
Octanuclear Cu(II) cluster–tungstosilicate composite as efficient electrocatalyst for oxygen evolution reaction at near-neutral pH†
Junqi
Lin
 *,
Xin
Chen
,
Nini
Wang
,
Shanshan
Liu
,
Yanmei
Chen
,
Zhengfang
Tian
and
Zhijun
Ruan
*
*,
Xin
Chen
,
Nini
Wang
,
Shanshan
Liu
,
Yanmei
Chen
,
Zhengfang
Tian
and
Zhijun
Ruan
*
Hubei Key Laboratory of Processing and Application of Catalytic Materials, College of Chemistry and Chemical Engineering, Huanggang Normal University, Huanggang, 438000 China. E-mail: linjunqi@hgnu.edu.cn; ruanzhijun@hgnu.edu.cn
First published on 20th November 2021
Abstract
Transition metal clusters have been used for water oxidation with high catalytic efficiency. However, they are water-soluble and often used homogeneously rather than heterogeneously. Herein, a water-soluble octanuclear Cu(II) cluster was heterogenized by binding it with α-undecatungstosilicate [α-SiW11O39]8−via the electrostatic effect. Octanuclear cluster [Cu8(dpk·OH)8(OAc)4]4+ (dpk·OH = monoanion of the hydrated, gem-diol form of di-2-pyridyl ketone) was transformed into insoluble [Cu8(dpk·OH)8(OAc)4]2@[α-SiW11O39] (1) composite. This composite catalyzes water electro-oxidation efficiently with an onset potential of only 370 mV and attains a catalytic current density of 1 mA cm−2 at a low overpotential of 580 mV with a Tafel slope value of 62 mV per decade at near-neutral pH. Electrochemical measurements and spectroscopy characterization indicate that this composite remains stable and active after prolonged electrolysis experiments. This work reveals that the electrostatic interaction between transition metal complexes and polyoxoanions is a feasible and convenient method to synthesize efficient heterogenized homogeneous water oxidation catalysts (WOCs).
Introduction
Hydrogen energy is regarded as a potential alternative to fossil energy. The utilization of hydrogen is of great significance for mitigating the greenhouse effect and environmental issues.1–3 Massive efforts have been made on electrocatalytic water splitting to produce hydrogen. The bottleneck of water splitting is water oxidation that requires high activation energy for the four-electron oxidation process.4–6 To boost the electrochemical oxygen evolution reaction, numerous catalysts were developed and tested for water oxidation, and most of them were heterogeneous materials.7–13 Additionally, the catalysts that are based on transition-metal-containing complexes have attracted much attention because of their high catalytic efficiency and structural flexibility.14–20Compared with electrocatalysts that are based on heterogeneous materials, heterogenized homogeneous water oxidation catalysts (WOCs) based on complexes of earth-abundant transition metals are much more desirable.21 It is because the latter can be readily modified by ligand design for better performance.22 Homogeneous WOCs mainly contain polyoxometalates and transition metal complexes with organic ligands. In the catalysis of water oxidation, some polyoxometalate molecules were heterogenized by electrostatic attraction using charged carriers such as chemically modified graphene oxide or molecular sieve.23–27 In addition, water-soluble polyoxometalates were electrostatically bound to large alkali cations or alkaline cations, directly becoming water-insoluble to serve as stable electrocatalysts for water oxidation in acidic media.28–30 Different from polyoxometalates, homogeneous WOCs based on mononuclear metal complexes were heterogenized on the surface of a carrier by chemical bonding or π-stacking methods.31–33 These methods involve the modification of ligand structure, which could be rather complicated, and the resulting complexes may decompose under oxidative conditions.34 It is noteworthy that more and more water-soluble multinuclear complexes have been reported to exhibit outstanding catalytic activity toward water oxidation.35–37 Nevertheless, so far there is no effective and convenient method for the heterogenization of those homogeneous complexes.
Among the multitudinous catalysts for water oxidation, those Cu-based materials are widely studied because they are non-toxic, environmentally friendly, and inexpensive. Among them are Cu-containing metal–organic frameworks, copper oxide, copper sulfide, copper phosphide and copper selenide.38–47 To the best of our knowledge, there are only a few examples of Cu-based WOCs that are resulting from heterogenization of homogeneous complexes by π-stacking between graphene carriers and ligands with an electron-conjugated structure.22
Herein, by direct electrostatic binding between octanuclear Cu(II) cluster [Cu8(dpk·OH)8(OAc)4]4+ (dpk·OH = the monoanion of the hydrated, a gem-diol form of di-2-pyridyl ketone) and an antioxidative all-inorganic α-undecatungstosilicate [α-SiW11O39]8−, we readily produced [Cu8(dpk·OH)8(OAc)4]2@[α-SiW11O39] (1) composite as heterogenized homogeneous WOCs. The chemical composition of composite 1 was investigated by Fourier transform infrared (FTIR), Raman spectra and elemental analysis. When tested in a phosphate buffer solution (PBS) at near-neutral pH of 6.5, composite 1 efficiently catalyzes electrochemical water oxidation, giving a current density of 1 mA cm−2 at an overpotential of 580 mV with an onset overpotential of only 370 mV. Collective experiments including controlled potential electrolysis, FTIR, cyclic voltammetry (CV), scanning electron microscope (SEM), transmission electron microscope (TEM) and X-ray photoelectron spectroscopy (XPS) demonstrated that composite 1 is a robust catalyst. Combined with [α-SiW11O39]8−, [Cu8(dpk·OH)8(OAc)4]4+ in composite 1 exhibits outstanding activity and remains intact during electrocatalytic water oxidation. It is worth noting that some other water-soluble complex based WOCs, such as [Co4(dpk·OH)4(OAc)2(H2O)2]2+ and [Ni(THA)]2+ (NHA = 16-membered 1,3,6,9,11,14-hexaazatricyclo[12.2.1.16,9] octadecane), can also be heterogenized by binding with counter polyoxoanions. In addition, synthesis of analogous composite material from [Cu8(dpk·OH)8(OAc)4]4+ and other polyoxoanions, such as [SiW12O40]4− is also feasible. This work manifests that the direct binding of transition metal complexes with tungstosilicates by electrostatic effects can be a new strategy to obtain heterogenized homogeneous WOCs with high efficiency.
