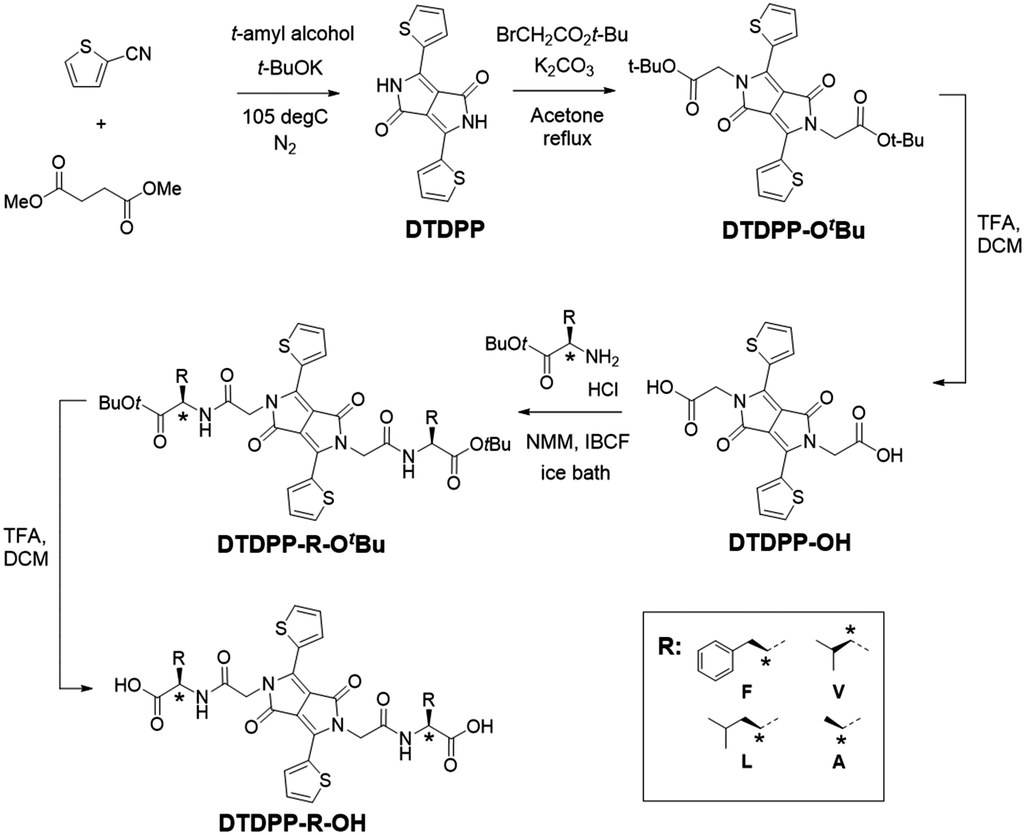 | ||
| Scheme 1 Synthetic route to the DTDPPs used here. | ||
The DTDPP core is an intensely coloured pigment that is highly insoluble in most organic solvents. The solubility of DTDPP-OtBu is improved as expected with alkylation of the –NH site. In this work, the DTDPP-R-OH's were analysed in the solution phase alongside the DTDPP-R-OtBu's since the -tBu group increases solubility and allows a wider range of solvents to be used for analysis of photophysical properties. By doing so, shifts in the optical spectra can be accurately depicted as changes in solvent polarity or changes in aggregation. All solutions were prepared at a ten-fold dilution concentration series. Table S1 (ESI†) shows the solvents and the concentrations of the stock solutions prepared, with example absorption spectra shown in Fig. 2.
