
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Charging of drops impacting onto superhydrophobic surfaces†
Diego
Díaz
 a,
Diana
Garcia-Gonzalez
ab,
Pravash
Bista
a,
Stefan A. L.
Weber
ac,
Hans-Jürgen
Butt
a,
Amy
Stetten
*a and
Michael
Kappl
*a
a,
Diana
Garcia-Gonzalez
ab,
Pravash
Bista
a,
Stefan A. L.
Weber
ac,
Hans-Jürgen
Butt
a,
Amy
Stetten
*a and
Michael
Kappl
*a
aMax Planck Institute for Polymer Research, Ackermannweg 10, 55128 Mainz, Germany. E-mail: kappl@mpip-mainz.mpg.de; stetten@mpip-mainz.mpg.de
bPhysics of Fluids Group, Max Planck Center Twente for Complex Fluid Dynamics, Department of Science and Technology, University of Twente, P.O. Box 217, Enschede 7500 AE, The Netherlands
cDepartment of Physics, Johannes Gutenberg University, Staudingerweg 10, 55128 Mainz, Germany
First published on 24th January 2022
Abstract
When neutral water drops impact and rebound from superhydrophobic surfaces, they acquire a positive electrical charge. To measure the charge, we analyzed the trajectory of rebounding drops in an external electric field by high-speed video imaging. Although this charging phenomenon has been observed in the past, little is known about the controlling parameters for the amount of drop charging. Here we investigate the relative importance of five of these potential variables: impact speed, drop contact area, contact line retraction speed, drop size, and type of surface. We additionally apply our previously reported model for sliding drop electrification to the case of impacting drops, suggesting that the two cases contain the same charge separation mechanism at the contact line. Both our experimental results and our theoretical model indicate that maximum contact area is the dominant control parameter for charge separation.
1 Introduction
Drop impact has been extensively studied for more than 100 years, including collision of drops with other liquids and different solid surfaces.1,2 This phenomenon is present in nature3,4 and is important for a variety of industrial applications.5–7 Recently, the impact and motion of water drops has been proposed as a possible source to generate electricity.8–18 In addition, since charging may influence dynamic wetting on surfaces,19–21 there is also fundamental interest in such processes.It is known that impacting drops on hydrophobic surfaces may lead to charge separation and consequent electrification of the liquid and surface. A rebounding drop usually leaves the surface negatively charged, while the drop charges positively.22–25 The existence of an electrical double layer and the ions naturally present in water could explain the charge separation process. One example of charge separation occurs when drops slide down a hydrophobic tilted plane. After sliding, drops usually acquire a positive charge and deposit a surface charge of opposite sign on the solid.26–30 In a similar way, water drops that condense and coalesce on a superhydrophobic surface jump-off with a positive charge.31 Applications using such charging include designing surface charge gradients to transport drops on superamphiphobic surfaces.20
Although many such drop charging phenomena have been observed, charge separation processes that involve the movement of the three-phase contact line are not well understood due to difficulties in performing quantitative experiments. Furthermore, charge separation caused by flowing liquids involves processes far from equilibrium. As a result, it is difficult to derive a quantitative thermodynamic theory to describe them. Different fundamental effects may lead to net drop charging and it is challenging to recognize which ones contribute or dominate.
We will focus on the case of impacting drops that acquire some charge during contact with a surface. The charging of such drops has never been systematically quantified. The magnitude of drop charge after impact and the dependency on parameters like drop size, contact area, impact speed, and the retraction motion of the contact line is still unclear. Knowledge of such dependencies is a prerequisite for the understanding of the charging mechanisms and controlling the charge of impacting drops.
In this article, we address the issue of systematic measurement and modeling of impacting drop charge. We use an external electric field to reproducibly quantify the charge of rebounding drops on different superhydrophobic surfaces. When a water drop impacts these surfaces, it bounces off with a net charge and then deflects due to the electrostatic force. Our measurements allow us to isolate the influence of diverse drop parameters. We propose an extension of a previous theoretical model for electrification of sliding drops,30 through which we can quantitatively describe the charging process. We show that the maximum spreading contact area is the dominant parameter controlling the amount of charge acquired.
2 Experimental details
In our experiments, we used five types of superhydrophobic surface coatings on glass microscope slides (26 × 76 mm2, 1 mm thick). Four were based on silicone nanofilaments (SN): pure SN, SN silanated with perfluorodecyltrichlorosilane (SN-FDTS), octadecyltrichlorosilane modified SN (SN-OTS) and polydimethylsiloxane brush-coated SN (SN-PDMS brush). The fifth surface used was a candle soot-templated fluorinated superhydrophobic surface (see ESI† for preparation details). These superhydrophobic surfaces were selected because water drops rebound fully off of them at a wide range of impact speeds.We characterized the surface topography of silicone nanofilaments and candle soot by scanning electron microscopy (Fig. 1). Candle soot consists of carbon particles of up to 40 nm, forming a loose fractal-like network (Fig. 1a). We coated the particles with a silica shell of 20 nm.32 The static contact angles of 4 μL drops on these surfaces is 162° ± 2. Silicone nanofilaments form a dense network on the glass substrate, showing a tangled, bent and hooked distribution, with diameters between 20 and 50 nm (Fig. 1b).