Results and discussion
Structure of Cu cluster
The acquired X-ray diffraction pattern of the as-synthesized Cu complex is in good agreement with the simulated pattern from the reported CIF of [Cu8(dpk·OH)8(OAc)4](ClO4)4·9H2O (Fig. S1†). The infrared spectrum of the Cu complex shows all the characteristic vibrations described by literature. (Fig. S2†).48,49 It is noteworthy that the IR signals at 626 cm−1 and around 1000 cm−1 are assigned to v(ClO4−).50 The structure of K8[α-SiW11O39] was verified from the FTIR spectrum, which is in good agreement with the reported data (Fig. S3†).51 Alike the natural Mn4CaO5 cluster center for oxygen evolution, there are two structurally similar but independent [Cu4O4] cubanes in the octanuclear Cu(II) cluster of [Cu8(dpk·OH)8(OAc)4](ClO4)4.48,49 Each [Cu4O4] cubane is completed by four Cu(II) atoms and four deprotonated oxygen atoms from dpk·OH− ligands. Doubly bridged by two anti-syn acetate ligands via the two Cu atoms, the two cubanes are centro-symmetrically connected in the tetracation (Fig. 1).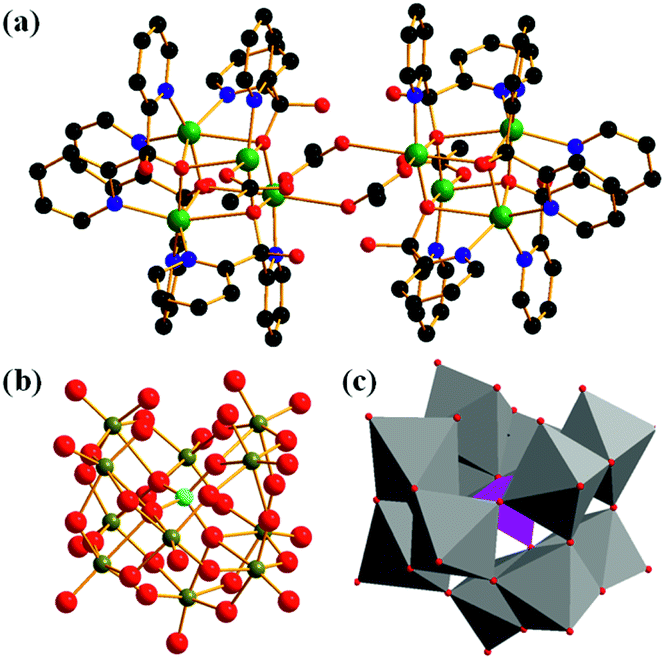 | ||
| Fig. 1 (a) Molecular structure of [Cu8(dpk·OH)8(OAc)4]4+ and (b) [α-SiW11O39]8− (green, Cu; black, C; red, O; blue, N; light blue, Si; deep yellow, W, H atoms and counter cations were omitted for clarity). (c) Polyhedral representation of [α-SiW11O39]8− (gray octahedron, WO6 unit; purple tetrahedron, SiO4 unit). | ||
Fig. S5† shows that there was no obvious precipitation when an aqueous solution of [α-SiW11O39]8− was mixed with a Cu2+ solution, which is different from that of mixing [Cu8(dpk·OH)8(OAc)4]4+ and [α-SiW11O39]8− (Fig. S4†), discarding the possibility that composite 1 is a product of [α-SiW11O39]8− and Cu2+ (Fig. S5†). The chemical composition and structure of composite 1 were first investigated by FTIR analysis. As shown in Fig. 2, the FTIR spectrum of composite 1 shows that all vibration signals of [Cu8(dpk·OH)8(OAc)4]4+. However, the v(ClO4−) bands at 626 cm−1 and around 1000 cm−1 in the FTIR spectrum of [Cu8(dpk·OH)8(OAc)4](ClO4)4 were not observed in that of composite 1, confirming that ClO4− has been displaced. Raman spectra of composite, Cu cluster and tungstosilicate are shown in Fig. S6,† and spectra display the characteristic peaks of Cu cluster and K8[α-SiW11O39] and the composite, clearly. The Raman spectrum of composite 1 retains almost all the characteristics of K8[α-SiW11O39], most importantly, the position of the characteristic peaks of K8[α-SiW11O39] in the Raman spectrum is almost identical to that in composite 1. Additionally, composite 1 also retains the characteristic peaks of the octanuclear Cu cluster except for the Raman signal of ClO4− at 937 cm−1, confirming the structure of [α-SiW11O39]8− in composite 1. XPS was used to characterize composite 1 and K8[α-SiW11O39]. As shown in Fig. S7–S9,† the W 4f and Si 2p binding energy of K8[α-SiW11O39] is similar to that of composite 1. The similar peak shapes and identical separation between the main peak and the satellite peak in both K8[α-SiW11O39] and composite 1 clearly illustrate that there are no changes in valence states and chemical environments of W and Si elements before and after binding with [Cu8(dpk·OH)8(OAc)4]4+. The results of the elemental analysis indicate that [Cu8(dpk·OH)8(OAc)4]4+ and [α-SiW11O39]8− are intact during the synthetic process, and the mole ratio of [Cu8(dpk·OH)8(OAc)4]4+ and [α-SiW11O39]8− in composite 1 is 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, in agreement with the charge numbers of the [Cu8(dpk·OH)8(OAc)4]4+ cation and [α-SiW11O39]8− anion. The Thermogravimetric (TG) test of composites indicates that the uncoordinated water content of composite 1 is 0.23% (Fig. S10†). Therefore, there is all most no impurity of water in the composite. Accordingly, the chemical composition of composite 1 is described as [Cu8(dpk·OH)8(OAc)4]2@[α-SiW11O39].
:1, in agreement with the charge numbers of the [Cu8(dpk·OH)8(OAc)4]4+ cation and [α-SiW11O39]8− anion. The Thermogravimetric (TG) test of composites indicates that the uncoordinated water content of composite 1 is 0.23% (Fig. S10†). Therefore, there is all most no impurity of water in the composite. Accordingly, the chemical composition of composite 1 is described as [Cu8(dpk·OH)8(OAc)4]2@[α-SiW11O39].
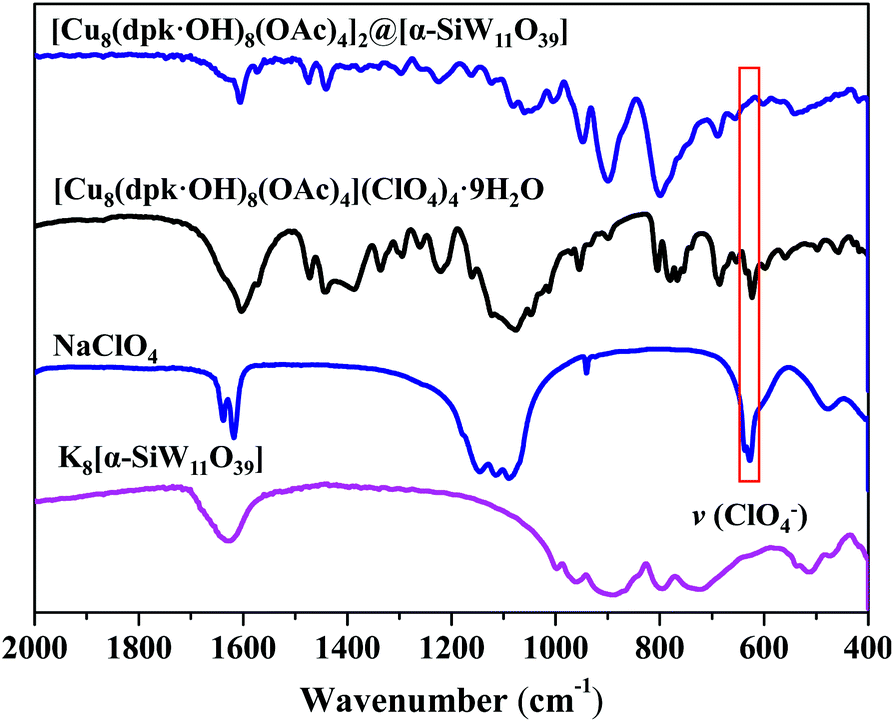 | ||
| Fig. 2 FTIR spectra of composite 1 (blue), [Cu8(dpk·OH)8(OAc)4] (ClO4)4 (black), NaClO4 (cyan) and K8[α-SiW11O39] (pink). | ||
The microstructure and morphology of the as-synthesized composite 1 were measured using SEM. Fig. 3a shows the low-magnified SEM images of the as-synthesized composite 1. The size of composite 1 is on the micron scale (Fig. S11†). The highly magnified SEM image (Fig. 3b) suggests the irregular shapes of composite 1. The TEM image presented in Fig. 3c further confirms its morphology. As expected, no obvious lattice fringes can be observed from the HRTEM image (Fig. 3d). Fig. S12 and S13† display the EDS analysis of the as-synthesized sample, revealing the coexistence and homogeneous distribution of Cu, O, N, Si and W elements in composite 1. Confirming that composite 1 exists in the form of direct electrostatic binding rather than the physical blend of [Cu8(dpk·OH)8(OAc)4](ClO4)4 and K8[α-SiW11O39]. By measuring N2 adsorption–desorption isotherms, the BET surface area of composite 1 was determined to be 28.8 m2 g−1 (Fig. S14†).
