
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrofabrication of large volume di- and tripeptide hydrogels via hydroquinone oxidation†
Courtenay
Patterson
a,
Bart
Dietrich
 a,
Claire
Wilson
a,
Andrew R.
Mount
b and
Dave J.
Adams
*a
a,
Claire
Wilson
a,
Andrew R.
Mount
b and
Dave J.
Adams
*a
aSchool of Chemistry, University of Glasgow, G12 8QQ, UK. E-mail: dave.adams@glasgow.ac.uk
bEastCHEM, School of Chemistry, University of Edinburgh, EH9 3FJ, UK
First published on 3rd January 2022
Abstract
The fabrication of protected peptide-based hydrogels on electrode surfaces can be achieved by employing the electrochemical oxidation of hydroquinone to benzoquinone, liberating protons at the electrode–solution interface. The localised reduction in pH below the dipeptide gelator molecules pKa initiates the neutralisation, self-assembly and formation of self-supporting hydrogels exclusively at the electrode surface. Previous examples have been on a nanometre to millimetre scale, using deposition times ranging from seconds to minutes. However, the maximum size to which these materials can grow and their subsequent mechanical properties have not yet been investigated. Here, we report the fabrication of the largest reported di- and tri-peptide based hydrogels using this electrochemical method, employing deposition times of two to five hours. To overcome the oxidation of hydroquinone in air, the fabrication process was performed under an inert nitrogen atmosphere. We show that this approach can be used to form multilayer gels, with the mechanical properties of each layer determined by gelator composition. We also describe examples where gel-to-crystal transitions and syneresis occur within the material.
Introduction
Hydrogels are a class of soft materials characterised by both their elastic and viscous-like properties.1,2 Hydrogels can be subcategorized into supramolecular or chemical hydrogels.3 Chemical hydrogels self-assemble through covalent irreversible crosslinks and display much higher mechanical strength than supramolecular gels, which are held together by weaker intermolecular forces such as hydrogen bonding and π–π interactions.4,5Low molecular weight hydrogelators (LMWGs) are small molecules that self-assemble in a hierarchal fashion to form supramolecular hydrogels.6–9 Upon applying a suitable trigger, the gelator molecules self-assemble to form one-dimensional structures such as fibres. These then entangle to form a self-supporting three-dimensional network. There are many classes of LMWG; one interesting sub-class is the aromatic functionalised dipeptide.6,10–13 These gelators form supramolecular hydrogels using a wide variety of physical and chemical gelation triggers.14 Commonly used triggers include pH changes,15 solvent switches,16 use of enzymes,17 temperature changes,18 UV irradiation,19 and electrical signals.20
Electrochemical gelation employs the use of electrical control to trigger the self-assembly.21,22 Electrochemical gelation techniques can be split into two categories; an indirect electrochemical approach and a direct electrochemical approach.23 A direct electrochemical approach to fabricating hydrogels commonly employs an applied electric field, which results in gel formation through processes such as electrophoresis, dielectrophoresis (DEP) and electrotaxis.24,25 The gels are formed far from the electrode surface.
An indirect electrochemical approach sees the production of target chemical species at the electrode surface through faradaic reaction, which are subsequently responsible for the self-assembly of the gelator molecules.26 This technique affords excellent spatiotemporal control and high localisation of gelation to the electrode surface. Spatiotemporal control is the major benefit of indirect electrofabrication as the rate of gel growth, size, and thickness can be easily altered to suit the application, simply by changing the current and applied deposition time. The electrode material, size and geometry can also be varied. The most common hydrogels fabricated using this approach include biologically derived polysaccharides such as chitosan,27–29 silk30 and alginate.31
Aromatic functionalised dipeptide hydrogels can be fabricated on electrode surfaces, offering a synthetic alternative to materials derived from biological sources.32–34 Aromatic functionalised dipeptide gelators have free carboxylic acid groups on the C-terminus and self-assemble via non-covalent interactions. Hydrogels are formed when the pH drops below the pKa of the terminal carboxylic acid in aqueous conditions, removing electrostatic repulsion through protonation and neutral acid formation.35 Protonation has previously been achieved electrochemically. The electrochemical oxidation of hydroquinone (HQ) to benzoquinone (BQ) generates protons at the electrode–solution interface (Fig. 1), creating a localised drop in pH at the electrode surface below the pKa of the dipeptide gelator.36,37 Therefore, gelation can be achieved exclusively at the electrode surface, leaving the pH of the bulk solution unaffected.
