
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOperando isotope selective ammonia quantification in nitrogen reduction studies via gas chromatography-mass spectrometry†
Davide
Ripepi
 ,
Riccardo
Zaffaroni
,
Martin
Kolen
,
Joost
Middelkoop
and
Fokko M.
Mulder
*
,
Riccardo
Zaffaroni
,
Martin
Kolen
,
Joost
Middelkoop
and
Fokko M.
Mulder
*
Department of Chemical Engineering, Faculty of Applied Sciences, Delft University of Technology, 2629HZ Delft, The Netherlands. E-mail: F.M.Mulder@tudelft.nl
First published on 3rd March 2022
Abstract
Rapid advances in electrocatalytic ammonia synthesis are impeded by laborious detection methods commonly used in the field and by constant risk of external contaminations, which generates misleading false positives. We developed a facile real-time GC-MS method for sensitive isotope NH3 quantification, requiring no external sample manipulations. This method ensures high detection reliability paramount to accelerate (electro-)catalyst screening.
Ammonia (NH3) is an essential commodity at the base of modern agriculture, as well as a potential high energy density and carbon free energy carrier compound (22.5 MJ kg−1).1 Currently, the Haber–Bosch process produces NH3 at large scale from dinitrogen (N2) and hydrogen (H2) derived from steam-methane reforming. The overall ammonia synthesis process demands high pressure and temperature operating conditions and it has a significant impact on the worldwide energy consumption (1–2%) and carbon dioxide emissions (1.4%).2 In this context, direct electrochemical nitrogen reduction to ammonia, when powered by renewable energy sources, would have negligible carbon footprint and may represent a more flexible and decentralised option. In a future characterised by a large number of distributed renewable energy generation sites, such electrochemical approaches may provide an environmental friendly solution to the growing demand for green ammonia. The electrochemical conversion of N2 to NH3 involves, among other challenges,3,4 the arduous activation and stepwise hydrogenation of the N–N triple bond (941 kJ mol−1) and competition with the kinetically more favoured hydrogen evolution reaction (HER). This results in sluggish ammonia production rates (usually of the order of 10–10 mol NH3 cm−2 s−1) and poor faradaic efficiency (FE) around 10%.5 Values that are still far from the commercially relevant threshold of 10–6 mol NH3 cm−2 s−1 at 90% FE,6 thus revealing the relatively low maturity of current electrochemical ammonia synthesis technology and highlighting the urgent need for faster electro-catalyst development.
In recent years, the topic of electrochemical nitrogen reduction reaction (NRR) to ammonia has witnessed an increasing number of published scientific reports that have been later proven to be false positives, causing misspending of time and resources in pursuing misleading results.7,8 As such, due to the limited amount of ammonia produced in typical electrocatalytic experiments, rigorous protocols must be followed to categorically exclude false positive results that might arise from extraneous NH3 contaminations or from labile nitrogen-containing compounds9–14 (e.g. NOx). Recognised sources of contaminations in nitrogen reduction experiments include carrier gas,10 catalyst materials,15,16 electrolyte,17 membranes,18 human breath,19 ambient air20 and laboratory ware.21 Consequently, considerable effort and time are constantly dedicated to perform multiple control measurements and careful background quantifications. Reported ammonia synthesis results must include blank tests under an argon atmosphere, and under purified dinitrogen in an open circuit, as well as electroreduction of purified 15N2 gas. Purification is necessary to remove any 14/15NH3 or 14/15NOx from the feed gasses. The 15NH3 produced from 15N2, when reported over time and in quantitative agreement with the ammonia obtained with 14N2, represents the ultimate validation to confirm dinitrogen reduction to ammonia. Therefore, reliable and rapid detection methods, capable of quantitative discrimination of NH3 isotopologues, are paramount for high throughput catalyst testing and the advancement of this research field.