 | ||
| Fig. 2 (a) Normalised absorption spectra and (b) normalised fluorescence spectra (excitation wavelength = 540 nm) for DTDPP-V-OtBu both at a concentration of 2 × 10−3 mg mL−1 in different solvents. (c) Normalised absorption spectra for DTDPP-F-OH at 2 × 10−3 mg mL−1. (d) Normalised absorption spectra for DTDPP-R-OHs in water (pH 10.5) at ≈2 × 10−3 mg mL−1 highlighting the difference between aliphatic and aromatic amino acids in the peak maxima. | ||
The absorption and emission spectra of the solutions give a characteristic dual band structure that is seen for DPP donor–acceptor–donor architectures, with the high energy band corresponding to the π–π* transition while the low energy band is attributed to the intramolecular charge transfer (ICT) from donor (thiophene) to acceptor (DPP).1,2,8,29 All samples in organic solvents show vibronic features (as seen in Fig. 2a–c), even across a concentration range (Fig. S22, ESI†). Additionally, the different derivatives give similar data in the same organic solvent (Fig. S23, ESI†). Both these observations show that the samples are molecularly dissolved in organic solvents, and the optical properties here are governed mainly by solvent effects. The λmax is found around 540 nm for all the samples, with a blue shift in this absorption maxima when solvent polarity is increased (seen as well in the emission studies). This negative solvatochromism implies that the ground state of the chromophore is more polar than the excited state, which is indicative of the charge transfer that is occurring between the donor and the acceptor units at ground state without the need for electron excitation. This argument is also made by Szabadai et al., where via computational calculations they showed that during the S1–S0 transition the DPP core (as the electron acceptor) has an overall negative charge, and the thiophene units have a positive charge (as the electron donors).30
As seen in Fig. 2c, the DTDPP-R-OH samples in water at high pH (2 × 10−3 mg mL−1, pH 10.5) exhibit a blue shift of λmax and broadening of peaks (in comparison to samples in organic solvents), characteristic of aggregates in solution. Broad peaks are seen across the concentration range (Fig. S24, ESI†). There is a difference in absorption maxima between the samples with an aliphatic amino acid side chain and the samples with an aromatic side chain, which can be seen in Fig. 2d. This shift can be explained by differences in aggregation due to the different interplanar interactions. This shows that an aqueous environment influences the packing and orientation of the molecules most significantly.
The emission spectra of the concentration series (Fig. S25, ESI†) show the highest intensities for the 2 × 10−2 mg mL−1 and 2 × 10−3 mg mL−1 samples. The higher concentration samples (2 mg mL−1 and 2 × 10−1 mg mL−1) show the lowest emission intensity due to fluorescence quenching. Samples were irradiated at every peak seen in the corresponding absorbance profiles and emission peaks are seen at every irradiation wavelength which is an indication of ICT (Fig. S26, ESI†). The spectra for DTDPP-R-OtBu and DTDPP-R-OH samples consistently show two bands (at around 550 nm and at around 600 nm) for all excitation wavelengths used.
The DTDPP-R-OH derivatives were dissolved in water with two equivalents of 1 M NaOH, starting at a concentration of 5 mg mL−1. These samples were stirred overnight until a clear, dark red solution, indicating full dissolution, was achieved at around a pH of 10.5 (Fig. 3a). The viscosity measurements in Fig. 3c show shear-thinning behaviour characteristic of micellar structures in solution that confirms the presence of aggregates as seen in Fig. 2c. All DTDPP-R-OH examples here form gels (as seen in Fig. 3b) when the pH is decreased by hydrolysis of GdL to gluconic acid.25,26 The absorption spectra for solutions and gels at the gelation concentration show that the peaks for the gel phase are broader than the solution bands which indicates a greater degree of aggregation between both materials (Fig. S27, ESI†).
 | ||
| Fig. 3 (a) DTDPP-V-OH pH 10.5 sol (b) DTDPP-V-OH gel (c) viscosity measurements for DTDPP-F-OH 5 mg mL−1, DTDPP-V-OH 10 mg mL−1, DTDPP-L-OH 15 mg mL−1, and DPP-A-OH 5 mg mL−1 (all in water at pH 10.5). | ||
The minimum gelation concentrations (mgc) for the hydrogels are outlined in Table 1. The amount of added GdL was increased in line with the gelator concentration to ensure the pH decreases sufficiently. The pH values of the resulting gels were ≈3.5 and these resulting materials could be inverted without flow, which is suggestive of gel formation. Rheology showed that the values of G′ and G′′ are at least one order of magnitude between both values (Fig. 4a and bB). As is typical of such gels, these samples break at relatively low strain and G′ and G′′ are frequency independent.
