
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA pressure-jump study on the interaction of osmolytes and crowders with cubic monoolein structures†
Göran
Surmeier
,
Michael
Paulus
,
Eric
Schneider
,
Susanne
Dogan
 *,
Metin
Tolan
and
Julia
Nase
*,
Metin
Tolan
and
Julia
Nase
Fakultät Physik/DELTA, Technische Universität Dortmund, 44221 Dortmund, Germany. E-mail: susanne.dogan@tu-dortmund.de
First published on 11th January 2022
Abstract
Many vital processes that take place in biological cells involve remodeling of lipid membranes. These processes take place in a milieu that is packed with various solutes, ranging from ions and small organic osmolytes to proteins and other macromolecules, occupying about 30% of the available volume. In this work, we investigated how molecular crowding, simulated with the polymer polyethylene glycol (PEG), and the osmolytes urea and trimethylamine-N-oxide (TMAO) affect the equilibration of cubic monoolein structures after a phase transition from a lamellar state induced by an abrupt pressure reduction. In absence of additives, swollen cubic crystallites form after the transition, releasing excess water over several hours. This process is reflected in a decreasing lattice constant and was monitored with small angle X-ray scattering. We found that the osmotic pressure exerted by PEG and TMAO, which are displaced from narrow inter-bilayer spaces, accelerates the equilibration. When the radius of gyration of the added PEG was smaller than the radius of the water channels of the cubic phase, the effect became more pronounced with increasing molecular weight of the polymers. As the release of hydration water from the cubic structures is accompanied by an increasing membrane curvature and a reduction of the interface between lipids and aqueous phase, urea, which has a slight affinity to reside near membrane surfaces, stabilized the swollen crystallites and slowed down the equilibration dynamics. Our results support the view that cellular solutes are important contributors to dynamic membrane processes, as they can accelerate dehydration of inter-bilayer spaces and promote or counteract membrane curvature.
Introduction
Lipid membranes envelope biological cells and divide their interior into compartments and organelles.1,2 They do not only act as static barriers but undergo remodeling events like fusion and budding to regulate the mass transfer between the separated areas.3,4 Obviously, membrane composition and various proteins play an important role in such processes.5,6 It is also known that hydration and curvature of lipid bilayers can strongly be affected by non-specific interactions with solutes in the aqueous environment of membranes.7–10 The cellular environment contains high amounts of solutes, ranging from ions and organic osmolytes to macromolecules, like proteins, ribosomes, nucleic acids and carbohydrates, occupying about 30% of the available cell volume.11–13 The presence of a crowded environment shifts chemical equilibria and interaction constants due to the excluded volume effect.14–17 It was found that crowding can be a driving force of vesicle fusion or budding when background molecules are placed either outside or inside of vesicles.18–20It has long been assumed that organic osmolytes are “background molecules” with the sole purpose to provide the adaptation of a cell to osmotic stress in a hypertonic environment, and have no relevance to cellular processes acting as compatible solutes.21 In fact, their influence on cellular processes is small compared to other cellular solutes like e.g. inorganic ions, which do not interfere with the metabolism only in low concentration ranges.22 However, it became evident that the compatibility of organic osmolytes is restricted. The influence of different organic osmolytes on the structure and interaction of proteins has been a focal point of research in the last decade.23–25 While some osmolytes stabilize native states of macromolecules, others act as denaturants. A widely studied couple are trimethylamine-N-oxide (TMAO) and urea, which occur in a characteristic ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in many marine species that are exposed to high osmotic and hydrostatic pressures.26–30 The interplay of TMAO, which shifts the equilibrium of proteins towards folded states, and urea, which promotes unfolding, is crucial for preserving the functionality of proteins at extreme conditions. TMAO shows strong interactions with water that prevent the molecule from residing in proximity of macromolecules,31 while interactions of urea with protein backbones and amino acid side chains are favorable.32,33 Therefore, the presence of these molecules promotes protein structures with either small or large surfaces exposed to the solvent. It was found that the counteracting behavior of TMAO and urea also translates into different interactions with lipid structures and recent studies suggest that the preferential exclusion of molecules like TMAO from interbilayer spaces provides an important contribution to processes of membrane remodeling.7,8,34–36
:1 in many marine species that are exposed to high osmotic and hydrostatic pressures.26–30 The interplay of TMAO, which shifts the equilibrium of proteins towards folded states, and urea, which promotes unfolding, is crucial for preserving the functionality of proteins at extreme conditions. TMAO shows strong interactions with water that prevent the molecule from residing in proximity of macromolecules,31 while interactions of urea with protein backbones and amino acid side chains are favorable.32,33 Therefore, the presence of these molecules promotes protein structures with either small or large surfaces exposed to the solvent. It was found that the counteracting behavior of TMAO and urea also translates into different interactions with lipid structures and recent studies suggest that the preferential exclusion of molecules like TMAO from interbilayer spaces provides an important contribution to processes of membrane remodeling.7,8,34–36
In biological systems, membranes are complex structures built of a variety of different molecules and active fusion processes are driven by proteins. However, this study focusses on basic interactions of lipid layers with solutes and thus a simple model system is applied to identify fundamental effects of additives on hydration and curvature of membranes. Membrane remodeling like fusion and budding involves changes of curvature and the occurrence of non-lamellar lipid structures. Therefore, lipids that form, e.g., bicontinuous cubic or hexagonal phases are a powerful tool to study membrane processes.37–41 The lipid monoolein forms bicontinuous cubic phases spontaneously in an aqueous medium42,43 and is a widely studied molecule due to its potential in fields like drug delivery,44–47 nucleic acid delivery,48–52 medical imaging,46,53,54 membrane protein crystallization,55–59 and biosensing.60–63 For monoolein/water mixtures, detailed temperature composition phase diagrams exist43 and also the pressure-dependent behavior was investigated before.37,64 At ambient pressure and a temperature of 25 °C, monoolein forms the cubic Pn3m phase in excess water. By the application of hydrostatic pressure, a transition into a lamellar crystalline phase can be triggered.
