
Modeling the drying of polymer coatings
Venugopala Swami
Punati
 and
Mahesh S.
Tirumkudulu
*
and
Mahesh S.
Tirumkudulu
*
Department of Chemical Engineering, Indian Institute of Technology Bombay, Powai, Mumbai, 400076, India. E-mail: mahesh@che.iitb.ac.in
First published on 15th November 2021
Abstract
We investigate the drying phenomenon in polymer coatings by developing a model that accounts for the polymer-lean phase (liquid) and the polymer-rich phase (solid), while predicting the stress in the coating. The governing equations are developed for the two phases separately. In the dilute polymer region, the effect of polymer diffusion on its concentration distribution is considered. The polymer-rich phase is modeled as a porous structure, with entangled polymer chains whose diffusive motion is arrested. We employ Biot's theory to model the porous skin and to determine the stress in the coating. The governing equations are solved by employing a finite difference scheme. The effects of polymer diffusion and the skin's permeability on the coating stress are studied. We find good agreement between the predictions of the polymer concentration profile and that obtained from measurements.
1 Introduction
Polymer coatings are ubiquitous and find applications in the manufacture of automobiles, drugs and pharmaceuticals, electronics and several other products.1–7 The polymer coatings typically contain one or more film forming polymers along with additives that impart colour, gloss, enhanced wetting characteristics, anti-microbial property and other special properties to the final film. The coating solution is applied as a thin liquid film on a substrate and the film is allowed to dry. The loss of solvent leads to an increase in the polymer concentration and this process is accompanied by shrinkage of the liquid film. The process continues until reaching the gelation concentration, when the polymer chains form a space-filling network to give the coating a solid-like consistency. Further evaporation results in shrinkage of the film in the thickness direction while adhesion to the substrate restricts the coating from shrinking in the plane of the film. The latter gives rise to transverse stress, also known as drying stress or coating stress. When the stress value exceeds a threshold value, the coating may rupture or, if the adhesion with the substrate is weak, it may debond from the substrate.The study of drying stresses has a long history though Croll was the first to systematically investigate the drying stress in polymer coatings.8–10 Croll measured the drying stress in polymer coatings (binary mixture) by applying them on a cantilever and measuring the deflection at the cantilever's endpoint during the drying process.8 He found that the final stress is independent of the coating's initial thickness and initial polymer volume fraction. Next, he presented a simple mathematical model relating stress to the polymer volume fraction, where the latter was assumed to be constant through the thickness of the coating.9 The drying stress develops once the polymer volume fraction exceeds the polymer gelation volume fraction, which corresponds to the concentration at which the film starts to exhibit solid-like mechanical properties. Experiments with two different polymer solutions showed that the fraction of solvent remaining in the film at the end of the drying process was small (about 0.1) thereby allowing the application of linear elasticity theory for determination of stress. The drying stress value obtained from linear elasticity matched well with those obtained from experiments. Experiments were also performed to determine the critical stress required for peel-off from the substrate.10 Increasing the coating thickness increases the elastic energy, leading to delamination at lower stress.
While the simple model of Croll gives a good estimate of the final stress, the drying phenomenon is more complex. The stress in drying coatings depends on the constituent materials, their mechanical properties, the drying rate and the gelation concentration of the coating material. Prediction of drying stress requires the knowledge of the coating's mechanical properties as a function of polymer concentration, the spatial distribution of the polymer concentration in all three dimensions, and spatial and temporal variation in drying rates over the surface of the film. Mathematical models were initially developed to describe the evolution of drying stress in biological materials,11,12 ceramics and clays,13–15 which were later extended to explain the drying stress in polymer coatings. Haghighi and Segerlind developed a finite element (FE) procedure to determine drying stress in agricultural products, like seeds, for a given moisture and temperature distribution in the material.11,12 They assumed a linear viscoelastic response with material properties being functions of temperature and moisture content. Later, by employing the same FE procedure, Hasatani and coworkers13–15 studied the drying-induced stresses in clays and ceramics. They assumed clay to be a linear viscoelastic material, which was approximated using a Maxwell model. They solved the heat and mass transfer equations using the finite element procedure of Haghighi and Segerlind,11,12 to determine both temperature and moisture distribution in clay. The moisture content was found to be less near surfaces exposed to drying leading to large gradients in moisture content at the surfaces. This causes differential shrinkage in the material producing tensile stress near the surfaces exposed to drying and compressive stress near the core. As drying proceeds, the moisture content at the core decreases, and the compressive stresses slowly convert into tensile stresses. Increasing the drying rate increases the maximum tensile and compressive stresses, respectively, in the outer and core portions of the film. Finally, increasing the non-uniformity in the drying rates across different surfaces warps drying clays/ceramics.15
Tam et al.16 investigated drying stress in polymer films with elasto-viscoplastic rheology, where the polymer/solvent concentration was obtained by solving the diffusion–convection equation. They determined the shrinkage in film due to change in solvent concentration and employed the finite strain theory of Lee and Liu17 to obtain stresses in drying coatings. Their one-dimensional model predicts a non-uniform polymer concentration across the thickness of the coating with the maximum found at the drying (top) surface and the minimum near the substrate. Consequently, the maximum stress in the coating is at the top surface. The peak stress in the coating decreased by decreasing the mass transfer rate at the top surface and by increasing the diffusion in the coating. They also observed that in the two-dimensional drying problem, the edge effects were negligible at a distance of four thickness lengths from the coating edge. Later, Lei et al.18,19 developed a model to predict stress in drying coatings by assuming large elastic and viscoplastic deformations.16 Their model assumes that coatings develop stress once the polymer volume fraction exceeds the gelation volume fraction. The polymer volume fraction in the coating is obtained by solving the diffusion equation for the entire film and no distinction was made between the dilute polymer regions and the gelled regions. Their model predicts that the initial solvent concentration or the final coating thickness does not influence the final drying stress in the coating. Low evaporation rates reduce the final drying stress in the coating due to stress relaxation. Depending upon the coating's relaxation time, the model allows for the coating stress to exceed the yield stress. Christodoulou et al.20 studied the drying coatings by employing conservation laws, mass transport, and mixture theory to predict the stress. The polymer network was assumed to exhibit either a nonlinear elastic or hypoelastic response. For uniform thickness coatings, it was found that drying stress in the coatings is higher if the modulus of the film is allowed to increase with polymer concentration and when yielding is arrested. Rigid spherical particles embedded in coatings over soft substrates penetrate easily into the substrate on decreasing the modulus ratio of the overcoat to the substrate, leading to the starry night effect known in the photographic industry.
Some of the models discussed above, for example in Lei et al.,18 identified that the stress in the coating originates from the skin region where the polymer volume fraction is greater than or equal to the gelation polymer volume fraction. However, the aforementioned models ignore changes in the transport properties upon skin formation, such as the rapid decrease in polymer diffusivity, which drops by orders of magnitude in the skin region.21 In contrast, Ozawa et al.22 and Okuzono et al.23 developed a model for drying in polymer coatings where such variations were accounted for. They solved the convection–diffusion equations for the polymer concentration with the diffusion coefficients for the dilute polymer region being different from the gel domain, and obtained the spatial distribution of polymer concentration in the coatings. However, their model does not predict the stress generated during drying.
Although the focus of the present work is on modeling of the drying process, measurement of drying stress plays an important role in understanding the phenomenon. Francis et al.24 provide an overview of the stress measurement studies on polymer coatings due to both drying and film curing processes. It is observed that decreasing the solvent amount in the coating at gelation reduces shrinkage after gelation; hence the drying stress in the coating decreases. This can be achieved either by decreasing the polymer glass transition temperature or by increasing the drying temperature. The stress in the coatings may also be lowered by changing the solvent type or the humidity. Coatings with slow-drying solvents and dried at high relative humidity experience lower drying stress. In such coatings, Young's modulus of the coating increases slowly during the drying process enabling the coating to relax during drying.
A recent study of direct relevance to the present investigation is that of Tomar et al.,25 who investigated the drying of polymer coatings by measuring the stress and the solvent evaporation in silicone coatings. They found that the final stress in the coating is independent of its thickness, which is consistent with earlier investigations.8 Using micro-Raman spectroscopy, they measured the polymer concentration and showed that while thin films dry uniformly, thick films form a skin at the surface. They showed that there exists a critical thickness above which the film cracks with the critical thickness decreasing with increasing stiffness of the substrate.
Despite the detailed models, to the best of our knowledge, no study has reported quantitative comparison of the predicted concentration profile and stress evolution with measurements. The work reported here aims to reduce this gap, by modeling the drying phenomenon of a binary mixture of polymer and solvent and comparing the predicted concentration profile with measurements.25 We restrict ourselves to one-dimensional drying, where all variations are only in the thickness direction. We distinguish three stages during the drying process. During the first stage, the polymer concentration everywhere is less than the gelation polymer concentration, and hence it is in a liquid form. Also, there is no resistance from the substrate to the shrinking liquid coating. Therefore, there is no stress in the coating. We solve the diffusion equation and obtain the evolution of the polymer concentration. The first stage ends when the polymer concentration at the top surface reaches the gelation polymer concentration. In the second stage, the coating contains a skin region at the top surface, where the polymer concentration is greater or equal to the gelation concentration, with the dilute polymer solution region below it. The skin thickness increases with drying, and the liquid region decreases as the solvent evaporates from the top surface. Although the skin rests over the liquid, it is assumed to be pinned to the substrate at film edges, which are far from the central region considered here. Loss of solvent leads to volume shrinkage, which in turn leads to stress development. We model the stress development by employing Biot's formulation for a porous material. The assumption of a linear model stems from Croll's measurements, where small strain (∼0.03) was recorded at the end of the drying process. The third stage of drying begins when the entire film has a polymer volume fraction greater than the gelation volume fraction with the top surface polymer volume fraction remaining less than one. Otherwise, the drying phenomenon ends with the second stage when the skin region at the top surface reaches a volume fraction of one and shuts off the drying process. In the final stage of drying, the coating contains only the skin region.