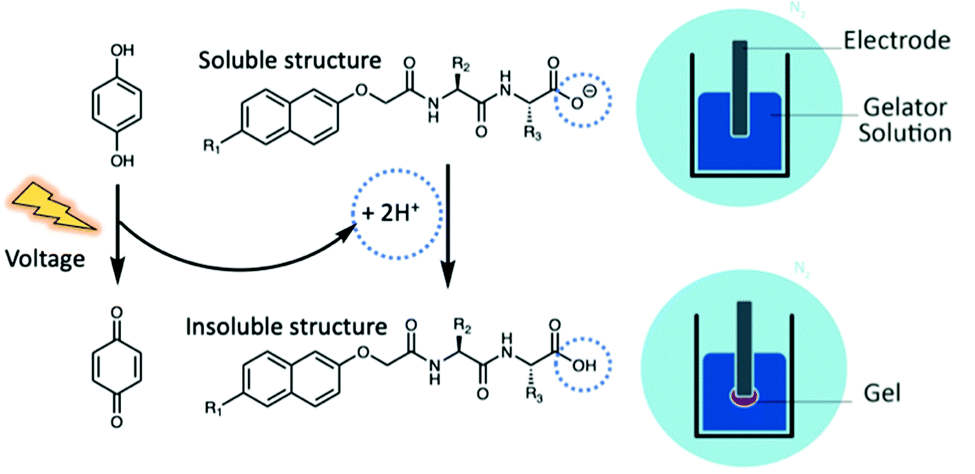 | ||
| Fig. 1 Schematic representation of electrochemical gelation via HQ oxidation. To prevent the oxidation of HQ in air, the fabrication process is performed in an inert atmosphere using nitrogen. | ||
A variety of peptide and dipeptide hydrogels have been fabricated using the electrochemical oxidation of HQ to form soft materials of various morphologies and sizes. Cameron and co-workers14 first reported the surface nucleated growth of dipeptide gel membranes using this method, forming nanometre thick hydrogel films of the N-protected dipeptide Fmoc-Leu-Gly-OH. Using galvanostatic (constant current) conditions, the gel volume increased with deposition time and the rate of gel growth from the electrode surface could be controlled by modifying the applied current. Cameron and co-workers33 later used this method to grow thin dipeptide layers on electrode surfaces, placing seeding layers containing HQ in contact with dipeptide solutions for 24 hours. Later, it was shown that direct polymerisation of electrochemically fabricated hydrogels to form microporous electrochromic polymers was possible.34 Raeburn et al. grew millimetre thick dipeptide hydrogels of various chemical composition and size with excellent spatiotemporal control.32 Sequential assembly of two dipeptide LMWGs was also achieved. A variety of patterned hydrogels were also created on the electrode surfaces, further showing the versatility and practicality of this technique.
The electrochemical oxidation of HQ has also been used to successfully create peptide hydrogel-coated nanoelectrode biosensors, with the hydrogel allowing small biomarker molecules to diffuse through the gel matrix while simultaneously preventing biofouling.38 Payne and co-workers39 used this electrochemical method to form peptide hydrogels that act as temporary scaffolds for agarose hydrogels, which are widely used materials in biological sciences. A further study by Payne and co-workers40 showed this could be performed within microfluidic channels, extending the potential of these materials to animal-on-a-chip applications.
All of the work in this area has involved the formation of hydrogels on the nanometre to millimetre scales. Larger gel structures and their material properties have not yet been explored or reported. It has also been generally assumed that peptide hydrogels formed using this method have uniform mechanical properties and pH throughout, but due to the small scale on which these materials have been grown to date, this has not been determined. To investigate the maximum size to which these materials can grow, much longer deposition times are required. One of the most commonly encountered problems with longer deposition times is the slow oxidation of HQ to BQ in air, as well as the formation of other quinhydrone species from various side reactions. This means some of the HQ is oxidised in solution before it can be consumed electrochemically, which inhibits the HQ electrochemical reaction and reduces gelation efficiency. This can be identified by the gelator/HQ solution turning a brown/red colour, indicating that the HQ has been oxidised to BQ or another quinhydrone species.
Here, we report the formation of large gels (around 3 cm3 in size) and discuss the effect of size and the time taken to grow the gels on their mechanical properties. In order to grow these electrochemically fabricated hydrogels to their maximum size, while simultaneously preventing the oxidation of HQ, we have used deposition times ranging from two to five hours under an inert nitrogen atmosphere. We also describe examples where syneresis and gel-to-crystal transitions occur during gelation and report the resulting effect of these phenomenon on the mechanical properties.