Conventional spectrophotometric techniques, like the Berthelot method, are not able to discriminate ammonium isotopes and suffer from serious interferences from solvents and other species present in solution, extensive sample manipulation and instability of derivatives. Recently, other analytical methods have been proposed with the aim to overcome the limitations of spectrophotometric ammonia detection;22,23 however, only nuclear magnetic resonance (NMR) and ultrahigh performance liquid chromatography-mass spectrometry (UPLC-MS) techniques allow selective detection of 15NH3 in labeling experiments.24,25
Quantitative 1H NMR is by far the most widely used isotope-sensitive technique for NH3 detection.26 Ammonia is detected in its acidified form, i.e. ammonium in solution (NH4+), and isotopic discrimination is based on the different nuclear spins of 14NH4+ and 15NH4+, which interact with the four nearby protons yielding respectively a triplet and a doublet in the proton NMR spectrum. With this technique, Nielander et al. reached high sensitivity (1 μM NH4+) with a state of the art cryoprobe equipped 900 MHz NMR spectrometer, but at the expense of a prohibitive data acquisition time of about one hour.24 However, a practical detection limit of about 10 μM has been reported for more accessible NMR spectrometers with magnetic fields ranging from 300 MHz to 400 MHz.24,27,28 Alternatively, UPLC-MS offers the advantage of a reduced acquisition time. However, this method relies on a chemical derivatization reaction of ammonia with dansyl chloride, which is susceptible to variations in pH.
Despite being powerful analytical methods, both these isotope-sensitive techniques are indirect, via acid traps or chemical derivatization, requiring different degrees of sample manipulation, and characterised by time-consuming procedures. Each step in the preparation of the analyte solution required for NMR, or ammonia derivatization with dansyl chloride, extends the overall analysis time and it represents a potential source of contamination and external errors. Real-time data acquisition is prohibitive and unpractical. Moreover, NMR and UPLC-MS measure the concentration of ammonia present in the electrolyte. As such, an extensive reaction time is required to accumulate sufficient ammonia to reach the lower limit of quantification (LOQ), above possible background contaminations, which also results in a larger consumption of expensive 15N2 gas (>500€ per L) during isotope labeling experiments. A single experiment can require up to several hours for reliable multiple point measurements (ESI – Section S.2†), representing one of the main bottlenecks in current research on electrocatalytic dinitrogen reduction. A direct gas phase NH3 detection able to distinguish 14NH3 from 15NH3, overcomes such limitations; moreover, it becomes particularly relevant when testing high current density electrochemical devices, such as gas diffusion electrodes, where the product of the reaction evolves in the gas stream.29–31
In this communication, we report a gas chromatography-mass spectrometry (GC-MS) method for quantitative isotope-sensitive ammonia detection in real-time and without any external sample manipulation, aiming to boost catalyst screening in the NRR. Ammonia is detected directly in the gas phase, with less than 2 min chromatographic elution time, by continuous sampling of the electrochemical cell headspace and regardless of the electrolyte nature.32 The proposed GC-MS method can quantify 15NH3/14NH3, down to 1 ppm in the gas phase, and simultaneously detect other gaseous species, giving a complete set of relevant information to better asses reaction selectivity and the presence of contaminants. We further compare the results obtained from GC-MS with those from 1H NMR measurements for the same mixed 15N and 14N ammonia solution, confirming the consistent quantification of mixed ammonia isotope samples with the two methods.
