
Plasma enhanced anti-coking performance of Pd/CeO2 catalysts for the conversion of methane†
Xiucui
Hu
 a,
Yadi
Liu
a,
Liguang
Dou
a,
Cheng
Zhang
ab,
Shuai
Zhang
ab,
Yuan
Gao
a,
Xin
Tu
c and
Tao
Shao
*ab
a,
Yadi
Liu
a,
Liguang
Dou
a,
Cheng
Zhang
ab,
Shuai
Zhang
ab,
Yuan
Gao
a,
Xin
Tu
c and
Tao
Shao
*ab
aBeijing International S&T Cooperation Base for Plasma Science and Energy Conversion, Institute of Electrical Engineering, Chinese Academy of Sciences, Beijing 100190, China. E-mail: st@mail.iee.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
cDepartment of Electrical Engineering and Electronics, University of Liverpool, Liverpool L69 3GJ, UK
First published on 10th November 2021
Abstract
The direct nonoxidative conversion of methane (CH4) to valuable chemicals has attracted increasing interest. However, the carbon deposition will inevitably occur due to CH4 decomposition at high temperature. Here, we report the conversion of CH4 assisted by non-thermal plasma into olefins and H2 over a Pd/CeO2 catalyst. The addition of the plasma could effectively enhance the anti-coking performance of the catalyst, facilitating the conversion of CH4. The interaction between plasma and catalyst was explored in detail. The energized electron and ions generated by plasma could interact with the adsorbed CH3 species to efficiently suppress the consequent dehydrogenation of the adsorbed CH3, accelerating the desorption of the CH3 species from the surface of the catalyst, thus reducing the amount of carbon deposition on the catalyst surface. The highly efficient catalyst assisted by plasma is effective in enhancing the CH4 conversion and suppressing the carbon deposition, which deserves to be widely applied in catalysis.
1. Introduction
The continuous fossil energy consumption and the increasingly emerging environmental problems have demanded widespread attention toward alternative renewable and sustainable energy resources.1 In particular, CH4 as a primary component of natural gas is considered one of the alternatives to nonrenewable coal and crude oil due to its huge reserves, and its upgrading to valuable hydrocarbon feedstocks (such as aromatics, olefins, oxygenates) and hydrogen.2–4 However, excessive cracking of CH4 can lead to carbon deposition and catalyst deactivation. Thus, strategies to minimize the excessive cracking of CH4 are important in CH4-containing reactions, such as the conversion of CH4 and biogas reforming technology (CH4/CO2 conversion).5The CH4 conversion can be conventionally achieved either by indirect methods concerning multiple catalytic transformations, such as through synthesis gas (a mixture of CO and H2)6,7 and methanol,8 or by direct methods consisting of the oxidative coupling of methane (OCM),9,10 nonoxidative dehydroaromatization of methane (MDA)11,12 and nonoxidative conversion of methane to olefins, aromatics, and hydrogen (MTOAH).13–15 Among them, the MTOAH route is a promising approach for CH4 conversion to C2 products (C2H4 and C2H2) and hydrogen due to zero emissions of carbon dioxide and maximum carbon atom utilization.13,14 However, it still remains a grand challenge to achieve the CH4 activation at low temperature because of the highest C–H bond strength (434 kJ mol−1), the high ionization energy, the low electron affinity and polarizability.16,17
It has been reported that single Fe atoms embedded in the SiO2 matrix (Fe@SiO2) at 950–1090 °C possessed high catalytic selectivity for the nonoxidative conversion of CH4 to ethylene (C2H4), aromatics (benzene, naphthalene) and H2.13 The conversion of CH4 reached 48.1% and the selectivity of ethylene exceeded 48% at 1090 °C. The C–H of CH4 was activated over atomic Fe sites to form methyl (CH3) radicals. The CH3 radicals diffused easily from the catalyst surface into the gas phase to trigger a chain reaction (CH3 + CH3 → C2H6), which subsequently underwent a series of gas-phase reactions to generate the target products. No coke deposition was observed because the absence of Fe ensembles suppressed the C–C coupling and carbon coking under the high-temperature catalytic conditions.13 The development of the stable single-atom catalyst at high temperature during the CH4 conversion reaction provides a new route to inhibit carbon deposition. Dipu et al. reported that the CH4 was activated over the Ni–P/SiO2 catalysts,15 which showed the high selectivity (99.9%) of ethane and ethylene at 850 °C with the low selectivity (0.1%) of the carbon (C) deposition, although the conversion of methane was only 0.08%. The activation of CH4 first generated ethane (C2H6) via two CH3 radicals coupled to each other as the primary product on the catalyst surface, and then the thermal conversion of C2H6 to other target products (olefins and benzene) in the gas phase.15 The above results showed that for the CH4 conversion, it is crucial to suppress the carbon deposition through tuning the generation and desorption of the CH3 species over catalysts and guiding the subsequent series of reactions in the gas phase. However, the active metal species not only aggregate inevitably into larger nanoparticles, but also are easily inactivated at high temperatures (ca. 1000 °C). Therefore, an innovative technique for the direct conversion of the CH4 process is highly desirable.
The low-temperature activation of CH4 with highly reactive catalysts assisted by non-thermal plasma (NTP) is a promising strategy to achieve the CH4 conversion more efficiently and selectively because NTP can generate extremely active electrons with a mean electron energy of 1–10 eV,18–20 which can activate inert molecules (e.g., CH4 or CO2) at atmospheric pressure and low temperatures into the reactive species, such as excited atoms, molecules, ions and radicals (CH3 radicals and H radicals).21,22 Until now, much effort has concentrated on the direct nonoxidative conversion of methane using various NTPs with or without a catalyst, including dielectric barrier discharge (DBD),23,24 pulsed discharge,25,26 spark discharge,27 radio-frequency discharge,28 corona discharge29 and microwave discharge.30 In particular, the nanosecond pulsed discharge (NPD) has attracted much attention because of its unique features, such as higher energy efficiency,31 the extreme non-equilibrium character,32 massive high energy electrons,33 and the short pulse duration suppressing the transition from plasma to thermal-equilibrium state.34 However, the high conversion of CH4 and selectivity of the olefins still remain a grand challenge. Thus, combining both advantages of the catalyst and plasma was a promising route.
For the efficient catalyst materials, ceria (CeO2) as a well-known functional rare earth material has been extensively used as a catalyst support in a variety of catalytic reactions owing to its unique oxygen storage and release capacity.35 In addition, ceria can greatly overcome the sintering of the deposited metal to disperse and stabilize the metals as small-size species, even ultra-fine clusters or single atoms at high temperature, by tuning the interaction between the active metal and CeO2 support.36,37 Ceria-supported Pd catalysts have shown some promising properties for CH4 activation,35,38 while the formation of carbon deposition is disadvantaged for its catalytic applications.
Herein, we reported Pd/CeO2 with low loading for the direct nonoxidative conversion of CH4 into light hydrocarbons and H2 assisted by nanosecond pulsed DBD at atmospheric pressure to reduce the amount of carbon deposition. A notable enhancement on the CH4 conversion for the plasma-catalysis is compared with the catalysis-only. The introduction of plasma strongly increased the coke-resistance of the catalyst by the electron-impact with the catalyst surface. The possible reaction mechanisms in the conversion of CH4 assisted by plasma with or without a catalyst were proposed by a range of catalyst characterizations, product analysis and the plasma kinetic modeling.