Results and discussion
We focus on four naphthalene-protected di- and tri-peptide gelators; 6Br2NapAV, 6Br2NapVF, 2NapAA and 2NapVVV (Fig. 2). 6Br2NapAV and 2NapAA have been reported previously by our group and were chosen as they exhibit unusual gelation behaviour in the bulk (specifically, syneresis and a gel-to-crystal transition, see below).32,35,42 Stock solutions of all gelators were prepared at a concentration of 5 mg mL−1 and adjusted to pH 8. To grow the hydrogels, 5 mg mL−1 of HQ was added to the gelator solutions immediately prior to gel deposition. Once the HQ had fully dissolved, the gelator/HQ solution was transferred to the dish containing the three-electrode set up within the desiccator. It is important to note that the procedure was performed in a desiccator as it could be fully degassed prior to running the experiment. In theory, any degassable container can be used. A constant current was then applied to the working electrode, resulting in gel formation at the electrode surface (Fig. 3). As current is proportional to production of protons at the electrode surface, increasing the current sees an increased rate of gel growth. When the current is applied the local pH drops due the oxidation of hydroquinone, generating protons that diffuse away from the electrode surface into solution. The growth of the low pH region surrounding the electrode surface is likely governed by proton diffusion, with the size of this low pH zone limited to how far the protons have diffused with time. The initial linear increase in gel area shows diffusion controls how large the gels grow with time in this stagnant system. After ∼100 minutes, the gel area can be seen to begin to plateau (Fig. 3d), with no further increase in gel area after 200 min, which indicates no further growth in this low pH zone.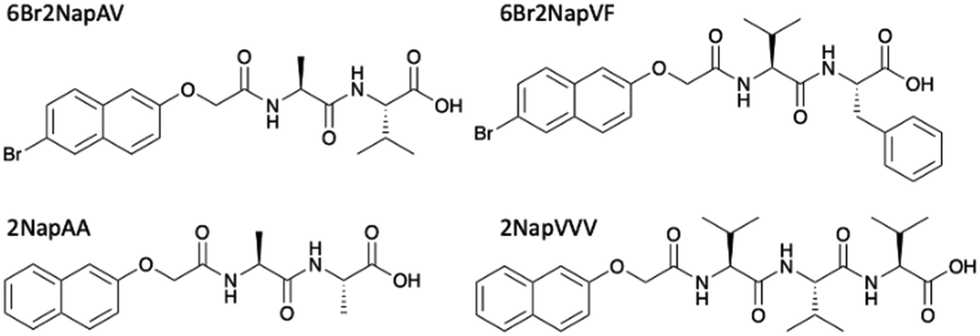 | ||
| Fig. 2 Chemical structure and names used throughout of gelator molecules; the gelation of 6Br2NapAV and 2NapAA have been described previously.32,35,42 | ||
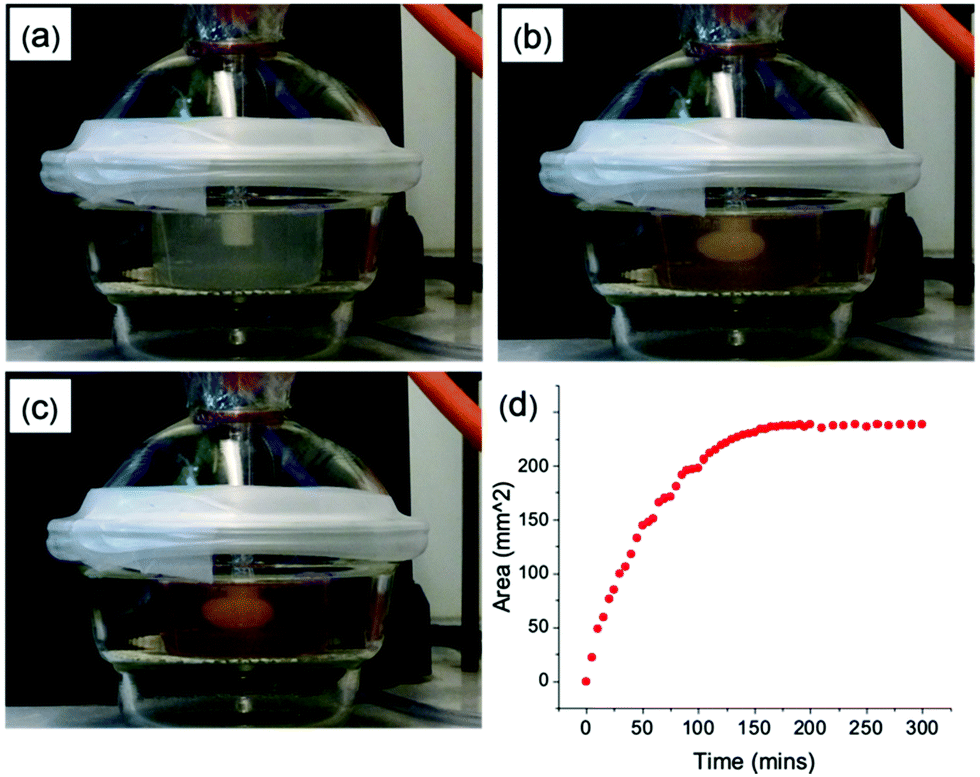 | ||
| Fig. 3 Series of photographs showing gel growth in real time (a) 0 hours (b) 2 hours (c) 5 hours (d) rate of gel growth with time. Initial conditions [pH] = 8, [2NapVVV] = 5 mg mL−1, [HQ] = 5 mg mL−1, [NaCl] = 0.1 M, [current] = 1 mA. | ||
It is reassuring that the slope of the linear portion of this graph is ∼2.5 × 10−4 cm2 s−1. Assuming a growing near hemispherical gel of cross-sectional area πr2/2 and 3-dimensional diffusional controlled growth for which r2 = 6Dt, this results in a sensible value of D ∼ 3 × 10−5 cm2 s−1. To determine the appropriate current value for the growing of each gel, a cyclic voltammogram (CV) (0.2 V s−1) was taken of the HQ/gelator solution. These can be found in the ESI† (Fig. S6a–d). To ensure a satisfactory rate of gel growth by maintaining good gelation kinetics, a current value from the CV was chosen to grow the gels using fast chronopotentiometry. Using this approach, a constant current of 2 mA was chosen for 6Br2NapAV, 6Br2NapVF and 2NapAA. For 2NapVVV, a current of 1 mA was applied. Potential time transients can be found in the ESI† (Fig. S7).
The hydrogels of 6Br2NapAV, 2NapAA and 2NapVVV all showed the same growth trend, linearly increasing in area with deposition time before plateauing in size (Fig. S4, ESI†). Unlike the other gelators, 6Br2NapVF does not form a transparent solution at 5 mg mL−1, meaning we were unable to monitor gel growth in situ with time. For the purposes of this study, we assume 6Br2NapVF follows the same trend as 6Br2NapAV and 2NapAA, plateauing after two hours as the same current value of 2 mA was used. In all cases, after five hours, gels of at least 3.35 cm3 were fabricated (volume of gels was calculated manually immediately after gel deposition).