Mass spectrometry is a widely used approach in real-time trace gas analysis, capable of detecting multiple species even at ppb concentration levels.33 In a mass spectrometer, the ionization source converts the incoming molecules in detectable charged fragments. Ions with different mass-to-charge (m/z) ratios are then separated by the mass analyser and measured by the detector, thus allowing isotopic discrimination. The ionised fragments of a molecule produce a distinctive pattern in the m/z spectrum, which can be used for the identification of complex mixtures. Ammonia detection by MS at low concentrations is notoriously challenging, due to the overlapping m/z ratios of water and ammonia ionised fragments. Consequently, even small variations of the water background in the system impede reliable ammonia quantification. To circumvent this issue, we combined chromatographic separation with mass spectrometry. With the proposed system, aliquots of the gaseous analyte are directly collected from the reactor headspace by flowing a carrier gas, i.e. Ar, 14N2 or 15N2, avoiding any external sample manipulations. The ammonia concentration in equilibrium in the gas phase is strongly correlated with the ammonia present in the electrolyte, allowing a complete quantification of the produced NH3 in both liquid and gas phases.23 A small sample volume (500 μL) is then automatically injected into the GC column, via a set of switching diaphragm valves (ESI Fig. 1†). An Agilent Select Low Ammonia column, with suitable temperature and pressure settings (ESI Fig. 2†), offers adequate separation between ammonia and water. The two components of interest are rapidly eluted at different retention times and clearly discernible from the chromatographs in Fig. 1. Once eluted from the GC column, the analyte is partitioned between a pulse discharge detector (PDD) and a single quadrupole mass spectrometer (ISQ™ from Thermo Fisher Scientific), which simultaneously analyse the sample with matching retention times. Therefore, a mass spectrum is assigned to each peak in the chromatograph (Fig. 1). The optimisation of the split ratio of the analyte between the PDD and the mass spectrometer is a key aspect affecting the sensitivity of each detector. Restriction lines of suitable internal diameter and length were used to adjust the split ratio. In the present work, two 1 m long capillary lines of 0.15 mm and 0.25 mm internal diameter were connected to the MS and the PDD respectively. This configuration ensures a MS detection limit of 1 ppm of ammonia in the gas phase, allowing real time quantification of reaction rates down to a lower limit of 10−13 mol cm−2 s−1 (assuming 1 cm2 catalyst area and a constant carrier gas flowrate of 1 mL min−1). This detection range is already relevant for studies on the nitrogen reduction reaction. However, users