In this work, we examined the influence of background molecules on membrane processes by studying how crowders and the osmolytes TMAO and urea affect curvature and hydration of cubic monoolein structures. We conducted pressure-jumps across the lamellar to cubic phase boundary at excess water conditions to trigger the formation of a swollen cubic phase. Subsequently, we monitored the diffusion of water from hydration to excess volume by recording the decrease of the cubic lattice constant as a function of time with small angle X-ray scattering (SAXS). Molecular crowding was simulated by adding the polymer polyethylene glycol (PEG) as it was already done in previous studies.14,23,65,66 We found that the addition of PEG and TMAO reduces the lattice constant of the cubic phase and tends to accelerate the equilibration after a pressure-jump. When the radius of gyration of PEG was smaller than the radius of the water channels of the lipid phase, both effects became more pronounced with increasing molecular weight of the polymers. In contrast, urea slowed the decrease of the lattice constant down, stabilizing lipid structures with large surface areas.
Experimental setup
The pressure-jumps were conducted with a high hydrostatic pressure sample cell designed for X-ray studies.67 The cell is made of the high tensile strength alloy Inconel 718 and is sealed with diamond windows along the beam path, resisting pressures of up to 5 kbar. The sample liquid is filled into an inner cell that is separated from the pressure transmitting liquid by polyimide windows. The sample temperature was controlled by a circulating water flow and set to 298 K.Monoolein and buffer solution were poured into the inner sample cell. The substances were mixed mechanically, and the samples were allowed to equilibrate for at least 12 hours. For PEG samples, 10 mM pressure stable BisTris buffer at pH 5 was used and added in concentrations of 80 wt%. PEG of molecular weights between 200 g mol−1 and 35000 g mol−1 was dissolved in the buffer solution in concentrations of 150 g L−1 before it was mixed with monoolein. For urea and TMAO samples, 10 mM BisTris buffer at pH 7 was used and added in concentrations of 90 wt%. The osmolytes were dissolved in the buffer solution in concentrations of 1 M. For mixtures of urea and TMAO in a 2:1 ratio, 0.67 M urea and 0.33 M TMAO were added. All chemicals were purchased from Sigma Aldrich and used as received.
The SAXS experiments were performed at the beamlines BL268 and BL969 of DELTA (Dortmund, Germany) with photon energies of 12 keV (λ = 1.033 Å) and 13 keV (λ = 0.954 Å) using MAR345 image plate detectors. The beamsize was 0.5 × 0.5 mm2 at BL2 and 1 × 1 mm2 at BL9. The setups were calibrated with silver behenate.70 Pressure-jumps were conducted from between 1 and 2 kbar down to 50 bar. The reduction of pressure took approximately 10 s. After the pressure-jump, SAXS scans were taken at four-minute intervals for several hours. In the cubic phase regime, the exposure time was 150 s and in the lamellar regime, the exposure time was 30 s. The two-dimensional SAXS patterns were integrated azimuthally to obtain the scattering intensity as a function of the wave vector transfer q and an interpolated background was subtracted. Phase behavior and lattice constant a of the sample can then be determined based on Bragg reflections that occur at characteristic ratios of the reciprocal lattice constant 2π/a.42 The positions of the Bragg reflections were extracted by fitting Gaussian functions.
Results
Fig. 1 shows the pressure-dependent phase behavior of monoolein at excess water conditions upon pressurization and pressure release (left). Moreover, selected integrated SAXS patterns are shown with the obtained fits (right). The Bragg reflections are labelled with the corresponding Miller indices. Exemplary SAXS curves and fits of samples containing additives are presented in the ESI.† In agreement with the results of Czeslik et al.,71 we observed the cubic Pn3m phase at 50 bar and a transition into a lamellar phase at high pressures. The inset provides a more detailed view on the behavior of the cubic phase at increasing pressures. Repetitive measurements at four-minute intervals showed that the Pn3m phase is not at equilibrium at pressures above 0.5 kbar, as the cubic structures are slowly swelling. Additional measurement series (data not shown) showed that the swelling carries on for many hours or even days at constant pressure until a phase transition into a lamellar phase occurs. Upon pressure release from 4 kbar, a lamellar phase is observed in the corresponding pressure regime, confirming that the cubic phase is not the equilibrium state. However, an instantaneous collapse of the metastable cubic phase could be reliably induced by increasing the pressure above 2.5 kbar. | ||
| Fig. 1 Left: Phase behavior and lattice parameters a of monoolein in excess water upon pressurization (red arrows) and pressure release (green arrows). The inset provides a closer look on the lattice constant of the Pn3m phase during pressurization, revealing that it increases as a function of time even at constant pressure. The scans were taken at four-minute intervals and the data points are connected to indicate their chronological order. Right: Exemplary integrated SAXS data (colored dots) and fits (black lines) of scans from each phase regime. The Miller indices of the lattice planes that correspond to the observed Bragg reflections are designated. | ||
After the pressure-induced phase transition, a lamellar phase with a spacing of approximately 43 Å formed. During the subsequent release of pressure, a phase transition occurred at 1.75 kbar, as the spacing of the lamellar structures abruptly increased to approximately 49 Å. Since the spacing of both phases barely changed as a function of pressure, it is likely that they exhibit a crystalline chain order. Czeslik et al.71 observed an intermediate phase with a spacing of 43 Å at 7.5 °C upon a pressure-induced cubic Pn3m to lamellar transition at 1.4 kbar, before a stable lamellar phase with a spacing of 47 Å formed. They confirmed the crystalline character of the latter phase with wide angle scattering. Qiu and Caffrey43 found a metastable and a stable lamellar crystalline phase at low temperatures with similar spacings. Consistent with the behavior observed in these studies, we found that the phase with the higher spacing is the equilibrium state in the entire pressure range from 0.5 to 4 kbar, while the phase with the lower spacing is metastable. In the following, we refer to the stable phase as Lc and to the metastable phase as Lm phase. The moment and pressure at which a transition into the stable phase occurred was found to be largely random. However, a reduction of the pressure from more than 2.5 kbar to 1.5 kbar or less reliably induced a transition into the stable Lc phase.