2 Mathematical model
Consider a binary mixture of solvent and polymer applied to a rigid substrate at time t = 0. The densities of solvent and polymer are assumed to be identical and constant throughout the drying process. The coating's initial thickness is h0. We assume that the length l and the width b of the coating are large compared to its thickness. The solvent dries from the top surface of the film (one-dimensional drying). At t = 0, the polymer volume fraction ϕ is constant (ϕ0) throughout the coating. Therefore,| ϕ(z, t = 0) = ϕ0. | (1) |
As the drying proceeds, the evaporation rate is constant initially but with the development of the gel region at the top, the drying rate drops and solvent loss occurs via diffusion of solvent molecules from the interior of the film. Experiments of Tomar et al.25 suggest that the changing evaporation rate for the entire drying period can be captured using an exponential function,
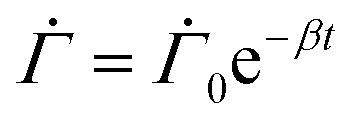 | (2) |
 represents the solvent evaporation rate at time t = 0 and β depends on the initial evaporation rate, the initial polymer volume fraction and the initial thickness of the coating25 (see Appendix A). The exponent β represents how quickly the solvent evaporation rate decreases. Higher the value of β, higher the decay rate in evaporation. Matching the rate of solvent loss from the coating to the decreasing rate in thickness, we obtain
represents the solvent evaporation rate at time t = 0 and β depends on the initial evaporation rate, the initial polymer volume fraction and the initial thickness of the coating25 (see Appendix A). The exponent β represents how quickly the solvent evaporation rate decreases. Higher the value of β, higher the decay rate in evaporation. Matching the rate of solvent loss from the coating to the decreasing rate in thickness, we obtain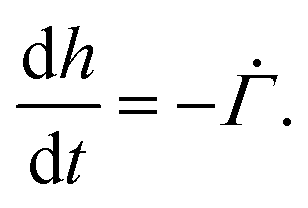 | (3) |
2.1 Liquid phase drying
As the solvent evaporates from the coating, the volume fraction of the polymer increases at the top surface. Due to the resulting gradient in the polymer volume fraction, the polymer diffuses inward away from the coating surface, i.e., towards the substrate. In the present problem, the coordinate system is placed at the top surface so that, in the new reference frame, the substrate moves towards the top surface. The governing partial differential equation for polymer transport in the liquid coating is given by (see the detailed derivation in Appendix B) | (4) |
The solvent evaporates but the particles do not leave the top surface, implying a zero flux condition for the particle phase at that location. In the new reference frame, the drying problem is transformed into a filtration problem, where the substrate moves toward the top surface with a velocity 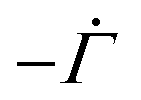 . Also, the substrate is impermeable to the solvent. Hence, near the substrate, the liquid and polymer velocities are equal to that of the substrate. The boundary conditions for the liquid region become
. Also, the substrate is impermeable to the solvent. Hence, near the substrate, the liquid and polymer velocities are equal to that of the substrate. The boundary conditions for the liquid region become
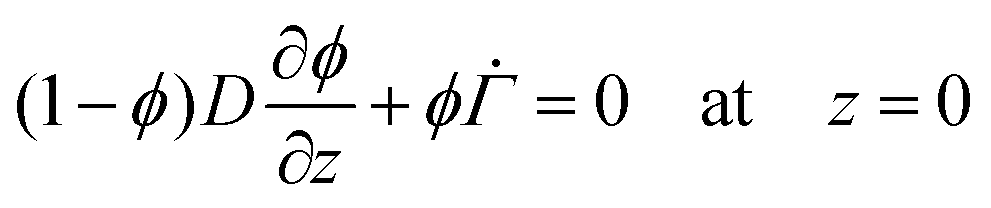 | (5) |
 | (6) |
Following Ozawa et al.,22 we model the diffusion coefficient D in the dilute polymer region (ϕ < ϕg) as
| D = D0(1 − ϕ)ϕm, | (7) |
2.2 Liquid-skin phase drying
With increasing polymer volume fraction at the top surface, the polymer chains come in contact and entangle, and at the polymer gelation volume fraction (ϕg), the polymer chains are no longer free to diffuse in the film.21,26 Experiments of Rauch and Köhler21 show that the polymer diffusivity drops by orders of magnitude close to the gelation volume fraction. In the present model, the region of the film with ϕ ≥ ϕg is referred to as the skin and is treated as a poroelastic solid. Alternatively, previous models such as those of Ozawa et al.22 assume the skin region as a concentrated polymer mixture with a different expression for diffusivity in this region.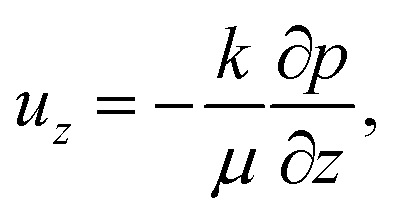 | (8) |
Equating the rate of change of solvent in the porous network to Darcy's equation provides the governing partial differential equation for the pore pressure (p) in the skin (see Appendix C for detailed derivation),
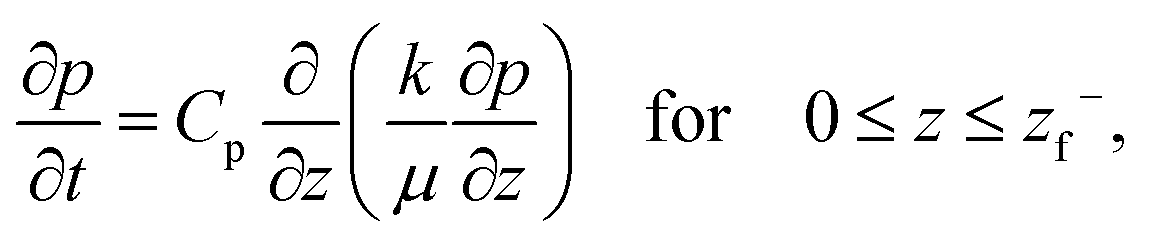 | (9) |
 | (10) |
 | (11) |
By obtaining the pressure p in the skin, the stress and strain in the skin are given by
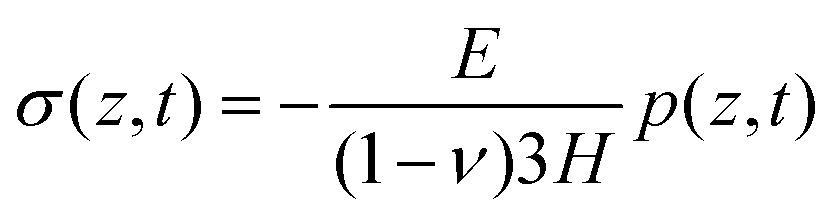 | (12) |
 | (13) |
The solvent flux at the top surface is equal to the evaporation rate. At the bottom surface of the skin, the polymer volume fraction is equal to ϕg. Since the polymer forms a gel at ϕg, the stress at the bottom surface of the skin is zero. The boundary conditions for the top and bottom surface of the skin become
 | (14) |
and
| p = 0 at z = zf−, | (15) |
respectively (see Fig. 1(c)).
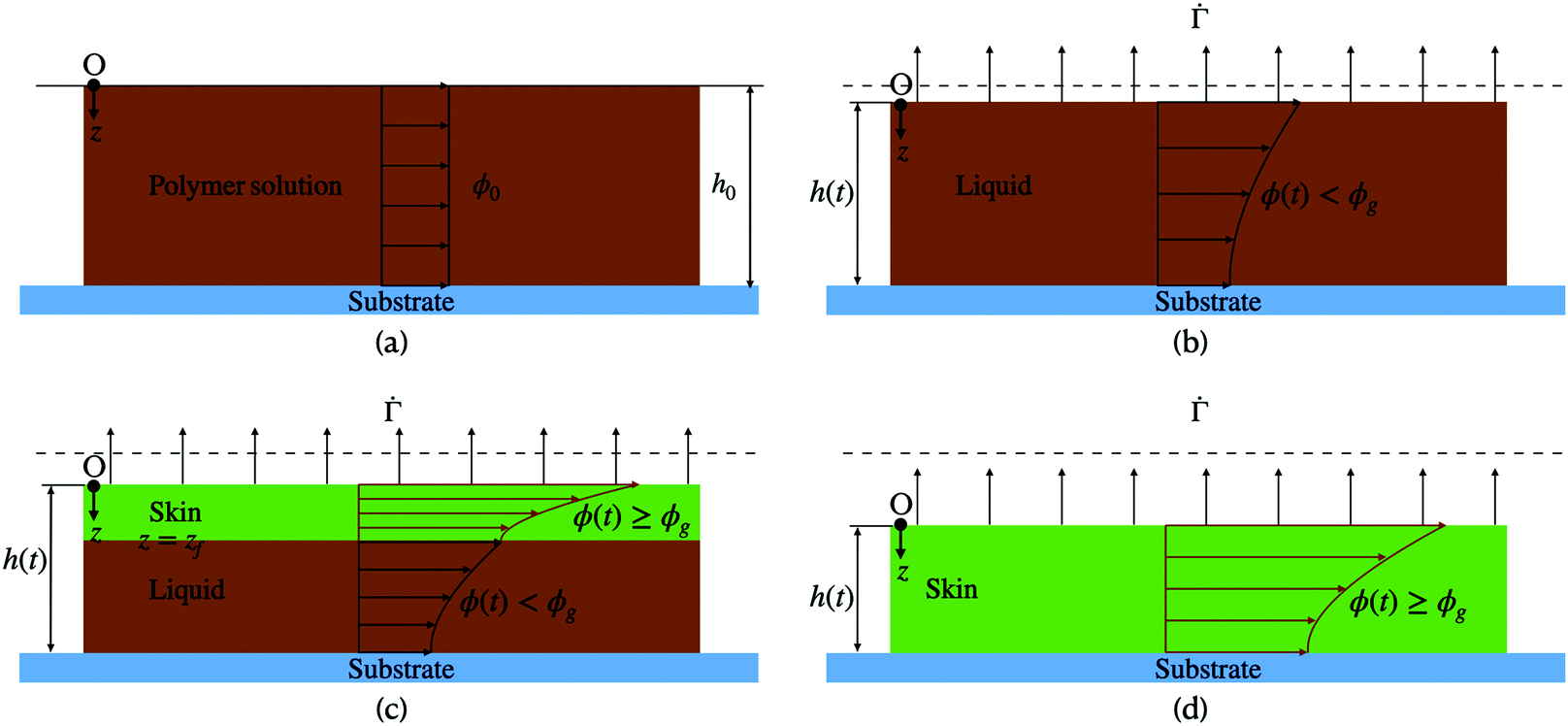 | ||
| Fig. 1 (a) A liquid polymer coating of height h = h0 is deposited onto the substrate at time t = 0. The polymer volume fraction ϕ = ϕ0 in the coating. (b) Liquid phase drying: ϕ < ϕg, where ϕg is the polymer gelation volume fraction. (c) Liquid-skin phase drying: In the skin, ϕ ≥ ϕg. In the liquid ϕ < ϕg. (d) Skin phase drying: ϕ > ϕg everywhere in the coating. The dotted line represents the initial thickness of the coating. | ||
 | (16) |
 | (17) |
2.3 Skin phase drying
When the liquid–skin interface reaches the substrate, ϕ ≥ ϕg everywhere in the coating. The governing partial differential equation for the pressure in the solid phase is the same as that of (9) for the entire film (0 ≤ z ≤ h(t)), with the boundary conditions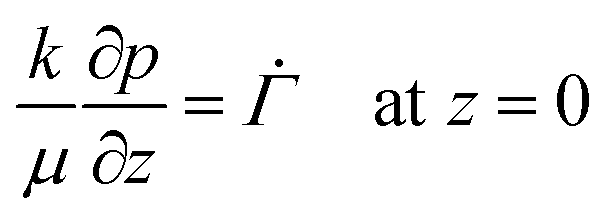 | (18) |
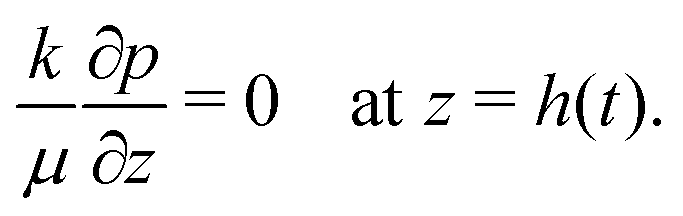 | (19) |
By solving (9) with the above boundary conditions (18) and (19), one may obtain the pressure distribution during drying of the solid phase. Substituting this pressure distribution in (12) and (13) one may evaluate the stresses and strains in the coating, respectively.