have the option to tune, to a large extent, the ratio of MS and PDD flows to achieve even higher sensitivity either on the MS or PDD. Compared to previous ammonia detection methods, the presented GC-MS method can directly and simultaneously measure other species in the analyte with the MS detector or with dedicated extra channels usually installed in GC instruments (e.g. flame ionization detector, thermal conductivity detector, etc.). The advantage of a single analytical method that is not exclusively ammonia sensitive allows researchers to directly assess reaction selectivity by measuring reaction by-products, as well as the presence of gaseous/volatile contaminants in the experimental setup. For instance, gaseous NOx do not suffer from severe overlapping m/z fragments with other species commonly present in electrochemical nitrogen reduction experiments. Therefore NOx could be potentially discriminated by their characteristic mass spectrum, without requiring a strict chromatographic separation.
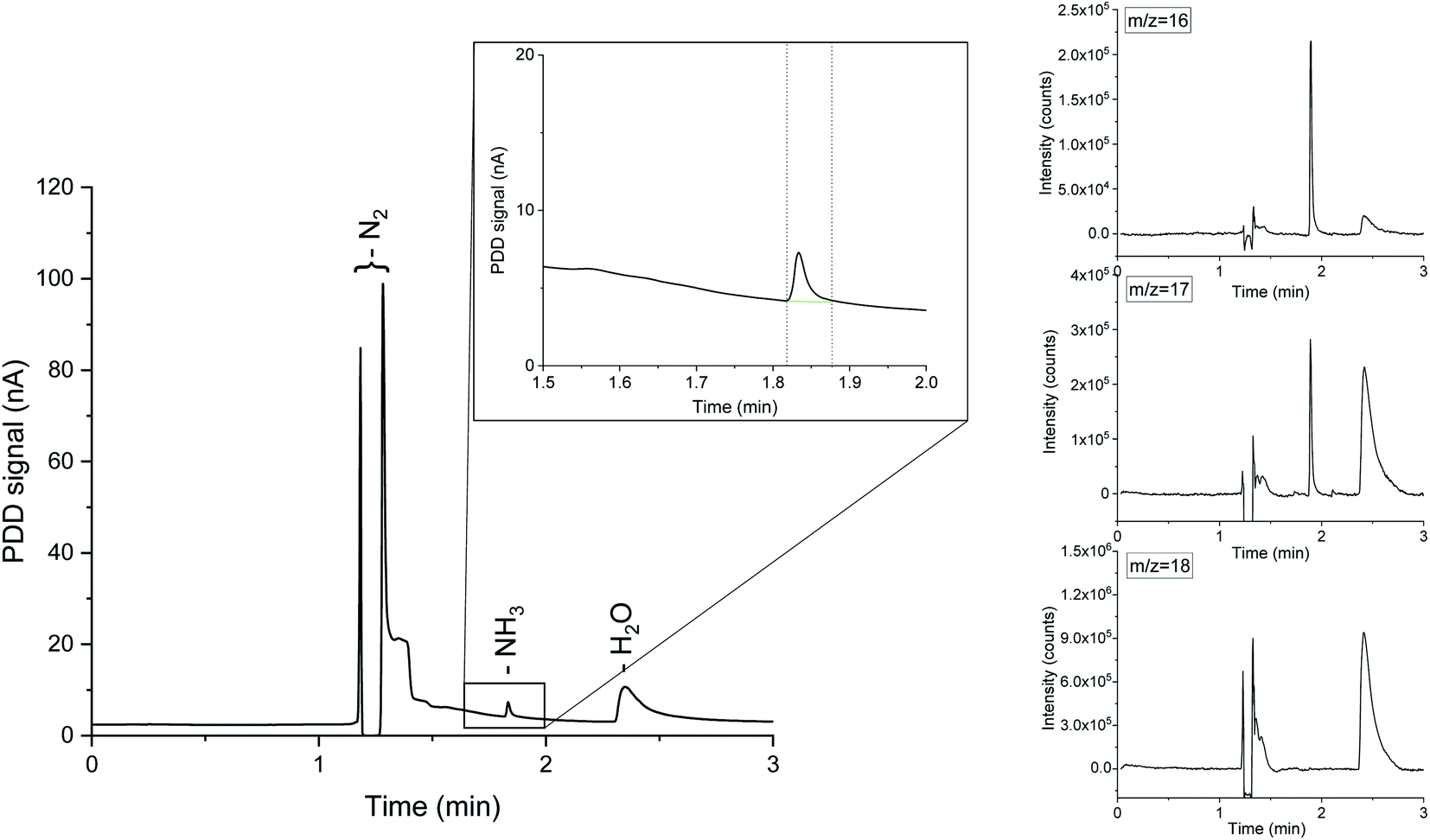 | ||
| Fig. 1 On the left, the full chromatograph corresponding to 13.8 ppm of NH3 in nitrogen. Ammonia is eluted at 1.83 minutes, while water appears at about 2.30 minutes. The enclosure highlights the region of the chromatograph around the ammonia elution time. On the right, the MS signal corresponding to the m/z 18, 17 and 16. At the ammonia retention time m/z 17 and 16 are the only contributions, demonstrating the absence of water interference. | ||
Instrument calibration was conducted by carefully diluting gaseous NH3 from certified calibration gas cylinders, with starting concentrations of 13.8 ppm and 2.2 ppm in N2. Ammonia detection is confirmed by the appearance of a peak in the chromatograph at a retention time of 1.83 min and separated from the water vapour present in the sample, which is eluted at about 2.30 min, as shown in Fig. 1. The electron ionization mass spectrum at the corresponding retention time validates the presence of ammonia with two main m/z fragments, 17 and 16, with relative intensities of 100% and 80.1% respectively (ESI Table 2†).34 On the other hand, the ionization of water generates fragments at m/z equal to 17 (21.2%) and 18 (100%) at 2.30 min. A constant background of the m/z 18 and 17 at the NH3 retention time (1.83 min) clearly indicates the absence of water interferences in the ammonia quantification (Fig. 1 and ESI Fig. 3†). The integrated MS signal of both m/z ratio 16 and 17 at ammonia retention time shows a linear behaviour in the range 1 ppm to 13.8 ppm (Fig. 2 and ESI Table 1†). As such, mass-sensitive ammonia detection can be achieved accurately with a coupled GC-MS, even in the presence of water vapour (ESI Fig. 3–6†), resolving the challenge of overlapping water/ammonia ionization fragments encountered in mass spectrometers alone.