Upon pressure release, the phase transition into the cubic phase occurred between 0.5 and 0.25 kbar. At this point, the cubic phase formed with an increased lattice constant compared to equilibrium. Subsequently, the lattice constant slowly decreased as the cubic structures release excess water.
Based on the described behavior, we applied the following measurement protocol. For each sample, we determined the equilibrium lattice constant of the cubic phase at 50 bar. Then, we increased the pressure to 3.5 kbar to induce a transition into the lamellar phase. After that, we reduced the pressure again, typically to 1 or 1.5 kbar, to trigger the formation of the Lc phase. From this starting point, we performed a pressure-jump back to 50 bar and monitored the decrease of the Pn3m lattice constant as a function of time.
The equilibrium lattice constant of monoolein in the Pn3m phase at 50 bar upon addition of PEG with different molecular weights is shown in the inset of Fig. 2. The measurement error of the SAXS experiments is below the symbol sizes. The variation of the lattice parameters between the individual samples (circles) is due to the sensitivity of the monoolein samples to external conditions and slow equilibration times. The typical error range for the cubic phase is ±2.5 Å and for the lamellar ±0.3 Å.
 | ||
| Fig. 2 Decrease of the Pn3m lattice constant after a pressure-induced lamellar-to-cubic transition in presence of PEG of different molecular weight as a function of time t (colored symbols) and fits (grey lines). The pressure-jumps were conducted at t = 0. The inset shows the Pn3m lattice constant at 50 bar and the lamellar spacing at 1500 bar before the pressure-jumps as a function of the radius of gyration RG of the added PEG. The results for pure monoolein are shown at RG = 0 for comparison. The circles represent the obtained values of different samples to illustrate the variance. The crosses mark the mean values. | ||
If the water content of the lipid phases is known, the length of the lipid molecules in the structure and the size of the water channels can be determined.42 For monoolein/water mixtures at 25 °C and ambient pressure, the maximum water capacity of the Pn3m phase at equilibrium is 44.5 wt%.43 Based on this value and with the observed mean Pn3m lattice constant of monoolein in excess water at 50 bar of 102.8 Å, the length of the lipid molecules is determined as approximately 16.1 Å. Assuming that the length of the molecules depends essentially on pressure and temperature, but is barely affected by the addition of PEG, this value can be used to estimate the radii of the water channels rw of the cubic structures in presence of PEG. Fig. 3 shows the ratio of rw to the radius of gyration RG of the added PEG as a function of RG. With increasing size of the polymers, the cubic structures are more and more compressed. It is known that the osmotic pressure exerted by PEG decreases as a function of its molecular weight at constant mass concentration.72 However, the osmotic pressure between the water channels of the cubic phase and the excess water regime is determined by the concentration gradient between both regions. Fig. 3 demonstrates that the dimensions of water channels and polymers are in a similar order of magnitude. Therefore, the increasing level of compression with increasing size of the polymers can be explained by an increasing exclusion of PEG from the cubic structures. The larger the PEG molecules are, the more unfavorable the confinement in the cubic structure becomes due to the restriction of the configuration entropy. Once the radius of gyration was larger than the radius of the water channels, we observed the cubic Ia3d phase instead of the Pn3m phase. In the Ia3d regime, no further compression of the lipid phases as a function of RG occurred, and it can be assumed that the polymers are completely displaced from the water channels.