3 Non-dimensionalisation
We render all the equations dimensionless using the following scaling: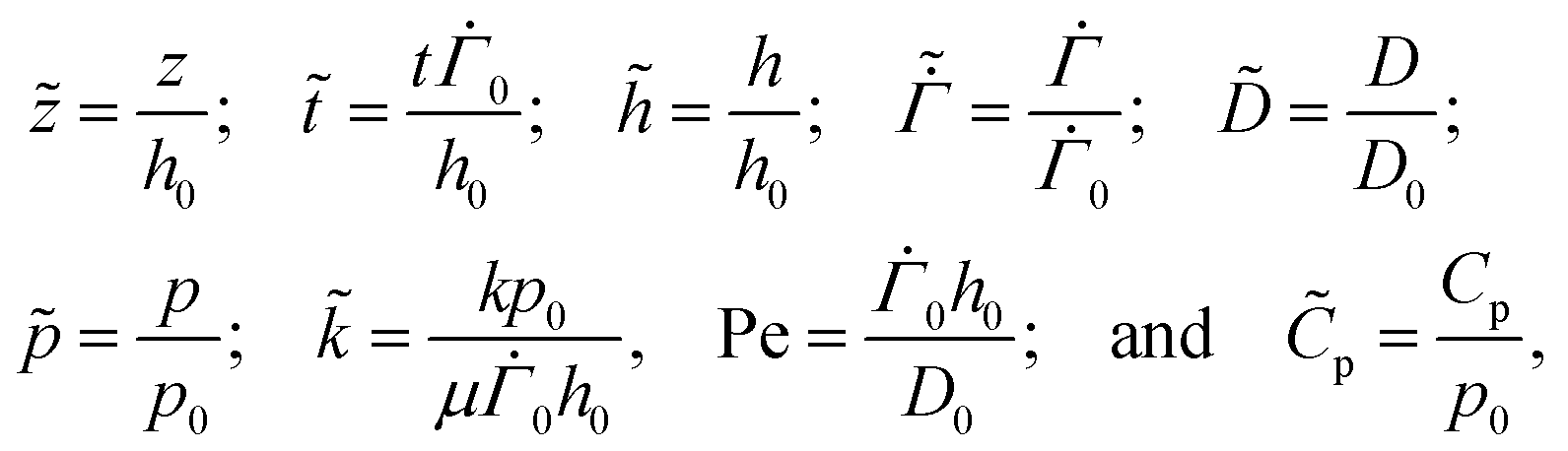 | (20) |
 .
.
For the liquid phase drying, by employing the dimensionless terms (20), the governing partial differential equation (4) for the polymer volume fraction ϕ becomes
 | (21) |
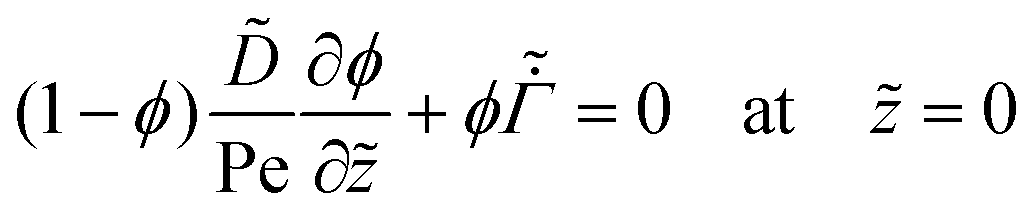 | (22) |
 | (23) |
On substituting the dimensionless variables (20) in (9), (14) and (15), for the skin region of the liquid–skin phase drying, we have
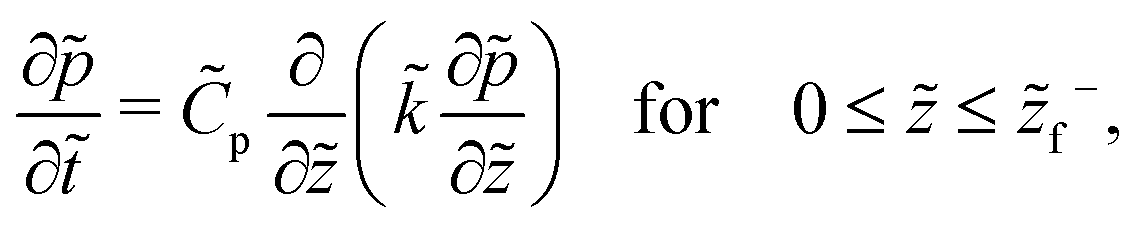 | (24) |
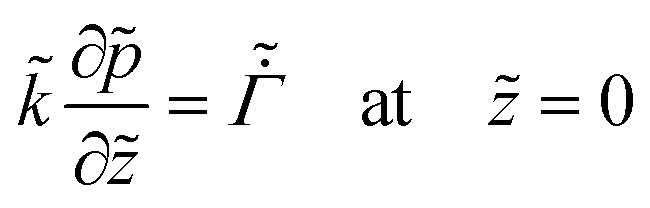 | (25) |
and
![[p with combining tilde]](https://www.rsc.org/images/entities/i_char_0070_0303.gif) = 0 at = 0 at ![[z with combining tilde]](https://www.rsc.org/images/entities/i_char_007a_0303.gif) = f−, = f−, | (26) |
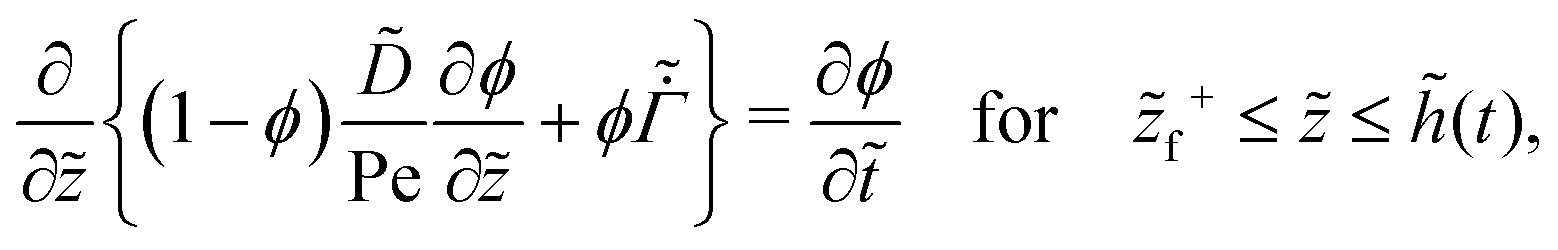 | (27) |
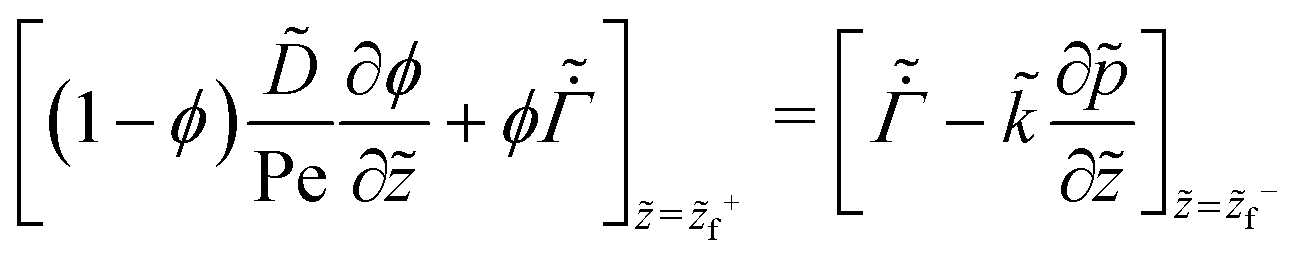 | (28) |
 | (29) |
For the skin phase drying, the non-dimensional form of (9) to find pressure in the skin is
 | (30) |
 | (31) |
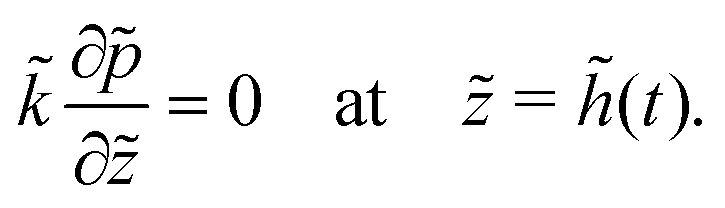 | (32) |
The stress (12) and the strain (13) in their dimensionless form are written as
 | (33) |
 | (34) |
Finally, the dimensionless forms for the evaporation rate (2), the diffusion coefficient (7), and the permeability (11), respectively, become
 | (35) |
![[D with combining tilde]](https://www.rsc.org/images/entities/i_char_0044_0303.gif) = (1 − ϕ)ϕ5, = (1 − ϕ)ϕ5, | (36) |
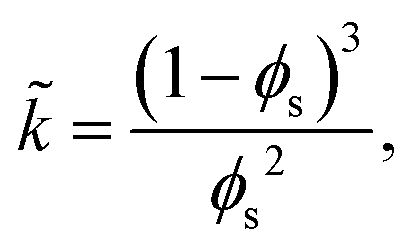 | (37) |
![[small beta, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e10e.gif) = (1 − ϕ0)−1.
= (1 − ϕ0)−1.
We solve the dimensionless governing equations for each phase by employing an implicit finite volume approach. The procedure is described in Appendix D. Care must be taken while solving the equations as the positions of the nodes change on the moving boundaries of the substrate surface and the liquid–skin interface. It is to be noted that computations are halted either when the polymer volume fraction at the top surface reaches one or when the evaporation rate is negligible.