 | ||
| Fig. 1 (a) Scanning electron microscopy image of a candle soot surface after being coated with a silica shell. Inset shows a 4 μL drop deposited on the surface with a static contact angle of 162° ± 2. (b) Scanning electron microscopy image of silicone nanofilaments deposited on glass. Inset shows a 4 μL drop deposited on the surface with a static contact angle of 162° ± 1. | ||
We carried out three types of experiments: (1) 4 μL drops of deionized water were released from heights between 0.4 and 5 cm onto all the surfaces mentioned above, (2) volumes between 3.5 and 13 μL onto SN-FDTS from 3 cm height, and (3) 6 μL drops released onto SN-PDMS brush and candle soot surfaces from 1.5 cm height. We recorded the impacting process using a high-speed camera in front of the sample (Photron FastCam Mini UX100, 5000 fps) to track the motion of the drop mass centre (Fig. S1, ESI†). Two copper plates were placed vertically over a Teflon plate to act as electrodes (Fig. 2a) and hence generate a horizontal electrical field E = V/d = 55 kV m−1, where V is the applied voltage and d the separation distance of the copper plates (18 mm). The sample surfaces were placed between the electrodes on the Teflon plate. The Teflon served as a non-conductive surface to isolate copper plates and sample. A grounded syringe pump generated neutral water drops. We shielded drop and needle with an aluminum tube to prevent polarization effects. We controlled the maximum spreading radius Rmax by the Weber number We = ρv02D0/γ, where ρ, v0, D0 and γ are the density, impact speed (0.2 m s−1 < v0 < 1 m s−1), diameter and surface tension of the drop. We will consider the impact speed in terms of We in the next sections. Sample surfaces were neutralized by an Ionizing Air Blower (Aerostat PC Ionizing Air Blower, Pennsylvania, USA) for 2 minutes before each drop. We observed complete rebounds with conservation of volume in the range of 1 < We < 13, and drop break up for We > 13. We can assure then that drops remain in the Cassie–Baxter state.33
 | ||
| Fig. 2 (a) Schematic of the experimental setup. (b–d) 4 μL drop bouncing from a SN-FDTS surface in three cases: no electric field (b); electric field oriented from right to left (c); electric field oriented from left to right (d). The blue markers represent the movement of the drop mass centre. Each image shows an overlay of three drops moments: 25%, 50% and 75% of the total duration of the recorded video. x1 and x2 represent the positions of the centre of mass when the drop leaves the surface, and the moment it contacts the surface again, respectively. (e) Verification that the detected charge was independent of the applied electric field. The horizontal line corresponds to Q = 20 pC. Drop volume: 4 μL. | ||
3 Results
In this section, we will analyze the experimental results of drops that rebound and are deflected by a horizontal electric field. We will explain the drop charge calculation method and discuss the influence of the surface coating, retraction speed, impact speed, drop volume, and contact area in the charge separation process.3.1 Deflection of drops and drop charge calculation
In the absence of an electric field, our rebounding drops followed a vertical trajectory (Fig. 2b). When an electric field was applied, rebounding drops followed a trajectory including lateral movement (x-direction) in the direction of the electric field (Fig. 2c and d). This indicates that drops acquired a positive charge when rebounding from the surface.The distribution of the electric field along several directions between our two metal plates was simulated using COMSOL Multiphysics 5.5 (Section III, ESI†). The simulations indicated that the electric field varies with height less than ± 5% around the average value (Fig. S4a, ESI†) in the range of the rebounding heights of our drops, which were always below the height of the plates. Within the range of the drop lateral movement (maximum of 2 mm) around the center, the lateral change in field (Fig. S4b and c, ESI†) is less than 1 kV m−1 (∼2% error), leading to an error of less than 10% in the charge calculation. Therefore, according these small and systematic deviations, we assume for our experiments an homogeneous electric field in all directions.
The homogeneity of the electric field implies that drops laterally move as  , where Δx = |x2–x1|, with x1 = x(t1) as the lateral position of the drop mass centre at which the drop bounces from the surface and x2 = x(t2) is the lateral position where the drop impacts the surface again after bouncing (Fig. S1, ESI†). t = t2–t1 is the time interval between these events. Since the electrostatic force is FE = QE = max (Q is the drop charge and m is the drop mass), we calculated the drop charge as Q = 2ρVΔx/Et2. In our experiments, V is the initial and constant drop volume as we ensure that the drops rebound completely. For the case of drop break up, we calculated the charge of the primary drop subtracting the volume of the secondary ejected drops (Fig. S1g, ESI†). We only considered Weber numbers, where the volume of the main drop after break up is just slightly smaller (3.7–3.8 μL) than the original drop (4 μL). Only for higher Weber numbers, we observed also deflection of the secondary drops in the electric field, indicating partial charge transfer to the secondary drop as well. For the drop charge calculations, we did not measure directly the charge distribution in the drop. However, since the electric field is homogeneous, the exact charge distribution does not affect our measurements because the total force on the drop will equal to that on a point charge at the drop center.