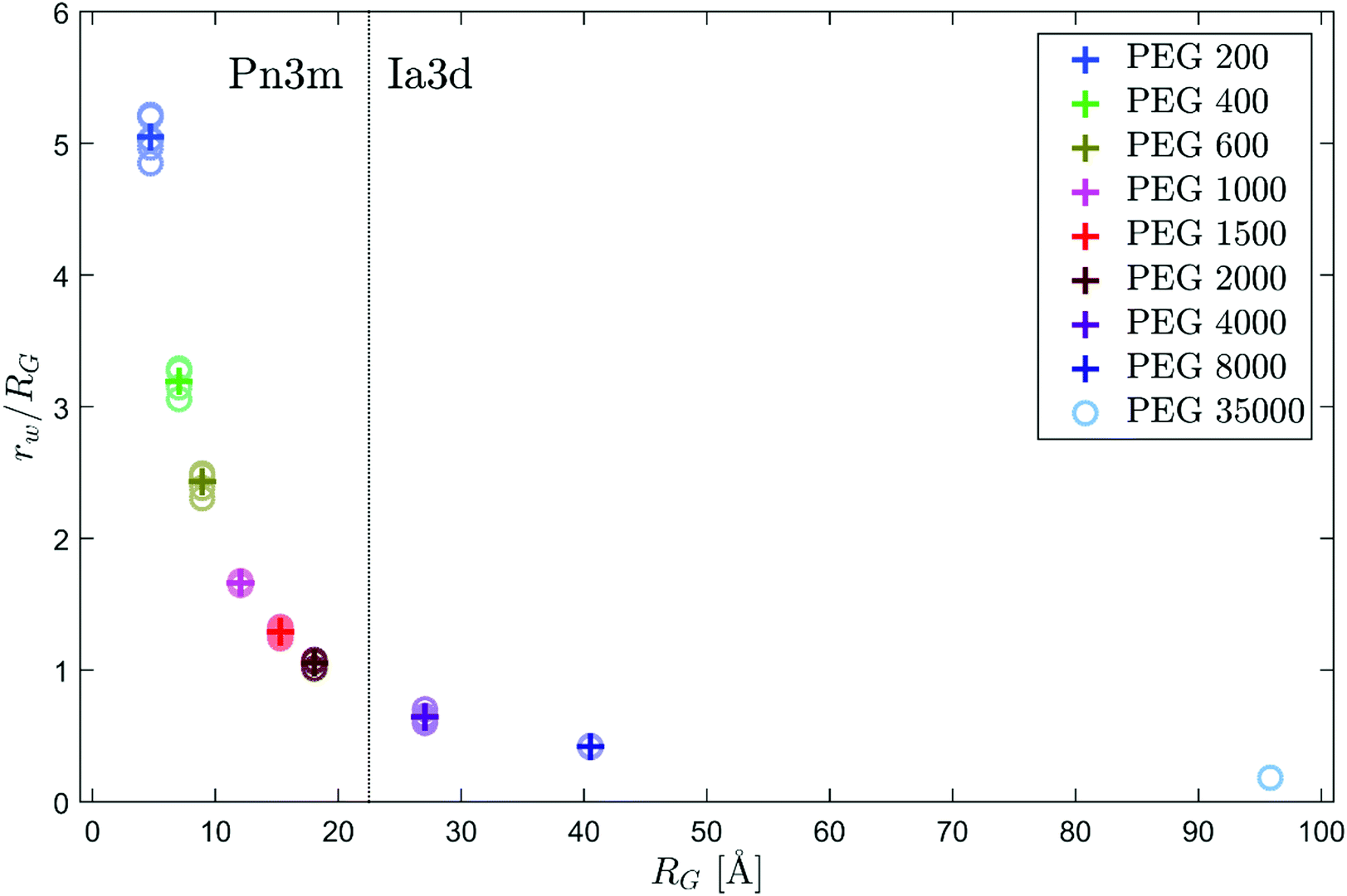 | ||
| Fig. 3 Ratio of the water channel radius rw of the Pn3m phase at 50 bar to the radius of gyration RG of the added PEG as a function of RG. The circles represent the obtained values of different samples to illustrate the variance. The crosses mark the mean values. | ||
The inset of Fig. 2 shows that the spacing of the lamellar crystalline phase at 1500 bar is largely unaffected by the presence of PEG. Qiu and Caffrey found that the maximum water capacity of the lamellar phase at ambient pressure is approximately 4 wt%.43 Based on this value, the hydrated inter-bilayer space can be estimated to be approximately 2 Å thick. Thus, all water molecules that are embedded in the lipid structure are in close proximity to the monoolein headgroups and therefore have limited mobility.73 Due to the lack of free hydration water, a displacement of water molecules from the lipid structures by osmotic pressure is barely possible. Therefore, the Lc phase is, in contrast to the Pn3m phase, not compressed by PEG.
Especially with PEG of higher molecular weights, we observed a formation of cubic structures already at 1000 bar in some cases. Therefore, different starting pressures between 1 and 2 kbar were used for the pressure-jumps. Fig. 2 shows the reduction of the lattice constant of the Pn3m phase of monoolein in presence of PEG of different molecular weights after pressure-jumps from the lamellar regime as a function of time. The integrated intensity of the observed Bragg reflections was constant from the second scan on for all samples (see ESI†), indicating that the formation of the Pn3m phase was completed in less than four minutes and that the crystallites did not grow considerably during the water release process. Moreover, the width of the reflections at equilibrium was similar for all samples so that it can be assumed that the mean size of the crystallites was largely unaffected by the addition of the polymers.
The equilibration after a pressure-jump accelerated with increasing molecular weight of the added PEG. To compare the speed of the process for different samples, we determined the equilibration times as the time that elapsed after the pressure-jump until the released water volume per second and unit cell fell permanently below a threshold of 30 Å3 s−1. For this purpose, a smoothed and continuous representation of the data was generated by fitting. We superposed two exponential functions to reproduce the convex decrease of the lattice parameter and a hyperbolic tangent to take into account an intermediate increase of the reduction rate that occurred mainly with pure monoolein. The resulting curves are shown in Fig. 2 as grey lines. With the length of the lipid molecules stated above, the water content of the unit cells was determined and by calculating the time derivative, the water release rate was obtained.
Fig. 4 shows the resulting equilibration times as a function of the radius of gyration of the added PEG. The osmotic pressure exerted by the polymers accelerated the equilibration process strongly. For pure monoolein, a water release rate of the Pn3m unit cells of 30 Å3 s−1 is reached after approximately three hours on average. This value reduces to less than 20 minutes in presence of PEG 2000. While the compression of the lipid structures at equilibrium increases steadily with the size of the polymers, the equilibration time reduces strongly already for PEG 400 and remains on a similar level for larger PEGs showing only a slight downward trend.