2. Experimental
2.1 Synthesis of catalysts
CeO2 supports and the Pd/CeO2 catalysts were synthesized by the reported hydrothermal method39 and deposition–precipitation method,40 respectively. The detailed synthesis process can be seen in the ESI.†2.2 Characterization of catalysts
Powder X-ray diffraction (XRD) was performed on a D8 ADVANCE (Bruker, Germany) diffractometer with Cu Kα radiation. The Raman spectra were obtained using a Raman microscope (inVia, RENISHAW, England) system by excitation of the sample at 532 nm with a measurement range from 100 to 4000 cm−1. High-resolution transmission electron microscopy (HR-TEM) was carried out on a JEM-2100F microscope (Japan) at 200 kV. The high-angle annular dark-field scanning transmission electron micrograph (HAADF-STEM) images and the corresponding elemental mappings were performed on the same instrument. The field-emission scanning electron microscopy (FE-SEM) combined with the energy dispersive X-ray analysis (EDS) was operated on a SIGMA microscope (Zeiss Merlin Compact, Germany). Before the test, the samples were dispersed in the ethanol and then dripped onto aluminum foil to accurately determine the carbon content. All analyzed elements were normalized and the carbon content was tested three times. The N2 adsorption–desorption measurement was operated at −196 °C on an ASAP 2020HD88 unit (Micromeritics, America) to obtain the specific surface area (SBET) values of each sample. X-ray photoelectron spectroscopy (XPS) was operated with Al Kα radiation (ESCALab-250Xi). The binding energy of all spectra was calibrated using the C 1s signal at 284.6 eV. Temperature programmed reduction by hydrogen (H2-TPR) was carried out on a Micromeritics Autochem II 2920 analyzer with a thermal conductivity detector (TCD). The fresh 0.5Pd/CeO2 sample (50 mg) was pretreated in air at 300 °C before the test. Then, the catalyst was heated in 10% H2/Ar (30 mL min−1) gas mixture from room temperature to 600 °C. Temperature programmed oxidation by oxygen (O2-TPO) was performed at the same analyser (Micromeritics Autochem II 2920) with a TCD. First, the used samples (100 mg) were activated at 300 °C in He. Then, the samples were heated in 2% O2/He (30 mL min−1) gas mixture from room temperature to 800 °C. Thermogravimetric (TG) analysis was carried out on a TG-DTA6300 instrument. The sample was treated in air from room temperature to 800 °C (10 °C min−1). The optical emission spectroscopy (OES) from the plasma in the CH4 DBD reactor was recorded using a spectrograph via an optical fiber equipped with a Princeton Instruments ICCD camera (Andor DH334T) in the range of 300–700 nm. During the test, the optical fiber was placed close to the ground electrode of the DBD reactor. For the spectrometer, the slit width was fixed at 50 μm and the grating groove density was specified to 1200 mm−1.2.3 Catalytic tests
The nonoxidative conversion of CH4 was performed in a DBD reactor, as shown in Fig. 1, in which the catalytic tests were performed at atmospheric pressure with a catalyst bed in the discharge zone. The reactor was a typical cylindrical DBD quartz tube (14 mm o.d. × 10 mm i.d.) reactor, which used a stainless-steel foil covering outside of the quartz tube as the grounding electrode and a stainless-steel rod (6 mm o.d.) placed along the axis of the quartz tube as the high voltage electrode. The quartz tube also served as the dielectric between the high voltage electrode and the grounding electrode. In the reactor, a sieve plate was embedded and used to fix the high voltage electrode. The discharge length of the DBD reactor was 130 mm. The discharge gap was 2 mm. The DBD reactor was connected to a nanosecond pulse power supply (Smart Maple HV-2015, China), which provided a peak voltage of 13 kV and an adjustable frequency. The current and voltage of the external capacitor and the actual applied voltage were detected by a digital oscilloscope (Tektronix, DPO 2024). In the plasma-catalysis test, 0.5 g of catalyst (20–40 mesh) was packed into the discharge area and pure CH4 was introduced into the DBD reactor with a gas hourly space velocity (GHSV) of 9800 mL g−1 h−1. Prior to the reaction, the catalyst was activated at 980 °C for 30 min in 10% H2/Ar gas mixture. For the temperature-dependent CH4 conversion test, the reactor temperature was controlled from 800 to 980 °C. At each temperature, the temperature is constant and the same as the set value. Each reaction was allowed to equilibrate for 1 h, and then the concentrations of outlet gases were analyzed. Meanwhile, the experiments were repeated three times and the average of the three measurements was taken. The gaseous products were analyzed via a gas chromatograph (Thermo-Fisher, Trace 1300) equipped with a thermal conductivity detector (TCD), as well as a flame ionized detector (FID), and calculated by an internal standard method. The TCD with a TDX-01 packed column was used to quantify the CH4 and H2. The light hydrocarbons including C2H6, C2H4, C3H8, C3H6, C4H10 and C2H2 were quantified through the FID with a HPPLOT Al2O3 capillary column.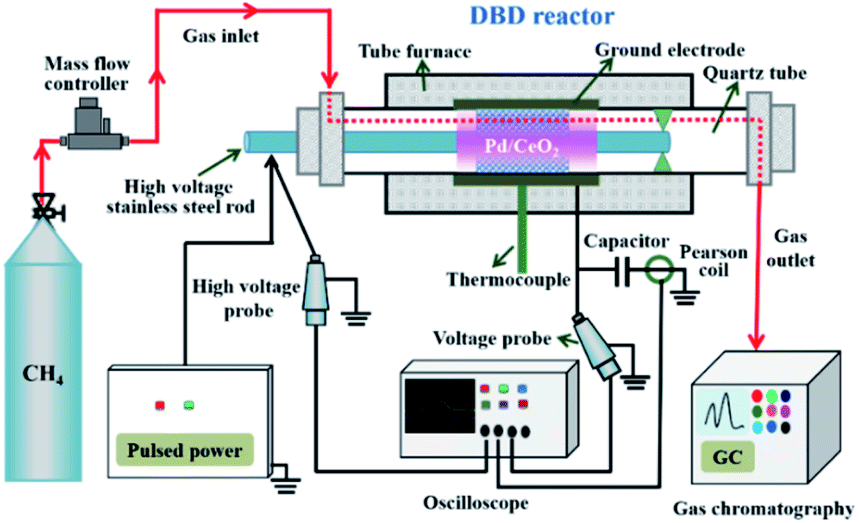 | ||
| Fig. 1 Schematic diagram of the fixed bed DBD reactor setup for the nonoxidative conversion of CH4. | ||
The calculations on CH4 conversion (CCH4), hydrocarbon products selectivity (SCxHy) and yields (YCxHy), H2 selectivity (SH2) and yield (YH2), carbon balances (BC), hydrogen balances (BH) and energy consumption (EC) of the discharge are shown in the ESI.†
2.4 Density functional theory calculation methods
Density functional theory (DFT) calculation was applied with the Castep program package with the GGA and PW91 functional in the package of Material Studio to simulate the conversion of CH4.41,42 Because of the nonmagnetic properties of Pd, the spin polarization effect was not considered. The convergence criterion for the energy, maximum force, stress and energy cutoff were set at 2 × 10−5 eV per atom, 0.05 eV Å−1, 0.1 GPa and 400 eV, respectively. A density mixing electronic minimizer with a mixing scheme of Pulay was used, and the convergence criteria for the self-consistent field (SCF) was set as 1.0 × 10−5 eV per atom. The Brillouin zone was sampled by a k-point of 3 × 3 × 1. The transition state (TS) was determined using the LST/QST method.The Pd (111) surface was modelled by a three-layer slab within a (3 × 3) super cell (27 Pd atoms in the cell), and the positions of all atoms (except for those in the bottom layer) were fully relaxed. A vacuum of 10 Å along the Z-direction was applied to avoid interactions between the periodic images.
The adsorption energy was calculated as:
| Eads = Eadsorbates/slab − Eslab − Eadsorbates | (1) |
The activation barrier Ea and reaction energy ΔE are defined as:
| Ea = E(TS) − E(IS) | (2) |
| ΔE = E(FS) − E(IS) | (3) |
2.5 Plasma kinetic modelling
A zero-dimensional (0D) plasma kinetic modelling was performed to elucidate the main reaction mechanism in the plasma for the conversion of CH4 without 0.5Pd/CeO2. The plasma chemistry consisting of 23 species (i.e., neutrals, radicals and charged species) and 132 reactions together with the corresponding rate coefficients are listed in Table S1 (see the ESI†). The rate coefficients related to the electron-impact reactions were calculated by a Boltzmann solver Bolsig+,43 according to the energy-dependent collision cross sections44,45 between the electrons and molecules. The rate coefficients of the electron–ion, ion–neutral and neutral–neutral reactions were usually a function of the gas temperature Tg. All species were assumed to be uniform in the reactor. The initial electron density was set to 107 cm−3, which is the sum of positive ion densities satisfying the electroneutrality constraint. Moreover, the initial densities of the radicals were set to 102 cm−3.The time evolution of the density for all species was formulated as:
 | (4) |
The time evolution of the electron energy equation is expressed as:
 | (5) |
The operating parameters of the simulation used here were the same as those of the experiment (see the Catalytic tests section).