, where Δx = |x2–x1|, with x1 = x(t1) as the lateral position of the drop mass centre at which the drop bounces from the surface and x2 = x(t2) is the lateral position where the drop impacts the surface again after bouncing (Fig. S1, ESI†). t = t2–t1 is the time interval between these events. Since the electrostatic force is FE = QE = max (Q is the drop charge and m is the drop mass), we calculated the drop charge as Q = 2ρVΔx/Et2. In our experiments, V is the initial and constant drop volume as we ensure that the drops rebound completely. For the case of drop break up, we calculated the charge of the primary drop subtracting the volume of the secondary ejected drops (Fig. S1g, ESI†). We only considered Weber numbers, where the volume of the main drop after break up is just slightly smaller (3.7–3.8 μL) than the original drop (4 μL). Only for higher Weber numbers, we observed also deflection of the secondary drops in the electric field, indicating partial charge transfer to the secondary drop as well. For the drop charge calculations, we did not measure directly the charge distribution in the drop. However, since the electric field is homogeneous, the exact charge distribution does not affect our measurements because the total force on the drop will equal to that on a point charge at the drop center.
The drag force can be neglected during the entire process and is defined by the Stokes' law FD = 6πrηv (r is the drop radius), η the viscosity of the air and v the drop velocity. For instance, a 4 μL drop impacting at We = 5, will experience a drag force of 1 nN when is laterally deflected by the electric field (see ESI,† Section IV) and an electrostatic force of FE = 9.81 μN. However, the drag force could become larger for the highly deformed drop shapes, where its velocity dependence would be in the worst case: 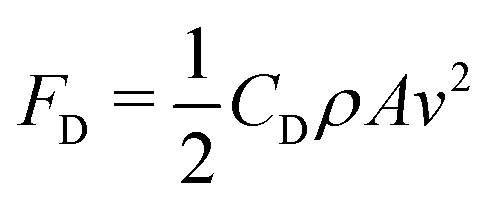 , with CD the drag coefficient, A the cross section area and vx the lateral drop velocity. Since the lateral drop velocity is close to zero at the spreading/retraction phase and at the maximum deformation moments upon bouncing, the lateral drag force will remain negligible at this point. When drops move close to the maximum lateral speed measured in the experiments (0.03 m s−1, see Fig. S5, ESI†), then the lateral drag force reaches less than 16% of the electrostatic force in the worst case. Thus, the drag force does not affect significantly our experiments.
, with CD the drag coefficient, A the cross section area and vx the lateral drop velocity. Since the lateral drop velocity is close to zero at the spreading/retraction phase and at the maximum deformation moments upon bouncing, the lateral drag force will remain negligible at this point. When drops move close to the maximum lateral speed measured in the experiments (0.03 m s−1, see Fig. S5, ESI†), then the lateral drag force reaches less than 16% of the electrostatic force in the worst case. Thus, the drag force does not affect significantly our experiments.
We performed control experiments assuring that drop charge is independent of the magnitude of the external electric field (Fig. 2e). This ruled out the possibility of induced electrification or dielectrophoretic effects. Thus, the acquired charge is the result of the interaction between the drop and the superhydrophobic surface. During a rebound without electric field, drops can exhibit a horizontal displacement of 0.03 ± 0.01 mm (We < 2). This displacement sets the limit of our detection method, which is equivalent to less than 3 pC.
Our detection technique of drop charge was calibrated using a similar method to Stetten et al.30 We detected the current signal of a rebounding drop touching an electrode probe prior impact and after the rebound from a superhydrophobic surface (Section II.I, ESI†). Simultaneously, we recorded the process by high-speed video imaging (Fig. S2a–d, ESI†). From the current signals (Fig. S2e and f, ESI†), we obtained values of drop charge very close to those obtained by our electric field method and not charge for the falling drop prior impact.
Ions, electrons, among other possibilities could be responsible for the positive charge of drops that interact with hydrophobic surfaces. Nevertheless, it has not been proven yet. We attribute the positive charge of rebounding drops to an accumulation of hydronium ions. When a hydrophobic surface interacts with water, it charges negatively most likely due to the absorption of hydroxyl ions.34–38 Accordingly, our superhydrophobic surfaces tend to absorb negative charges in form of hydroxyl ions from the impacting water drop. Hydronium ions form the complementary part of the electric double layer. When the drop rebounds positively charged, some of these ions accumulate in the drop.