 | ||
| Fig. 3 SEM (a and b), TEM (c and d) images of the composite 1. | ||
Electrocatalytic tests
The α-undecatungstosilicate K8[α-SiW11O39] is reported to be stable in aqueous solutions between pH 4.5 and 7.0.51 As shown in Fig. S14,† the electrocatalytic activity of [Cu8(dpk·OH)8(OAc)4](ClO4)4 decreases as the pH of the electrolyte decreases, which is reasonable, since high pH is kinetically favorable for water oxidation. To ensure the stability of [α-SiW11O39]8− and obtain better activity of the Cu cluster in composite 1, the catalytic activity of composite 1 was tested at near neutral pH of 6.5. In the cyclic voltammetry (CV) experiments of electrochemical water oxidation, the electrochemical properties of composite 1 were determined in PBS at pH = 6.5. The CV curve shows that the catalytic current arises at around 1.60 V (vs. RHE) and finally reaches ca. 2.5 mA cm−2 at 1.88 V (vs. RHE).For a blank test using a bare GC electrode, the catalytic current is negligible. The results indicate that composite 1 is essential for electrochemical water oxidation (Fig. 4). In the tests of Cu8(dpk·OH)8(OAc)4](ClO4)4, K8[α-SiW11O39, and Cu2+ (Fig. S16†), [α-SiW11O39]8− and Cu2+ are inactive while [Cu8(dpk·OH)8(OAc)4]4+ is active under the same conditions. The overall results confirm that the active species in composite 1 is [Cu8(dpk·OH)8(OAc)4]4+ and the inactive [α-SiW11O39]8− just functions as a carrier for the catalyst.
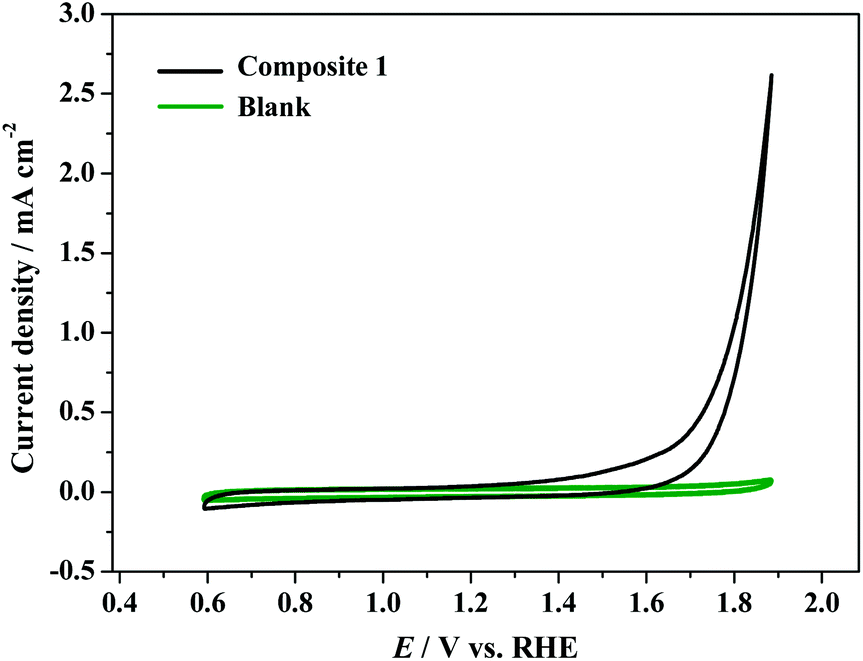 | ||
| Fig. 4 Cyclic voltammograms of composite 1 in 0.1 M PBS at a pH of 6.5. Scan rate = 50 mV s−1. | ||
To further investigate the electrocatalytic activity of composite 1, the catalytic current density (j) was obtained as a function of the overpotential at near-neutral pH of 6.5. As shown in Fig. 5, the Tafel plot can be fitted with a linear relationship in the overpotential range of 330–540 mV, and the logj versus overpotential plot gives a slope of 62 mV per decade, which is consistent with the mechanism involving a one-electron chemical pre-equilibrium step preceding the rate-limiting chemical step.52,53 An appreciable catalytic current can be observed at an overpotential of 370 mV. To achieve a catalytic current of 0.1 mA cm−2 and 1 mA cm−2, overpotentials of 490 mV and 580 mV are required, respectively. As depicted in Table S1,† the overpotential of composite 1 is comparable to that of some reported Cu-based materials capable of catalysing electrochemical water oxidation in near-neutral to basic solutions.54–63 Additionally, compared with some polyoxometalate-supported transition metal coordination complexes, composite 1 also shows moderate overpotential for water oxidation.64–67
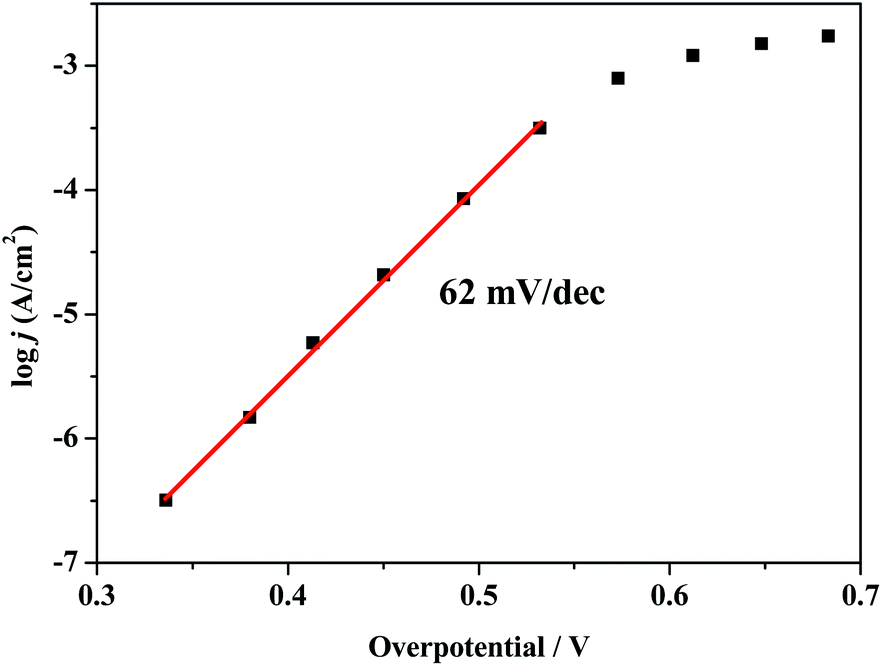 | ||
| Fig. 5 Tafel plot of composite 1 at pH 6.5. | ||
The catalytic stability of composite 1 coated on the GC electrode was monitored in continuous CV cycles. Compared to the CV curve of the second scan, that of the 1000th scan shows imperceptible change (Fig. S17†), revealing the stability of composite 1. To measure the faradaic efficiency and study the stability by the spectroscopic method, a controlled potential electrolysis (CPE) test of composite 1 coated on the FTO electrode was carried out. As shown in Fig. 6, during the CPE experiment of 10 h, current density decreased in the initial induction period and was then maintained at ca. 0.7 mA cm−2 when a potential of 1.78 V (vs. RHE) was applied. The stable catalytic current verifies that composite 1 was intact during electrolysis.