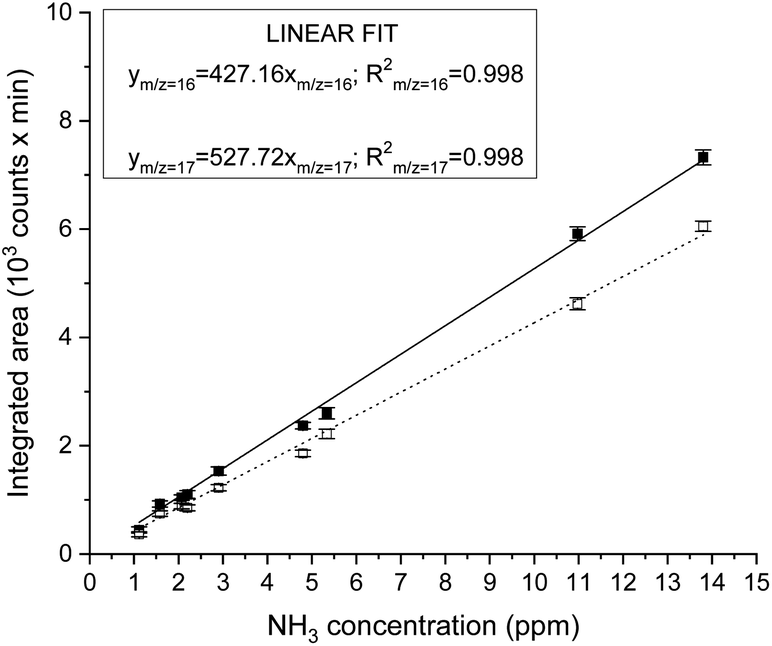 | ||
| Fig. 2 Ammonia calibration based on the integrated MS signal at m/z 17 (closed symbol) and 16 (open symbol), at 1.83 elution time. The calibration was carried out by the dilution of 13.8 ppm or 2.2 ppm certified NH3 calibration gases in nitrogen with purified N2. | ||
N-labeled control experiments in the field electrochemical ammonia synthesis require analytical methods to quantitatively discriminate between 15N and 14N ammonia. To demonstrate the capability of the presented GC-MS method in an environment comparable to a NRR experiment in aqueous electrolytes and in the presence of both isotopologues of NH3, we prepared a solution by mixing 28% 14N (BASF) and 3M 15N (Isotec, 98+ % 15N) ammonia aqueous solutions, to achieve a final 14NH3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15NH3 concentration of 0.21:0.68 mM. This mixed ammonia solution was then enclosed in a sealed vial equipped with a gas inlet and outlet, where 2.5 mL min−1 of N2 (6N) were allowed to flow at the headspace of the vial (Fig. 3a). The gas outlet was directly connected to the GC-MS. The chromatograph corresponding to the PDD detector shows only a single NH3 peak (Fig. 3b); therefore it is unable to distinguish between the ammonia derived from the two nitrogen isotopes. Yet, the contributions from 15NH3 and 14NH3 can be determined with the MS detector connected in parallel (Fig. 3c). The electron ionization mass spectrum of 15NH3 is characterised by three main contributions at m/z 18, 17 and 16, with relative intensities of 100, 80.1 and 7.5% respectively (ESI Table 2 and Fig. 4†).34 Therefore, 15N and 14N ammonia can be quantified from the integrated intensities corresponding to their ionised fragments (Fig. 3e and ESI Table 3†), as reported in the calculations available in the ESI (eqn (2)–(5)†). As such, the 15NH3/14NH3 ratio resulting from direct headspace analysis via GC-MS is equal to 3.30 ± 0.06, which is in close agreement with the 1H NMR analysis of an aliquot of the same solution (3.25 ± 0.04, Fig. 3d). The details of the NMR method used in this contribution have been presented elsewhere27 and in the ESI (Section S.5 and Table 4†) together with the data processing of Fig. 3d.