3.2 Surface coating
For the surfaces chosen in our trials, the difference in drop charging for different surface coatings is not as significant as the dependence of drop charging on other variables such as contact area. We performed experiments on five surfaces under identical conditions (same drop size and height) and the maximum difference in charge was ∼15 pC. When varying other parameters in the system, we see changes in the charge as large as 40 pC. This indicates that, while the type of surface coating does play some role in charging, it cannot explain the full range of drop charge that we observe. Dependencies observed in the following sections were consistent across all different surface coatings. Note that, considering how new these types of experiments are, we have only tried a limited number of surface coatings, so it might be possible that coatings with completely different surface chemistry could show a greater effect on drop charging.3.3 Influence of the retraction and spreading speed
In the following, we discuss the influence of the speed during the retraction and spreading phase in the charging process. Experiments with drop break up are not included since drop charge remains constant after reaching a saturation point. This aspect will be discussed in the next sections. To identify which parameters are essential in drop charging, we analyze the drop impact kinetics, specifically the contact radius R(t) and the spreading and retraction motion of the contact line. Plotting the scaled contact radius R(t)/Rmaxversus We reveals that all spreading curves of surfaces based on SN collapse onto a single curve (Fig. 3a and Fig. S5, ESI†) as previously reported for hydrophobic surfaces.39 This means that contact time for these surfaces remains constant when varying the impact speed. Candle soot is the exception due to the protrusions, which may lead to more friction of the moving contact line and higher energy dissipation, as also reflected by the lowest values of restitution coefficient (Fig. S1, ESI†). Drops may also impale in these protrusions, affecting the duration of contact time. Thus, contact time depends on the surface type but not on the Weber number. | ||
Fig. 3 (a) Time dependence of the scaled contact radius R(t)/Rmax upon impact onto SN-PDMS brush coating (4 μL drops). (b) Drop charge as a function of the average retraction speed ![[V with combining macron]](https://www.rsc.org/images/entities/i_char_0056_0304.gif) ret at different Weber numbers 1 < We < 13. (c) Drop charge vs. the average retraction speed for SN-FDTS, with the volumes: 3, 4, 6, 8 and 13 μL. ret at different Weber numbers 1 < We < 13. (c) Drop charge vs. the average retraction speed for SN-FDTS, with the volumes: 3, 4, 6, 8 and 13 μL. | ||
Extracting Ṙ(t) from the retracting curves (Fig. 3a and Fig. S5, ESI†), we observed a speed fluctuating around a central value during the drop recoil (Fig. S6a, ESI†). Therefore, we define an average retraction speed ret = Rmax/tr as a characteristic speed of the retraction phase. Here, tr is the time of the entire retraction movement until drop detachment. This parameter is relevant because charge deposition is most likely the result of a non-equilibrium process. If we assume an electric double layer formed at the coating/liquid interface, its disruption and subsequent charge separation is most likely influenced by the speed of the retraction motion.
We found that the charge for 4 μL drops tends to increase with ret (Fig. 3b). The increasing ret is a result of a further spreading as We is greater (Fig. S7, ESI†). Consequently, one might be tempted to conclude that the faster the contact line moves, the higher the charge. However, the opposite trend is seen when decreasing the drop volume (Fig. 3c). When drop volume decreases, drops spread at smaller Rmax and rebounds occur at shorter contact times40 and tr. The lower values of tr are sufficient to produce a faster retracting motion, so decreasing the volume increases ret. And yet, decreasing drop volume leads to decreased drop charge. This means that there is no direct correlation between ret and Q. Thus, we can exclude ret as a dominant parameter in the charge separation process.
Similarly, we can use the same argument for an average spreading speed s = Rmax/ts, where ts is the time at which the drop reaches Rmax from the impact on the surface. Taking s of the impact for the cases of Fig. 3b and c, we found that drop charge varies with ret and s following the same trend (Fig. S8, ESI†). Thus, s is also excluded as a controlling parameter.
3.4 Influence of Weber number, drop volume and maximum contact area
In our analysis, we must be careful to disentangle the effects of Weber number, drop volume, and contact area. Our experimental results show that drop charge increases with an increase in each of these three variables, however the three variables are not independent of one another, as will be discussed here.Drop charge increased linearly with the Weber number in the region where drop break up does not occur (Fig. 4a). We can write the relationship as Q = β(We–We0) with β ∈ [2,5]. The parameter β depends on the coating and drop volume, and We0 corresponds to the lowest Weber number to observe a complete rebound. We demonstrated this linear increase by varying drop speed at a constant volume of 4 μL. The linear trend is robust, as varying the drop volume while keeping constant its falling height reproduces the same behaviour (Fig. 4a, white triangles). Thus, with higher We, we find higher drop charge. The data in the shaded region of drop break up shows the existence of a saturation zone where the drop charge values tend to be unaltered with further increase of We. Besides, the difference in the saturation points of the surface coatings suggests that the maximum charge acquired by a drop depends on the surface chemistry of the impacted sample.