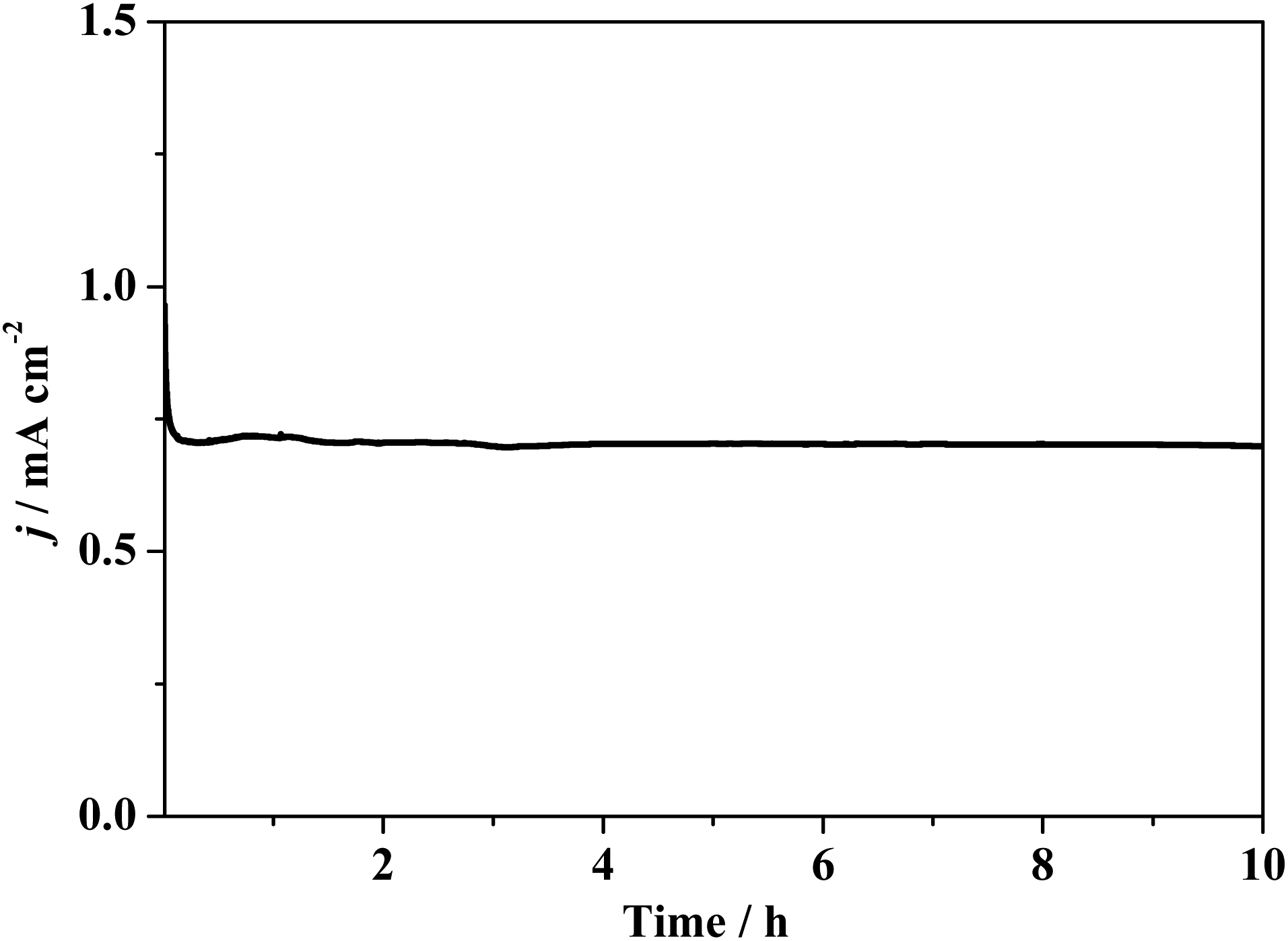 | ||
| Fig. 6 The current density curve obtained using CPE at 1.78 V (vs. RHE) with FTO electrode (2.0 cm × 1.0 cm, 1.0 cm2 coated with composite 1 and immersed in the electrolyte) as the working electrode in 0.1 M PBS at pH of 6.5. | ||
The faradaic efficiency for electrocatalytic water oxidation was also obtained by controlled potential electrolysis of composite 1 at pH = 6.5. The theoretical amount of O2 produced during electrolysis at 1.78 V (vs. RHE) is calculated from the total amount of charge passed through the electrolysis cell. After CPE of about 1.5 h, the amount of O2 detected was 8.9 μmol, implying a faradaic efficiency of 98% (Fig. 7). This value is comparable to heterogeneous materials such as metal oxide or metal hydroxide.38–41 The outcomes of present study demonstrate that the combination of homogeneous complex molecules with tungstosilicates is a feasible method to construct an efficient heterogenized homogeneous catalyst for water oxidation.
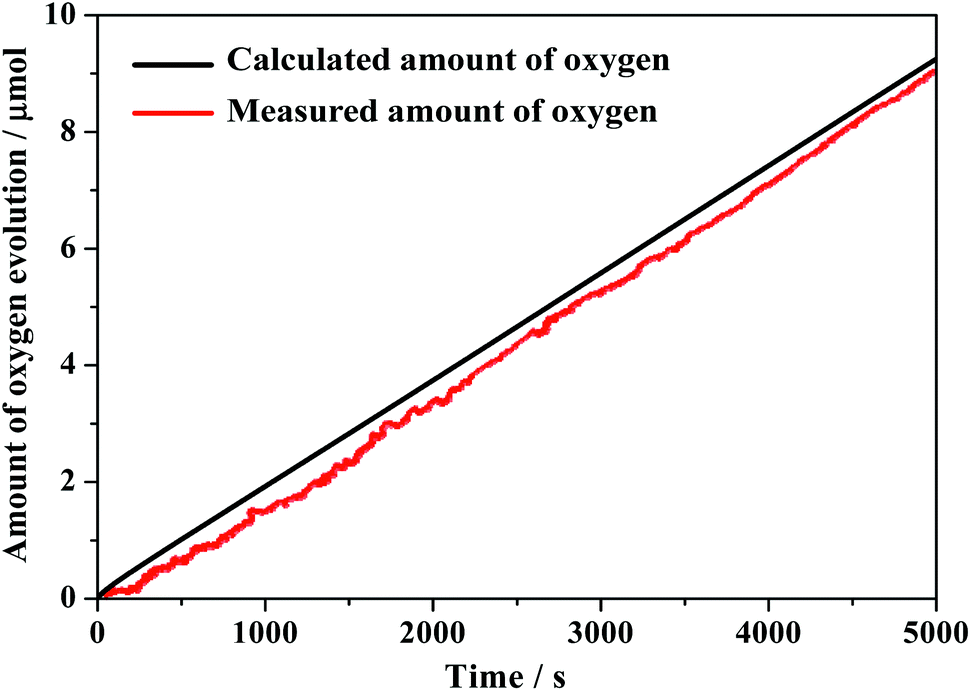 | ||
| Fig. 7 Faradaic efficiency of composite 1 at 1.78 V vs. RHE. The amount of evolved oxygen during electrolysis and the theoretical amount of evolved oxygen (assuming a faradaic efficiency of 100% and four electrons are needed to produce one oxygen molecule) are represented by the red line and black line, respectively. | ||
After the CPE test of 10 h, composite 1 on the surface of the FTO electrode was characterized by FTIR SEM, TEM and XPS analyses. As shown in Fig. 8, the catalyst used for 10 h in the CPE experiment (named herein as post-catalytic composite 1) displays all the characteristic peaks of pristine composite 1, confirming the structural integrity of composite 1 during electrolysis.