3. Results and discussion
3.1 Catalytic performance of the catalysts with the plasma for CH4 conversion
First, we studied the CH4 conversion performance and product selectivity of 0.5Pd/CeO2 at 980 °C. As shown in Fig. 2a, for the catalysis-only condition, CH4 conversion can reach 12.9% at 980 °C, which is much higher than that (4%) for the blank reactor, indicating the crucial catalytic CH4 activation of the 0.5Pd/CeO2 catalyst. The selectivity of the light olefins for 0.5Pd/CeO2 were obviously lower than those for the blank reactor, while the H2 selectivity was much higher, indicating the presence of the carbon deposition at high temperature. Under the plasma-catalysis condition, the CH4 conversion of Plasma + 0.5Pd/CeO2 (1 kHz) was similar to that of 0.5Pd/CeO2 without plasma when the discharge power was ca. 13.6 W, indicating that the weak discharge was not efficient for the removal of the adsorbed species on the catalyst surface. However, when the discharge power reached ca. 25.1 W, the CH4 conversion over the Plasma + 0.5Pd/CeO2 (3 kHz) was almost doubled from 12.9% to 23.6% (an enhancement of ca. 10.7%) compared with that under the catalysis-only. The CH4 conversion (4.7% vs. 4%) over the blank reactor with or without plasma was almost unchanged (Fig. 2a). The activity difference over 0.5Pd/CeO2 and the blank reactor suggests that the role of plasma could not only be related with the gas-phase reaction, but there could also be the existence of plasma-assisted surface reactions, promoting the conversion of CH4.46,47 The waveforms of the discharge voltage (Utotal), gas voltage (Ugas), dielectric voltage (Udiele) and discharge current (Itotal) are shown in Fig. S1.† The similar voltage–current waveforms indicated the same discharge properties, suggesting the stability of the controlled experiment. At the same discharge parameters, the EC for the Catalyst + Plasma is much lower than that for the Blank + Plasma (Table 1), exhibiting the key role of the Pd/CeO2 catalysts in improving the reaction performance. In addition, the stability tests of the catalysts at 980 °C under both thermal and plasma conditions were performed. As shown in Fig. S2,† the conversion of CH4 slightly decreased during the 6 h test under both thermal and plasma conditions, indicating the fast formation of carbon under catalysis-only conditions, and the collision between plasma and catalyst can ensure the desorption of the CH3 species from the catalyst surface to suppress the catalyst deactivation in a certain period of time. | ||
| Fig. 2 (a) CH4 conversion, product selectivity and (b) product yield over the 0.5Pd/CeO2 catalyst in comparison to that over a blank reactor with or without plasma at 980 °C (discharge voltage: 13 kV; frequency: 1 kHz or 3 kHz; rising time: 300 ns; falling time: 500 ns). | ||
| Samples | P (W) | EC (J mmol−1) |
|---|---|---|
| a Discharge parameter: discharge voltage: 13 kV; frequency: 1 kHz or 3 kHz; rising time: 300 ns; falling time: 500 ns. | ||
| Blank + Plasma (1 kHz) | 8.4 | 7.6 |
| Blank + Plasma (3 kHz) | 17.4 | 16.9 |
| Catalyst + Plasma (1 kHz) | 13.6 | 4.3 |
| Catalyst + Plasma (3 kHz) | 25.1 | 4.6 |
The selectivity of each product was similar to that without plasma (Fig. 2a), which suggested that the plasma was mainly involved in homogeneous CH4 activation, and as the initial step of activating CH4 to form CH3 radicals, without influence on the subsequent reaction pathway.46 Notably, the selectivity and the C & H balance for 0.5Pd/CeO2 with plasma were slightly lower than that for the blank reactor with plasma (Fig. 2a and S3a†), suggesting the presence of the high carbon hydrocarbons gas (C4+) resulting from further chain growth and cyclization, which were not detected by FID or TCD. Some high carbon hydrocarbon solidifications in the wall of the tube were observed during the test. The yield of each product under the plasma-catalysis condition was more than that for catalysis-only (Fig. 2b). For the CeO2 support (Fig. S4†), the CH4 conversion was similar to that for the blank reactor (5.5% vs. 4%). Moreover, the CH4 conversion increased from 5.5% to 11.3% (an enhancement of ca. 5.8%) after the introduction of plasma, indicating that the contribution of plasma could only be in connection with the gas-phase reaction. It also implied the importance of the Pd species in the catalytic CH4 conversion.
To better understand the effect of the 0.5Pd/CeO2 catalyst with or without plasma on the CH4 conversion, we studied the CH4 conversion, product selectivity and yields at lower temperatures. As displayed in Fig. 3, CH4 conversions, the selectivity and yields of the C2 products under the plasma-catalysis condition increased obviously at 800 °C and 900 °C, which were much higher than those from the blank reactor with plasma (Fig. S5†). The C & H balance was nearly 100% for the 0.5Pd/CeO2 catalyst with or without plasma at different temperatures (Fig. S3b†), suggesting less carbon deposition at lower temperatures. The corresponding voltage–current waveforms with plasma at 800 °C and 900 °C are shown in Fig. S6.† The CH4 conversion under the plasma-catalysis condition reached 5.2% at 900 °C, which are quite higher compared with those for the 0.5Pd/CeO2 catalyst, indicating that the addition of the plasma can promote the conversion of CH4. The selectivity of C2H4 and H2 reached 15% and 50%, respectively.
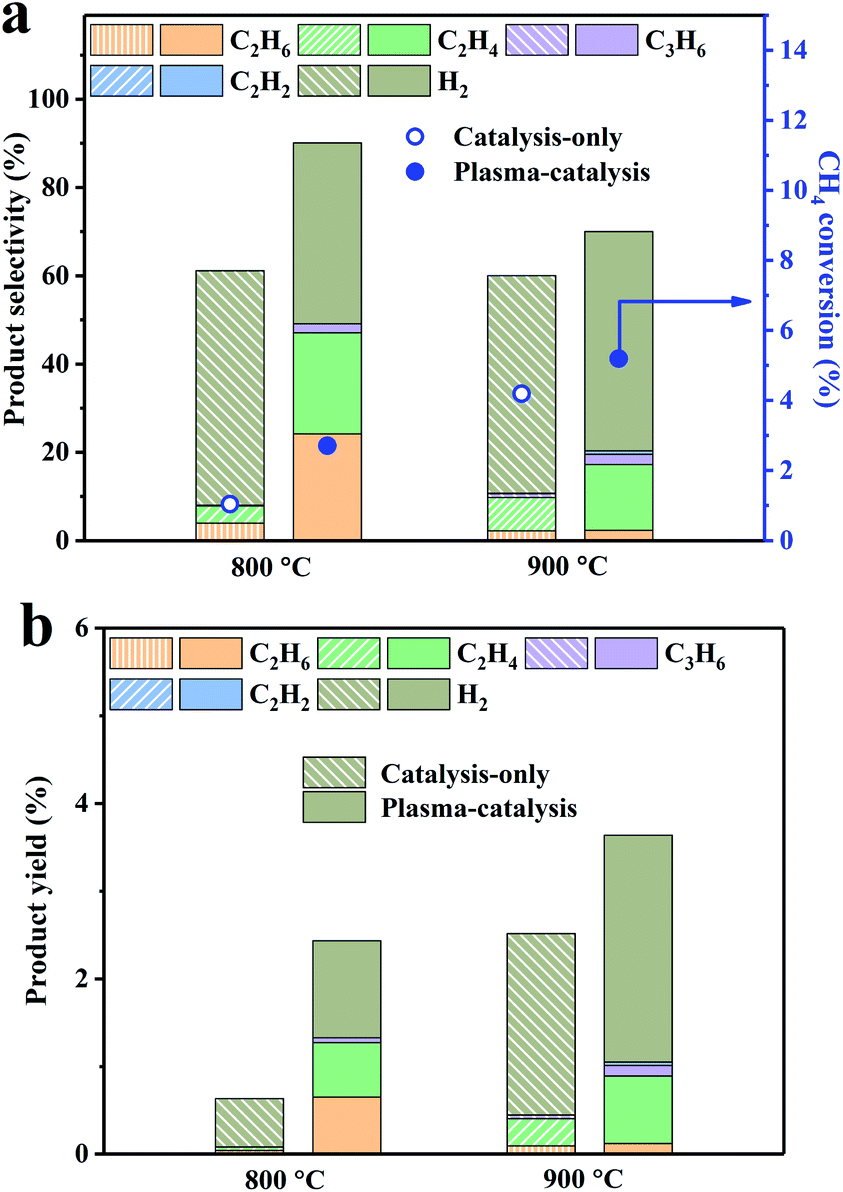 | ||
| Fig. 3 (a) Methane conversion, product selectivity and (b) product yield over the 0.5Pd/CeO2 catalyst with or without plasma at 800 °C and 900 °C (discharge voltage: 13 kV; frequency: 1 kHz; rising time: 300 ns; falling time: 500 ns). | ||
In addition, the 0.5Pd/CeO2 catalyst with plasma showed excellent low-temperature catalytic reactivity. At 800 °C, the CH4 conversion reached nearly 3%. The onset temperature of CH4 activation on the 0.5Pd/CeO2 catalyst with plasma is much lower than that previously reported for single-atom Fe@SiO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 13 and Pt1@CeO2.14 Table S2† shows the CH4 conversion and the selectivity of the C2 products (C2H6, C2H4 and C2H2) over various catalysts reported from the literature. Although the selectivity of the C2 products was slightly lower than those at higher temperature, the Pd/CeO2 catalyst with plasma in our work has the competitive CH4 conversion at lower temperature compared with that reported from the literature. This also provides a promising strategy to achieve the nonoxidative conversion of CH4 at lower temperatures.