The mechanical properties of the resulting gel structures were determined. Due to the irregular shape of the gels formed (see Fig. S9, ESI†), bulk rheology could not be used. Thus, cavitation rheology was used as a localized technique to probe the gels in their native environment.41 We have previously used cavitation rheology for a number of our gel systems,41 but not for examples grown on electrodes. Briefly, a syringe is lowered into the sample at a pre-defined depth. Air is then pumped through the syringe, forming a bubble within the sample. The critical pressure at which the bubble bursts can then be related to the elastic modulus of the gel network, with higher critical pressures indicating a stiffer network. As this technique is localised, multiple measurements can be taken from the same sample. This is achieved by lifting the syringe out of the sample and repeating the measurement in another area. In all cases, three measurements were collected from each sample. The error was then calculated to show the standard deviation between the measurements. The first series of measurements were collected from the gels when they reached their plateau sizes. For the gelators 6Br2NapAV, 6Br2NapVF and 2NapAA, this occurred after two hours. For 2NapVVV, three hours was needed to reach the plateau. The second measurements were then collected from gels grown for five hours. This was the case for all gelators. Fig. 4a shows that the mechanical properties of 6Br2NapVF and 2NapVVV gels were almost identical at their plateau time and after five hours, giving very similar critical pressures as well as displaying very small variation between measurements. This indicates that there is little to no difference in the structural and mechanical properties of the gels collected at these times. It also suggests that the network underpinning the plateau and five-hour gel is the same as well as being homogenous throughout the sample. However, 2NapAA and 6Br2NapAV show very different behaviour. In both cases, measurements taken from the five-hour samples show significantly higher critical pressure values when compared with the values collected at the plateau size. The three measurements collected from the five-hour gel also varied significantly, exhibiting much greater error within the sample. This indicates the gel network is inhomogeneous. These are interesting observations considering that when this electrochemical approach is used on macroelectrodes to form small volumes, no indication of unusual behaviour was observed.32 Despite both gelators showing the same trend, we believe this behaviour is due to two separate phenomena, unique to each gel network.
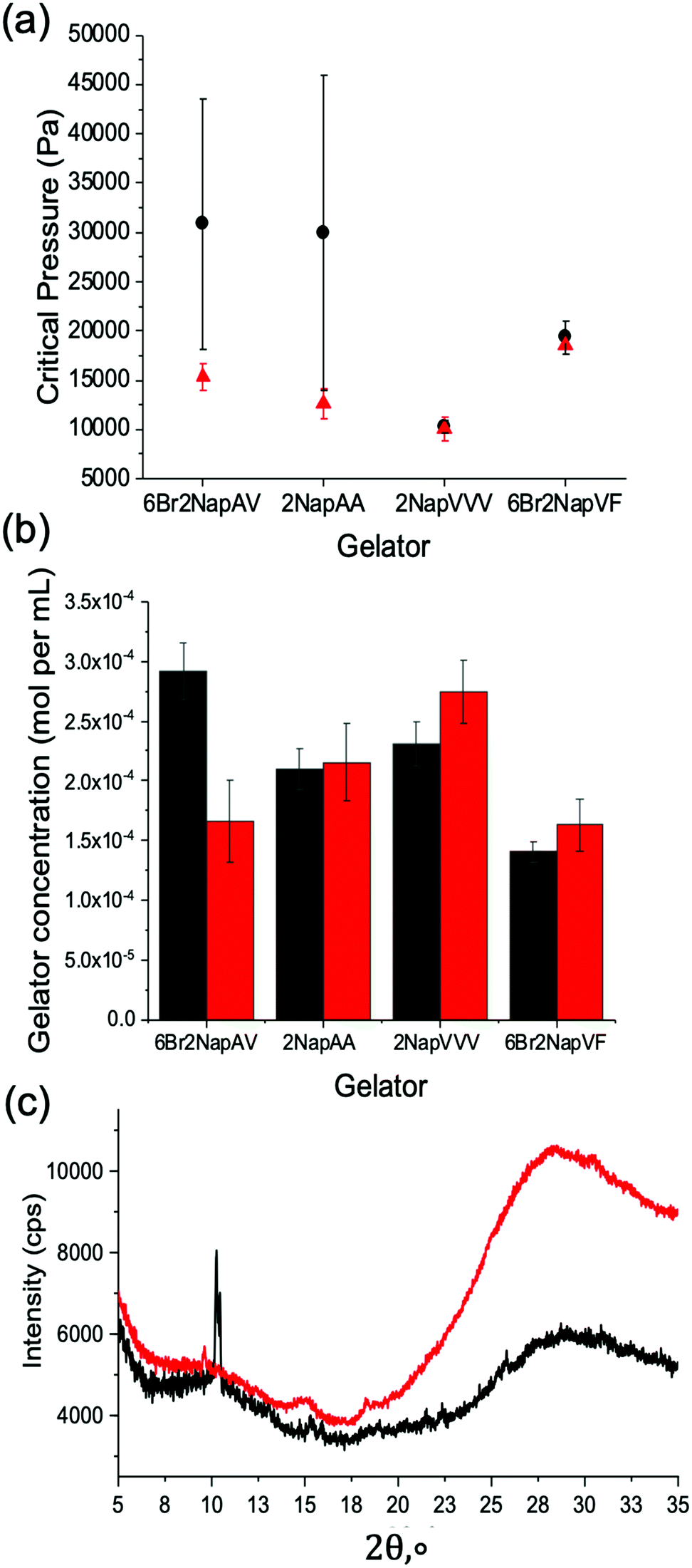 | ||
| Fig. 4 (a) Cavitation rheology measurements of electrochemically fabricated gels via HQ oxidation (b) relative gelator concentration measured by 1H NMR spectroscopy. (c) Powder X-ray diffraction patterns of 2NapAA gels grown for two hours and five hours. Measurements were taken from gels grown to their plateau size (red/triangle data) and gels grown for five hours (black/circle data). Measurements were performed in triplicate. Error bars show the standard deviation between measurements. | ||
Giuri et al.42 have recently shown that gels formed from 2NapAA exhibit unusual ageing properties, undergoing a gel-to-crystal transition when triggered by certain concentrations of glucono-δ-lactone (GdL). Their results show that the degree of crystallisation increases with decreasing pH, driven by the excess protonation of the carboxylic acid groups on the gel network. A full discussion as to the gel-to-crystal transition can be found elsewhere for this gelator.42 Whilst here we use a different method to lower the pH, we hypothesised that the higher critical pressure values and variability seen for 2NapAA were due to a gel-to-crystal transition. When the reaction was performed in the presence of bromophenol blue, the final gel was a uniform bright yellow, indicating that the material has a homogenous pH of <3. Giuri et al.42 report the gel-to-crystal transition occurs when the system drops below a pH of 4.1, placing our gels within this pH range.