 | ||
| Fig. 4 (a) Drop charge as a function of Weber number. Color markers: 4 μL drops on different surfaces, white markers: drops of 3, 4, 6, 8 and 13 μL impacting on SN-FDTS. The shaded region represents the Weber numbers where drop break up occurs. (b) Drop charge vs. the maximum contact area, Amax. Circular dashed lines enclose experiments with different Weber numbers, drop volumes, similar contact areas and drop charge. | ||
Higher We also brings with it a larger maximum contact area between the drop and surface, which could also be increased by increasing the drop volume. Then the maximum contact area arises as an implicit parameter influencing charging. Considering a symmetric spreading, we calculated the maximum contact area as Amax = πRmax2. We found that the drop charge increases with Amax (Fig. 4b) as well as with We and drop volume. However, note that for experiments with similar contact area and different Weber number and drop volume, the charge shows similar values (Fig. 4b, circular dashed lines). In particular, drops of 4 μL (We = 8.70, v0 = 0.75 m s−1) and 6 μL (We = 7.60, v0 = 0.55 m s−1) rebounding on SN-FDTS reach a maximum contact area of Amax ∼ 6 mm2, with charge values of ∼29.3 pC and ∼30.0 pC respectively. This suggests that We and drop volume are not the main control parameters of the charge separation process. In fact, Amax is a function of these two variables. Therefore, we can conclude that Amax is the governing parameter in the charging mechanism.
4 Theoretical model for charging of bouncing drops
We adapt the slide electrification theory developed by Stetten et al.30 to describe charging of drops quantitatively. The theory describes a water drop sliding across a surface and depositing charge at its tail. An electric double layer forms at the drop-surface interface. It is assumed that a fraction α of the total charge density in the Debye Layer, σL, is left behind on the surface after the drop has passed: σS = ασL. As the drop slides, it accumulates counter charge equivalent to the total deposited charge.In our case, drops are impacting and then retracting radially off the surface. During the impacting process, the contact line moves in two phases: spreading and retraction. We consider the formation of an electrical double layer as the drop spreads on the surface. At the end of the spreading, the drop reaches the maximum contact diameter. This is comparable to a drop that wets a hydrophobic surface prior sliding, until it reaches its maximum width or contact diameter. The formation time of the electrical double layer is estimated to be 6 μs,30 which occurs during the wetting phase (2 ms, Fig. 3a). Accordingly, the retracting movement of an impacting drop should lead to charge separation by disruption of the electrical double layer in a similar way to a sliding contact line.
In this model, we assume that the charge is only deposited at the rear of the receding contact line, so we only need to consider the retracting motion. It could be, of course, that charges already deposit on the solid surface during the spreading, where the electrical double layer forms. However, since the drop is still in contact with the surface during spreading, counter charges are expected to exist close by and the sum of all charges in the electrical double layer is zero. So true charge separation is most likely not to occur until the drop retracts, which breaks the electrical equilibrium at the liquid/solid interface as the surface and liquid are no more in contact. As the drop retracts after reaching Rmax, it deposits charge along its outer radius. This causes that drops gain the opposite net charge like a charging capacitor. Since the drop has a limited electrical capacitance, charge separation will be highest during the initial phase of the retraction and will become more and more hindered with increasing drop charge. Therefore, the amount of charge left on the surface should decrease in density with decreasing radius. We assume that the infinitesimal charge collected as the contact line slides over an area, 2πr′dr′ (Fig. 5a), equals α times the charge in the electric double layer dQ(r′) = −σs(2πr′)dr′. Here, r′ is the retracted position of the drop on the surface.
 | ||
| Fig. 5 (a) Schematic of charge deposition for a retracting drop. (b) Drop charge versus the retracted position r′ from eqn (1). The constants σ0S = 14 μC m−2 and λr = 0.6 mm were taking fitting eqn (1) into Fig. 4 at r′ = Rmax for SN-FDTS. | ||
Assuming that the drop starts neutrally charged at its maximum spreading radius Q(r′ = Rmax) = 0, (see ESI,† Section XI), we find the charge accumulated in the drop after it has retracted to radius r′:
 | (1) |
 | (2) |
The surface charge density left behind starts at its highest value, σ0 (at r′ = Rmax), and decreases with decreasing radius due to the increasing potential between drop and surface (Fig. S9, ESI†). For larger drops (Rmax = 10 mm), most of the charge left behind by a rebounding drop is concentrated at the outer ring of the deposition pattern (Fig. 6a). The drop fully charges before finishing its retraction, leaving a bare area in the centre of the deposition area because the drop capacitance limits further charge deposition on the surface. Smaller drops do not charge fully (Rmax < 5 mm) and there is charge left across the entire deposition area. This pattern is quite similar to that measured by Nie et al.41 as the charge pattern left behind by a drop pressed between two surfaces.