13 and Pt1@CeO2.14 Table S2† shows the CH4 conversion and the selectivity of the C2 products (C2H6, C2H4 and C2H2) over various catalysts reported from the literature. Although the selectivity of the C2 products was slightly lower than those at higher temperature, the Pd/CeO2 catalyst with plasma in our work has the competitive CH4 conversion at lower temperature compared with that reported from the literature. This also provides a promising strategy to achieve the nonoxidative conversion of CH4 at lower temperatures.
3.2 The effect of plasma on carbon deposited over the catalyst surface
The morphology and corresponding elemental mapping analysis of the fresh (named as 0.5Pd/CeO2-fresh) and used 0.5Pd/CeO2 catalyst without or with plasma (named as 0.5Pd/CeO2-used and Plasma + 0.5Pd/CeO2-used, respectively) were characterized. The fresh 0.5Pd/CeO2 catalyst calcined at 980 °C underwent severe sintering compared with the rod-like CeO2 supports subjected to the calcination at 400 °C (Fig. 4a, b, S7 and S8a†). The 0.5Pd/CeO2 catalyst were sintered into irregular shapes with a size of 200 nm or more. No palladium-containing phases were observed, indicating the stable Pd species disperse uniformly on CeO2. Only the clear interplanar spacing of 0.32 nm was detected (Fig. 4a and S8d†), corresponding to the (111) lattice fringes of CeO2.48 In addition, the elemental mapping analysis confirmed the homogeneous distribution of both Pd and Ce species (Fig. 4b and c). Notably, a small quantity of carbon species was also detected (6.8%, Table 2), which mainly resulted from the surface-adsorption carbonaceous species.35 | ||
| Fig. 4 (a, d, g) HR-TEM images, (b, e, h) SEM images and (c, f, i) the corresponding EDS mappings for the 0.5Pd/CeO2 catalysts: (a–c) the fresh catalyst; (d–f) the catalyst after catalysis-only; (g–i) the catalyst after plasma-catalysis. The collection zones of the STEM-EDS elemental mapping images corresponded to the SEM areas. | ||
Under catalysis-only condition, except the (111) and (002)48 lattice fringes of CeO2, large amounts of carbon species over the catalyst were observed (Fig. 4d–f and S8b, e†). Furthermore, only weak Ce, O and Pd signals were detected (Fig. 4e and f). The carbon species were typical of amorphous carbon and had limited order due to the absence of the observable graphitic reflections in XRD or the HR-TEM.49 However, under plasma-catalysis condition, besides the distribution of the carbon element, the signals of Ce, O and Pd elements were obvious (Fig. 4g–i), indicating that the introduction of plasma can effectively inhibit the degree of the carbon deposition. The quantification of the carbon element from the EDS results also verified this point (Table 2). The carbon content (31.0%) under the plasma-catalysis condition was much lower than that (100.0%) under the catalysis-only condition (Table 2). To better exhibit the distribution of the Pd species, HAADF-STEM images and the corresponding elemental mappings were performed. The active Pd species were approximately isolated and monodispersed on CeO2 (Fig. S9†). In a word, the addition of the plasma can effectively reduce the amount of the carbon deposition. Furthermore, CeO2 as support can effectively suppress the aggregation of the Pd species during the high-temperature catalytic reaction, showing the outstanding heat resistance and structure stability.
The specific surface areas (SBET) of the samples were measured by N2 adsorption–desorption isotherms and the corresponding results are summarized in Table 2. The CeO2 support showed relatively high SBET at ca. 77 m2 g−1. However, the SBET value decreased seriously to 0.19 m2 g−1 for the fresh 0.5Pd/CeO2 calcined at 980 °C, mainly due to the increasing CeO2 crystallite size, as verified by the SEM images. After the CH4 conversion, the SBET value (3.49 m2 g−1) evidently increased, and was much higher than that (0.31 m2 g−1) for the used Plasma + 0.5Pd/CeO2, which was mainly attributed to the greater carbon deposition on the used 0.5Pd/CeO2.
XRD results showed that all of the diffraction peaks for the measured samples could be assigned to the face-centered cubic CeO2 (JCPDS 34-394) phase (Fig. 5a). No diffraction peaks were consistent with the Pd species, possibly due to the low loading of the Pd species. It also suggested the small particle size and high dispersion of the Pd species on the CeO2 support, which was in line with the HAADF-STEM results. In addition, the mean crystallite sizes (d) of the samples were calculated according to the Scherrer equation, which are summarized in Table S3.† The 0.5Pd/CeO2-freshly calcined at 980 °C became much larger compared with the CeO2 supports subjected to the calcination at 400 °C (24.9 nm vs. 8.8 nm). However, the mean crystallite sizes of CeO2 obviously decreased for the used 0.5Pd/CeO2 with or without plasma. Moreover, the size of CeO2 for the used Plasma + 0.5Pd/CeO2 was apparently larger compared with that for the used 0.5Pd/CeO2 (Table S3†). This can be due to the lower carbon deposition and greater exposure to CeO2 after the introduction of the plasma. These phenomena supported the interpretation of the SEM images well.
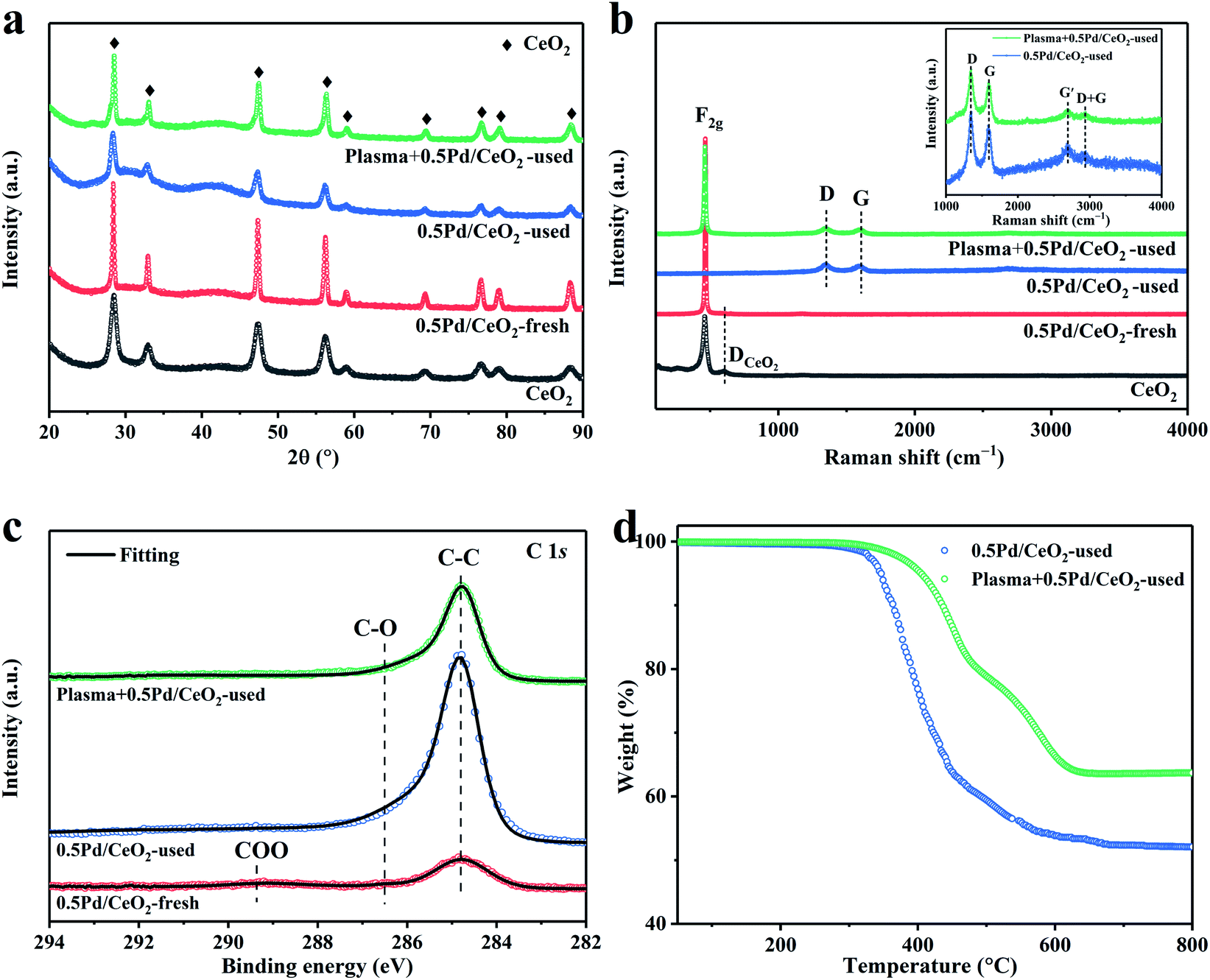 | ||
| Fig. 5 (a) XRD patterns and (b) Raman spectra of the CeO2 support, the fresh and used 0.5Pd/CeO2 catalyst with or without plasma. (c) XPS spectra of C 1s for the fresh 0.5Pd/CeO2, the used 0.5Pd/CeO2 and Plasma + 0.5Pd/CeO2. (d) TGA profiles of the used 0.5Pd/CeO2 and Plasma + 0.5Pd/CeO2 following the long-term stability tests. | ||
Raman spectral tests were used to investigate the surface specific structure of the catalysts. As shown in Fig. 5b, for the fresh 0.5Pd/CeO2, except for the F2g mode (462 cm−1) and the defect-induced (DCeO2, 601 cm−1) mode of the cubic CeO2 fluoride phase,35,48 no peaks related to the Pd species were detected, suggesting the small size and low loading of the Pd species. For the used 0.5Pd/CeO2 catalyst, there was the presence of four new peaks. The strong peak at 1349 cm−1 was assigned to the disorder in graphitic carbon denoted as D.50 The G-band at 1596 cm−1 and the G′-band at 2694 cm−1 were ascribed to the graphitic in-plane stretching vibrations from the sp2-bonded carbon and 2D vibrations,51,52 respectively. The presence of the G-band demonstrated the existence of graphitic carbon.53 The weak G′-band represented the short-range honeycomb structure, indicating the partially ordered carbon structure.49 Another weak peak at 2932 cm−1 was assigned to the combination of G and D.52 However, no peaks were assigned to the CeO2 fluoride phase and Pd species, suggesting that the deposition of the carbon species covered the catalyst signal. Surprisingly, the peak related to the F2g mode of ceria was detected for the used Plasma + 0.5Pd/CeO2, indicating that the degree of the carbon deposition can be effectively weakened after the introduction of plasma. The similar ID/IG ratios (Table 2) between the used 0.5Pd/CeO2 and Plasma + 0.5Pd/CeO2 suggested the same graphitic structure, indicating that the addition of plasma did not change the structure of the 0.5Pd/CeO2 catalyst.