:15NH3 concentration of 0.21:0.68 mM. This mixed ammonia solution was then enclosed in a sealed vial equipped with a gas inlet and outlet, where 2.5 mL min−1 of N2 (6N) were allowed to flow at the headspace of the vial (Fig. 3a). The gas outlet was directly connected to the GC-MS. The chromatograph corresponding to the PDD detector shows only a single NH3 peak (Fig. 3b); therefore it is unable to distinguish between the ammonia derived from the two nitrogen isotopes. Yet, the contributions from 15NH3 and 14NH3 can be determined with the MS detector connected in parallel (Fig. 3c). The electron ionization mass spectrum of 15NH3 is characterised by three main contributions at m/z 18, 17 and 16, with relative intensities of 100, 80.1 and 7.5% respectively (ESI Table 2 and Fig. 4†).34 Therefore, 15N and 14N ammonia can be quantified from the integrated intensities corresponding to their ionised fragments (Fig. 3e and ESI Table 3†), as reported in the calculations available in the ESI (eqn (2)–(5)†). As such, the 15NH3/14NH3 ratio resulting from direct headspace analysis via GC-MS is equal to 3.30 ± 0.06, which is in close agreement with the 1H NMR analysis of an aliquot of the same solution (3.25 ± 0.04, Fig. 3d). The details of the NMR method used in this contribution have been presented elsewhere27 and in the ESI (Section S.5 and Table 4†) together with the data processing of Fig. 3d.
 | ||
| Fig. 3 (a) Schematic representation of the headspace measurement of a 15NH3/14NH3 (0.21/0.68 mM) aqueous solution. (b) Full chromatograph. (c) Closed up, around the ammonia elution time (1.83 min) of the MS signal related to the m/z 18, 17 and 16. (d) 1H NMR measurement of the 15NH3/14NH3 aqueous solution, in agreement with the GC-MS quantification. Line fitting is shown as blue dotted lines. (e) Relative intensity of the integrated m/z at 1.83 min. | ||
Conclusions
The aim of this study is to present an isotope sensitive method for the determination of NH3 at a low concentration level, typically encountered in electrochemical ammonia synthesis applications. In this fast-growing field, the detection of NH3 is of primary relevance due to the increased concern over the need for more reliable experiments. Compared with previously reported NH3 detection methods, which rely on extensive NH3 accumulation in a liquid medium (electrolyte or acid trap) and subsequent indirect quantification, the presented GC-MS method detects ammonia in less than 2 minutes directly from the gas phase with 1 ppm detection limit (equals to rates as low as 10−13 molNH3 cm−2 s−1 at a carrier gas flowrate of 1 mL min−1). The proposed approach requires no external sample manipulations, resulting in the lowest risk of external contaminations and reduced analysis time. GC-MS detection is mass sensitive and therefore suitable for 15N-labelled control experiments. As such, the direct, fast and sensitive ammonia quantification reduces the usage of expensive 15N2, cutting down the, sometimes prohibitive, experimental costs both in terms of time and reagents. Contrary to conventional MS detection, the accurate quantification of a mixture of 14NH3 and 15NH3 with GC-MS is not influenced by background water, which is eluted at a different time. Importantly, this gas analysis method is not exclusively ammonia sensitive and therefore it can also provide additional information on by-products and the level of contaminants.In summary, we provide a fast and highly reliable mass and isotope sensitive NH3 detection method with unique capabilities. This approach accelerates the testing and discovery of new active catalysts for the NRR and therefore it should become a standard method essential for publications involving ammonia synthesis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is part of the Open Technology research program with Project No. 15234 which is (partly) financed by The Netherlands Organisation for Scientific Research (NWO).Notes and references
- F. M. Mulder, J. Renewable Sustainable Energy, 2014, 6, 033105 CrossRef.