 | ||
| Fig. 4 Period t after a pressure-jump until the released water volume per time and Pn3m unit cell decreased below a limit of 30 Å3 s−1 as a function of the radius of gyration RG of the added PEG as a measure for the speed of the equilibration dynamics. The results for pure monoolein are shown at RG = 0 for comparison. The circles represent the obtained values of different samples to illustrate the variance. The crosses mark the mean values. | ||
In contrast to PEG, urea and TMAO are small molecules with few configurational degrees of freedom. Nevertheless, they also have a strong influence on monoolein structures due to their interactions with water and lipid interfaces. The inset of Fig. 5 shows the lattice constant of the Pn3m phase at 50 bar and the spacing of the Lc phase at 1000 bar in presence of urea and TMAO before a pressure-jump. In agreement with current knowledge, urea increases the lattice constant as its favorable interactions with the lipid headgroups counteract the negative curvature of the bilayers, whereas TMAO is displaced from the cubic crystallites and compresses the structure due to the resulting osmotic pressure.7,9,58 The values observed for urea are presumably higher than in equilibrium, as the lattice constant converges to lower values after a pressure-jump. Urea strongly slows down the equilibration dynamics of the cubic phase, so that the waiting time of 12 h between sample preparation and experiment might not be sufficient. From the behavior of the urea samples after a pressure-jump, an equilibrium lattice constant of approximately 110 Å is determined. In a 2:1 ratio, the effects of urea and TMAO on the Pn3m lattice constant compensate each other and within the range of variance, no difference to pure monoolein is observed. Similar to PEG, TMAO does not affect the structure of the Lc phase, while urea slightly increases the spacing. An even less pronounced expansion is visible when urea and TMAO are added in a 2:1 ratio. In both cases, the expansion is presumably due to urea molecules that enter the narrow interbilayer spaces of the Lc phase, since urea has an affinity to reside at lowly hydrated membrane interfaces.35,74
 | ||
| Fig. 5 Decrease of the Pn3m lattice constant after a pressure-induced lamellar-to-cubic transition in presence of TMAO and urea as a function of time t (colored symbols) and fits (grey lines). The inset shows the Pn3m lattice constant at 50 bar and the lamellar spacing at 1000 bar before the pressure-jumps. The results for pure monoolein are shown at RG = 0 for comparison. The circles represent the obtained values of different samples to illustrate the variance. The crosses mark the mean values. At 50 bar, for urea, the value of the equilibrium lattice constant determined from the convergence after a pressure-jump is indicated with an 'X' instead. | ||
Fig. 5 shows how the addition of urea and TMAO affected the equilibration dynamics of the Pn3m phase after a pressure-jump from the lamellar regime. In all cases, the increase of the integrated intensity of the observed Bragg reflections occurred on a much shorter time scale than the reduction of the lattice constant (see ESI†) and the width of the reflections at equilibrium was similar. Thus, it can be assumed that the cubic crystallites formed fast and reached similar dimensions for all samples.
The different interaction mechanisms of the two osmolytes with lipid membranes induced counteracting effects on the water release process. In absence of additives, the equilibration process took five hours on average. Urea strongly reduced the speed of the water-release and after more than 10 hours, the lattice constant was still considerably decreasing. In contrast, for three of four TMAO samples, the reduction of the lattice parameter essentially occurred during the first hour after the pressure-jump. In all cases, TMAO caused an acceleration of the dynamics. The addition of urea and TMAO in a 2:1 ratio caused equilibration times that are similar to pure monoolein. However, due to the large fluctuations between the individual samples, it is not possible to conclude whether this ratio provides an optimum compensation of the counteracting effects.
Conclusions
In this work, we investigated the influence of solutes on the equilibration dynamics of cubic monoolein phases. The effect of the size of background molecules on the dehydration of the lipid structures was examined using PEG of different molecular weight. For polymers with a radius of gyration that was smaller than the radius of the Pn3m water channels, a linear reduction of the lattice parameter with increasing size was found. Larger PEG induced a phase transition into the Ia3d phase. While PEG 200 had only a minor effect on the equilibration dynamics after a pressure-jump, larger polymers strongly accelerated the release of hydration water from swollen cubic crystallites. A faster decrease of the lattice constant was also observed when TMAO was added. In both cases, a preferred displacement of the solutes from the lipid structures induces an osmotic pressure between hydration and excess water regime. Residing in the confined hydration volume of the cubic crystallites is unfavorable for PEG due to the restriction of the configuration entropy that increases with the size of the polymers. In contrast, TMAO is a small and rigid molecule that is displaced from membrane interfaces due to its dipole moment and strong interactions with water.7 The dehydration of inter-bilayer spaces is a prerequisite for membrane fusion. Therefore, the observed ability of PEG and TMAO to accelerate the dehydration of lipid structure indicates that the presence of both substances promotes fusion.Urea has an affinity to interact with bilayer surfaces.32,33 In this work, we demonstrated that it stabilizes swollen Pn3m structures and reduces the speed of the equilibration dynamics after pressure-jumps across the lamellar-to-cubic phase boundary. Intermediate states of membrane fusion are characterized by high negative curvature. Our experiments show that urea counteracts the formation of such structures. In a 2:1 ratio, the effects of urea and TMAO compensated each other to a large extend.
Our results support the view that cellular solutes contribute to membrane processes that involve changes of the curvature of lipid layers or require dehydration of inter-bilayer spaces.7,8 This work shows that background molecules can either increase or reduce barriers for membrane remodeling.
Author contributions
G. S., J. N. and M. P. designed research. G. S., E. S., S. D. and M. P. performed research. G. S. analyzed data. G. S. and S. D. wrote the paper with contributions from the co-authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the DELTA machine group for providing synchrotron radiation and the Deutsche Forschungsgemeinschaft (FOR 1979) for financial support. This work was supported by RESOLV, funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – EXC 2033 – 390677874 – RESOLV.References
- D. Casares, P. V. Escribá and C. A. Rosselló, Membrane lipid composition: effect on membrane and organelle structure, function and compartmentalization and therapeutic avenues, Int. J. Mol. Sci., 2019, 20, 2167 CrossRef CAS PubMed.
- S. Cohen, A. M. Valm and J. Lippincott-Schwartz, Interacting organelles, Curr. Opin. Cell Biol., 2018, 53, 84–91 CrossRef CAS PubMed.
- T. Farmer, N. Naslavsky and S. Caplan, Tying trafficking to fusion and fission at the mighty mitochondria, Traffic, 2018, 19, 569–577 CrossRef CAS PubMed.
- V. Haucke and M. M. Kozlov, Membrane remodeling in clathrin-mediated endocytosis, J. Cell Sci., 2018, 131, jcs216812 CrossRef PubMed.
- S. Halldorsson, S. Li, M. Li, K. Harlos, T. A. Bowden and J. T. Huiskonen, Shielding and activation of a viral membrane fusion protein, Nat. Commun., 2018, 9, 1–9 CrossRef CAS PubMed.
- R. B. Lira, T. Robinson, R. Dimova and K. A. Riske, Highly efficient protein-free membrane fusion: a giant vesicle study, Biophys. J., 2019, 116, 79–91 CrossRef CAS PubMed.