4 Results
First, we discuss the predictions of our model by varying the dimensionless parameters Pe and![[C with combining tilde]](https://www.rsc.org/images/entities/i_char_0043_0303.gif) p while keeping constant the evaporation rate
p while keeping constant the evaporation rate  , the diffusion coefficient , and the permeability
, the diffusion coefficient , and the permeability ![[k with combining tilde]](https://www.rsc.org/images/entities/i_char_006b_0303.gif) . Later, we present the model predictions by holding constant the values of dimensionless parameters Pe and p while allowing
. Later, we present the model predictions by holding constant the values of dimensionless parameters Pe and p while allowing  , , and to vary during drying. Finally, we compare the predicted volume fraction distributions with the measurements of Tomar et al.25
, , and to vary during drying. Finally, we compare the predicted volume fraction distributions with the measurements of Tomar et al.25
We set the initial parameters in the drying as ϕ0 = 0.43, h0 = 120 × 10−6 m, 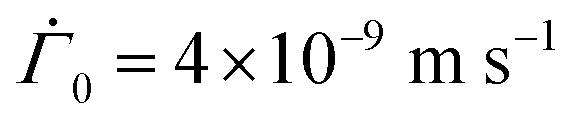 , and μ = 4 × 10−3 Pa s. The values correspond to those observed in previously reported experiments,25 where the silicone polymer, trimethylsiloxysilicate resin, was dispersed in decamethylcyclopentasiloxane. The solidification volume fraction may be determined using the Fox equation,21 which requires the glass transition temperature of both the polymer and solvent. In the absence of reliable data of either, the gelation volume fraction was fixed at ϕg = 0.85 following Croll.9 To vary Pe and p in our computation, we varied the diffusion coefficient D0 and the permeability constant K0, respectively. Lower the diffusivity D0, higher the Peclet number Pe. And, higher the permeability K0, higher the non-dimensional number p.
, and μ = 4 × 10−3 Pa s. The values correspond to those observed in previously reported experiments,25 where the silicone polymer, trimethylsiloxysilicate resin, was dispersed in decamethylcyclopentasiloxane. The solidification volume fraction may be determined using the Fox equation,21 which requires the glass transition temperature of both the polymer and solvent. In the absence of reliable data of either, the gelation volume fraction was fixed at ϕg = 0.85 following Croll.9 To vary Pe and p in our computation, we varied the diffusion coefficient D0 and the permeability constant K0, respectively. Lower the diffusivity D0, higher the Peclet number Pe. And, higher the permeability K0, higher the non-dimensional number p.
First, we choose four different combinations of Pe and p and plot the polymer volume fraction ϕ(, ![[t with combining tilde]](https://www.rsc.org/images/entities/i_char_0074_0303.gif) ), see Fig. 2. In all plots, the top represents the film surface and the bottom end represents the substrate. As expected, ϕ increases in the coating with solvent evaporation, and it is maximum at the top surface ( = 0) and minimum at the substrate ( =
), see Fig. 2. In all plots, the top represents the film surface and the bottom end represents the substrate. As expected, ϕ increases in the coating with solvent evaporation, and it is maximum at the top surface ( = 0) and minimum at the substrate ( = ![[h with combining tilde]](https://www.rsc.org/images/entities/i_char_0068_0303.gif) ). As expected, the coating height () decreases as drying proceeds. Comparing Fig. 2(a) with Fig. 2(b) (or Fig. 2(c) with Fig. 2(d)), an increase in Pe causes a decreased diffusion of the polymer towards the substrate leading to a steeper concentration gradient.
). As expected, the coating height () decreases as drying proceeds. Comparing Fig. 2(a) with Fig. 2(b) (or Fig. 2(c) with Fig. 2(d)), an increase in Pe causes a decreased diffusion of the polymer towards the substrate leading to a steeper concentration gradient.
 | ||
Fig. 2 Evolution of the polymer volume fraction in the coatings with different Pe and p as indicated in the plots. In each plot, the polymer volume fraction distributions in the coating are plotted at different non-dimensional times and these are noted at the bottom of their associated curves. The dashed vertical lines in the plots represent the polymer gelation volume fraction ϕg. Here, h0 = 120 × 10−6 m, 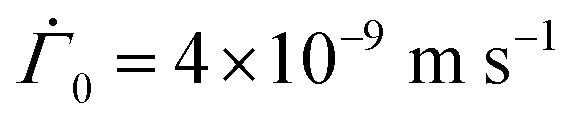 and μ = 4 × 10−3 N s m−2 while D0 and K0 are varied to obtain various Pe and p. and μ = 4 × 10−3 N s m−2 while D0 and K0 are varied to obtain various Pe and p. | ||
As drying proceeds, ϕ reaches ϕg at the surface leading to skin formation. The vertical dashed lines in the plots represent the gelation volume fraction ϕg. On any ϕ curve, the portion to the left of ϕg-line represents the liquid region, and the portion to the right of ϕg-line represents the skin domain. At the end of liquid phase drying, the coatings with a high Pe (due to large variation in ϕ) contain a small skin region at the top and a large liquid region beneath it. As the solvent evaporates from the coating, the height of the skin region increases and the height of the liquid region decreases simultaneously, see Fig. 2(b and d). Comparing Fig. 2(b and d), one may observe that changing the permeability of the skin (p) changes evolution of ϕ during liquid-skin phase drying. Initially, when the skin forms, i.e., at ϕ = ϕg, the solvent pore pressure in the skin is g = 0. The solvent flux in the skin, the evaporation rate, and the permeability set the pressure gradient in the porous network. For a high solvent flux or evaporation rate, the pressure gradient across the skin will be large. For high permeability of the porous network, the liquid can easily flow through the skin. Hence it requires a lower pressure gradient across the skin thickness. The pressure gradient across the skin causes to vary in the skin and changes away from g. Due to the pressure in the porous network, the skin deforms to generate stress in the coating. These deformations in the porous network also cause the polymer volume fraction ϕ to change in the skin. Higher the deformation, greater is the change in ϕ of the skin. For high permeability, the pressure gradient across the skin is small; hence we see small gradients of ϕ during the liquid-skin phase drying. Consequently, increasing p leads to small deviations of ϕ from ϕg. At low Pe, Fig. 2(a and c), a large skin region forms at the top at the beginning of the liquid-skin phase drying. Due to the small liquid region, there is hardly any change in the evolution of ϕ even when p is varied over an order of magnitude during the liquid-skin phase drying. Also, the shape of the ϕ-profiles is closer to the one observed at the end of liquid phase drying. Overall, the Pe influences the concentration profile much more than the permeability.
Once the liquid-skin phase drying is over, there is only a porous network over the substrate. At this stage, the solvent evaporation from the coating's top surface changes significantly the pore pressure in the skin, which increases the polymer volume fraction everywhere in the coating. Since the properties of the skin, i.e., permeability, have been assumed to be constant in Fig. 2, the polymer volume fraction profiles in the skin do not deviate much from the one observed at the end of liquid-skin phase drying. We stop computations when the polymer volume fraction at the top surface reaches one. Because of this, some solvent remains trapped in the coating, which is in agreement with previous experiments.9
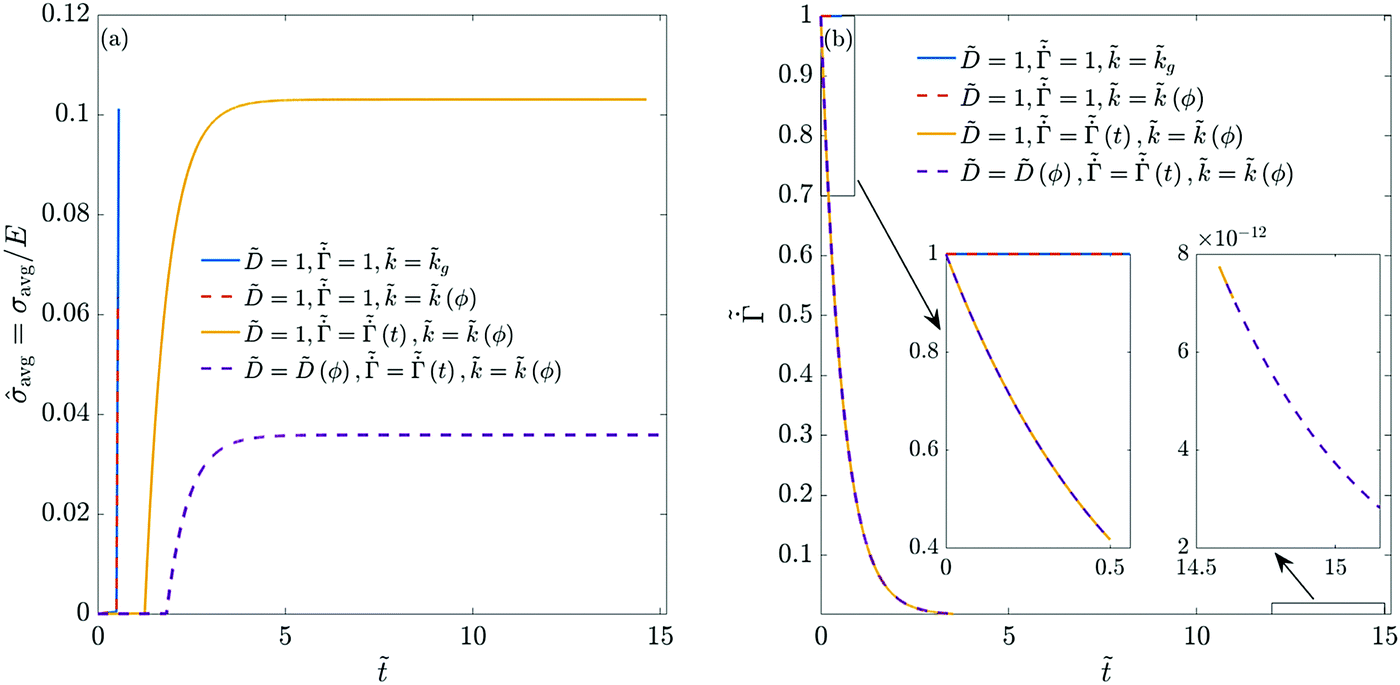
Fig. 3 plots the final stress and the thickness-averaged stress for the previous four different combinations of Pe and p. Note that the final stress value corresponds to the time when the polymer volume fraction at the top becomes one. We observe that with increasing Pe, the maximum final stress in the coating decreases. At high Pe, the skin thickness formed at the end of liquid phase drying is small (see Fig. 2). Further, the concentration at the top surface reaches one even when much of the liquid is trapped in the film. This decreases the stress (both the final maximum stress and the average stress) in coatings dried at high Pe. These results are in qualitative agreement with the observations of Tomar et al.,25 who noted low stresses for very thick films.§ Increasing the permeability p reduces the pressure gradient across the skin during drying. Therefore, the final maximum stress is reduced in magnitude but the average stress is high due to more spatial uniformity in the polymer concentration.