Fig. 5c and S10† show the XPS spectra of C 1s, Pd 3d, Ce 3d and O 1s for the 0.5Pd/CeO2 samples. For the Pd 3d XPS spectrum of the fresh 0.5Pd/CeO2 (Fig. S10a†), two symmetric peaks at 337.5 eV and 342.8 eV corresponded to the highly dispersed Pd species in the Pd2+ state.54 However, for the used Plasma + 0.5Pd/CeO2 (Fig. S10a†), the accurate confirmation of the Pd species was difficult due to the poor noise resulting from the lower Pd content covered partially by the carbon. From the H2-TPR results (Fig. S11†), the highly dispersed PdO species were easily reduced below 200 °C,55 while the reduction of the PdO species interacting strongly with CeO2 can be reduced below 400 °C.55 Thus, the active state of 0.5Pd/CeO2 was the metallic Pd phase.
To explore the coordination structure of the carbon species, C 1s XPS spectra were studied (Fig. 5c). For the fresh sample, the peak at 284.8 eV can be assigned to the C–C bond from graphitized carbon.56 Two weak peaks at 286.5 eV and 289.2 eV were related to the C–O bond and COO bond,56,57 respectively. These bonds may result from the contaminant carbon of the measurement system58 or the adsorption of CO2 and H2O in the air. For the used 0.5Pd/CeO2 and Plasma + 0.5Pd/CeO2, they showed the same coordination structure of carbon. The peak intensity related to the C–C bond increased, confirming the deposition of the carbonaceous species during CH4 conversion. The C–O bond still existed, primarily from the CO2 adsorption in air. Notably, the peak intensity of the C–C bond for the used 0.5Pd/CeO2 was much higher compared with the used Plasma + 0.5Pd/CeO2. Moreover, for the XPS spectra of Pd 3d, Ce 3d and O 1s (Fig. S10†), the peak intensity of the fresh 0.5Pd/CeO2 was much stronger than that of the used Plasma + 0.5Pd/CeO2. These observations are in accordance with the Raman and SEM results. The amount of carbon deposition can be effectively reduced after the addition of plasma.
The contents and types of the deposited carbon were characterized by TGA and O2-TPO. As summarized in Table 2, the TGA results showed that the used Plasma + 0.5Pd/CeO2 possessed a total weight reduction of 36.3 wt%, in agreement with the SEM-EDS data (Table 2). Meanwhile, more carbon content (48.0 wt%) for the used 0.5Pd/CeO2 were detected, while this value was much lower than the SEM-EDS results (ca. 100%). This is due to the large amounts of carbon covered over the catalyst, and the thickness of carbon reached the lower detection limit of the SEM instrument. Thus, weak catalyst information was detected, leading to higher carbon content. In addition, they showed the similar TGA and O2-TPO profiles (Fig. 5d and S12†), confirming the same types of the deposited carbon. The weight loss between 380 and 520 °C was ascribed to amorphous carbon and the weight loss between 520 and 720 °C corresponded to filamentary carbon.59,60 The weight loss at more than 720 °C resulted from the burn-off of graphitic carbon, which was difficult to gasify or removed by regeneration.59 These carbon species were responsible for the low catalytic activation of 0.5Pd/CeO2.
3.3 Reaction mechanism of the catalysts with the plasma for CH4 conversion
To identify the active species of the plasma-catalysis condition during the conversion of CH4, the OES measurement was performed. As shown in Fig. S13,† the excited CH (C2Σ+ → X2Π), CH (A2Δ → X2Π) and C2 (d3Π → a3Π) bands were identified in the OES spectra,61 demonstrating the existence of CH radicals. This also indicates the presence of CH3 and CH2 radicals because the CH radicals are generated via the progressive dehydrogenation of CH4 under the plasma condition.62 Two CH3 radicals coupled and further reacted with other active species to generate other products. The rest of the peaks were assigned to the vibration of N2 and N2+,63,64 which mainly resulted from the discharge between the ground electrode and the quartz tube. The bands assigned to CH and C2 were very weak compared with those from N2 and N2+. It was mainly because the size (20–40 mesh) and voids of the catalyst were very small, leading to the light from the CH4 discharge being obscured.The DFT calculations were carried out to explore the reaction mechanism and understand the reason of carbon deposition on 0.5Pd/CeO2 without plasma. The Pd (111) surface was constructed to adsorb CH4, which was easily activated to generate adsorbed CH3 (0.88 eV, Fig. 6). However, the consequent dehydrogenation of the adsorbed CH3 on the catalyst surface is much more favorable compared with the desorption (1.02 eV vs. 2.15 eV). The adsorbed CH4 mainly occurred in consecutive dehydrogenation steps on the Pd-site, finally forming the carbon deposition. Therefore, the conversion of CH4 on the Pd-site tends to form carbon-coking, leading to a large amount of carbon covering the catalyst surface, lowering the catalytic performance. Notably, the desorption of the adsorbed CH3 was easier than that of the adsorbed CH2 and CH.
on the catalyst surface is much more favorable compared with the desorption (1.02 eV vs. 2.15 eV). The adsorbed CH4 mainly occurred in consecutive dehydrogenation steps on the Pd-site, finally forming the carbon deposition. Therefore, the conversion of CH4 on the Pd-site tends to form carbon-coking, leading to a large amount of carbon covering the catalyst surface, lowering the catalytic performance. Notably, the desorption of the adsorbed CH3 was easier than that of the adsorbed CH2 and CH.
 | ||
| Fig. 6 CH4 consecutive dehydrogenation pathways (blue lines) and desorption energy of the reaction intermediates (orange lines) on the Pd-site based on DFT simulations. | ||
After the introduction of plasma, CH4 molecules were cracked to generate the abundant CH3 radicals, and two CH3 radicals coupled easily to generate C2H6. If the CH3 radicals can readily react with the adsorbed CH3 to generate C2H6, we also simulated the steps via DFT. As shown in Fig. S14,† the energy for the reaction of the CH3 radicals with the adsorbed CH3 to generate the C2H6 was ca. 2.76 eV, suggesting the difficult process. It also indicated that the conversion of CH4 to hydrocarbons could occur first through desorption of adsorbed CH3 from the surface of the catalyst to form CH3 radicals, and then the CH3 radical reacted with the gaseous molecules.