- D. R. MacFarlane, P. V. Cherepanov, J. Choi, B. H. R. Suryanto, R. Y. Hodgetts, J. M. Bakker, F. M. Ferrero Vallana and A. N. Simonov, Joule, 2020, 4, 1186–1205 CrossRef CAS.
- A. R. Singh, B. A. Rohr, J. A. Schwalbe, M. Cargnello, K. Chan, T. F. Jaramillo, I. Chorkendorff and J. K. Nørskov, ACS Catal., 2017, 7, 706–709 CrossRef CAS.
- L. Hu, Z. Xing and X. Feng, ACS Energy Lett., 2020, 5, 430–436 CrossRef CAS.
- B. Yang, W. Ding, H. Zhang and S. Zhang, Energy Environ. Sci., 2021, 14, 672–687 RSC.
- U. S. DOE, Renewable Energy to Fuels Through Utilization of Energy-Dense Liquids (REFUEL) Program Overview, 2016 Search PubMed.
- J. Choi, H.-L. Du, C. K. Nguyen, B. H. R. Suryanto, A. N. Simonov and D. R. MacFarlane, ACS Energy Lett., 2020, 5, 2095–2097 CrossRef CAS.
- S. Licht, B. Cui, B. Wang, F.-F. Li, J. Lau and S. Liu, Science, 2020, 369, 780 CrossRef CAS PubMed.
- S. Z. Andersen, V. Čolić, S. Yang, J. A. Schwalbe, A. C. Nielander, J. M. McEnaney, K. Enemark-Rasmussen, J. G. Baker, A. R. Singh, B. A. Rohr, M. J. Statt, S. J. Blair, S. Mezzavilla, J. Kibsgaard, P. C. K. Vesborg, M. Cargnello, S. F. Bent, T. F. Jaramillo, I. E. L. Stephens, J. K. Nørskov and I. Chorkendorff, Nature, 2019, 570, 504–508 CrossRef CAS PubMed.
- J. Choi, B. H. R. Suryanto, D. Wang, H.-L. Du, R. Y. Hodgetts, F. M. Ferrero Vallana, D. R. MacFarlane and A. N. Simonov, Nat. Commun., 2020, 11, 5546 CrossRef CAS PubMed.
- Y. Zhao, R. Shi, X. Bian, C. Zhou, Y. Zhao, S. Zhang, F. Wu, G. I. N. Waterhouse, L.-Z. Wu, C.-H. Tung and T. Zhang, Adv. Sci., 2019, 6, 1802109 CrossRef PubMed.
- Y. Zhao, F. Wu, Y. Miao, C. Zhou, N. Xu, R. Shi, L.-Z. Wu, J. Tang and T. Zhang, Angew. Chem., Int. Ed., 2021, 60, 21728–21731 CrossRef CAS PubMed.
- C. Tang and S.-Z. Qiao, Joule, 2019, 3, 1573–1575 CrossRef.
- U. B. Shahid, Y. Chen, S. Gu, W. Li and M. Shao, Trends Chem., 2022, 4, 142–156 CrossRef.
- Y. Chen, H. Liu, N. Ha, S. Licht, S. Gu and W. Li, Nat. Catal., 2020, 3, 1055–1061 CrossRef CAS.
- W. Yu, P. Buabthong, C. G. Read, N. F. Dalleska, N. S. Lewis, H.-J. Lewerenz, H. B. Gray and K. Brinkert, Sustainable Energy Fuels, 2020, 4, 5080–5087 RSC.
- L. Li, C. Tang, D. Yao, Y. Zheng and S.-Z. Qiao, ACS Energy Lett., 2019, 4, 2111–2116 CrossRef CAS.
- H. Liu, Y. Zhang and J. Luo, J. Energy Chem., 2020, 49, 51–58 CrossRef.