 | ||
| Fig. 3 (a) Final drying stress in the coatings with different Pe and p. (b) Evolution of the thickness-averaged drying stress in the coatings with different Pe and p. The parameters used in the computations are the same as that of Fig. 2. We stop computations when the polymer volume fraction at the top surface reaches one. The final stress distribution corresponds to this time. | ||
We next allow the coating parameters to vary during the drying process and plot ϕ(, ) in Fig. 4 and the average stress and the evaporation rate in Fig. 5. First, we set and 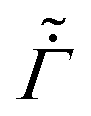 as constants and allow the permeability to vary as a function of ϕ. Based on the ϕ distribution, this leads to a maximum at the bottom surface of the skin and a minimum at the top surface. Also, the solvent flux decreases as we move away from the top surface. Thus, we see a higher pressure gradient near the top surface and a lower pressure gradient near the bottom surface of the skin. When the volume fraction at the top surface reaches unity, the polymer volume fraction at the substrate is small compared to that obtained for constant . This reduces the average stress in the coatings when compared to coatings with constant , see Fig. 5(a). When ϕ reaches one, the permeability becomes zero, and the drying stops.
as constants and allow the permeability to vary as a function of ϕ. Based on the ϕ distribution, this leads to a maximum at the bottom surface of the skin and a minimum at the top surface. Also, the solvent flux decreases as we move away from the top surface. Thus, we see a higher pressure gradient near the top surface and a lower pressure gradient near the bottom surface of the skin. When the volume fraction at the top surface reaches unity, the polymer volume fraction at the substrate is small compared to that obtained for constant . This reduces the average stress in the coatings when compared to coatings with constant , see Fig. 5(a). When ϕ reaches one, the permeability becomes zero, and the drying stops.
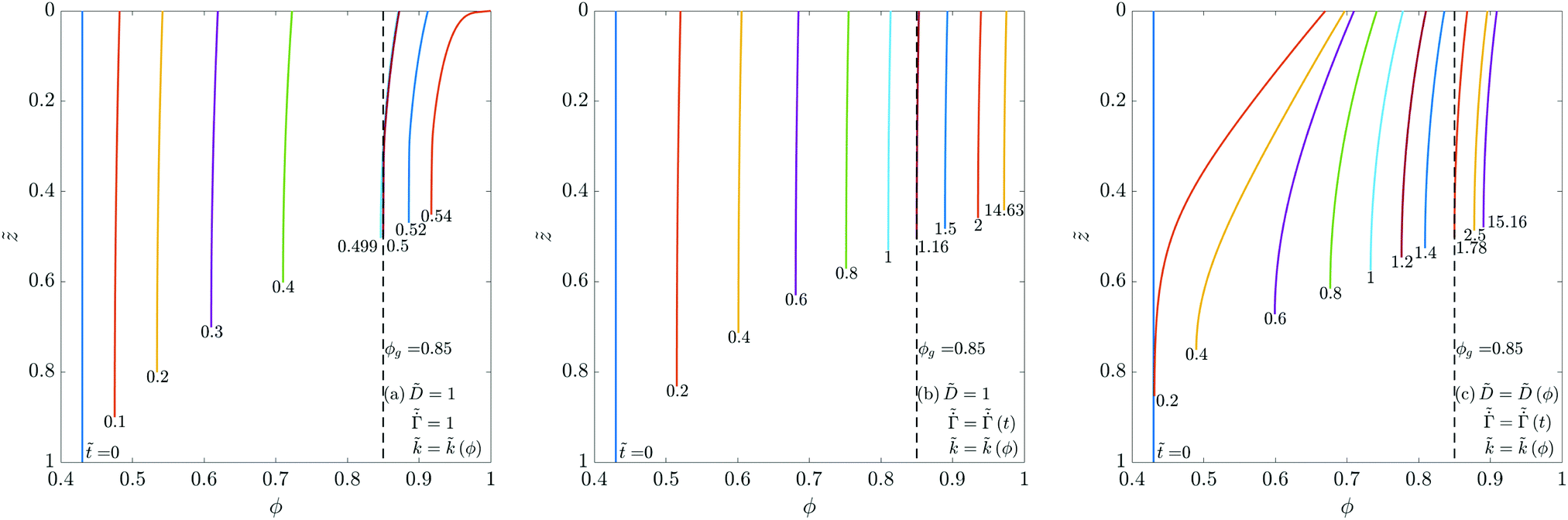 | ||
Fig. 4 Evolution of the polymer volume fraction in the coatings: (a) keeping and  as constants while varying , (b) keeping as a constant while varying as constants while varying , (b) keeping as a constant while varying 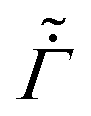 and , and (c) varying , and , and (c) varying ,  and . The function forms for and . The function forms for  , (ϕ) and (ϕ) are given by (35), (36) and (37), respectively. In each plot, the polymer volume fraction is plotted at different non-dimensional times and the latter are noted at the bottom of the curves. The dashed vertical lines in the plots represent the polymer gelation volume fraction ϕg. Computations are obtained taking Pe = 0.017 and p = 23 , (ϕ) and (ϕ) are given by (35), (36) and (37), respectively. In each plot, the polymer volume fraction is plotted at different non-dimensional times and the latter are noted at the bottom of the curves. The dashed vertical lines in the plots represent the polymer gelation volume fraction ϕg. Computations are obtained taking Pe = 0.017 and p = 23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 558. 558. | ||
 | ||
| Fig. 5 We plot (a) the thickness-averaged coating stress and (b) the evaporation rate in the coating, while allowing its properties to change during the drying process. In (b), the left- and right-hand side insets show the evaporation rates at small and large , respectively. The parameters used in the computations are the same as that in Fig. 4. For a constant evaporation rate, the polymer volume fraction at the top surface reaches one. | ||
Next, the value of is held constant while  and are varied as per (35) and (37), respectively. The polymer concentration variation is plotted in Fig. 4(b). As the evaporation rate decreases over time, the solvent flux in the coating decreases. Therefore, we see a smaller variation in ϕ during the liquid phase drying. In the computations, the liquid phase drying ends after satisfying ϕ = ϕg near the substrate. Thus the entire coating enters the skin phase drying without any liquid-skin phase drying. During skin phase drying, the pressure gradient across the skin is small due to the low solvent flux in the skin. Therefore, the variation in ϕ is also small. With increasing time, the evaporation rate drops to a very small value (see the right-hand side inset in Fig. 5(b)), and we hardly notice any change in the coating's properties. Therefore, the simulations are stopped when ϕ ≈ 0.98 at the top surface. As ϕ is close to unity with a uniform distribution across the coating thickness, the average stress in the coating is high compared to the previous case.
and are varied as per (35) and (37), respectively. The polymer concentration variation is plotted in Fig. 4(b). As the evaporation rate decreases over time, the solvent flux in the coating decreases. Therefore, we see a smaller variation in ϕ during the liquid phase drying. In the computations, the liquid phase drying ends after satisfying ϕ = ϕg near the substrate. Thus the entire coating enters the skin phase drying without any liquid-skin phase drying. During skin phase drying, the pressure gradient across the skin is small due to the low solvent flux in the skin. Therefore, the variation in ϕ is also small. With increasing time, the evaporation rate drops to a very small value (see the right-hand side inset in Fig. 5(b)), and we hardly notice any change in the coating's properties. Therefore, the simulations are stopped when ϕ ≈ 0.98 at the top surface. As ϕ is close to unity with a uniform distribution across the coating thickness, the average stress in the coating is high compared to the previous case.
Finally, we allow all the coating parameters to vary during the drying process and plot the ϕ variation in Fig. 4(c). Since the diffusivity is an increasing function of ϕ, the diffusivity is low at initial times. Further, the evaporation rate is high at initial times. Consequently, the polymer volume fraction at the top surface increases rapidly leading to a steep concentration gradient. Once the diffusivity increases to a significant value and the evaporation rate falls, the gradient reduces and the concentration becomes more uniform across the thickness. After the coating enters the skin phase drying, and when ϕ ≈ 0.92 at the top surface, we hardly observe any change in the coating properties (because of the very low evaporation rate at long times). From the right hand-side inset in Fig. 5(b), we see that the non-dimensional evaporation rate is of the order of 10−12, which is very small. As a result, the solvent flux in the coating is negligible, and there is no change in pressure inside the skin. Therefore, the simulations are stopped at this stage and the average stress in the coating is plotted, see Fig. 5(a). The stress behavior in these coatings is similar to the previous case, i.e., it increases over time and reaches a constant value. Compared to the previous case, the final ϕ value at the top surface is smaller, with higher variation across the film thickness. This leads to a lower stress at the end of the drying period.
4.1 Comparison with experiments
We now compare the model predictions with measurements. Tomar et al.25 measured the polymer volume fraction at different depths of drying coating. The thickness of the coating was measured at the end of the drying process and the initial and the final height are related by a simple mass balance| h0(1 − ϕ0) = hf(1 − ϕf), | (38) |
 are set to the values reported in Tomar et al.25 The permeability coefficient K0 is chosen as discussed earlier in Section 2.2.1. We allow the parameters D,
are set to the values reported in Tomar et al.25 The permeability coefficient K0 is chosen as discussed earlier in Section 2.2.1. We allow the parameters D,  and k to vary during the drying process (as described in Section 2). It is to be noted that in Tomar et al.,25 the ratio of the solvent intensity to the polymer intensity is presented instead of the polymer volume fraction. In the experiments, intensities are calibrated against their respective weight fractions. As the initial polymer volume fraction is known, first, we convert the intensity ratios to weight fraction ratios using a multiplication factor. Later, using these weight fraction ratios and the densities of solvent and polymer, one may obtain the polymer volume fractions as a function of time and depth.
and k to vary during the drying process (as described in Section 2). It is to be noted that in Tomar et al.,25 the ratio of the solvent intensity to the polymer intensity is presented instead of the polymer volume fraction. In the experiments, intensities are calibrated against their respective weight fractions. As the initial polymer volume fraction is known, first, we convert the intensity ratios to weight fraction ratios using a multiplication factor. Later, using these weight fraction ratios and the densities of solvent and polymer, one may obtain the polymer volume fractions as a function of time and depth.