- S. Sukenik, S. Dunsky, A. Barnoy, I. Shumilin and D. Harries, TMAO mediates effective attraction between lipid membranes by partitioning unevenly between bulk and lipid domains, Phys. Chem. Chem. Phys., 2017, 19, 29862–29871 RSC.
- M. Manisegaran, S. Bornemann, I. Kiesel and R. Winter, Effects of the deep-sea osmolyte TMAO on the temperature and pressure dependent structure and phase behavior of lipid membranes, Phys. Chem. Chem. Phys., 2019, 21, 18533–18540 RSC.
- H. Takahashi, A. Matsuo and I. Hatta, Effects of chaotropic and kosmotropic solutes on the structure of lipid cubic phase: Monoolein-water systems, Mol. Cryst. Liq. Cryst. Sci. Technol., Sect. A, 2000, 347, 231–238 CrossRef CAS.
- S. R. Al-Ayoubi, P. K. Schinkel, M. Berghaus, M. Herzog and R. Winter, Combined effects of osmotic and hydrostatic pressure on multilamellar lipid membranes in the presence of PEG and trehalose, Soft Matter, 2018, 14, 8792–8802 RSC.
- S. B. Zimmerman and S. O. Trach, Estimation of macromolecule concentrations and excluded volume effects for the cytoplasm of Escherichia coli, J. Mol. Biol., 1991, 222, 599–620 CrossRef CAS PubMed.
- R. J. Ellis and A. P. Minton, Join the crowd, Nature, 2003, 425, 27–28 CrossRef CAS PubMed.
- A. B. Fulton, How crowded is the cytoplasm?, Cell, 1982, 30, 345–347 CrossRef CAS PubMed.
- K. Julius, J. Weine, M. Gao, J. Latarius, M. Elbers, M. Paulus, M. Tolan and R. Winter, Impact of Macromolecular Crowding and Compression on Protein–Protein Interactions and Liquid–Liquid Phase Separation Phenomena, Macromolecules, 2019, 52, 1772–1784 CrossRef CAS.
- H.-X. Zhou, G. Rivas and A. P. Minton, Macromolecular crowding and confinement: biochemical, biophysical, and potential physiological consequences, Annu. Rev. Biophys., 2008, 37, 375–397 CrossRef CAS PubMed.
- A. P. Minton, The influence of macromolecular crowding and macromolecular confinement on biochemical reactions in physiological media, J. Biol. Chem., 2001, 276, 10577–10580 CrossRef CAS PubMed.
- P. H. Schummel, M. Gao and R. Winter, Modulation of the Polymerization Kinetics of α/β-Tubulin by Osmolytes and Macromolecular Crowding, ChemPhysChem, 2017, 18, 189–197 CrossRef CAS PubMed.
- H. Terasawa, K. Nishimura, H. Suzuki, T. Matsuura and T. Yomo, Coupling of the fusion and budding of giant phospholipid vesicles containing macromolecules, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 5942–5947 CrossRef CAS PubMed.
- B. R. Lentz, PEG as a tool to gain insight into membrane fusion, Eur. Biophys. J., 2007, 36, 315–326 CrossRef CAS PubMed.
- S. Hui, T. Kuhl, Y. Guo and J. Israelachvili, Use of poly (ethylene glycol) to control cell aggregation and fusion, Colloids Surf., B, 1999, 14, 213–222 CrossRef CAS.
- P. H. Yancey, Organic osmolytes as compatible, metabolic and counteracting cytoprotectants in high osmolarity and other stresses, J. Exp. Biol., 2005, 208, 2819–2830 CrossRef CAS PubMed.
- P. H. Yancey, Water stress, osmolytes and proteins, Am. Zool., 2001, 41, 699–709 CAS.
- K. Julius, S. R. Al-Ayoubi, M. Paulus, M. Tolan and R. Winter, The effects of osmolytes and crowding on the pressure-induced dissociation and inactivation of dimeric LADH, Phys. Chem. Chem. Phys., 2018, 20, 7093–7104 RSC.
- N. Smolin, V. P. Voloshin, A. V. Anikeenko, A. Geiger, R. Winter and N. N. Medvedev, TMAO and urea in the hydration shell of the protein SNase, Phys. Chem. Chem. Phys., 2017, 19, 6345–6357 RSC.
- A. Rani and P. Venkatesu, Changing relations between proteins and osmolytes: a choice of nature, Phys. Chem. Chem. Phys., 2018, 20, 20315–20333 RSC.
- V. Voloshin, N. Smolin, A. Geiger, R. Winter and N. N. Medvedev, Dynamics of TMAO and urea in the hydration shell of the protein SNase, Phys. Chem. Chem. Phys., 2019, 21, 19469–19479 RSC.
- P. Ganguly, J. Polák, N. F. A. van der Vegt, J. Heyda and J.-E. Shea, Protein Stability in TMAO and Mixed Urea–TMAO Solutions, J. Phys. Chem. B, 2020, 124, 6181–6197 CrossRef CAS PubMed.
- D. R. Canchi and A. E. García, Cosolvent effects on protein stability, Annu. Rev. Phys. Chem., 2013, 64, 273–293 CrossRef CAS PubMed.
- J. R. Treberg, B. Speers-Roesch, P. M. Piermarini, Y. K. Ip, J. S. Ballantyne and W. R. Driedzic, The accumulation of methylamine counteracting solutes in elasmobranchs with differing levels of urea: a comparison of marine and freshwater species, J. Exp. Biol., 2006, 209, 860–870 CrossRef CAS PubMed.
- T.-Y. Lin and S. N. Timasheff, Why do some organisms use a urea-methylamine mixture as osmolyte? Thermodynamic compensation of urea and trimethylamine N-oxide interactions with protein, Biochemistry, 1994, 33, 12695–12701 CrossRef CAS PubMed.