To confirm this gel-to-crystal transition, powder X-ray diffraction (PXRD) measurements were performed on 2NapAA gels grown for two hours (i.e., at the point the gel reaches a plateau in volume) and after five hours. The PXRD data in Fig. 4c show that after two hours an amorphous material is formed, with some weak peaks suggesting a small amount of crystalline material. The peak positions correlate with our previous data for 2NapAA, showing that the crystals have the same structure when grown here using an electrochemical trigger as compared to in the bulk using GdL.42 In contrast to the two-hour sample, gels grown for five hours showed much sharper diffraction peaks, confirming substantially higher levels of crystalline material within the five-hour gel. As the degree of crystallisation increases with decreasing pH,42 this is to be expected. The application of current for longer times sees the prolonged production of protons at the electrode-solution interface, exposing the gel to a low pH environment for longer. As a result, the material has more time to undergo the gel-to-crystal transition. For the five-hour gels, these findings explain the high error and increased critical pressure values seen in the cavitation rheology data, with the increased level of crystalline material disrupting the homogenous gel network. The formation of crystals throughout the gel network would also result in substantially higher critical pressure values when compared to the weak supramolecular network.
For 6Br2NapAV, a different ageing phenomenon is responsible for the increased critical pressure values and greater variability observed in the cavitation rheology data. Previous reports have shown that 6Br2NapAV undergoes syneresis.43 Syneresis causes the gel network to contract and shrink with time.44 This is thought to be due to the removal of negative charge from the self-assembled structures, leading to hydrophobic interactions between the neutral fibres. The contraction of the gel network results in the expulsion of water, leading to a denser, more compact network. This behaviour can usually be monitored using bulk rheology. However, as above, due to the irregular shape of the gels formed using this technique, bulk rheology data could not be collected. The tendency for this material to behave in this way would explain the higher critical pressure values reported for the five-hour gels, as a denser, stiffer network would be present. As the gels grow, more gelator is needed to form the gels. After reaching a plateau in size, further changes may occur. Conceptually, this could be due to more gelator self-assembling within the pre-formed gel or by network stiffening as the pH changes, with no further gel formation. To understand this, we freeze-dried the gels at different times and determined the amount of the gelator present using quantitative 1H NMR data (Fig. 4b). The data show that for 2NapAA, 2NapVVV and 6Br2NapVF, the gelator concentration within the plateau and five-hour gels are very similar. The small difference is potentially due to the method used to select the plateau time. To track the rate of gel growth and determine the plateau time, a camera was used to capture an image every 30 seconds in front of the electrode. The outline of the gel in each image is then traced using ImageJ. This is used to determine the time the gel reaches a plateau in size.
For 6Br2NapAV, there is a notable difference in concentration between the two gels, with the five-hour gel exhibiting a notably higher concentration of gelator in comparison with its plateau size gel. As mentioned previously, 6Br2NapAV has been shown to undergo syneresis with time.43 This suggests that the gel network may be contracting and expelling water as it grows, causing it to shrink. As the gel does not visibly shrink in size, gel growth must continue in order to maintain its plateau size. This again shows that gel size is determined by proton diffusion, with the low pH region limited to a certain distance from the electrode surface. Consequently, this would account for the higher gelator concentration observed in the five-hour gel as the gel network becomes denser and more concentrated as it synereses, while simultaneously growing to maintain its plateau size.
An additional level of complexity was added by investigating multi-layered gels (Fig. 5a and b). As the gelators 6Br2NapVF and 2NapVVV showed no unusual ageing properties, they were chosen to grow the layers. As mentioned previously, gel area cannot be monitored in situ for 6Br2NapVF as the solution at 5 mg mL−1 is not transparent. This meant gel growth with time could not be monitored for the multi-layered gels. To achieve this multi-layered gel structure, a gel core of 6Br2NapVF was grown for one hour at a current of 2 mA. The electrode with the gel attached was then removed from the 6Br2NapVF solution, placed in a solution of 2NapVVV/HQ, and a current of 2 mA applied for a further hour. Two distinct gel layers were formed within the structure, with the 2NapVVV gel growing outwards from the surface of the pre-existing 6Br2NapAV gel. This was visualised by adding universal indicator to the 2NapVVV solution. It is important to note that universal indicator was not used when measuring the mechanical properties of the gel layers. A similar approach has been shown previously by Raeburn et al.,32 but with substantially smaller amounts of gel. Here, we form much larger multi-layered gels.