According to product analysis and a range of catalyst characterizations, it can be concluded that there is serious carbon deposition over only 0.5Pd/CeO2. However, the amount of carbon deposition is greatly reduced after the addition of plasma. The influence of plasma on the carbon deposition has been discussed in detail. First, the intense electric field and the diffusion of the active species generated by plasma could modify the surface charge transfer and work function of catalysts to promote the desorption of CH3.65,66 Secondly, the energized electron and ions generated by plasma could impact the adsorbed CH3 species, which could accelerate the desorption of the CH3 species from the catalyst surface, reducing the amount of carbon deposition. The density of electron averaged over one period was ca. 1011 cm−3 (Fig. 7a). The desorption of the adsorbed CH3 species needed at least 2.15 eV (Fig. 6). Thus, the electron ratio in the range from 2 eV to 9 eV (the minimum energy threshold of CH4 dissociation and ionization collision) was calculated, which enabled the adsorbed CH3 to desorb. As shown in Fig. 7b, there was about 55% of the electrons in the 2–9 eV energy range when the reduced field intensity was more than 100 Td. Although not all of the electrons participated in the desorption of CH3, a part of electrons >2 eV provided the possibility in the CH3 desorption from the catalyst surface into the gaseous phase, inhibiting the progressive dehydrogenation of adsorbed CH3.
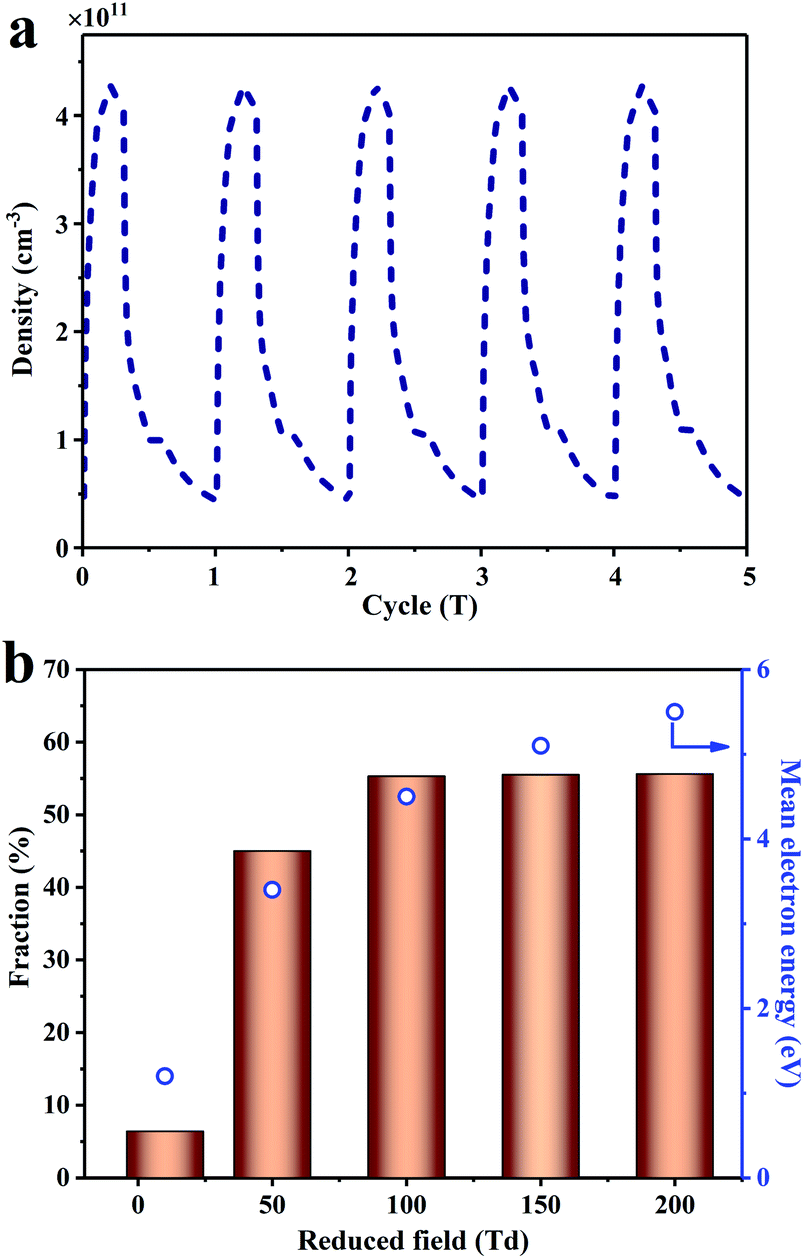 | ||
| Fig. 7 (a) The density of the electron as a function of time for the CH4 conversion assisted by plasma without catalyst. (b) The ratio of the electron and mean electron energy as a function of reduced field under the plasma-only conditions. | ||
Combining the above analysis with a previous study,13 we infer that the reaction mechanism of CH4 conversion assisted by plasma in the nonoxidation condition is that CH4 was first activated by the Pd-site to form the adsorbed CH3. Partially adsorbed CH3 bombarded by plasma can desorb from the Pd-site to form the CH3 radical. Two CH3 radicals were coupled to form C2H6. The forming C2H6 species subsequently underwent dehydrogenation and recombination reactions in the gas phase to generate C2H4 and other hydrocarbons. The proposed reaction mechanisms on the Pd/CeO2 surface for the direct conversion of CH4 into olefins and H2 with the thermal catalysis and plasma catalysis methods are displayed in Fig. 8.
 | ||
| Fig. 8 Proposed reaction mechanisms on the Pd/CeO2 surface for the direct conversion of CH4 with the thermal catalysis and plasma catalysis methods. | ||
3.4 Plasma kinetic modeling
A plasma kinetic modeling based on a zero-dimensional model was performed to study the possible reaction pathways in the plasma for the CH4 conversion without the 0.5Pd/CeO2 catalyst. The operating parameters of the simulation used here were the same as those of the experiment (see Catalytic tests section). The simulation results were obtained under the condition of 800 °C and 760 torr. Fig. 9 shows the densities of radicals as a function of time for five periods. The densities of the radicals exhibited similar periodic behaviours. The density of each radical continuously increased during the pulse duration and gradually decreased during the pulse-off period, indicating that the generation of hydrocarbons and H2 mainly occurred in the gas phase through the recombination of the radicals during the pulse-off period. Moreover, the densities of radicals averaged over one period were calculated and the most abundant radical was CH3 (1013 cm−3), followed by H, CH2, C2H5, CH, and C2H3.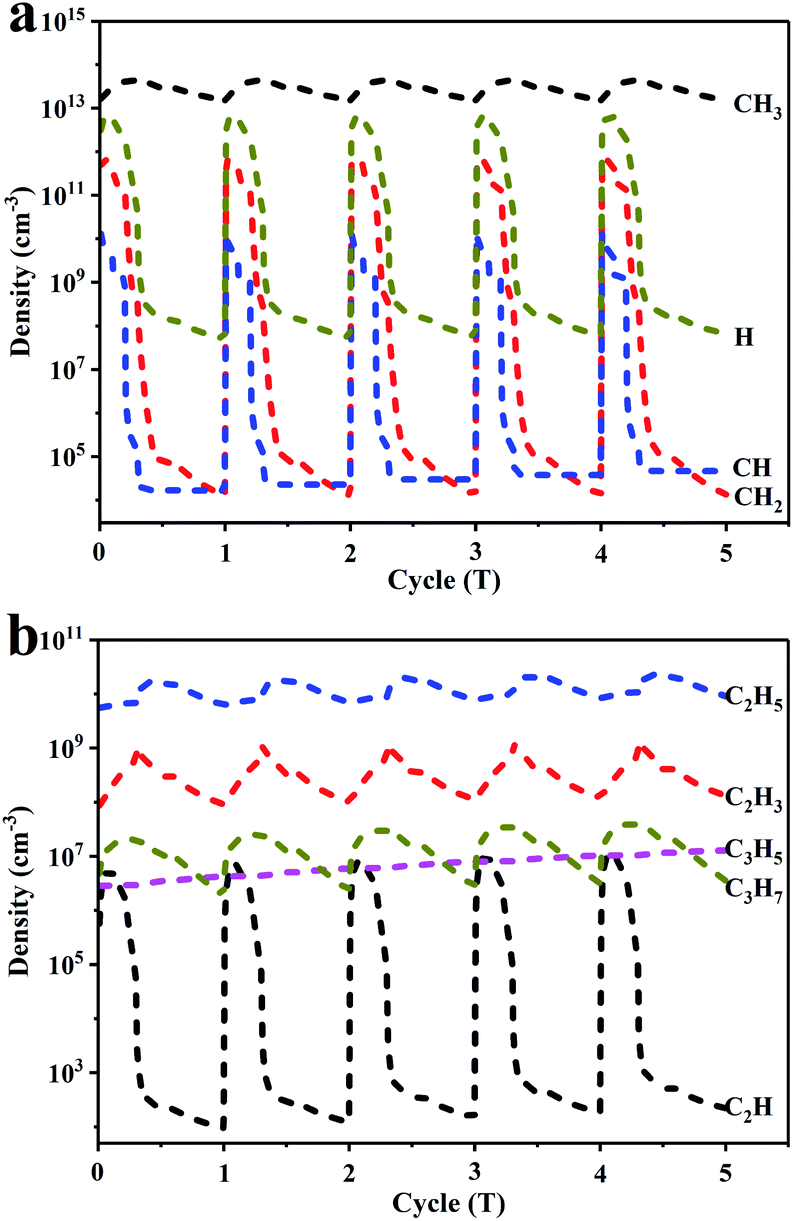 | ||
| Fig. 9 The density of each radical as a function of time during five periods for the CH4 conversion assisted by plasma without catalyst. | ||
Fig. S15† displays the densities of CH4 and the gas products as a function of time. The density of CH4 was initially 6.84 × 1018 cm−3, which decreased gradually due to the dissociation and ionization reactions to generate various radicals. Subsequently, hydrocarbons such as C2H6, C2H4, C2H2, C3H6 and H2 formed via the recombination of the radicals. The rapid growth of the densities for the products appeared within the first 0.5 s. Subsequently, the growth slowed and finally did not significantly change. A residence time of 4.8 s was calculated. H2 was formed with the highest density at 4.8 s. The densities of C2H6 and C2H4 were in the order of 1016 cm−3, which is one order of magnitude higher than that for C2H2 and C3H6.