Following the experiments of Tomar et al.,25 the initial polymer volume fraction is taken to be ϕ0 = 0.43 in our computations. We set the initial coating thickness h0 = 80 μm, which at the end of drying gives hf = 40 μm. The strains reported in Tomar et al.25 during the drying are very small. Hence we consider the gelation polymer volume fraction for these coatings as ϕg = 0.9. Note that the assumed value is close to that measured by Croll8 for polystyrene in toluene (0.83) and poly(isobutyl methacrylate) (0.84). The predicted polymer volume fraction at different z is compared with the measurements for hf = 40 μm taken at the centre of the film and far from the edges of the film, see Fig. 6. The polymer volume fraction in the coating increases at all locations as the solvent evaporates from the coating. The polymer volume fraction is maximum at the top surface of the coating. As we move away from the top surface, i.e., towards the substrate, the polymer volume fraction decreases. The model predictions match well with the measured values at different z justifying the methodology adopted here. The model captures not only the initial rapid increase but also the subsequent slower increase in concentration. The transition between the two rates also occurs at about the same time as predicted.
 | ||
Fig. 6 Variation of the polymer volume fraction at three different locations in the coating. The solid lines represent the model predictions, while the dashed lines with symbols represent the measurements of Tomar et al.25 The inset shows the comparison for small t. We set ϕ0 = 0.43, ϕg = 0.9, h0 = 80 μm, D0 = 2.9 × 10−11 m2 s−1 and  in our simulations. in our simulations. | ||
Since the polymer concentration is below the gelation concentration, the model does not predict any stress at these times. However, experiments do show small values of stress at these early times. This is mainly because of the spatial variation of film thickness in experiments. The polymer volume fraction near the edges approach the gelation volume fraction faster than the rest of the coating due to lower thickness in these regions. The resulting skin formed at the coating edges results in stress that bends the cantilever. Such effects can only be captured when the full three dimensional problem is solved.
5 Conclusions and future scope
We have developed a three-phase drying model for one-dimensional drying and investigated the effect of various parameters on the drying phenomenon in polymer coatings. Solvent evaporation from the coating causes the polymer volume fraction to increase near the top surface leading to skin formation. For coatings with a high Peclet number Pe, the thickness of the skin formed at the end of liquid phase drying is small due to the large gradient in the polymer volume fraction. Increasing the permeabilityp for the coatings decreases the final stress in the coating. The average stress is high when the skin formation is delayed, which allows the entire film to reach gelation. Of all the various parameters, the evaporation rate and the diffusion coefficient were found to influence the stress evolution significantly. Allowing the diffusion coefficient to vary during the drying reduces the final stress significantly. The predicted concentration profile as a function of time and depth was compared with measurements, and a good agreement was observed.
Future work would focus on developing a full three-dimensional drying model that accounts for the yielding and plastic deformation of the film so as to obtain a more accurate stress evolution in polymer coatings. This would require reliable measurements of the coating properties such as diffusion coefficient D, permeability k, and Young's modulus E during drying.
Conflicts of interest
There are no conflicts to declare.Appendix A: evaporation rate
In Tomar et al.,25 the height h of the coating with time t is obtained by measuring the amount of solvent lost from the coating during drying. It is observed that the height of the polymer coating decreases with time as| h(t) = hf + (h0 − hf)e−βt | (A.1) |
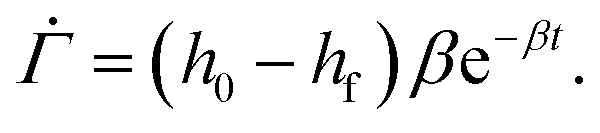 | (A.2) |
 | (A.3) |
 | (A.4) |
= (1 − ϕ0)−1.
Appendix B: liquid domain equations
The total flux J in the liquid region is given by the summation of the polymer flux Jp and the solvent flux Jl. Therefore, we may write| J = Jp + Jl. | (B.1) |
| Jp = ϕvp | (B.2) |
| Jl = (1 − ϕ)vl, | (B.3) |
| Jp = ϕ(vp − vl) + ϕvl. | (B.4) |
 | (B.5) |
 | (B.6) |
 | (B.7) |
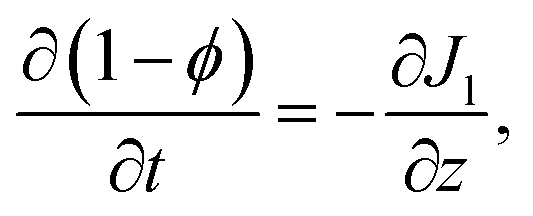 | (B.8) |
 | (B.9) |
| Jp + Jl = C, | (B.10) |
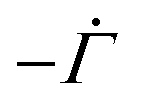 . Therefore,
. Therefore, 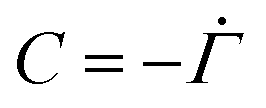 . Substituting (B.6) and (B.3) in (B.10), and solving for the liquid velocity one may obtain
. Substituting (B.6) and (B.3) in (B.10), and solving for the liquid velocity one may obtain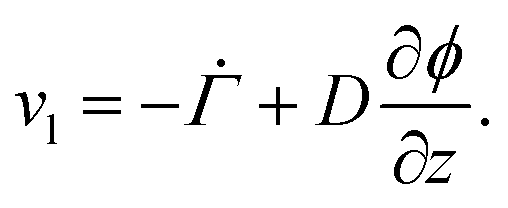 | (B.11) |
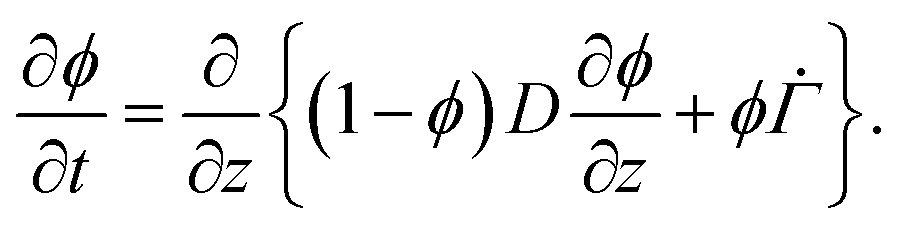 | (B.12) |
Appendix C: skin equations
We model the skin as a poroelastic material. The stress–strain relationships for a general three dimensional poroelastic material are27 | (C.1) |
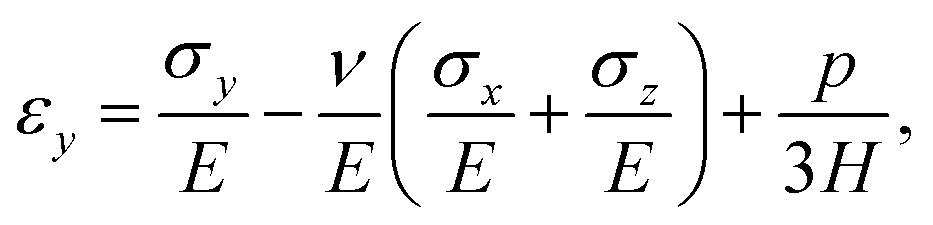 | (C.2) |
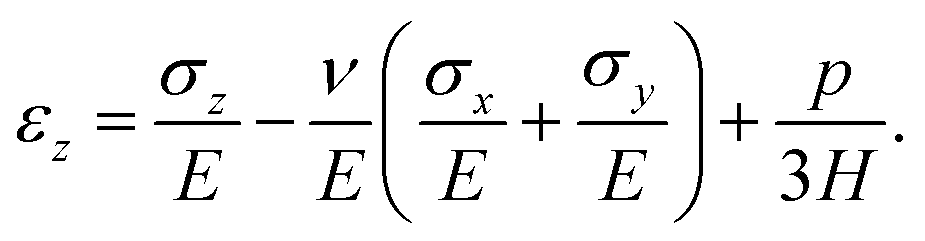 | (C.3) |
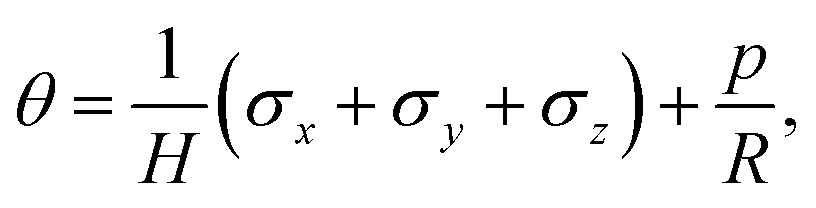 | (C.4) |
During drying, the skin is not allowed to shrink due to the constraints imposed in the transverse direction. Therefore, we may write
| εx = 0 and εy = 0. | (C.5) |
| σx = σy = σ. | (C.6) |
| σz = 0. | (C.7) |
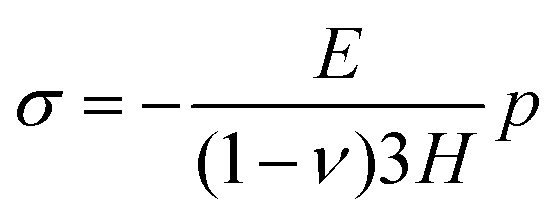 | (C.8) |
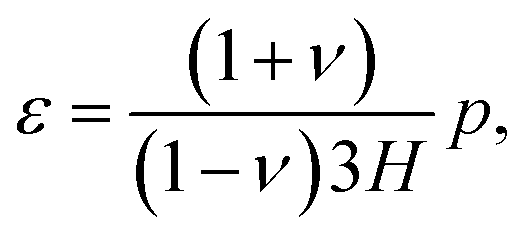 | (C.9) |
On substituting (C.6)–(C.8) in (C.4), and determining the rate of liquid change in a porous material from the resulting equation one obtains
 | (C.10) |
 | (C.11) |
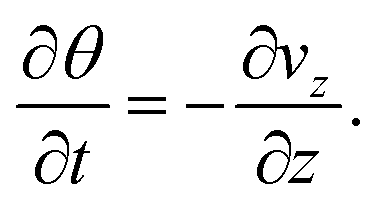 | (C.12) |
 | (C.13) |
Assuming equal densities for the liquid and the polymer phases, the change in liquid content in a unit volume should equal the change in the volume of the porous material,27
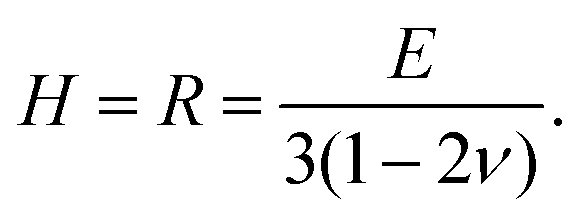 | (C.14) |
Appendix D: numerical solution procedure
We solve the governing equations of the entire drying process by employing the procedures described below. We now discuss the discretization scheme used to solve the governing equations. We divide the region betweenstart and end into N (or n) equal segments with N + 1 (or n + 1) faces and N (or n) grid points. Here, N and n represent the number of grid points in the liquid region and the skin region, respectively. The locations of the faces and the grid points from the coating's top surface are given by = fi and = gi, respectively. For the liquid region, fi = f1, f2,…,fN+1 and gi = g1, g2,…,gN, and for the skin region, fi = f1, f2,…,fn+1 and gi = g1, g2,…,gn. We assume that the grid point gi is always at the center of two faces, i.e. fi and fi+1. The properties at the grid points are obtained after solving the corresponding governing equations. Then the face values are obtained either by interpolation or by extrapolation. For the computations, we calculate the particle flux (superscript p) in the liquid region and the solvent flux (superscript l) in the skin region. These fluxes are evaluated at the faces by employing the properties at the grid points. Here, we present the discretized forms for fluxes, governing partial differential equations and boundary conditions.