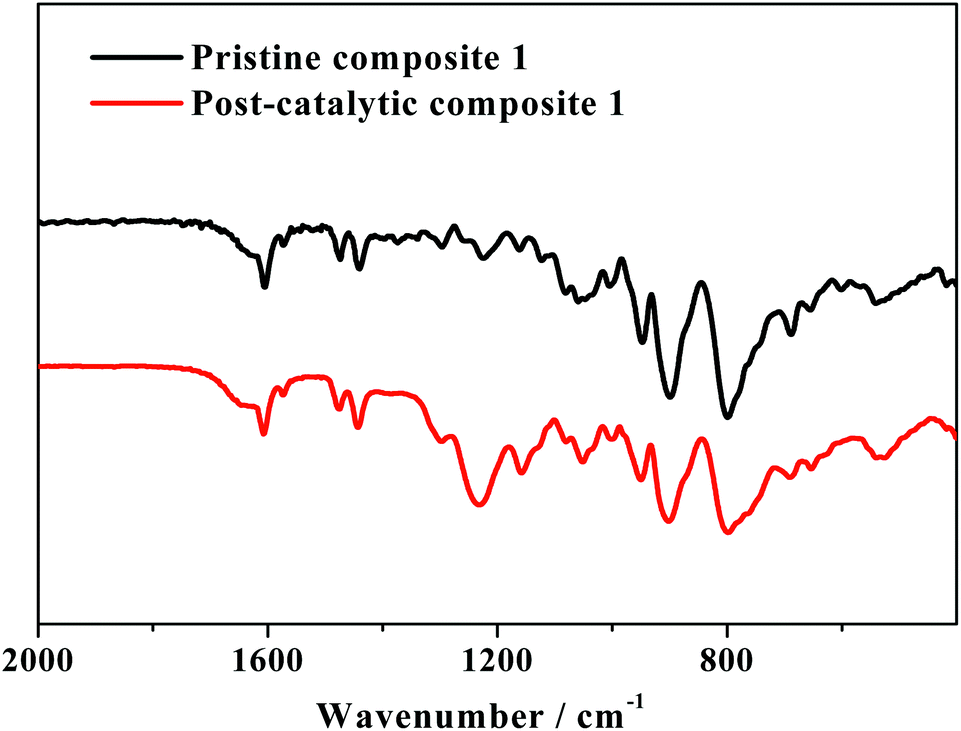 | ||
| Fig. 8 FTIR spectra of pristine composite 1 and post-catalytic composite 1 after controlled potential electrolysis at 1.78 V (vs. RHE). (Note that the strong infrared vibrations between 900 cm−1 and 1250 cm−1 are due to the added Nafion).13 | ||
As shown in SEM (Fig. 9a and b) and TEM (Fig. 9c and d) images of the sample on the surface of the FTO electrode tested after CPE, composite 1 kept its basic scale and structure. The EDS analysis of the post-catalytic composite 1 also indicates the homogeneous distribution of Cu, O, N, Si and W elements (Fig. S18 and S19†). The morphology observation and elemental analysis together confirm the stability of the composite.
 | ||
| Fig. 9 SEM (a and b), TEM (c and d) images of the post-catalytic composite 1. | ||
The XPS scans of pristine and post-catalytic composite 1 are shown in Fig. 10 and S19–S21.† The Cu 2p spectrum of the fresh catalyst sample gives two peaks at 933.9 eV and 954.1 eV with two satellite peaks at 943.0 eV and 962.6 eV, which are assignable to Cu 2p3/2 and Cu 2p1/2, respectively (Fig. 10). The W 4f spectra show peaks at 37.1 eV and 35.0 eV ascribable to [α-SiW11O39]8− (Fig. S21†). In the XPS spectrum of the post-catalytic catalyst, Cu 2p and W 4f binding energies were similar to those of the pristine sample, the identical separation between the main peak and the satellite peak in both samples and the consistency of peak shapes clearly illustrates that there are no changes in the valence states and chemical environments of Cu, W during electrochemical water oxidation.68 Compared with Cu 2p and W 4f peaks, the Si 2p peak is much lower in intensity (Fig. S22†). It is understandable because the content of the Si element is extremely low (ca. 0.4 atom% according to the chemical composition of [Cu8(dpk·OH)8(OAc)4]2@[α-SiW11O39]).
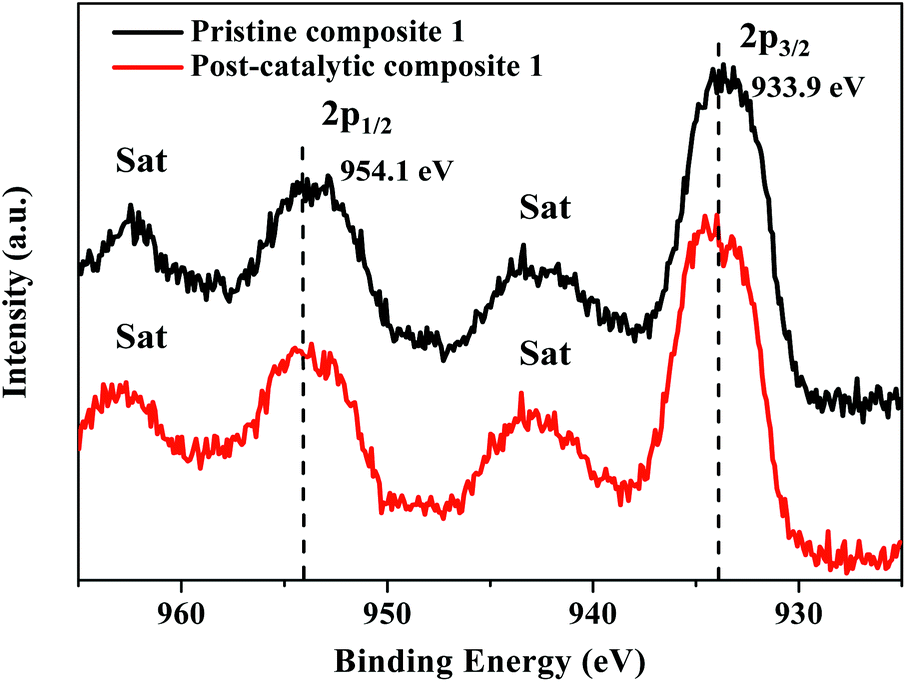 | ||
| Fig. 10 High-resolution XPS Cu 2p spectra of pristine and post-catalytic composite 1. | ||
Electrostatic binding is a convenient method to synthesize robust heterogeneous electrocatalysts by heterogenization of other water-soluble complexes, such as tetranuclear Co complex [Co4(dpk·OH)4(OAc)2(H2O)2]2+ (Fig. S23 and S24†) and [Ni(NHA)]2+ (Fig. S25 and S26†). The Co-containing composite and Ni-containing composite were characterized by elemental analysis, FTIR spectra and TG tests (Fig. S27–S30†). They were confirmed to contain metal complex cation and polyoxoanion and are named as [Co4(dpk·OH)4(OAc)2(H2O)2]4@[α-SiW11O39] and [Ni(NHA)]4@[α-SiW11O39], respectively. Most importantly, they are capable of catalyzing water oxidation at pH 6.5 (Fig. S31†). The onset overpotential of [Co4(dpk·OH)4(OAc)2(H2O)2]4@[α-SiW11O39] required for water oxidation at neutral pH of 6.5 is only 400 mV, which is lower than that reported in some previous reports for other cobalt materials.69–74 Besides, synthesis of composite [Cu8(dpk·OH)8(OAc)4]@[SiW12O40] containing octanuclear Cu clusters and non-lacunary polyoxometalate [SiW12O40]4− was further confirmed to be feasible (Fig. S32 and S33†). As shown in Fig. S34,† composite 1 shows higher activity than [Cu8(dpk·OH)8(OAc)4]@[SiW12O40], probably because of the lower Cu cluster content in [Cu8(dpk·OH)8(OAc)4]@[SiW12O40] compared with composite 1. In a word, it is indeed feasible to obtain efficient WOCs by combining transition metal complexes with polyoxoanions through electrostatic interaction. In the next step, electrocatalysts with lower overpotential and higher efficiency for water oxidation are expected to be achieved by the synthesis of composites containing different kinds of transition metal complexes and polyoxoanions.