 | ||
| Fig. 5 (a) The electrochemical gelation of multi-layered di- and tri-peptide hydrogels via HQ oxidation (b) formation of multi-layered gel (i) 6Br2NapVF inner layer (ii) 2NapVVV outer layer. Universal indicator added to 2NapVVV solution to help visualise the layers. (iii) Multi-layered gel of 6Br2NapVF and 2NapVVV detached from electrode (iv) cross section of multi-layered gel (c) cavitation rheology measurements of the gel layers. Inner layer: 6Br2NapVF, outer layer: 2NapVVV (black/circle data). Inner layer: 2NapVVV, outer layer: 6Br2NapVF (red/triangle data). Measurements were performed in triplicate. Error bars show the standard deviation between measurements. | ||
As shown previously, there is a maximum distance over which protons can diffuse from the electrode surface, meaning there is a limit to how large these gels can grow. When the 6Br2NapVF gel is removed from its corresponding gelator solution and placed within the 2NapVVV solution, the acidic environment within the gel is retained. This is confirmed by the stability of the gel when placed in the 2NapVVV solution. It is possible that there is some gel dissolution around the outside of the gel upon placing it in the pH 8 2NapVVV solution.
However, turning on the current quickly re-establishes the acidic environment. Once the current is switched on protons can again diffuse away from the electrode surface, through the pre-existing gel and into the new gelator solution. This sees the formation of an outer gel layer which grows from the edge of the preformed 6Br2NapVF gel core. To probe the chemical composition of the multi-layered gel, 1H NMR spectra were generated from freeze-dried sections of both layers (Fig. 6).
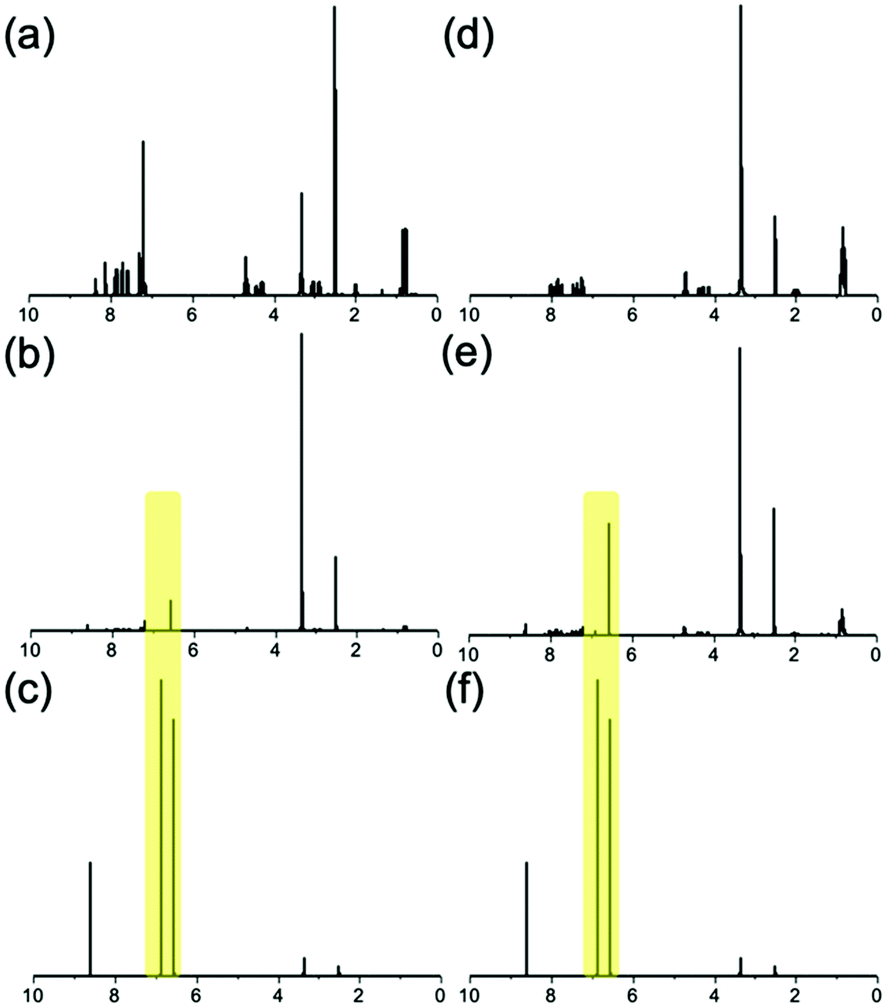 | ||
| Fig. 6 1H NMR spectra of (a) pure 6Br2NapVF (b) 6Br2NapVF gel layer (c) quinhydrone (d) pure 2NapVVV (e) 2NapVVV gel layer (f) quinhydrone. HQ and BQ peaks from fabrication process highlighted. | ||
The spectra confirmed that each layer consisted of only their respective gelator as well as HQ and BQ generated during the fabrication process. This shows there is a well-defined interface between the layers. We hypothesise this is due to the micellar aggregates of the second gelator solution being too large to diffuse into the gel core. However, due to their much smaller size, protons can diffuse through the gel network and into the second gelator solution, allowing gel growth to continue. This means the second gelator cannot augment the inner layer. The mechanical properties of the gel layers were again investigated using cavitation rheology. This was achieved by cutting a section from the gel to expose the layers, followed by lowering the needle into the target layer. Further explanation of this method can be found in the ESI† (Section 7.1). It is important to highlight that the multi-layered material must be cut for the mechanical properties to be measured. Therefore, we focus on the trends observed rather than absolute values.