First, we present the discretized forms of the particle flux and the polymer volume fraction evolution equation for the liquid region. The particle flux at the face fi of the liquid region is found from
 | (D.1) |
 | (D.2) |
 | (D.3) |
Later, we present the liquid flux and the pore pressure evolution equations in their discretized forms. The solvent flux in the skin region at face fi is found from
 | (D.4) |
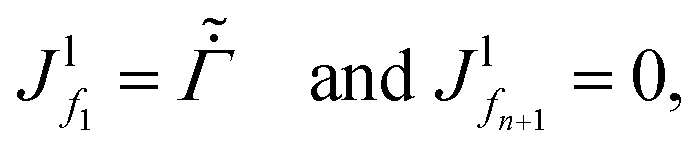 | (D.5) |
in the skin by employing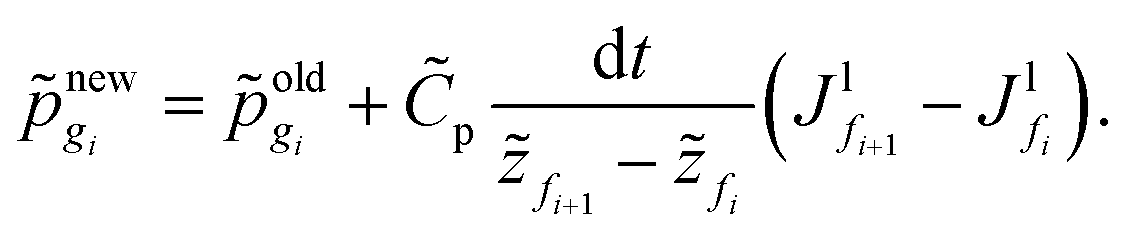 | (D.6) |
| fn+1 = 0. | (D.7) |
fM+1 and gM. For the liquid region, the top boundary condition changes to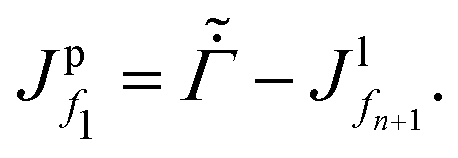 | (D.8) |
D.1 Liquid phase drying algorithm
For the liquid phase drying, we start with time = 0, polymer volume fraction ϕ = ϕ0 and = 0. We solve (21)–(23) to obtain the evolution of the polymer volume fraction in the coating. For this we employ the following algorithm.
Step 1. Set.
(a) = 0 and new = 1.
(b) f = [0, d, 2d,…,Nd].
(c) g = [d/2, 3d/2,…,(2N − 1)d/2],
(d) new = [f1, g1, f2, g2,…,gN, fN+1].
(e) ϕnew = [ϕf1,ϕg1,ϕf2,ϕg2,…,ϕgN,ϕfN+1], where ϕfi = ϕgi = ϕ0.
(f) new = [f1, g1, f2, g2,…,gN, fN+1], where fi = gi = 1.
Step 2. Set dl for the computations.
Step 3. Assign ϕold = ϕnew, old = new and old = new.
Step 4. Calculate the particle flux at the faces Jfi for fi = f2, f3,…,fN.
Step 5. From the particle fluxes obtain 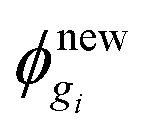 .
.
Step 6. Next obtain the size of the new element, i.e.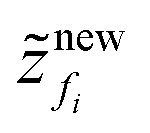 .
.
Step 7. Obtain the particle volume fraction at the new face coordinates, i.e. , either by interpolation or by extrapolation.
, either by interpolation or by extrapolation.
Step 8. Now evaluate the diffusion coefficient new at all the grid points and face points.
Step 9. Now update the time new = old + dt and the coating height  .
.
Step 10. Now divide the new region into 2N + 1 equispaced points new and obtain ϕnew and new at these points using linear interpolation.
Step 11. Save new, new, new, ϕnew and new to files for every N1 iterations.
Step 12. Now repeat from Step 3 until  (or)
(or) 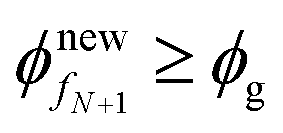 .
.
D.2 Liquid-skin phase drying algorithm
During the liquid-skin phase drying, we solve (24)–(26) and (27)–(29), respectively, to obtain the evolution of the polymer volume fraction in the skin and liquid regions. We follow the procedure given below to obtain the solutions.Step 1. If the liquid phase drying ends by satisfying 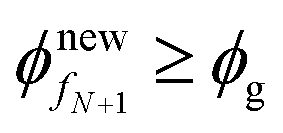 , then go to step 2. Otherwise go to step 3.
, then go to step 2. Otherwise go to step 3.
Step 2. In this case, the entire coating becomes the skin and we do the following:
(a) We assign the values of ϕnew from the liquid region directly to the skin.
(b) We find the permeability of the skin.
(c) We know the solvent flux in the coating during the liquid phase drying. We assign this solvent flux to the skin. This sets the solvent pore pressure (, ) in the skin.
(d) By employing , we obtain stress ![[small sigma, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e10d.gif) and strain ε in the coating.
and strain ε in the coating.
(e) We stop the liquid-skin phase drying and proceed to skin phase drying.
Step 3. In this case, the coating consists of both the skin and the liquid regions. We separate the skin and liquid regions to solve the corresponding governing equations. Before proceeding to solve the respective governing equations for liquid and skin regions, we do the following.
(a) Take the properties of the coating , , , ϕ, and from the end of liquid phase drying.
(b) Find the location in the coating, where the polymer volume fraction ϕ becomes ϕg, and assign the value to I, i.e. ϕ( = I, ) = ϕg. Here I represents the thickness of the skin (or) the location of the liquid and skin interface from the coating's top surface.
(c) Assign and ϕ of 0 ≤ ≤ I to s and ϕs, respectively.
(d) By employing ϕs, we obtain the permeability of the skin.
(e) Also, assign the solvent flux of 0 ≤ ≤ I to the skin's solvent flux, and find the pressure in the skin.
(f) Using the pressure, we obtain the stress and the strain ε in the skin region.
(g) Obtain news by dividing the skin region into 2n + 1 equal points. We now obtain ϕnews, new, new, new and εnew using interpolation. Also, obtain the solvent flux at new faces using interpolation. Here, n is the number of grid points in the skin region.
(h) Assign , ϕ and of I ≤ ≤ to l, ϕl, and l, respectively.
(i) Obtain newl by dividing the liquid region into 2N + 1 equal points. We now find ϕnewl and new using interpolation. We also find the solvent flux at new faces using interpolation. Here, N is the number of grid points in the liquid region.
(j) Finally, assign and to new and new, respectively.
(k) Save new, new, news, new, ϕnews, new, new, εnew, newl, ϕnewl and new to files.
Step 4. Set the time increment dls for the liquid-skin phase drying.
Step 5. Also, set old = new, old = new, olds = news, old = new, ϕolds = ϕnews, old = new, old = new, εold = εnew, oldl = newl, ϕoldl = ϕnewl and old = new.
Step 6. We solve the skin governing equations. For this we follow Steps 4–9 described in the next section, see Section D.3. Only difference is in the bottom boundary condition. This gives us news, new, ϕnews, new, new and εnew for the skin region. We also obtain the skin compression Δs = news,2n+1 − olds,2n+1.
Step 7. First, we shift oldl by Δs and assign it to oldl. We then calculate the solvent and particle fluxes at the new interface. By employing this particle flux, we solve the governing equations for the liquid region. For this, we follow Steps 4–8 in Section D.1 and obtain newl, ϕnewl and new. We also obtain the change in the height of the liquid region Δl = newl,2N + 1 − oldl,2N + 1.
Step 8. In the liquid region, we find out the location , where ϕl becomes ϕg. This gives us the location of the new interface znewI. Now we add this region (new skin), i.e. the region between newl,1 and znewI, to the existing skin. The properties in the new skin newns, newns, ϕnewns, newns, newns and εnewns are set by employing a procedure similar to that followed in Steps 3(c–f).
Step 9. We divide the skin (0 ≤ ≤ znewI) into 2n + 1 equal points and obtain news,f, new,f, ϕnews,f, new,f, new,f and εnew,f using interpolation.
Step 10. We divide the remaining liquid region into 2N + 1 equispaced points and obtain new,fl, ϕnew,fl and new,f using interpolation.
Step 11. Assign news = new,fs, new = new,f, ϕnews = ϕnew,fs, new = new,f, new = new,f, εnew = εnew,f, newl = new,fl, ϕnewl = ϕnew,fl and new = new,f.
Step 12. We update the time and the height of the coating by employing new = new + dls and new = old + Δs + Δl, respectively.
Step 13. Save new, new, news, new, ϕnews, new, new, εnew, newl, ϕnewl and new to files for every N2 iterations.
Step 14. If the height of the liquid region l(= new − newI) ≤ δl, we stop the iterations and save the coating properties to the files. Otherwise, we repeat the above from Step 5. Here δl is a very small value.
D.3 Skin phase drying algorithm
Step 1. Takenew, new, news, new, ϕnews, new, new and εnew at the end of liquid-skin phase drying.
Step 2. Set ds for the simulations.
Step 3. Assign old = new, old = new, news, olds = news, old = new, ϕolds = ϕnews, old = new, old = new and εold = εnew.
Step 4. Obtain new by solving the finite difference equations of pressure.
Step 5. Find the polymer volume fraction ϕnew in the skin by employing the procedure described later in Appendix E, i.e. (E.4).
Step 6. Evaluate the permeability of the skin region new = (1 − ϕnew)3/(ϕnew)2.
Step 7. Evaluate the size of the elements (or  ) by conserving the total amount of polymer.
) by conserving the total amount of polymer.
Step 8. Obtain news.
Step 9. Calculate stress new and strain εnew in the coating using new.
Step 10. Obtain new,fs by dividing the skin into 2n + 1 equal segments. We then find the properties new,f, ϕnew,f, new,f, new,f, and εnew,f by employing either interpolation or extrapolation.