Conclusions
For the first time, a water-soluble octanuclear cluster [Cu8(dpk·OH)8(OAc)4]4+ was successfully heterogenized via electrostatic binding with [α-SiW11O39]8−. The as-synthesized composite [Cu8(dpk·OH)8(OAc)4]2@[α-SiW11O39] shows excellent electrocatalytic performance toward water oxidation at near-neutral pH in terms of overpotential and long-term stability. Compared with the reported processes for the immobilization of homogeneous WOCs that are based on transition metal complexes, the strategy adopted for the synthesis of composite 1 bypasses the tedious procedures of structural modification. This work provides insightful guidelines for the design and synthesis of heterogenized homogeneous catalysts, especially those that are based on homogeneous transition metal complex complexes for water oxidation reaction or other electrocatalytic systems.Experimental section
Material
All chemicals involved in this work, except [Cu8(dpk·OH)8(OAc)4](ClO4)4, [Co4(dpk·OH)4(OAc)2(H2O)2](ClO4)2, [Ni(NHA)](ClO4)2 and K8[α-SiW11O39], were purchased from a chemical company and used without further purification. Ultrapure water (18.25 MΩ cm) used for the preparation of catalyst and buffer solution was from a Water Purifier.Instrumentation
Powder X-ray diffraction (PXRD) data of complex were collected using a Bruker D8 advance powder X-ray Cu Kα radiation diffractometer (λ = 0.154 06 nm) at 40 kV and 40 mA. Infrared spectra (1 wt% of sample in KBr pellet) were acquired using a ThermoFisher scientific Nicolet iS5 FTIR spectrometer. Raman spectra were acquired over a Renishaw inVia Reflex, LabRam HR Evolution (laser emitting at 785 nm). Elemental analysis for the metal element was performed using a TJA ICP-atomic emission spectrometer (IRIS Advantage ER/S). The TG test was conducted on SDT-Q600 analyser, elemental analysis for C, H, N elements was performed using Elementar Vario EL, Thermo Fisher using the CNH mode. The corresponding sample was heated to 1150 °C under a pure oxygen environment to produce CO2, H2O, NOxet al. Then, all the samples were reduced in the reduction tubes at 850 °C by deoxidized copper to obtain CO2, H2O, N2, etc. which are separated by adsorption–desorption column and detected by gas chromatography with a thermal conductivity detector and the element content of C, H and N was obtained. The microstructural profiles of composite 1 were examined using a scanning electron microscope (SEM, Carl Zeiss Sigma 300) combined with energy-dispersive X-ray spectroscopy (EDS). A transmission electron microscope (TEM, jeol2100f) was used to obtain detailed structural information on composite 1. The specific surface area of composite 1 was computed from the N2 physisorption data acquired at 77 K (Micromeritics ASAP 2460 3.01) by using the Brunauer–Emmett–Teller BET method. XPS analysis was performed over a Thermo Scientific K-Alpha spectrometer. Quantitative detection of O2 was done using a fluorescence-based oxygen sensor (Ocean Optics).Synthesis of catalyst
The octanuclear Cu(II) complex [Cu8(dpk·OH)8(OAc)4](ClO4)4 was synthesized in an aqueous medium by reacting Cu(ClO4)2·9H2O with di-2-pyridyl ketone according to literature.48,49 K8[α-SiW11O39] was synthesized according to the method reported by Tézé et al.51 Composite 1 was obtained by adding 10 mL aqueous solution of 2 mM [Cu8(dpk·OH)8(OAc)4](ClO4)4 to 10 mL aqueous solution of 1 mM K8[α-SiW11O39]. There was the immediate formation of composite 1 in the form of a light green precipitate. The precipitate was collected by centrifugation and washed with ultrapure water and ethanol several times, followed by vacuum drying.[Co4(dpk·OH)4(OAc)2(H2O)2](ClO4)2 was synthesized according to the reported literature.36 [Ni(NHA)](ClO4)2 was prepared by the one-pot template method.75 Co-containing composites and Ni-containing composite were synthesized by adding 10 mL aqueous solution of 2 mM corresponding complex to 10 mL aqueous solution of 0.5 mM K8[α-SiW11O39], respectively. Composite in the form of red precipitate ([Co4(dpk·OH)4(OAc)2(H2O)2]4@[α-SiW11O39]) and light-yellow precipitate ([Ni(NHA)]4@[α-SiW11O39]) were collected by centrifugation and washed with ultrapure water and ethanol several times, followed by vacuum drying. Composite [Cu8(dpk·OH)8(OAc)4]@[SiW12O40] was synthesized by adding 10 mL aqueous solution of Cu cluster (2 mM) to 10 mL aqueous solution of H4[SiW12O40] (2 mM). The precipitate of [Cu8(dpk·OH)8(OAc)4]@[SiW12O40] formed immediately was collected by centrifugation and washed with ultrapure water and ethanol several times, followed by vacuum drying.
All composites were decomposed by treating with 2 M NaOH (transfer complex cation in composite into metal oxide or metal hydroxide, named as CuOx, CoOx and NiOx) and then treated with extra 2 M H2SO4 (transfer CuOx, CoOx or NiOx into soluble metal cation) to obtained solutions for elemental analysis, Elemental analysis of composite 1 found (%): Cu, 13.9; W, 27.0; C, 31.6; H, 2.3; N, 6.0; Si, 0.4. Calc. (%): Cu, 13.8; W, 27.4; C, 31.2; H, 2.3; N, 6.1; Si, 0.4. Elemental analysis value for K and Cl elemental is lower than 0.1. Elemental analysis of Co containing composite found (%): Co, 12.8; W, 27.4; C, 31.0; H, 2.5; N, 6.0; Si, 0.4. Calc. (%): Co, 12.7; W, 27.1; C, 30.9; H, 2.4; N, 6.0; Si, 0.4. Elemental analysis of Ni containing composite found (%): Ni, 6.0; W, 51.6; C, 14.7; H, 2.6; N, 8.6, Si, 0.7. Calc (%): Ni, 5.9; W, 51.5; C, 14.6; H, 2.6; N, 8.5; Si, 0.6. For these two composites, elemental analysis value for K and Cl elemental is lower than 0.1. Elemental analysis of [Cu8(dpk·OH)8(OAc)4]@[SiW12O40] found: Cu, 9.8; W, 41.8; C, 22.1; H, 1.6; N, 4.3, Si, 0.5. Calc. (%): Cu, 9.7; W, 42.2; C, 22.0; H, 1.6; N, 4.3; Si, 0.5. Elemental analysis value for K and Cl elemental is lower than 0.1.
Electrochemical tests of catalyst
The 0.1 M PBS was obtained by adjusting the pH of 0.1 M NaH2PO4 aqueous solution with 0.1 M Na2HPO4. All electrochemical tests were carried out in a standard three-electrode system and recorded using a CHI660E electrochemical analyzer with a glassy carbon (GC), Ag/AgCl (3.5 M KCl) and Pt wire as working, reference and auxiliary electrodes, respectively. The GC electrode (0.071 cm2) was polished with 300 nm and 50 nm Al2O3 slurry for at least 3 min, followed by sonication in ethanol and distilled water separately for about 15 s and rinsed with ultrapure water. Then, 5 mg of the composite was dispersed in 1 mL of mixed solvent (ultrapure water and ethanol, 3:1 v/v) containing 10 μL 5 wt% Nafion in lower aliphatic alcohol and water. The mixed solution was then sonicated for at least 30 min, forming a homogeneous ink. Then, 5 μL of the mixed solution was drop-cast onto the GC electrode (0.35 mg cm−2) to form a thin film. The GC electrode was then dried under infrared light and used for the electrochemical measurements. All potentials measured in this work were converted to the reversible hydrogen electrode (RHE) scale according to the Nernst equation: ERHE = EAg/AgCl + 0.208 V + pH × 0.059 V. The current density was calculated from the geometric area (0.071 cm2) of the GC electrode. The overpotential was calculated according to the following formula: overpotential = ERHE − 1.23 V. For the controlled potential electrolysis experiment, fluorine-doped tin oxide (FTO) electrode (2 cm × 1 cm, 1 cm2 coated with the composite) were used as the working electrode in 0.1 M PBS at pH of 6.5. The FTO electrode was cleaned by sonication in acetone, ethanol, and ultrapure water for 30 min, separately. For electrochemical tests, 70 μL of the mixed solution of composite 1 was drop-cast onto the FTO electrode (0.35 mg cm−2) to form a thin film.