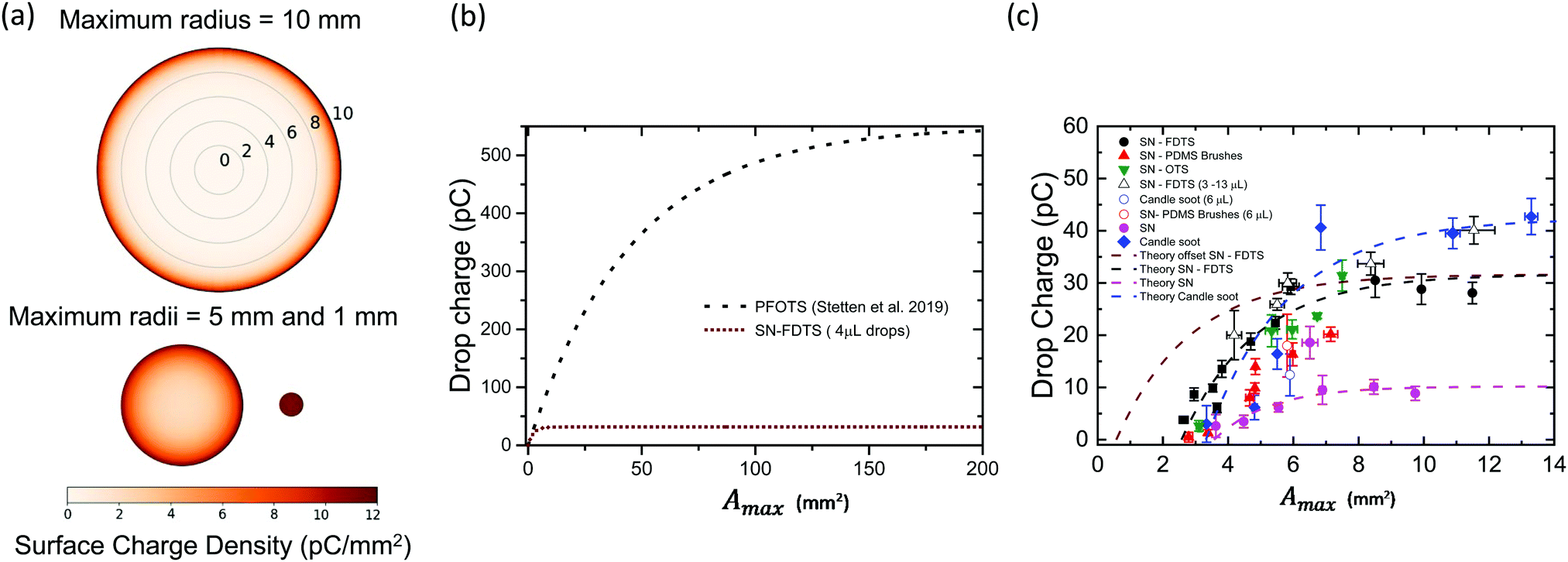 | ||
| Fig. 6 (a) Visualization of the predicted surface charge distribution left behind by three drops with the same volume but different maximum spreading areas. (b) Total drop charge (retracted to r′ = 0) versus maximum spreading area from eqn (3) for PFOTS coated surfaces (σ0S = 12 μC m−2; λr = 2.7 mm, black dotted line, from30) and our SN-FDTS surfaces for 4 μL (σ0S = 14 μC m−2; λr = 0.6 mm, brown dashed line). (c) Experimental data for drop charge versus maximum spreading area. The theoretical prediction for SN-FDTS (shown in (b), brown dashed line), SN and candle soot is plotted on the same axes. The parameters used for the fitting were: σ0S = 5 μC m−2 and λr = 0.52 mm for SN; σ0S = 17 μC m−2 and λr = 0.63 mm for candle soot. All the surface coatings have the fits starting from A0 as the minimum measured area. SN-FDTS data is fitted with an additional curve using A0 = 0.57 mm for a resting drop, as predicted by the model (brown dashed line). | ||
To compare our drop charge measurements with the proposed theory, we set r′ = 0 because the drop has fully retracted and when it leaves surface. Inserting r′ = 0 into eqn (1), we find the total charge on the drop as a function of Amax:
 | (3) |
5 Discussion
Note that for small Amax values in eqn (3), the surface charge density σ0S can be approximated to dQ/dAmax. This means that drop charge is proportional to Amax and Rmax at low and large contact areas respectively, with a transition at λr2. Hence, σ0S sets the slope of the initial points in Fig. 4b, while λr determines the saturation value. Eqn (3) predicts the drop charge saturation when Amax > 125 mm2 for a PFOTS coated glass surface30 and our superhydrophobic SN-FDTS surfaces, with σ0S = 14 μC m−2 and λr = 0.6 mm (Fig. 6b). Plotting the theoretical model in the same range of our experimental data, we get a similar trend with the different surface coatings (Fig. 6c), using the following parameters: σ0S = 6 μC m−2 and λr = 0.52 mm for SN; σ0S = 17 μC m−2 and λr = 0.63 mm for candle soot. According our model, we used A0 = 0.57 ± 0.04 mm2 as the area of a resting drop to set the minimum Amax for SN-FDTS (measured for 4 μL drops onto SN surfaces). The rest of fits consider A0 as the minimum area in the data as a comparison. The fitting parameters change according to the surface coating. As σ0S and λr are greater, the saturation limit is higher and more charge is left on the surface upon impact. For instance, candle soot surfaces show the highest surface charge density and λr as well as the saturation charge. Hence, the saturation limit is determined not only by the total capacitance of the drop, but also on the surface chemistry of the coatings.Our experimental data in Fig. 6c are offset in terms of area compared to the theoretical prediction. Furthermore, charge dissipation processes not included in the theory could partially discharge the drop during extended contact times, reducing the net charge of the drop. This explains the difference in the slope when varying the drop volume compared to varying the impact speed. Since drops retract slower with an increase of volume, more time is available for discharging mechanisms. Another important issue that also explains the offset at low Amax is the surface structure of our substrates. The parameters σ0S and λr in30 were determined for flat fluorinated glass surfaces. These should have the same surface chemistry as our fluorinated superhydrophobic substrates. However, the nanoscale roughness of our substrates should lead to a much smaller effective liquid–solid contact area than the apparent contact area Amax, as the drops remain in the Cassie state during bouncing. In particular, we can estimate the effective contact area between the surface and drop using the Cassie–Baxter equation:33