Measurements taken from the 6Br2NapVF layer gave significantly higher critical pressure values than the 2NapVVV layer, producing a gel with two distinct regions of mechanical stiffness (Fig. 5c). For this material, the 6Br2NapVF core is mechanically stiffer than the outer region containing 2NapVVV. To show the mechanical and chemical properties of the gel layers are interchangeable, the fabrication process was repeated in reverse. To a solution of 2NapVVV, a current of 2 mA was applied for one hour, this time forming the core of the multi-layered gel. The 2NapVVV solution was then removed and replaced with 6Br2NapVF solution. Again, a current of 2 mA was applied for a further hour to fabricate the outer layer. The cavitation rheology data show the same trend, with higher critical pressure values collected from the 6Br2NapVF layer in comparison to the 2NapVVV layer. This highlights the rheological and chemical properties within the material can be finely tuned, simply by interchanging the gelator solution.
Conclusions
To conclude, we have shown that large hydrogels (of around 3–4 cm3) can be grown on electrode surfaces via the electrochemical oxidation of HQ. All hydrogels showed the same trend, linearly increasing in volume until they plateaued in size. This shows that maximum gel size is determined by proton diffusion. The mechanical properties of the plateau-sized gels and five-hour gels were investigated for all gelators. For the gelators 6Br2NapVF and 2NapVVV, the mechanical properties of the five-hour gels and the plateau-sized gels were almost identical. Therefore, once this maximum size is reached, the gel network and subsequent mechanical properties remain constant. For the gelators 2NapAA and 6Br2NapAV, the mechanical properties of the five-hour gels and the plateau-sized gels varied significantly. For the gelator 2NapAA, gel-to-crystal transitions occur within the gel when the sample is exposed to a pH below 4.1. As the five-hour sample is exposed to a low pH environment for longer, increased crystalline material builds up within the gel in comparison to the two-hour sample. This means the mechanical stiffness of the five-hour sample is increased but not homogenously, leading to large intra-sample variation. The same trend is also observed for the five-hour sample of 6Br2NapAV. However, a different aging phenomenon called syneresis is responsible. We have also shown that this approach can be extended to growing multi-layered gels with varying mechanical and chemical properties, allowing for the easy fabrication of single and multicomponent materials with excellent spatiotemporal control.Author contributions
Conceptualisation (DA, CP); data curation (DA, CW, CP, BD); formal analysis (DA, CP, CW, DA); funding acquisition (DA); investigation (all); methodology (all); project administration (DA); supervision (DA); writing (all).Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Leverhulme Trust for funding (RPG-2019-165).Notes and references
- E. M. Ahmed, J. Adv. Res., 2015, 6, 105–121 CrossRef CAS PubMed.
- K. Varaprasad, G. M. Raghavendra, T. Jayaramudu, M. M. Yallapu and R. Sadiku, Mater. Sci. Eng., 2017, 79, 958–971 CrossRef CAS PubMed.
- S.-k. Ahn, R. M. Kasi, S.-C. Kim, N. Sharma and Y. Zhou, Soft Matter, 2008, 4, 1151–1157 RSC.
- R. Parhi, Adv. Pharm. Bull., 2017, 7, 515–530 CrossRef CAS PubMed.
- R. Dong, Y. Pang, Y. Su and X. Zhu, Biomater. Sci., 2015, 3, 937–954 RSC.
- X. Du, J. Zhou, J. Shi and B. Xu, Chem. Rev., 2015, 115, 13165–13307 CrossRef CAS PubMed.
- P. Terech and R. G. Weiss, Chem. Rev., 1997, 97, 3133–3160 CrossRef CAS PubMed.
- L. A. Estroff and A. D. Hamilton, Chem. Rev., 2004, 104, 1201–1218 CrossRef CAS PubMed.
- R. Pérez-Pedroza, A. Ávila-Ramírez, Z. Khan, M. Moretti and C. A. E. Hauser, Adv. Polym. Technol., 2021, 2021, 8815006 CrossRef.
- S. Fleming and R. V. Ulijn, Chem. Soc. Rev., 2014, 43, 8150–8177 RSC.
- A. D. Martin and P. Thordarson, J. Mater. Chem. B, 2020, 8, 863–877 RSC.
- H. McEwen, E. Y. Du, J. P. Mata, P. Thordarson and A. D. Martin, J. Mater. Chem., 2017, 5, 9412–9417 RSC.
- T. N. Tikhonova, N. N. Rovnyagina, Z. A. Arnon, B. P. Yakimov, Y. M. Efremov, D. Cohen-Gerassi, M. Halperin-Sternfeld, N. V. Kosheleva, V. P. Drachev, A. A. Svistunov, P. S. Timashev, L. Adler-Abramovich and E. A. Shirshin, Angew. Chem., Int. Ed., 2021, 60, 25339–25345 CrossRef CAS PubMed.
- E. K. Johnson, D. J. Adams and P. J. Cameron, J. Mater. Chem., 2011, 21, 2024–2027 RSC.
- V. Jayawarna, M. Ali, T. A. Jowitt, A. F. Miller, A. Saiani, J. E. Gough and R. V. Ulijn, Adv. Mater., 2006, 18, 611–614 CrossRef CAS.
- A. Mahler, M. Reches, M. Rechter, S. Cohen and E. Gazit, Adv. Mater., 2006, 18, 1365–1370 CrossRef CAS.
- Z. Yang, G. Liang and B. Xu, Acc. Chem. Res., 2008, 41, 315–326 CrossRef CAS PubMed.
- R. Vegners, I. Shestakova, I. Kalvinsh, R. M. Ezzell and P. A. Janmey, J. Pept. Sci., 1995, 1, 371–378 CrossRef CAS PubMed.