Step 11. Assign news = new,fs, new = new,f, ϕnew = ϕnew,f, new = new,f, new = new,f and εnew = εnew,f.
Step 12. Update the time and height of the coating using new = old + ds and  , respectively.
, respectively.
Step 13. Save new, new, news, new, ϕnews, new, new and εnew to files for every N3 iterations.
Step 14. If ϕ1s < 1 or  (or |pnew − pold| > δp), repeat the above from step 3. Otherwise, terminate the simulations and save the final properties of the coating. Here δ and δp are small values.
(or |pnew − pold| > δp), repeat the above from step 3. Otherwise, terminate the simulations and save the final properties of the coating. Here δ and δp are small values.
Appendix E: calculation for skin element
Let us consider an element of thickness dzti (with unit width and depth), with the grid point gi, and faces fti and fti+1, at time t. The polymer volume fraction at the grid point (also within the element) is ϕi. The solvent fluxes at the faces fi and fi+1 are and
and 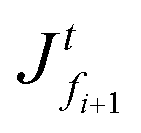 , respectively, as shown in Fig. 7. At time t, the total volumes of the element, polymer and solvent in the element are given by, respectively,
, respectively, as shown in Fig. 7. At time t, the total volumes of the element, polymer and solvent in the element are given by, respectively,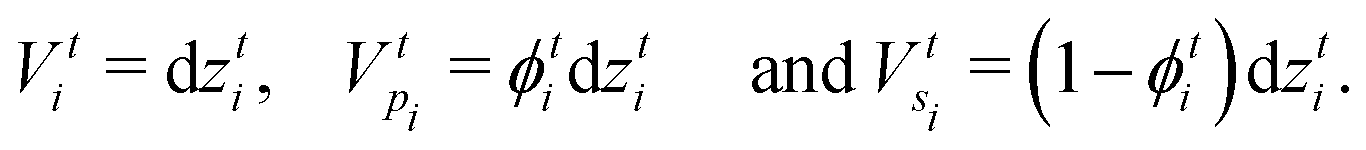 | (E.1) |
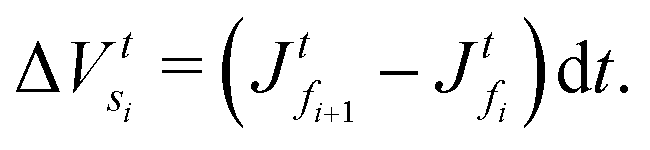 | (E.2) |
 . Hence, the element size (thickness) also changes by the same amount (represented as Δ(dzti)). Therefore, the total volumes of the element, polymer and solvent at time t + dt are
. Hence, the element size (thickness) also changes by the same amount (represented as Δ(dzti)). Therefore, the total volumes of the element, polymer and solvent at time t + dt areand
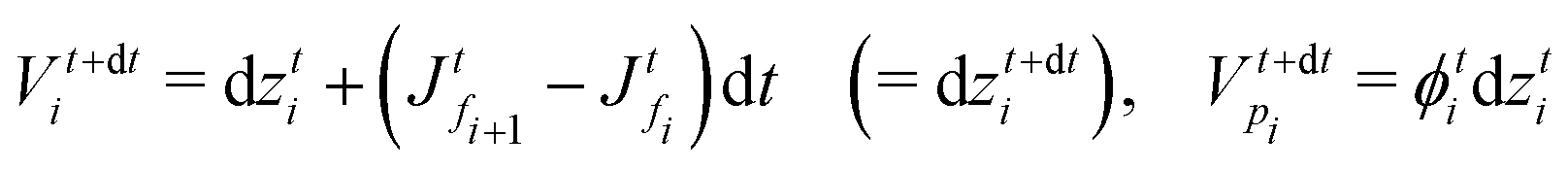
 | (E.3) |
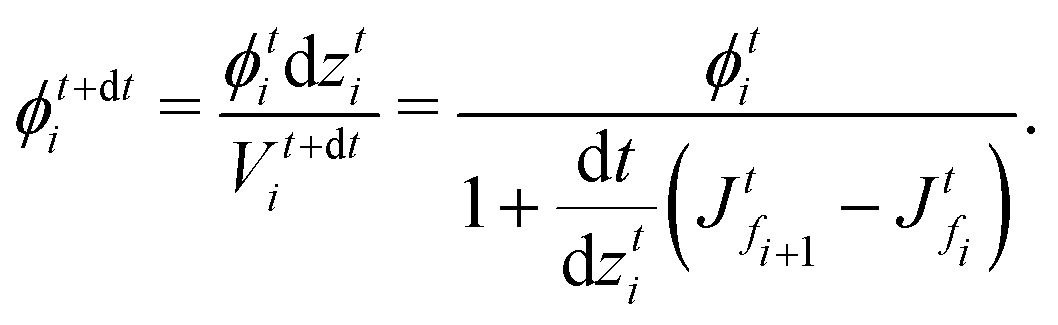 | (E.4) |
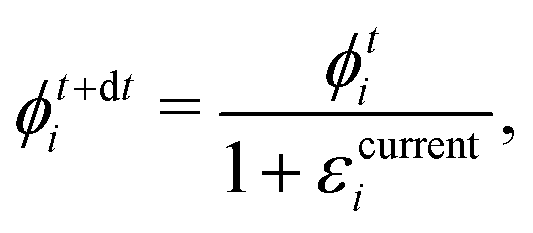 | (E.5) |
 | (E.6) |
 | ||
| Fig. 7 Sketch for skin element while drying. The red solid lines indicate the faces and the black dashed line indicates the grid point. The black arrows at the faces indicate the flux at those faces. | ||
Acknowledgements
VSP thanks the Department of Chemical Engineering, IIT Bombay, for financial support. MST acknowledges funding from SERB, Department of Science and Technology (Grant number: EMR/2017/001630).Notes and references
- K. Friedrich, Adv. Ind. Eng. Polym. Res., 2018, 1, 3–39 Search PubMed.
- K. D. Weiss, Prog. Polym. Sci., 1997, 22, 203–245 CrossRef CAS.
- Q. Wei and R. Haag, Mater. Horiz., 2015, 2, 567–577 RSC.
- L. Wu and J. Baghdachi, Functional polymer coatings: principles, methods, and applications, John Wiley & Sons, Hoboken, New Jersy, USA, 2015 Search PubMed.
- M. Hosseini and A. S. H. Makhlouf, Industrial applications for intelligent polymers and coatings, Springer, New York, USA, 2016 Search PubMed.
- W. Daughton and F. Givens, J. Electrochem. Soc., 1982, 129, 173–179 CrossRef CAS.
- J. J. Licari and L. A. Hughes, Handbook of polymer coatings for electronics: Chemistry, technology and applications, Noyes, Park Ridge, New Jersy, USA, 1990 Search PubMed.
- S. G. Croll, J. Coat. Technol., 1978, 50, 33–38 CAS.
- S. G. Croll, J. Appl. Polym. Sci., 1979, 23, 847–858 CrossRef CAS.
- S. G. Croll, J. Coat. Technol., 1980, 52, 35–43 CAS.
- K. Haghighi and L. Segerlind, Trans. ASAE, 1988, 31, 930–937 Search PubMed.
- K. Haghighi and L. Segerlind, Trans. ASAE, 1988, 31, 938–946 Search PubMed.
- M. Hasatani, Y. Itaya and K. Hayakawa, Drying Technol., 1992, 10, 1013–1036 CrossRef CAS.
- M. Hasatani, Y. Itaya, K. Muroie and S. Taniguchi, Drying Technol., 1993, 11, 815–830 CrossRef CAS.
- Y. Itaya, S. Mabuchi and M. Hasatani, Drying Technol., 1995, 13, 801–819 CrossRef CAS.
- S. Y. Tam, L. E. Scriven and H. K. Stolarski, Mater. Res. Soc. Symp. Proc., 1995, 356, 547–552 CrossRef CAS.
- E. H. Lee and D. T. Liu, J. Appl. Phys., 1967, 38, 19–27 CrossRef CAS.
- H. Lei, J. A. Payne, A. V. McCormick, L. F. Francis, W. W. Gerberich and L. E. Scriven, J. Appl. Polym. Sci., 2001, 81, 1000–1013 CrossRef CAS.
- H. Lei, L. Francis, W. Gerberich and L. Scriven, AlChE J., 2002, 48, 437–451 CrossRef CAS.
- K. N. Christodoulou, E. J. Lightfoot and R. W. Powell, AlChE J., 1998, 44, 1484–1498 CrossRef CAS.
- J. Rauch and W. Köhler, Phys. Rev. Lett., 2002, 88, 185901 CrossRef CAS.
- K. Ozawa, T. Okuzono and M. Doi, Jpn. J. Appl. Phys., 2006, 45, 8817–8822 CrossRef CAS.
- T. Okuzono, K. Ozawa and M. Doi, Phys. Rev. Lett., 2006, 97, 136103 CrossRef PubMed.
- L. F. Francis, A. V. McCormick, D. M. Vaessen and J. A. Payne, J. Mater. Sci., 2002, 37, 4717–4731 CrossRef CAS.
- B. S. Tomar, A. Shahin and M. S. Tirumkudulu, Soft Matter, 2020, 16, 3476–3484 RSC.
- P.-G. De Gennes, Scaling concepts in polymer physics, Cornell University Press, Ithaca, USA, 1979 Search PubMed.
- M. A. Biot, J. Appl. Phys., 1941, 12, 155–164 CrossRef.
- J. Kozeny, Royal Academy of Science, Vienna, Proc. Class I, 1927, vol. 136, pp. 271–306 Search PubMed.
- P. C. Carman, Trans. Inst. Chem. Eng., 1937, 15, 150–166 CAS.
- K. L. Buehler and J. L. Anderson, Ind. Eng. Chem. Res., 2002, 41, 464–472 CrossRef CAS.
- R. B. Bird, W. E. Stewart and E. N. Lightfoot, Transport Phenomena, John Wiley & Sons Inc., USA, 2002 Search PubMed.
Footnotes |
| † Changing the exponent to m = 3 in our computations does not change the predictions significantly. |
‡ We obtain K0 by equating the permeability obtained from (11) and eqn (6) and (7) of Buehler and Anderson30 at ϕ ≈ 0.43. At this ϕ, the value of  is close to one. The predicted trend matches their measurements over a wide range of ϕ. is close to one. The predicted trend matches their measurements over a wide range of ϕ. |
| § If the coating is assumed to be elasto-viscoplastic, the situation becomes complex as drying stress may decrease with decreasing Pe, since stress relaxes more quickly in presence of viscous dissipation.24 |
| This journal is © The Royal Society of Chemistry 2022 |