cos![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) (θ*) = f1cos(θ1) + f2cos(θ2), (θ*) = f1cos(θ1) + f2cos(θ2), | (4) |
Another aspect that may play a role in the charging process is the relative humidity (RH) of the environment. We performed our experiments under lab ambient conditions, so we did not control RH (measured values were 30–40%). Recently, Sosa et al.42 showed that RH affects the charging of sliding drops only when RH > 70%. Thus, we assume that relative humidity does not affect the charging process in this work.
6 Conclusions
Our results show that initially neutral water drops rebound off of superhydrophobic surfaces positively charged. We found experimentally that the maximum spreading area is the main parameter controlling the amount of charge. Hence, the charge depends implicitly on the drop volume and impact speed.Our model, based on a radial solution of the slide electrification theory, is in good agreement with the experimental results. This supports the idea that a drop deposits charge along the outer ring of the impact area during the retraction phase. Impacting drops occur universally in nature and industry. Understanding and being able to model these charge separation phenomena has broad-ranging impacts. It could allow the optimization of energy-harvesting applications using rebounding drops. It could aid in the electrostatic guidance of charged bouncing drops or it could help us understand the fundamentals of tribocharging in liquids. Future studies for a better understanding of the underlying charge separation mechanism might involve use of other liquids and probing the influence of salts and pH.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research leading to these results has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme, Grant agreement no 883631 (H. J. B., A. S.). D. D. acknowledges the support from the Bilateral Agreement of the Doctoral scholarships DAAD/BECAS Chile, 2018 (57395809). We would like to thank Jie Liu, Alfons Becker and Diego Farías for helpful discussions. Open Access funding provided by the Max Planck Society.References
- A. L. Yarin, I. V. Roisman and C. Tropea, Collision Phenomena in Liquids and Solids, Cambridge University Press, 2017 Search PubMed.
- S. Lin, B. Zhao, S. Zou, J. Guo, Z. Wei and L. Chen, J. Colloid Interface Sci., 2018, 516, 86–97 CrossRef CAS PubMed.
- J. C. Bird, R. Dhiman, H.-M. Kwon and K. K. Varanasi, Nature, 2013, 503, 385–388 CrossRef CAS PubMed.
- T. Gilet and L. Bourouiba, J. R. Soc., Interface, 2015, 12, 20141092 CrossRef CAS PubMed.
- L. Mishchenko, B. Hatton, V. Bahadur, J. A. Taylor, T. Krupenkin and J. Aizenberg, ACS Nano, 2010, 4, 7699–7707 CrossRef CAS PubMed.
- W. Jia and H.-H. Qiu, Exp. Therm. Fluid Sci., 2003, 27, 829–838 CrossRef CAS.
- A. Vallet and C. Tinet, J. Crop Prot., 2013, 48, 63–68 CrossRef.
- A. M. Duffin and R. J. Saykally, J. Phys. Chem. C, 2008, 112, 17018–17022 CrossRef CAS.
- T. Krupenkin and J. Taylor, Nat. Commun., 2011, 2, 448 CrossRef PubMed.
- Z. Yang, E. Halvorsen and T. Dong, Appl. Phys. Lett., 2012, 100, 213905 CrossRef.
- J. Moon, J. Jeong, D. Lee and H. Pak, Nat. Commun., 2013, 4, 1487 CrossRef PubMed.
- Z.-H. Lin, G. Cheng, L. Lin, S. Lee and Z. L. Wang, Angew. Chem., Int. Ed., 2013, 52, 12545–12549 CrossRef CAS PubMed.