The CH4 conversion, selectivity of major products and C & H balance based on the above calculation for particle densities are displayed in Fig. S16a.† The calculated CH4 conversion and product selectivity at 4.8 s matched well with the experimental results (Table S4†), further verifying the reliability of the model. The kinetic simulation for CH4 pyrolysis in the blank reactor was also studied. As displayed in Fig. S17,† the change trends based on CH4 conversion and product selectivity were in accordance with those from CH4 conversion assisted by plasma without catalyst (Fig. S16a†). The thermal cracking of CH4 was achieved via consecutive dehydrogenation processes. Each free radical reacted to form the final products.
To explore the underlying mechanism governing CH4 conversion activated by plasma, the generation and loss rates of each product are presented in Fig. S16b–f.† The reaction mechanism of CH4 conversion assisted by plasma was obviously different from that of CH4 pyrolysis. As summarized in Fig. S18,† first, CH4 molecules were dissociated to form CH3, CH2, CH and H radicals with the electron. Subsequently, two CH3 radicals combined or CH4 reacted with CH2 to form C2H6. C2H6 was readily dehydrogenative, followed by a series of gas-phase reaction to form various light hydrocarbons. Moreover, the CH3 radicals could activate CH4 homogeneously to enhance the CH4 conversion (R38, Table S1†) and CH4 could react with H radicals to produce the CH3 radicals (R37, Table S1†). Thus, the cycle process promoted the conversion of CH4, also explaining the reason of the highest density of CH3 radicals (Fig. 9).
4. Conclusions
In summary, we successfully prepared the stable and highly dispersed Pd species on CeO2. The Pd species interact strongly with the CeO2 support, efficiently preventing the Pd species from aggregation during the high-temperature catalytic test. The Pd/CeO2 catalyst assisted by NTP showed the obvious promotion effect for the conversion of CH4 compared with thermal-catalysis. The introduction of plasma is not only able to activate the C–H bond of CH4 to dehydrogenation, but also effectively enhance the coke-resistance of the catalyst. This combination of plasma and catalyst offers a new guidance for lowering the activation temperature of CH4 and the carbon deposition to achieve the conversion of methane into high value-added chemicals.Author contributions
Xiucui Hu: conceptualization; investigation; formal analysis; writing-original draft. Yadi Liu: software; resources; data curation; formal analysis; visualization. Liguang Dou: visualization; software; resources. Cheng Zhang: validation; visualization. Shuai Zhang: resources; data curation. Yuan Gao: visualization. Xin Tu: supervision; writing-review & editing. Tao Shao: project administration; funding acquisition; supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Science Fund for Distinguished Young Scholars [grant number 51925703], the National Natural Science Foundation of China [grant numbers 51637010, 51807190, 52007178]. We acknowledge the National Supercomputing Center in Shenzhen for providing the computational resources and the software. We thank Dr Xiaoyang Cui for help on the SEM characterizations.Notes and references
- Z. Li, Q. Lin, M. Li, J. Cao, F. Liu, H. Pan, Z. Wang and S. Kawi, Renewable Sustainable Energy Rev., 2020, 134, 110312 CrossRef CAS.
- S. Wu, X. Tan, J. Lei, H. Chen, L. Wang and J. Zhang, J. Am. Chem. Soc., 2019, 141, 6592–6600 CrossRef CAS PubMed.
- S. D. Senanayake, J. A. Rodriguez and J. F. Weaver, Acc. Chem. Res., 2020, 53, 1488–1497 CrossRef CAS.
- C. Zhang, C. Ren, S. Zhang, H. Xue, Y. Zhao, Z. Zhou, J. Chen, Z. Luo, W. Sang, L. Jing, Y. Teng, Q. Qiu, C. Zhang, M. Gong, G. Zhang, T. Shao and L. Xiao, Energy Fuels, 2021, 35, 13930–13936 CrossRef CAS.
- S. Mehla, A. E. Kandjani, R. Babarao, A. F. Lee, S. Periasamy, K. Wilson, S. Ramakrishna and S. K. K. Bhargava, Energy Environ. Sci., 2021, 14, 320–352 RSC.
- H. M. Torres Galvis, J. H. Bitter, T. Davidian, M. Ruitenbeek, A. I. Dugulan and K. P. de Jong, J. Am. Chem. Soc., 2012, 134, 16207–16215 CrossRef CAS.
- G. H. Torres, J. H. Bitter, C. B. Khare, M. Ruitenbeek, A. I. Dugulan and K. P. de Jong, Science, 2012, 335, 835–838 CrossRef.
- P. Tian, Y. Wei, M. Ye and Z. Liu, ACS Catal., 2015, 5, 1922–1938 CrossRef CAS.
- J. Xu, Y. Zhang, X. Xu, X. Fang, R. Xi, Y. Liu, R. Zheng and X. Wang, ACS Catal., 2019, 9, 4030–4045 CrossRef CAS.
- P. Chawdhury, K. V. S. S. Bhargavi and C. Subrahmanyam, Sustainable Energy Fuels, 2021, 5, 3351–3362 RSC.
- S. H. Morejudo, R. Zanón, S. Escolástico, I. Yuste-Tirados, H. Malerød-Fjeld, P. K. Vestre, W. G. Coors, A. Martínez, T. Norby, J. M. Serra and C. Kjølseth, Science, 2016, 353, 563–566 CrossRef CAS.
- I. Julian, M. B. Roedern, J. L. Hueso, S. Irusta, A. K. Baden, R. Mallada, Z. Davis and J. Santamaria, Appl. Catal., B, 2020, 263, 118360 CrossRef CAS.
- X. Guo, G. Fang, G. Li, H. Ma, H. Fan, L. Yu, C. Ma, X. Wu, D. Deng, M. Wei, D. Tan, R. Si, S. Zhang, J. Li, L. Sun, Z. Tang, X. Pan and X. Bao, Science, 2014, 344, 616–619 CrossRef CAS PubMed.
- P. Xie, T. Pu, A. Nie, S. Hwang, S. C. Purdy, W. Yu, D. Su, J. T. Miller and C. Wang, ACS Catal., 2018, 8, 4044–4048 CrossRef CAS.
- A. L. Dipu, S. Ohbuchi, Y. Nishikawa, S. Iguchi, H. Ogihara and I. Yamanaka, ACS Catal., 2019, 10, 375–379 CrossRef.
- P. Schwach, X. Pan and X. Bao, Chem. Rev., 2017, 117, 8497–8520 CrossRef CAS.
- Z. Liu, E. Huang, I. Orozco, W. Liao, R. M. Palomino, N. Rui, T. Duchoň, S. Nemšák, D. C. Grinter, M. Mahapatra, P. Liu, J. A. Rodriguez and S. D. Senanayake, Science, 2020, 368, 513–517 CrossRef CAS.
- T. Shao, R. Wang, C. Zhang and P. Yan, High Voltage, 2018, 3, 14–20 CrossRef.
- S. Xu, S. Chansai, C. Stere, B. Inceesungvorn, A. Goguet, K. Wangkawong, S. F. R. Taylor, N. Al-Janabi, C. Hardacre, P. A. Martin and X. Fan, Nat. Catal., 2019, 2, 142–148 CrossRef CAS.
- X. Chen, S. Zhang, S. Li, C. Zhang, J. Pan, A. B. Murphy and T. Shao, Sustainable Energy Fuels, 2021, 5, 787–800 RSC.