- M. E. Roth-Konforti, M. Comune, M. Halperin-Sternfeld, I. Grigoriants, D. Shabat and L. Adler-Abramovich, Macromol. Rapid Commun., 2018, 39, 1800588 CrossRef PubMed.
- K. Sipa, K. Kowalewska, A. Leniart, A. Walcarius, G. Herzog, S. Skrzypek and L. Poltorak, Electrochem. Commun., 2021, 123, 106910 CrossRef CAS.
- J. Li, S. Wu, E. Kim, K. Yan, H. Liu, C. Liu, H. Dong, X. Qu, X. Shi, J. Shen, W. E. Bentley and G. F. Payne, Biofabrication, 2019, 11, 032002 CrossRef CAS PubMed.
- E. R. Cross, SN Appl. Sci., 2020, 2, 397 CrossRef.
- K. Ino, F. Ozawa, N. Dang, K. Hiramoto, S. Hino, R. Akasaka, Y. Nashimoto and H. Shiku, Adv. Biosyst., 2020, 4, 1900234 CrossRef PubMed.
- S. Ahadian, S. Yamada, J. Ramón-Azcón, M. Estili, X. Liang, K. Nakajima, H. Shiku, A. Khademhosseini and T. Matsue, Acta Biomater., 2016, 31, 134–143 CrossRef PubMed.
- Y. Koizumi, N. Shida, M. Ohira, H. Nishiyama, I. Tomita and S. Inagi, Nat. Commun., 2016, 7, 10404 CrossRef CAS PubMed.
- T. Chen, D. A. Small, L.-Q. Wu, G. W. Rubloff, R. Ghodssi, R. Vazquez-Duhalt, W. E. Bentley and G. F. Payne, Langmuir, 2003, 19, 9382–9386 CrossRef CAS.
- M. Lei, X. Qu, H. Liu, Y. Liu, S. Wang, S. Wu, W. E. Bentley, G. F. Payne and C. Liu, Adv. Funct. Mater., 2019, 29, 1900065 CrossRef.
- S. Wu, K. Yan, Y. Zhao, C.-C. Tsai, J. Shen, W. E. Bentley, Y. Chen, H. Deng, Y. Du, G. F. Payne and X. Shi, Adv. Funct. Mater., 2018, 28, 1803139 CrossRef.
- B. Alfaro-González, D. Ulate, R. Alvarado and O. Argüello-Miranda, Biomed. Phys. Eng. Express, 2018, 4, 035011 CrossRef.
- J. E. Bressner, B. Marelli, G. Qin, L. E. Klinker, Y. Zhang, D. L. Kaplan and F. G. Omenetto, J. Mater. Chem. B, 2014, 2, 4983–4987 RSC.
- F. Ozawa, K. Ino, T. Arai, J. Ramón-Azcón, Y. Takahashi, H. Shiku and T. Matsue, Lab Chip, 2013, 13, 3128–3135 RSC.
- J. Raeburn, B. Alston, J. Kroeger, T. O. McDonald, J. R. Howse, P. J. Cameron and D. J. Adams, Mater. Horiz., 2014, 1, 241–246 RSC.
- E. K. Johnson, L. Chen, P. S. Kubiak, S. F. McDonald, D. J. Adams and P. J. Cameron, Chem. Commun., 2013, 49, 8698–8700 RSC.
- P. S. Kubiak, S. Awhida, C. Hotchen, W. Deng, B. Alston, T. O. McDonald, D. J. Adams and P. J. Cameron, Chem. Commun., 2015, 51, 10427–10430 RSC.
- L. Chen, S. Revel, K. Morris, L. C. Serpell and D. J. Adams, Langmuir, 2010, 26, 13466–13471 CrossRef CAS PubMed.
- A. Ucar, E. González-Fernández, M. Staderini, N. Avlonitis, A. F. Murray, M. Bradley and A. R. Mount, Analyst, 2020, 145, 975–982 RSC.
- E. R. Cross and D. J. Adams, Soft Matter, 2019, 15, 1522–1528 RSC.
- A. Piper, Ben M. Alston, D. J. Adams and A. R. Mount, Faraday Discuss., 2018, 210, 201–217 RSC.
- Y. Liu, Y. Cheng, H.-C. Wu, E. Kim, R. V. Ulijn, G. W. Rubloff, W. E. Bentley and G. F. Payne, Langmuir, 2011, 27, 7380–7384 CrossRef CAS PubMed.
- Y. Liu, E. Kim, R. V. Ulijn, W. E. Bentley and G. F. Payne, Adv. Funct. Mater., 2011, 21, 1575–1580 CrossRef CAS.
- A. M. Fuentes-Caparrós, B. Dietrich, L. Thomson, C. Chauveau and D. J. Adams, Soft Matter, 2019, 15, 6340–6347 RSC.
- D. Giuri, L. J. Marshall, B. Dietrich, D. McDowall, L. Thomson, J. Y. Newton, C. Wilson, R. Schweins and D. J. Adams, Chem. Sci., 2021, 12, 9720–9725 RSC.
- K. McAulay, L. Thomson, L. Porcar, R. Schweins, N. Mahmoudi, D. J. Adams and E. R. Draper, Org. Mater., 2020, 02, 108–115 CrossRef CAS.
- A. M. Castilla, M. Wallace, L. L. E. Mears, E. R. Draper, J. Doutch, S. Rogers and D. J. Adams, Soft Matter, 2016, 12, 7848–7854 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sm01626a |
| This journal is © The Royal Society of Chemistry 2022 |