- S.-H. Kwon, J. Park, W. K. Kim, Y. Yang, E. Lee, C. J. Han, S. Y. Park, J. Lee and Y. S. Kim, Energy Environ. Sci., 2014, 7, 3279–3283 RSC.
- Y. Xie, D. Bos, L. de Vreede, H. de Boer, M.-J. Meulen, M. Versluis, A. Sprenkels, A. Van den Berg and J. Eijkel, Nat. Commun., 2014, 5, 3575 CrossRef PubMed.
- L. Zheng, Z.-H. Lin, G. Cheng, W. Wu, X. Wen, S. Lee and Z. Wang, Nano Energy, 2014, 9, 291–300 CrossRef CAS.
- Q. Liang, X. Yan, G. Yousong, K. Zhang, M. Liang, S. Lu, X. Zheng and Y. Zhang, Sci. Rep., 2015, 5, 9080 CrossRef CAS PubMed.
- L. E. Helseth and X. D. Guo, Langmuir, 2015, 31, 3269–3276 CrossRef CAS PubMed.
- H. Wu, N. Mendel, D. van den Ende, G. Zhou and F. Mugele, Phys. Rev. Lett., 2020, 125, 078301 CrossRef CAS PubMed.
- I. Langmuir, J. Am. Chem. Soc., 1938, 60, 1190–1194 CrossRef CAS.
- Q.-Q. Sun, D. Wang, Y. Li, J. Zhang, S. Ye, J. Cui, L. Chen, Z. Wang, H.-J. Butt, V. Doris and X. Deng, Nat. Mater., 2019, 18, 936–941 CrossRef CAS PubMed.
- R. Digilov, Langmuir, 2000, 16, 6719–6723 CrossRef CAS.
- P. Lenard, Ann. Phys., 1892, 282, 584–636 CrossRef.
- J. J. Thomson, Philos. Mag. J. Sci., 1894, 37, 341–358 CrossRef.
- Z. Levin and P. Hobbs, Philos. Trans. R. Soc., A, 1971, 269, 555–585 CrossRef CAS.
- D. M. Chate and A. K. Kamra, Atmos. Res., 1993, 29, 115–128 CrossRef.
- K. Yatsuzuka, Y. Mizuno and K. Asano, J. Electrostat., 1994, 32, 157–171 CrossRef.
- L. E. Helseth and H. Z. Wen, Eur. J. Phys., 2017, 38, 055804 CrossRef.
- L. E. Helseth, Langmuir, 2019, 35, 8268–8275 CAS.
- A. Shahzad, K. R. Wijewardhana and J.-K. Song, Appl. Phys. Lett., 2018, 113, 023901 CrossRef.
- A. Z. Stetten, D. S. Golovko, S. A. L. Weber and H.-J. Butt, Soft Matter, 2019, 15, 8667–8679 RSC.
- N. Miljkovic, D. Preston, R. Enright and E. Wang, Nat. Commun., 2013, 4, 2517 CrossRef PubMed.
- X. Deng, L. Mammen, H.-J. Butt and D. Vollmer, Science, 2012, 335, 67–70 CrossRef CAS.
- A. B. D. Cassie and S. Baxter, Trans. Faraday Soc., 1944, 40, 546–551 RSC.
- V. Tandon, S. K. Bhagavatula, W. C. Nelson and B. J. Kirby, J. Electrophor., 2008, 29, 1092–1101 CrossRef CAS PubMed.
- T. Preočanin, A. Selmani, P. Lindqvist-Reis, F. Heberling, N. Kallay and J. Lützenkirchen, Colloids Surf., A, 2012, 412, 120–128 CrossRef.
- C. S. Tian and Y. R. Shen, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 15148–15153 CrossRef CAS PubMed.
- J. K. Beattie, Lab Chip, 2006, 6, 1409–1411 RSC.
- T. W. Healy and D. W. Fuerstenau, J. Colloid Interface Sci., 2007, 309, 183–188 CrossRef CAS PubMed.
- D. Bartolo, C. Josserand and D. Bonn, J. Fluid Mech., 2005, 545, 329–338 CrossRef.
- D. Richard, C. Clanet and D. Quéré, Nature, 2002, 417, 811 CrossRef CAS PubMed.
- J. Nie, Z. Ren, L. Xu, S. Lin, F. Zhan, X. Chen and Z. L. Wang, Adv. Mater., 2020, 32, 1905696 CrossRef CAS PubMed.
- M. D. Sosa, M. L. Martínez Ricci, L. L. Missoni, D. H. Murgida, A. Cánneva, N. B. D'Accorso and R. M. Negri, Soft Matter, 2020, 16, 7040–7051 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Sample preparation details, drop impact dynamics, full derivation of the theoretical model of charging for bouncing drops. See DOI: 10.1039/d1sm01725j |
| This journal is © The Royal Society of Chemistry 2022 |