| DTDPP-F-OH (mg mL−1) | DTDPP-V-OH (mg mL−1) | DTDPP-L-OH (mg mL−1) | DTDPP-A-OH (mg mL−1) | |
|---|---|---|---|---|
| mgc | 5 | 10 | 15 | 5 |
| GdL | 5 | 16 | 24 | 8 |
 | ||
| Fig. 4 (a) Strain and (b) frequency sweeps for DTDPP-F-OH 5 mg mL−1, DTDPP-V-OH 10 mg mL−1, DTDPP-L-OH 15 mg mL−1, and DTDPP-A-OH 5 mg mL−1. (c) Temperature sweep for DTDPP-F-OH 5 mg mL−1 gel showing G′ (dark blue) and G′′ (light blue), and temperature (yellow) and (d) thixotropy sweep for DTDPP-F-OH 5 mg mL−1 gel showing G′ (dark blue) and G′′ (light blue). | ||
Biswas et al. previously report the attempt to gel similar DTDPP-amino acid amphiphiles (with phenylalanine, tyrosine and leucine) from the methyl ester precursor via enzymatic hydrolysis.24 They reported a DTDPP-F-OH gel at higher concentration, that exhibited higher G′ and G′′ values. In their work, the DTDPP-L-OH does not form a gel which may have been due to the lower concentration used or the use of the enzymatic approach as opposed to a pH trigger. In addition, the previously reported DTDPP-F-OH hydrogel by Draper et al.23 prepared at the same mgc shows difference in rheological properties due to a lower initial pH prior to addition of GdL. Both aspects emphasise that for such systems the exact conditions and process used are extremely important aspects to consider.31,32
To explore this further with the DTDPP gelators here, the differences in preparation conditions (concentration and pH) and mechanical applications (temperature and mechano-sensitivity) were investigated. Lower gelator concentrations were tested for DTDPP-V-OH (5 mg mL−1 with 8 mg mL−1 GdL) and DTDPP-L-OH (5 mg mL−1 with 8 mg mL−1 GdL and 10 mg mL−1 with 16 mg mL−1 GdL), and despite these samples forming solids and the pH decreasing to 3.5, the rheological analysis in Fig. S28 in the ESI† shows that these are not true gels. Comparing all four gelators at a concentration of 5 mg mL−1, DTDPP-F-OH exhibits the greatest gel stiffness (G′ 2636 Pa, G′′ 406 Pa) and this is attributed to the additional π-interactions that arise from the aromatic phenylalanine group.33 The DTDPP-A-OH gel (G′ 2400 Pa, G′′ 414 Pa) is the second strongest gel. The branched and alkyl chains of valine and leucine result in DTDPP-V-OH (G′ 1096 Pa, G′′ 201 Pa) and DTDPP-L-OH (G′ 506 Pa, G′′ 82 Pa) being the weakest gels of the series.
The change in gel stability depending on the resultant pH was investigated by varying GdL concentration for the DTDPP-F-OH gelator as seen in Table S2 and Fig. S29 in the ESI.† Strain sweeps showed that the gel samples between pH 4.0 and pH 3.2 are stable materials with similar linear viscoelastic regions (LVER) and yield points. The samples at pH 4.9 and pH 6.2 are weaker materials, and the latter sample resembled a highly viscous solution rather than a gel on vial inversion. The DTDPP-F-OH hydrogel has a maximum stiffness at pH 3.2 (with 8 mg mL−1 GdL). However, for the remaining analysis of this paper, the gel at pH 3.5 (with 5 mg mL−1 GdL) is described to investigate all DTDPP-R-OH gels at the same pH.
A heat-cool rheology sweep was performed to test the thermal stability of the DTDPP-F-OH hydrogel as shown in Fig. 4c. When increasing the temperature to 80 °C, both G′ and G′′ values decrease with only G′ returning to the original value upon cooling back down to 25 °C. To analyse the recovery of the hydrogel after the application of high strain, a times sweep was run for DTDPP-F-OH hydrogel as shown in Fig. 4d. The values for G′ and G′′ show that the gel structure does recover after experiencing high strain albeit to slightly reduced G′ as compared to the original value.