- Y. Gao, S. Zhang, H. Sun, R. Wang, X. Tu and T. Shao, Appl. Energy, 2018, 226, 534–545 CrossRef CAS.
- S. Van Alphen, F. Jardali, J. Creel, G. Trenchev, R. Snyders and A. Bogaerts, Sustainable Energy Fuels, 2021, 5, 1786–1800 RSC.
- C. Xu and X. Tu, J. Energy Chem., 2013, 22, 420–425 CrossRef CAS.
- J. Kim, J. Jeoung, J. Jeon, J. Kim, Y. S. Mok and K. Ha, Chem. Eng. J., 2019, 377, 119896 CrossRef CAS.
- H. Sun, S. Zhang, Y. Gao, B. Huang, C. Zhang and T. Shao, Plasma Processes Polym., 2019, 16, 1900050 CrossRef.
- E. Delikonstantis, E. Igos, M. Augustinus, E. Benetto and G. D. Stefanidis, Sustainable Energy Fuels, 2020, 4, 1351–1362 RSC.
- X. Li, C. Lin, C. Shi, Y. Xu, Y. Wang and A. Zhu, J. Phys. D: Appl. Phys., 2008, 41, 175203 CrossRef.
- J. Bae, M. Lee, S. Park, M. Jeong, D. Hong, Y. D. Kim, Y. Park and Y. K. Hwang, Catal. Today, 2017, 293–294, 105–112 CrossRef CAS.
- A. Beloqui Redondo, E. Troussard and J. A. van Bokhoven, Fuel Process. Technol., 2012, 104, 265–270 CrossRef CAS.
- I. Julian, H. Ramirez, J. L. Hueso, R. Mallada and J. Santamaria, Chem. Eng. J., 2019, 377, 119764 CrossRef CAS.
- A. H. Khoja, M. Tahir and N. A. S. Amin, Energy Convers. Manage., 2019, 183, 529–560 CrossRef CAS.
- Z. Guo, Y. Yi, L. Wang, J. Yan and H. Guo, ACS Catal., 2018, 8, 10219–10224 CrossRef CAS.
- B. Huang, C. Zhang, H. Bai, S. Zhang, K. K. Ostrikov and T. Shao, Chem. Eng. J., 2020, 396, 125185 CrossRef CAS.
- M. Janda, V. Martišovitš, A. Buček, K. Hensel, M. Molnár and Z. Machala, J. Phys. D: Appl. Phys., 2017, 50, 425207 CrossRef.
- Z. Hu, X. Liu, D. Meng, Y. Guo, Y. Guo and G. Lu, ACS Catal., 2016, 6, 2265–2279 CrossRef CAS.
- X. Hu, X. Fu, W. Wang, X. Wang, K. Wu, R. Si, C. Ma, C. Jia and C. Yan, Appl. Catal., B, 2020, 268, 118424 CrossRef CAS.
- W. Yu, W. Wang, S. Li, X. Fu, X. Wang, K. Wu, R. Si, C. Ma, C. Jia and C. Yan, J. Am. Chem. Soc., 2019, 141, 17548–17557 CrossRef CAS PubMed.
- T. P. Senftle, A. C. T. van Duin and M. J. Janik, ACS Catal., 2017, 7, 327–332 CrossRef CAS.
- H. Mai, L. Sun, Y. Zhang, R. Si, W. Feng, H. Zhang, H. Liu and C. Yan, J. Phys. Chem. B, 2005, 109, 24380–24385 CrossRef CAS.
- H. Jeong, J. Bae, J. W. Han and H. Lee, ACS Catal., 2017, 7, 7097–7105 CrossRef CAS.
- L. Dou, M. Fu, Y. Gao, L. Wang, C. Yan, T. Ma, Q. Zhang and X. Li, Fuel, 2021, 283, 118984 CrossRef CAS.
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 6671–6687 CrossRef CAS PubMed.
- G. J. M. Hagelaar and L. C. Pitchford, Plasma Sources Sci. Technol., 2005, 14, 722–733 CrossRef CAS.
- R. K. Janev and D. Reiter, Phys. Plasmas, 2002, 9, 4071–4081 CrossRef CAS.
- R. K. Janev and D. Reiter, Phys. Plasmas, 2004, 11, 780–829 CrossRef CAS.
- J. Hao, P. Schwach, G. Fang, X. Guo, H. Zhang, H. Shen, X. Huang, D. Eggart, X. Pan and X. Bao, ACS Catal., 2019, 9, 9045–9050 CrossRef CAS.
- L. Wang, Y. Yi, H. Guo and X. Tu, ACS Catal., 2017, 8, 90–100 CrossRef.
- W. Wang, W. Yu, P. Du, H. Xu, Z. Jin, R. Si, C. Ma, S. Shi, C. Jia and C. Yan, ACS Catal., 2017, 7, 1313–1329 CrossRef CAS.
- H. A. Calderon, A. Okonkwo, I. Estrada-Guel, V. G. Hadjiev, F. Alvarez-Ramírez and F. C. Robles Hernández, Adv. Struct. Chem. Imaging, 2016, 2, 1–12 CrossRef.
- K. Han, W. Yu, L. Xu, Z. Deng, H. Yu and F. Wang, Fuel, 2021, 291, 120182 CrossRef CAS.
- A. K. Hill and L. Torrente-Murciano, Appl. Catal., B, 2015, 172–173, 129–135 CrossRef CAS.
- Y. A. Kim, K. Fujisawa, H. Muramatsu, T. Hayashi, M. Endo, T. Fujimori, K. Kaneko, M. Terrones, J. Behrends, A. Eckmann, C. Casiraghi, K. S. Novoselov, R. Saito and M. S. Dresselhaus, ACS Nano, 2012, 6, 6293–6300 CrossRef CAS PubMed.
- A. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14095–14107 CrossRef CAS.
- S. Hinokuma, H. Fujii, M. Okamoto, K. Ikeue and M. Machida, Chem. Mater., 2010, 22, 6183–6190 CrossRef CAS.
- G. Ercolino, P. Stelmachowski, G. Grzybek, A. Kotarba and S. Specchia, Appl. Catal., B, 2017, 206, 712–725 CrossRef CAS.
- X. Jia, Y. Dong, X. Li, H. Yu, T. Ma, W. Zhang and H. Dong, Appl. Surf. Sci., 2020, 517, 146207 CrossRef.
- Q. Yang, C. Zhou, J. Ni and X. Guan, Sustainable Energy Fuels, 2020, 4, 2768–2774 RSC.
- Q. Zhao, Y. Wang, Y. Wang, L. Li, W. Zeng, G. Li and C. Hu, Int. J. Hydrogen Energy, 2020, 45, 14281–14292 CrossRef CAS.
- Z. Xie, B. Yan, S. Kattel, J. H. Lee, S. Yao, Q. Wu, N. Rui, E. Gomez, Z. Liu, W. Xu, L. Zhang and J. G. Chen, Appl. Catal., B, 2018, 236, 280–293 CrossRef CAS.
- A. Wang, J. H. Harrhy, S. Meng, P. He, L. Liu and H. Song, Energy Convers. Manage., 2019, 191, 93–101 CrossRef CAS.
- X. Wang, Y. Gao, S. Zhang, H. Sun, J. Li and T. Shao, Appl. Energy, 2019, 243, 132–144 CrossRef CAS.
- Y. Yi, X. Wang, A. Jafarzadeh, L. Wang, P. Liu, B. He, J. Yan, R. Zhang, H. Zhang, X. Liu, H. Guo, E. C. Neyts and A. Bogaerts, ACS Catal., 2021, 11, 1765–1773 CrossRef CAS.
- Y. Li, D. Yang, J. Qiao, L. Zhang, W. Wang, Z. Zhao, X. Zhou, H. Yuan and W. Wang, Plasma Sources Sci. Technol., 2020, 29, 55004 CrossRef CAS.
- H. Wang, D. Yang, Q. Xu, H. Yuan, X. Zhou and W. Wang, J. Phys. D: Appl. Phys., 2021, 54, 025202 CrossRef CAS.
- L. Y. Jia, A. Farouha, L. Pinard, S. Hedan, J. D. Comparot, A. Dufour, K. Ben Tayeb, H. Vezin and C. Batiot-Dupeyrat, Appl. Catal., B, 2017, 219, 82–91 CrossRef CAS.
- X. Zhang, Y. Liu, M. Zhang, T. Yu, B. Chen, Y. Xu, M. Crocker, X. Zhu, Y. Zhu, R. Wang, D. Xiao, M. Bi, D. Ma and C. Shi, Chem, 2020, 6, 3312–3328 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1se01441b |
| This journal is © The Royal Society of Chemistry 2022 |