- D. Bolen and I. V. Baskakov, The osmophobic effect: natural selection of a thermodynamic force in protein folding, J. Mol. Biol., 2001, 310, 955–963 CrossRef CAS PubMed.
- M. C. Stumpe and H. Grubmüller, Interaction of urea with amino acids: implications for urea-induced protein denaturation, J. Am. Chem. Soc., 2007, 129, 16126–16131 CrossRef CAS PubMed.
- D. R. Canchi and A. E. García, Backbone and side-chain contributions in protein denaturation by urea, Biophys. J., 2011, 100, 1526–1533 CrossRef CAS PubMed.
- J. Valerio, S. Bernstorff and S. Funari, Effect of urea and tmao on lipid bilayers, Eur. Pharm. J., 2017, 64, 24–27 CrossRef CAS.
- Q. D. Pham, A. Wolde-Kidan, A. Gupta, A. Schlaich, E. Schneck, R. R. Netz and E. Sparr, Effects of Urea and TMAO on Lipid Self-Assembly under Osmotic Stress Conditions, J. Phys. Chem. B, 2018, 122, 6471–6482 CrossRef CAS PubMed.
- J. A. Mondal, Effect of trimethylamine N-oxide on interfacial electrostatics at phospholipid monolayer–water interfaces and its relevance to cardiovascular disease, J. Phys. Chem. Lett., 2016, 7, 1704–1708 CrossRef CAS PubMed.
- A. Levin, C. Jeworrek, R. Winter, K. Weise and C. Czeslik, Lipid Phase Control and Secondary Structure of Viral Fusion Peptides Anchored in Monoolein Membranes, J. Phys. Chem. B, 2017, 121, 8492–8502 CrossRef CAS PubMed.
- D. P. Siegel, Fourth-Order Curvature Energy Model for the Stability of Bicontinuous Inverted Cubic Phases in Amphiphile- Water Systems, Langmuir, 2010, 26, 8673–8683 CrossRef CAS PubMed.
- M. Chavarha, H. Khoojinian, L. E. Schulwitz Jr, S. C. Biswas, S. B. Rananavare and S. B. Hall, Hydrophobic surfactant proteins induce a phosphatidylethanolamine to form cubic phases, Biophys. J., 2010, 98, 1549–1557 CrossRef CAS PubMed.
- B. G. Tenchov, R. C. MacDonald and B. R. Lentz, Fusion peptides promote formation of bilayer cubic phases in lipid dispersions. An x-ray diffraction study, Biophys. J., 2013, 104, 1029–1037 CrossRef CAS PubMed.
- T.-Y. D. Tang, N. J. Brooks, C. Jeworrek, O. Ces, N. J. Terrill, R. Winter, R. H. Templer and J. M. Seddon, Hydrostatic Pressure Effects on the Lamellar to Gyroid Cubic Phase Transition of Monolinolein at Limited Hydration, Langmuir, 2012, 28, 13018–13024 CrossRef CAS PubMed.
- C. V. Kulkarni, W. Wachter, G. Iglesias-Salto, S. Engelskirchen and S. Ahualli, Monoolein: a magic lipid?, Phys. Chem. Chem. Phys., 2011, 13, 3004–3021 RSC.
- H. Qiu and M. Caffrey, The phase diagram of the monoolein/water system: metastability and equilibrium aspects, Biomaterials, 2000, 21, 223–234 CrossRef CAS PubMed.
- A. Ganem-Quintanar, D. Quintanar-Guerrero and P. Buri, Monoolein: a review of the pharmaceutical applications, Drug Dev. Ind. Pharm., 2000, 26, 809–820 CrossRef CAS PubMed.
- J. C. Shah, Y. Sadhale and D. M. Chilukuri, Cubic phase gels as drug delivery systems, Adv. Drug Delivery Rev., 2001, 47, 229–250 CrossRef CAS PubMed.
- X. Mulet, B. J. Boyd and C. J. Drummond, Advances in drug delivery and medical imaging using colloidal lyotropic liquid crystalline dispersions, J. Colloid Interface Sci., 2013, 393, 1–20 CrossRef CAS PubMed.
- E. Nazaruk, P. Miszta, S. Filipek, E. Górecka, E. M. Landau and R. Bilewicz, Lyotropic cubic phases for drug delivery: diffusion and sustained release from the mesophase evaluated by electrochemical methods, Langmuir, 2015, 31, 12753–12761 CrossRef CAS PubMed.
- I. Lopes, A. C. N. Oliveira, M. P. Sárria, J. P. Neves Silva, O. Gonçalves, A. C. Gomes and M. E. C. Real Oliveira, Monoolein-based nanocarriers for enhanced folate receptor-mediated RNA delivery to cancer cells, J. Liposome Res., 2016, 26, 199–210 CrossRef CAS PubMed.
- C. Leal, N. F. Bouxsein, K. K. Ewert and C. R. Safinya, Highly efficient gene silencing activity of siRNA embedded in a nanostructured gyroid cubic lipid matrix, J. Am. Chem. Soc., 2010, 132, 16841–16847 CrossRef CAS PubMed.
- H. Kim and C. Leal, Cuboplexes: Topologically active siRNA delivery, ACS Nano, 2015, 9, 10214–10226 CrossRef CAS PubMed.
- M. Kang and C. Leal, Soft Nanostructured Films for Actuated Surface-Based siRNA Delivery, Adv. Funct. Mater., 2016, 26, 5610–5620 CrossRef CAS.
- H. Kim, J. Sung, Y. Chang, A. Alfeche and C. Leal, Microfluidics synthesis of gene silencing cubosomes, ACS Nano, 2018, 12, 9196–9205 CrossRef CAS PubMed.
- A. Gupta, T. Stait-Gardner, L. De Campo, L. J. Waddington, N. Kirby, W. S. Price and M. J. Moghaddam, Nanoassemblies of Gd–DTPA–monooleyl and glycerol monooleate amphiphiles as potential MRI contrast agents, J. Mater. Chem. B, 2014, 2, 1225–1233 RSC.