Due to the slow pH change leading to gelation, the self-assembly process for the DTDPP-R-OH gelators can be tracked over time. The apparent pKa titrations (Fig. S30, ESI†) show that the pKa values for the derivatives are approximately 7 for DTDPP-F-OH, 5 for DTDPP-A-OH, 5 for DTDPP-V-OH, 7 for DTDPP-L-OH. Carrying out simultaneous time sweep rheology experiments and pH logging during the gelation process confirms that gelation starts at a pH below the apparent pKa (at approximately pH 7 for DTDPP-F-OH, pH 5 for DTDPP-A-OH, pH 5 for DTDPP-V-OH, and pH 7 for DTDPP-L-OH). This is further confirmed by monitoring the changes in assembly of the free molecules into solid-like fibres by NMR. This is done by tracking the reduction in intensity of the peaks from the gelator in solution as the free, dissolved molecules precipitate out of solution to form aggregates (as described in the methods section and in Fig. S1 in the ESI†). As exemplified for DTDPP-A-OH in Fig. 5, gelation starts after approximately 0.3 hours at pH 5 (orange data points), with an increase in the storage and loss moduli (dark and light blue data points respectively), and with NMR spectra showing that the percentage of the free, unassembled DTDPP-A-OH that can be detected decreases due to the molecule assembling into solid-like fibres (dark red data points). Similar observations are seen for the other derivatives with gelation starting after 0.2 hours for DTDPP-F-OH (Fig. S31, ESI†) and DTDPP-V-OH (Fig. S32, ESI†) and after 0.1 hours for DTDPP-L-OH (Fig. S33, ESI†).
 | ||
| Fig. 5 Time sweep analysis for DTDPP-A-OH with G′ (dark blue), G′′ (light blue), pH (orange) and % free molecule by NMR spectroscopy (dark red) shown with time as the gelation occurs after GdL addition. | ||
To understand the structures underpinning the solution and gel phases, small angle X-ray scattering (SAXS) was used on the DTDPP-R-OH samples. This technique has the advantage over imaging techniques of eliminating any drying requirements during sample preparation which can often lead to artefacts.34 Additionally, this provides a bulk measurement as opposed to the small amount of sample probed using microscopy. In all cases, the solution phase at high pH scatters weakly and the data are best fit to a power law (Table S3 and Fig. S34a–d, ESI†). This data indicates the presence of non-persistent micellar structures in the high pH solutions as was suggested by the viscosity measurements (Fig. 3c). In the gel phase, the scattering intensity increases significantly which is indicative of the formation of more persistent structures and the formation of a network (Fig. 6). The data for the DTDPP-A-OH, DTDPP-F-OH and DTDPP-V-OH are best fit to a flexible elliptical cylinder model combined with a power law (to take into account the excess scattering at low Q). The radii and axis ratio are similar in all cases despite the absorption spectra (Fig. 2d) suggesting different packing for the aliphatic and aromatic amino acids. The data for the DTDPP-L-OH system in comparison is best fit to flexible cylinder model combined with a power law. These structures have a higher radius than the other DTDPP-R-OH potentially due to the longer, branched alkyl side chain. Despite the differences in the aggregate models and dimensions, all cases show that long anisotropic structures underpin the gel phase. The absolute rheological values vary within the four gels despite the similarity within the SAXS data for three of the systems. In this SAXS experimental set up, 100 nm is the maximum length scale that can be probed, and so the differences in the rheology can be explained by differences in crosslinking and/or microstructure which are outside this length scale.
 | ||
| Fig. 6 SAXS data for DTDPP-R-OH gels (open black circles) with form factor fits (red line) for (a) DTDPP-F-OH; (b) DTDPP-V-OH; (c) DTDPP-L-OH; (d) DTDPP-A-OH. | ||
Conclusions
Despite the interest in diketopyrrolopyrroles, there are very few examples of gelators based on this structure. Here, we have shown that we can prepare hydrogels from a range of amino acid side chains linked to the DTDPP core. Interestingly we have an example that forms a gel by a pH switch that was previously reported not to form gels using a different trigger. The gels are formed at low pH by self-assembly of the molecules into a three-dimensional network made up of one-dimensional fibrous structures. There are interesting differences between the gelators from the perspective of minimum gelation concentration and rheological data, as well as the pKa of the gelators, all of which point to the opportunity to prepare a range of photocatalysts for example where the properties are tuned by the self-assembled structure as opposed to modifying the chemical structure.35–37Author contributions
Conceptualization – V. G., D. A.; data curation – V. G., A. S.; formal analysis – V. G., A. S., D. A.; funding acquisition – A. S., D. A.; investigation – V. G., A. S.; methodology – V. G., A. S., D. A.; project administration – D. A.; resources – D. A.; supervision – D. A.; writing – original draft – V. G., D. A.; writing – review & editing – all.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Leverhulme Trust (RPG-2019-165) for funding. We thank Bart Dietrich (University of Glasgow) for synthetic advice. We thank Daniel McDowall (University of Glasgow) for help with running the SAXS samples. This work benefitted from the SasView software, originally developed by the DANSE project under NSF award DMR-0520547. The Ganesha X-ray scattering apparatus was purchased under EPSRC Grant ‘Atoms to Applications’ (EP/K035746/1).Notes and references
- S. Qu and H. Tian, Chem. Commun., 2012, 48, 3039–3051 RSC.