- C. Turner, P. Španěl and D. Smith, Physiol Meas., 2006, 27, 321–337 CrossRef PubMed.
- W. H. Schlesinger and A. E. Hartley, Biogeochemistry, 1992, 15, 191–211 CrossRef CAS.
- T. Ishibashi, M. Himeno, N. Imaizumi, K. Maejima, S. Nakano, K. Uchida, J. Yoshida and M. Nishio, Nitric Oxide, 2000, 4, 516–525 CrossRef CAS PubMed.
- Y.-X. Lin, S.-N. Zhang, Z.-H. Xue, J.-J. Zhang, H. Su, T.-J. Zhao, G.-Y. Zhai, X.-H. Li, M. Antonietti and J.-S. Chen, Nat. Commun., 2019, 10, 4380 CrossRef PubMed.
- R. Zaffaroni, D. Ripepi, J. Middelkoop and F. M. Mulder, ACS Energy Lett., 2020, 5, 3773–3777 CrossRef CAS.
- A. C. Nielander, J. M. McEnaney, J. A. Schwalbe, J. G. Baker, S. J. Blair, L. Wang, J. G. Pelton, S. Z. Andersen, K. Enemark-Rasmussen, V. Čolić, S. Yang, S. F. Bent, M. Cargnello, J. Kibsgaard, P. C. K. Vesborg, I. Chorkendorff and T. F. Jaramillo, ACS Catal., 2019, 5797–5802, DOI:10.1021/acscatal.9b00358.
- W. Yu, N. S. Lewis, H. B. Gray and N. F. Dalleska, ACS Energy Lett., 2020, 5, 1532–1536 CrossRef CAS.
- As matter of fact, nitrogen has two NMR active nuclei; therefore N NMR could also be used to discriminate 14NH3 and 15NH3. However, both 15N and 14N NMR are significantly less sensitive than the heteronuclear coupling observed in 1H NMR, leaving the latter as a better option compared to N NMR..
- M. Kolen, W. A. Smith and F. M. Mulder, ACS Omega, 2021, 6, 5698–5704 CrossRef CAS PubMed.
- R. Y. Hodgetts, A. S. Kiryutin, P. Nichols, H.-L. Du, J. M. Bakker, D. R. Macfarlane and A. N. Simonov, ACS Energy Lett., 2020, 5, 736–741 CrossRef CAS.
- N. Lazouski, M. Chung, K. Williams, M. L. Gala and K. Manthiram, Nat. Catal., 2020, 3, 463–469 CrossRef CAS.
- T. Burdyny and W. A. Smith, Energy Environ. Sci., 2019, 12, 1442–1453 RSC.
- D. Ripepi, R. Zaffaroni, H. Schreuders, B. Boshuizen and F. M. Mulder, ACS Energy Lett., 2021, 3817–3823, DOI:10.1021/acsenergylett.1c01568.
- The proposed GC-MS method is suitable for quantification of gaseous NH3 evolving directly in the gas phase or from an electrolytic cell (with organic, inorganic, aqueous or non-aqueous electrolyte). However, the authors acknowledge that when ammonia is produced directly in the electrolyte this should have a pH above 9. On the other hand, at pH lower than 9 the chemical equilibrium is increasingly shifted towards the formation of ammonium ions (NH4+), which will remain in solution requiring a liquid analysis. This limitation could be overcome by externally shifting the pH at the end of the electrochemical experiment..
- C. D. Mowry, R. L. Jarek, J. Román-Kustas, A. C. Telles and A. S. Pimentel, in Materials Characterization, ASM International, 2019, vol. 10 Search PubMed.
- E. W. William, in NIST Chemistry WebBook, NIST Standard Reference Database Number 69, ed. P. J. Linstrom and W. G. Mallard, National Institute of Standards and Technology, Gaithersburg MD, p. 20899, DOI:10.18434/T4D303, retrieved January 27, 2022.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2se00123c |
| This journal is © The Royal Society of Chemistry 2022 |