- U. Bazylińska, J. Kulbacka, J. Schmidt, Y. Talmon and S. Murgia, Polymer-free cubosomes for simultaneous bioimaging and photodynamic action of photosensitizers in melanoma skin cancer cells, J. Colloid Interface Sci., 2018, 522, 163–173 CrossRef PubMed.
- A. Zabara, J. T. Y. Chong, I. Martiel, L. Stark, B. A. Cromer, C. Speziale, C. J. Drummond and R. Mezzenga, Design of ultra-swollen lipidic mesophases for the crystallization of membrane proteins with large extracellular domains, Nat. Commun., 2018, 9, 1–9 CrossRef CAS PubMed.
- C.-Y. Huang, V. Olieric, P. Ma, E. Panepucci, K. Diederichs, M. Wang and M. Caffrey, In meso in situ serial X-ray crystallography of soluble and membrane proteins, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2015, 71, 1238–1256 CrossRef CAS PubMed.
- V. Cherezov, Lipidic cubic phase technologies for membrane protein structural studies, Curr. Opin. Struct. Biol., 2011, 21, 559–566 CrossRef CAS PubMed.
- V. Cherezov, J. Clogston, M. Z. Papiz and M. Caffrey, Room to move: crystallizing membrane proteins in swollen lipidic mesophases, J. Mol. Biol., 2006, 357, 1605–1618 CrossRef CAS PubMed.
- V. Cherezov, J. Clogston, Y. Misquitta, W. Abdel-Gawad and M. Caffrey, Membrane protein crystallization in meso: lipid type-tailoring of the cubic phase, Biophys. J., 2002, 83, 3393–3407 CrossRef CAS.
- V. Grippo, S. Ma, R. Ludwig, L. Gorton and R. Bilewicz, Cellobiose dehydrogenase hosted in lipidic cubic phase to improve catalytic activity and stability, Bioelectrochemistry, 2019, 125, 134–141 CrossRef CAS PubMed.
- W. Sun, J. J. Vallooran, W.-K. Fong and R. Mezzenga, Lyotropic liquid crystalline cubic phases as versatile host matrices for membrane-bound enzymes, J. Phys. Chem. Lett., 2016, 7, 1507–1512 CrossRef CAS PubMed.
- J. J. Vallooran, S. Handschin, S. M. Pillai, B. N. Vetter, S. Rusch, H.-P. Beck and R. Mezzenga, Lipidic cubic phases as a versatile platform for the rapid detection of biomarkers, viruses, bacteria, and parasites, Adv. Funct. Mater., 2016, 26, 181–190 CrossRef CAS.
- E. Nazaruk, R. Bilewicz, G. Lindblom and B. Lindholm-Sethson, Cubic phases in biosensing systems, Anal. Bioanal. Chem., 2008, 391, 1569 CrossRef CAS PubMed.
- R. Winter, Effects of hydrostatic pressure on lipid and surfactant phases, Curr. Opin. Colloid Interface Sci., 2001, 6, 303–312 CrossRef CAS.
- N. F. Dupuis, E. D. Holmstrom and D. J. Nesbitt, Molecular-crowding effects on single-molecule RNA folding/unfolding thermodynamics and kinetics, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 8464–8469 CrossRef CAS PubMed.
- J. Tyrrell, K. M. Weeks and G. J. Pielak, Challenge of mimicking the influences of the cellular environment on RNA structure by PEG-induced macromolecular crowding, Biochemistry, 2015, 54, 6447–6453 CrossRef CAS PubMed.
- F. J. Wirkert, M. Paulus, J. Nase, J. Möller, S. Kujawski, C. Sternemann and M. Tolan, X-ray reflectivity measurements of liquid/solid interfaces under high hydrostatic pressure conditions, J. Synchrotron Radiat., 2014, 21, 76–81 CrossRef CAS PubMed.
- E. Schneider, M. Paulus, T. Witt, J. Bolle, H. Leif, M. Kowalski, W. Tillmann and M. Tolan, The new wide and small angle scattering setup at beamline BL2 of DELTA, DELTA Annu. Rep., 2019, 2019, 21–22 Search PubMed.
- C. Krywka, C. Sternemann, M. Paulus, N. Javid, R. Winter, A. Al-Sawalmih, S. Yi, D. Raabe and M. Tolan, The small-angle and wide-angle X-ray scattering set-up at beamline BL9 of DELTA, J. Synchrotron Radiat., 2007, 14, 244–251 CrossRef CAS PubMed.
- T. Huang, H. Toraya, T. Blanton and Y. Wu, X-ray powder diffraction analysis of silver behenate, a possible low-angle diffraction standard, J. Appl. Crystallogr., 1993, 26, 180–184 CrossRef CAS.
- C. Czeslik, R. Winter, G. Rapp and K. Bartels, Temperature-and pressure-dependent phase behavior of monoacylglycerides monoolein and monoelaidin, Biophys. J., 1995, 68, 1423–1429 CrossRef CAS PubMed.
- N. P. Money, Osmotic pressure of aqueous polyethylene glycols: relationship between molecular weight and vapor pressure deficit, Plant Physiol., 1989, 91, 766–769 CrossRef CAS PubMed.
- T. Yamada, N. Takahashi, T. Tominaga, S. Takata and H. Seto, Dynamical behavior of hydration water molecules between phospholipid membranes, J. Phys. Chem. B, 2017, 121, 8322–8329 CrossRef CAS PubMed.
- A. Nowacka, S. Douezan, L. Wadsö, D. Topgaard and E. Sparr, Small polar molecules like glycerol and urea can preserve the fluidity of lipid bilayers under dry conditions, Soft Matter, 2012, 8, 1482–1491 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sm01425k |
| This journal is © The Royal Society of Chemistry 2022 |