- G. S. Thool, K. Narayanaswamy, A. Venkateswararao, S. Naqvi, V. Gupta, S. Chand, V. Vivekananthan, R. R. Koner, V. Krishnan and S. P. Singh, Langmuir, 2016, 32, 4346–4351 CrossRef CAS PubMed.
- J. Humphreys, F. Malagreca, P. Hume, W. Lewis, E. S. Davies, S. P. Argent and D. B. Amabilino, Dyes Pigm., 2022, 197, 109836 CrossRef CAS.
- M. Pieczykolan, J. B. Derr, A. Chrayteh, B. Koszarna, J. A. Clark, O. Vakuliuk, D. Jacquemin, V. I. Vullev and D. T. Gryko, Molecules, 2021, 26, 4744 CrossRef CAS PubMed.
- A. Ruiz-Carretero, N. R. Ávila Rovelo, S. Militzer and P. J. Mésini, J. Mater. Chem. A, 2019, 7, 23451–23475 RSC.
- A. Iqbal, M. Jost, R. Kirchmayr, J. Pfenninger, A. Rochat and O. Wallquist, Bull. Soc. Chim. Belg., 1988, 97, 615–644 CrossRef CAS.
- M. Grzybowski and D. T. Gryko, Adv. Opt. Mater., 2015, 3, 280–320 CrossRef CAS.
- S. Ghosh, S. Shankar, D. S. Philips and A. Ajayaghosh, Mater. Today Chem., 2020, 16, 100242 CrossRef CAS.
- L. Huo, J. Hou, H.-Y. Chen, S. Zhang, Y. Jiang, T. L. Chen and Y. Yang, Macromolecules, 2009, 42, 6564–6571 CrossRef CAS.
- J. Podlesný, L. Dokládalová, O. Pytela, A. Urbanec, M. Klikar, N. Almonasy, T. Mikysek, J. Jedryka, I. V. Kityk and F. Bureš, Beilstein J. Org. Chem., 2017, 13, 2374–2384 CrossRef PubMed.
- C. Kim, J. Liu, J. Lin, A. B. Tamayo, B. Walker, G. Wu and T.-Q. Nguyen, Chem. Mater., 2012, 24, 1699–1709 CrossRef CAS.
- L. Dou, Y. Liu, Z. Hong, G. Li and Y. Yang, Chem. Rev., 2015, 115, 12633–12665 CrossRef CAS PubMed.
- E. R. Draper and D. J. Adams, Chemistry, 2017, 3, 390–410 CrossRef CAS.
- X. Du, J. Zhou, J. Shi and B. Xu, Chem. Rev., 2015, 115, 13165–13307 CrossRef CAS PubMed.
- L. A. Estroff and A. D. Hamilton, Chem. Rev., 2004, 104, 1201–1218 CrossRef CAS PubMed.
- O. P. Lee, A. T. Yiu, P. M. Beaujuge, C. H. Woo, T. W. Holcombe, J. E. Millstone, J. D. Douglas, M. S. Chen and J. M. J. Frechet, Adv. Mater., 2011, 23, 5359–5363 CrossRef CAS PubMed.
- B. Sun, W. Hong, H. Aziz and Y. N. Li, J. Mater. Chem., 2012, 22, 18950–18955 RSC.
- P. E. Hartnett, E. A. Margulies, C. M. Mauck, S. A. Miller, Y. Wu, Y.-L. Wu, T. J. Marks and M. R. Wasielewski, J. Phys. Chem. B, 2016, 120, 1357–1366 CrossRef CAS.