Author contributions
Yanmei Chen and Zhijun Ruan jointly supervised this work. Junqi Lin, Xin Chen, and Shanshan Liu designed and carried out the experiments. Junqi Lin, Xin Chen, Nini Wang and Zhengfang Tian analyzed the experimental data. Junqi Lin wrote the manuscript. All authors discussed the results and assisted during manuscript preparation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21704032 and 21501061), the Natural Science Foundation of Hubei Province (2017CFB223) and the Research Foundation of Huanggang Normal University (2042020043). The authors thank Prof. C. T. Au for helpful suggestions.Notes and references
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2015, 14, 2060–2086 RSC.
- H. Dotan, A. Landman, S. W. Sheehan, K. D. Malviya, G. E. Shter, D. A. Grave, Z. Arzi, N. Yehudai, M. Halabi, N. Gal, N. Hadari, C. Cohen, A. Rothschild and G. S. Grader, Nat. Energy., 2019, 4, 786–795 CrossRef CAS.
- L. Peng, L. Shang, T. Zhang and G. I. N. Waterhouse, Adv. Energy Mater., 2020, 10, 2003018 CrossRef CAS.
- K. Zhu, F. Shi, X. Zhu and W. Yang, Nano Energy, 2020, 73, 104761 CrossRef CAS.
- R.-Q. Yao, H. Shi, W.-B. Wan, Z. Wen, X.-Y. Lang and Q. Jiang, Adv. Mater., 2020, 32, 1907214 CrossRef CAS PubMed.
- L. An, C. Wei, M. Lu, H. Liu, Y. Chen, G. G. Scherer, A. C. Fisher, P. Xi, Z. J. Xu and C.-H. Yan, Adv. Mater., 2021, 2006328 CrossRef CAS PubMed.
- L.-A. Stern, L. Feng, F. Song and X. Hu, Energy Environ. Sci., 2015, 8, 2347–2351 RSC.
- K. Fan, Y. Ji, H. Zou, J. Zhang, B. Zhu, H. Chen, Q. Daniel, Y. Luo, J. Yu and L. Sun, Angew. Chem., Int. Ed., 2017, 56, 3289–3293 CrossRef CAS PubMed.
- X. Zhang, Y. Zhao, Y. Zhao, R. Shi, G. I. N. Waterhouse and T. Zhang, Adv. Energy Mater., 2019, 9, 1900881 CrossRef.
- J. Yin, J. Jin, M. Lu, B. Huang, H. Zhang, Y. Peng, P. Xi and C.-H. Yan, J. Am. Chem. Soc., 2020, 142, 18378–18386 CrossRef CAS PubMed.
- Y. Sun, X. Ren, S. Sun, Z. Liu, S. Xi and Z. Xu, Angew. Chem., Int. Ed., 2021, 60, 14536–14564 CrossRef CAS PubMed.
- Q. Xu, H. Jiang, X. Duan, Z. Jiang, Y. Hu, S. W. Boettcher, W. Zhang, S. Guo and C. Li, Nano Lett., 2021, 21, 492–499 CrossRef CAS PubMed.
- T. Tian, H. Gao, X. Zhou, L. Zheng, J. Wu, K. Li and Y. Ding, ACS Energy Lett., 2018, 3, 2150–2158 CrossRef CAS.
- M. Zhang, H.-T. Zhang, X.-J. Su, F. Xie and R. Liao, Angew. Chem., Int. Ed., 2021, 60, 12467–12474 CrossRef PubMed.
- M. K. Coggins, M.-T. Zhang, A. K. Vannucci, C. J. Dares and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 5531–5534 CrossRef CAS PubMed.
- H.-Y. Du, S.-C. Chen, X.-J. Su, L. Jiao and M.-T. Zhang, J. Am. Chem. Soc., 2018, 140, 1557–1565 CrossRef CAS PubMed.
- Y. Han, Y. Wu, W. Lai and R. Cao, Inorg. Chem., 2015, 54, 5604–5613 CrossRef CAS PubMed.
- J. Xin, L. Jian, Y. Bing, W. Xiang-Zhu, D. Bo-Wei, K. Yi, H. Mao-Yong, T. Chen-Ho and W. Li-Zhu, Angew. Chem., Int. Ed., 2018, 57, 7850–7854 CrossRef PubMed.
- K. J. Fisher, K. L. Materna, B. Q. Mercado, R. H. Crabtree and G. W. Brudvig, ACS Catal., 2017, 7, 3384–3387 CrossRef CAS.
- L. Wang, L. Duan, R. B. Ambre, Q. Daniel, H. Chen, J. Sun, B. Das and A. Thapper, J. Catal., 2016, 335, 72–78 CrossRef CAS.
- B. Zhang and L. Sun, Chem. Soc. Rev., 2019, 48, 2216–2264 RSC.
- P. Garrido-Barros, C. Gimbert-Suriñach, D. Moonshiram, A. Picón, P. Monge, V. S. Batista and A. Llobet, J. Am. Chem. Soc., 2017, 139, 12907–12910 CrossRef CAS PubMed.
- F. M. Toma, A. Sartorel, M. Iurlo, M. Carraro, P. Parisse, C. Maccato, S. Rapino, B. R. Gonzalez, H. Amenitsch, T. Da Ros, L. Casalis, A. Goldoni, M. Marcaccio, G. Scorrano, G. Scoles, F. Paolucci, M. Prato and M. Bonchio, Nat. Chem., 2010, 2, 826–831 CrossRef CAS PubMed.
- M. Zheng, Y. Ding, X. Cao, T. Tian and J. Lin, Appl. Catal., B, 2018, 237, 1091–1100 CrossRef CAS.
- Y. Choi, D. Jeon, Y. Choi, D. Kim, N. Kim, M. Gu, S. Bae, T. Lee, H.-W. Lee, B.-S. Kim and J. Ryu, ACS Nano, 2019, 13, 467–475 CrossRef CAS PubMed.
- S. Shahsavarifar, M. Masteri-Farahani and M. R. Ganjali, Langmuir, 2021, 37, 1925–1931 CrossRef CAS PubMed.
- G. Y. Lee, I. Kim, J. Lim, M. Y. Yang, D. S. Choi, Y. Gu, Y. Oh, S. H. Kang, Y. S. Nam and S. O. Kim, J. Mater. Chem. A, 2017, 5, 1941–1947 RSC.
- M. Blasco-Ahicart, J. Soriano-López, J. J. Carbó, J. M. Poblet and J. R. Galan-Mascaros, Nat. Chem., 2018, 10, 24–30 CrossRef CAS PubMed.
- X.-B. Han, D.-X. Wang, E. Gracia-Espino, Y.-H. Luo, Y.-Z. Tan, D.-F. Lu, Y.-G. Li, T. Wågberg, E.-B. Wang and L.-S. Zheng, Chin. J. Catal., 2020, 41, 853–857 CrossRef CAS.
- J. T. Arens, M. Blasco-Ahicart, K. Azmani, J. Soriano-López, A. García-Eguizábal, J. M. Poblet and J. R. Galan-Mascaros, J. Catal., 2020, 389, 345–351 CrossRef CAS.
- F. Li, B. Zhang, X. Li, Y. Jiang, L. Chen, Y. Li and L. Sun, Angew. Chem., Int. Ed., 2011, 50, 12276–12279 CrossRef CAS PubMed.
- Z. Chen, J. J. Concepcion, J. W. Jurss and T. J. Meyer, J. Am. Chem. Soc., 2009, 131, 15580–15581 CrossRef CAS PubMed.
- L. Xie, X. Li, B. Wang, J. Meng, H. Lei, W. Zhang and R. Cao, Angew. Chem., Int. Ed., 2019, 58, 18883–18887 CrossRef CAS PubMed.
- T.-T. Li, J. Qian, Q. Zhou, J.-L. Lin and Y.-Q. Zheng, Dalton Trans., 2017, 46, 13020–13026 RSC.
- G. Maayan, N. Gluz and G. Christou, Nat. Catal., 2018, 1, 48–54 CrossRef CAS.
- F. Song, R. Moré, M. Schilling, G. Smolentsev, N. Azzaroli, T. Fox, S. Luber and G. R. Patzke, J. Am. Chem. Soc., 2017, 139, 14198–14208 CrossRef CAS PubMed.