- C. Fu, P. J. Beldon and D. F. Perepichka, Chem. Mater., 2017, 29, 2979–2987 CrossRef CAS.
- M. Kaur and D. H. Choi, Chem. Soc. Rev., 2015, 44, 58–77 RSC.
- T. F. Abelha, G. Morris, S. M. Lima, L. H. C. Andrade, A. J. McLean, C. Alexander, J. Calvo-Castro and C. J. McHugh, Chem. – Eur. J., 2020, 26, 3173–3180 CrossRef CAS PubMed.
- A. Nyayachavadi, G. T. Mason, M. N. Tahir, M. U. Ocheje and S. Rondeau-Gagne, Langmuir, 2018, 34, 12126–12136 CrossRef CAS PubMed.
- E. R. Draper, B. Dietrich and D. J. Adams, Chem. Commun., 2017, 53, 1868–1871 RSC.
- S. Biswas, M. Kumar, A. M. Levine, I. Jimenez, R. V. Ulijn and A. B. Braunschweig, Chem. Sci., 2020, 11, 4239–4245 RSC.
- D. J. Adams, M. F. Butler, W. J. Frith, M. Kirkland, L. Mullen and P. Sanderson, Soft Matter, 2009, 5, 1856–1862 RSC.
- Y. Pocker and E. Green, J. Am. Chem. Soc., 1973, 95, 113–119 CrossRef CAS PubMed.
- S. Tang, E. H. Ghazvini Zadeh, B. Kim, N. T. Toomey, M. V. Bondar and K. D. Belfield, Org. Biomol., 2017, 15, 6511–6519 RSC.
- E. Heyer and R. Ziessel, Synlett, 2015, 2109–2116 CAS.
- J. Dhar, N. Venkatramaiah, A. A and S. Patil, J. Mater. Chem. C, 2014, 2, 3457–3466 RSC.
- R. S. Szabadai, J. Roth-Barton, K. P. Ghiggino, J. M. White and D. J. D. Wilson, Aust. J. Chem., 2014, 67, 1330–1337 CrossRef CAS.
- J. Raeburn, A. Zamith Cardoso and D. J. Adams, Chem. Soc. Rev., 2013, 42, 5143–5156 RSC.
- F. Tantakitti, J. Boekhoven, X. Wang, R. V. Kazantsev, T. Yu, J. Li, E. Zhuang, R. Zandi, J. H. Ortony, C. J. Newcomb, L. C. Palmer, G. S. Shekhawat, M. O. de la Cruz, G. C. Schatz and S. I. Stupp, Nat. Mater., 2016, 15, 469–476 CrossRef CAS PubMed.
- J. Zhou, X. Du, Y. Gao, J. Shi and B. Xu, J. Am. Chem. Soc., 2014, 136, 2970–2973 CrossRef CAS.
- L. L. E. Mears, E. R. Draper, A. M. Castilla, H. Su, Zhuola, B. Dietrich, M. C. Nolan, G. N. Smith, J. Doutch, S. Rogers, R. Akhtar, H. Cui and D. J. Adams, Biomacromolecules, 2017, 18, 3531–3540 CrossRef CAS PubMed.
- D. McDowall, B. J. Greeves, R. Clowes, K. McAulay, A. M. Fuentes-Caparros, L. Thomson, N. Khunti, N. Cowieson, M. C. Nolan, M. Wallace, A. I. Cooper, E. R. Draper, A. J. Cowan and D. J. Adams, Adv. Energy Mater., 2020, 10 Search PubMed.
- R. V. Kazantsev, A. J. Dannenhoffer, A. S. Weingarten, B. T. Phelan, B. Harutyunyan, T. Aytun, A. Narayanan, D. J. Fairfield, J. Boekhoven, H. Sai, A. Senesi, P. I. O’Dogherty, L. C. Palmer, M. J. Bedzyk, M. R. Wasielewski and S. I. Stupp, J. Am. Chem. Soc., 2017, 139, 6120–6127 CrossRef CAS.
- A. S. Weingarten, A. J. Dannenhoffer, R. V. Kazantsev, H. Sai, D. Huang and S. I. Stupp, J. Am. Chem. Soc., 2018, 140, 4965–4968 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sm00277a |
| This journal is © The Royal Society of Chemistry 2022 |