- F. Evangelisti, R. More, F. Hodel, S. Luber and G. R. Patzke, J. Am. Chem. Soc., 2015, 137, 11076–11084 CrossRef CAS PubMed.
- T. N. Huan, G. Rousse, S. Zanna, I. T. Lucas, X. Xu, N. Menguy, V. Mougel and M. Fontecave, Angew. Chem., Int. Ed., 2017, 129, 4870–4874 CrossRef.
- H. Chen, Y. Gao, Z. Lu, L. Ye and L. Sun, Electrochim. Acta, 2017, 230, 501–507 CrossRef CAS.
- M. Qian, X. Liu, S. Cui, H. Jia and P. Du, Electrochim. Acta, 2018, 263, 318–327 CrossRef CAS.
- J. Kang, J. Sheng, J. Xie, H. Ye, J. Chen, X.-Z. Fu, G. Du, R. Sun and C.-P. Wong, J. Mater. Chem. A, 2018, 6, 10064–10073 RSC.
- J. Song, C. Zhu, B. Z. Xu, S. Fu, M. H. Engelhard, R. Ye, D. Du, S. P. Beckman and Y. Lin, Adv. Energy Mater., 2017, 7, 1601555 CrossRef.
- X. Du, J. Huang and Y. Ding, Dalton Trans., 2017, 46, 7327–7331 RSC.
- C.-C. Hou, Q.-Q. Chen, C.-J. Wang, F. Liang, Z. Lin, W.-F. Fu and Y. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 23037–23048 CrossRef CAS PubMed.
- X. Wu, S. Li, J. Liu and M. Yu, ACS Appl. Nano Mater., 2019, 2, 4921–4932 CrossRef CAS.
- K. Karthick, S. Anantharaj, S. Patchaiammal, S. N. Jagadeesan, P. Kumar, S. R. Ede, D. K. Pattanayak and S. Kundu, Inorg. Chem., 2019, 58, 9490–9499 CrossRef CAS PubMed.
- K. Kannimuthu, K. Sangeetha, S. S. Sankar, A. Karmakar, R. Madhuab and S. Kundu, Inorg. Chem. Front., 2021, 8, 234–272 RSC.
- J. Lin, X. Liang, X. Cao, N. Wei and Y. Ding, Chem. Commun., 2018, 52, 12515–12518 RSC.
- V. Tangoulis, C. P. Raptopoulou, A. Terzis, S. Paschalidou, S. P. Perlepes and E. G. Bakalbassis, Inorg. Chem., 1997, 36, 3996–4006 CrossRef CAS.
- Y.-M. Li, J.-J. Zhang, R.-B. Fu, S.-C. Xiang, T.-L. Sheng, D.-Q. Yuan, X.-H. Huang and X.-T. Wu, Polyhedron, 2006, 25, 1618–1624 CrossRef CAS.
- A. Tézé, G. Hervéa, R. G. Finke and D. K. Lyon, Inorg. Synth., 1990, 85–96 Search PubMed.
- J. B. Gerken, J. G. McAlpin, J. Y. C. Chen, M. L. Rigsby, W. H. Casey, R. D. Britt and S. S. Stahl, J. Am. Chem. Soc., 2011, 133, 14431–14442 CrossRef CAS PubMed.
- R. Mishra, E. Ülker and F. Karadas, ChemElectroChem, 2017, 4, 75–80 CrossRef CAS.
- F. Yu, F. Li, B. Zhang, H. Li and L. Sun, ACS Catal., 2014, 5, 627–630 CrossRef.
- X. Liu, S. Cui, Z. Sun, Y. Ren, X. Zhang and P. Du, J. Phys. Chem. C, 2016, 120, 831–840 CrossRef CAS.
- X. Liu, S. Cui, Z. Sun and P. Du, Electrochim. Acta, 2015, 160, 202–208 CrossRef CAS.
- H. A. Younus, Y. Zhang, M. Vandichel, N. Ahmad, K. Laasonen, F. Verpoort, C. Zhang and S. Zhang, ChemSusChem, 2020, 13, 5088–5099 CrossRef CAS PubMed.
- S. Cui, X. Liu, Z. Sun and P. Du, ACS Sustainable Chem. Eng., 2016, 4, 2593–2600 CrossRef CAS.
- X. Liu, Z. Sun, S. Cui and P. Du, Electrochim. Acta, 2016, 187, 381–388 CrossRef CAS.
- J. Du, Z. Chen, S. Ye, B. J. Wiley and T. J. Meyer, Angew. Chem., Int. Ed., 2015, 54, 2073–2078 CrossRef CAS PubMed.
- T.-T. Li, S. Cao, C. Yang, Y. Chen, X.-J. Lv and W.-F. Fu, Inorg. Chem., 2015, 54, 3061–3067 CrossRef CAS PubMed.
- C. Lu, J. Du, X.-J. Su, M.-T. Zhang, X. Xu, T. J. Meyer and Z. Chen, ACS Catal., 2016, 6, 77–83 CrossRef CAS.
- C. Lu, J. Wang and Z. Chen, ChemCatChem, 2016, 8, 2165–2170 CrossRef CAS.
- M. V. Pavliuk, E. Mijangos, V. G. Makhankove, V. N. Kokozay, S. Pullen, J. Liu, J. Zhu, S. Styring and A. Thapper, ChemSusChem, 2016, 9, 2957–2966 CrossRef CAS PubMed.
- C. Singh, S. Mukhopadhyay and S. K. Das, Inorg. Chem., 2018, 57, 6479–6490 CrossRef CAS PubMed.
- Y. Li, X.-R. Chang, X.-J. Sang, J.-S. Li, Y.-H. Luo, Z.-Z. Zhu and W.-S. You, Eur. J. Inorg. Chem., 2019, 2019, 3597–3604 CrossRef CAS.
- C.-J. Wang, S. Yao, Y.-Z. Chen, Z.-M. Zhang and E.-B. Wang, RSC Adv., 2016, 6, 99010–99015 RSC.
- L. Yu, X. Du, Y. Ding and P. Zhou, Chem. Commun., 2015, 51, 17443–17446 RSC.
- S. Mukhopadhyay, J. Debgupta, C. Singh, A. Kar and S. K. Das, Angew. Chem., Int. Ed., 2018, 57, 1918–1923 CrossRef CAS PubMed.
- P. J. Zambiazi, G. O. Aparecido, T. V. B. Ferraz, W. S. J. Skinner, R. G. Yoshimura, D. E. B. Moreira, R. L. Germscheidt, L. L. Nascimento, A. O. T. Patrocinio, A. L. B. Formiga and J. A. Bonacin, Dalton Trans., 2020, 49, 16488–16497 RSC.
- M.-R. Liu, Y.-P. Lin, K. Wang, S. Chen, F. Wang and T. Zhou, Chin. J. Catal., 2020, 41, 1654–1662 CrossRef CAS.
- J. W. Tian, Y. P. Wu, Y. S. Li, J. H. Wei, J. W. Yi, S. Li, J. Zhao and D. S. Li, Inorg. Chem., 2019, 58, 5837–5843 CrossRef CAS PubMed.
- X. Wang, B. Li, Y. P. Wu, A. Tsamis, H. G. Yu, S. Liu, J. Zhao, Y. S. Li and D. S. Li, Inorg. Chem., 2020, 59, 4764–4771 CrossRef CAS PubMed.
- W. Zhou, D.-D. Huang, Y.-P. Wu, J. Zhao, T. Wu, J. Zhang, D.-S. Li, C. Sun, P. Feng and X. Bu, Angew. Chem., Int. Ed., 2019, 58, 4227–4231 CrossRef CAS PubMed.
- M. P. Suh, W. Shin, S. G. Kang, M. S. Lah and T. M. Chung, Inorg. Chem., 1989, 28, 1602–1605 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1se01221e |
| This journal is © The Royal Society of Chemistry 2022 |