
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical quantification of sulfamethoxazole antibiotic in environmental water using zeolitic imidazolate framework (ZIF)-derived single-atom cobalt catalyst in nitrogen-doped carbon nanostructures†
Qingyu
Gu
,
Zhongxue
Wang
,
Qianwen
Ding
,
Huiling
Li
,
Ping
Wu
 * and
Chenxin
Cai
*
* and
Chenxin
Cai
*
Jiangsu Key Laboratory of New Power Batteries, Jiangsu Collaborative Innovation Center of Biomedical Functional Materials, College of Chemistry and Materials Science, Nanjing Normal University, Nanjing 210023, P.R. China. E-mail: wuping@njnu.edu.cn; cxcai@njnu.edu.cn
First published on 10th August 2022
Abstract
An electrochemical sensor based on single-atom cobalt-anchored nitrogen-doped carbon catalysts (Co/N–C SACs), synthesized by pyrolyzing zeolitic imidazolate frameworks (Co-ZIF-8), was constructed to quantify sulfamethoxazole (SMX), a member of the sulfonamide family of antibiotics, in environmental water samples. The quantification depends on the electrocatalytic oxidation of the aromatic –NH2 group in the SMX molecule to the hydroxylamine group (–NHOH). The sensor was fabricated by depositing the Co/N–C SAC suspension (in a mixture of water, ethanol, and Nafion) on a glassy carbon (GC) electrode surface. The response of the sensor showed a linear relationship with SMX in a wide range of concentration (0.08–66.5 μM) with a limit of detection (LOD) of ∼15 nM at a signal-to-noise (S/N) ratio of 3. The sensor also had high repeatability, reproducibility, and stability. Moreover, it exhibited high selectivity and resistance to interference in the presence of other antibiotics (trimethoprim, ciprofloxacin, and norfloxacin selected randomly), endocrine disruptors (17β-estradiol), pesticides (carbaryl), common aqueous ions (Na+, K+, Fe3+, Cu2+, Ca2+, Cl−, Br−, CO32−, SO42−, and NO3−), and common surfactants (sodium dodecyl sulfate and Triton X-100). Furthermore, the sensor was used to quantify the SMX in environmental water samples (lake water, groundwater, and secondary wastewater) with high accuracy. This electrochemical method can be used for quantifying SMX in environmental water samples for various applications, including biomedical analysis, environmental pollutant detection, and water safety evaluation.
Introduction
Sulfamethoxazole (SMX, 4-amino-N-(5-methyl-3-isoxazolyl)benzenesulfonamide), a member of the sulfonamide family of antibiotics,1 exhibits a broad spectrum of antibacterial activity against both Gram-positive and Gram-negative aerobic bacteria.2 It is widely used for the treatment of bacterial infections of respiratory and urinary tracts and opportunistic infections associated with organ transplants.2,3 SMX is typically used in combination with trimethoprim,4 a dihydrofolate reductase inhibitor that suppresses folic acid metabolism by disturbing the generation of tetrahydrofolic acid from dihydrofolic acid in bacteria5 to produce a synergistic antibacterial activity. It has also been used in animal husbandry for enhancing the resistance of cows, pigs, and poultry against diseases and for increasing their production.6 SMX is widely used owing to its low cost, high efficacy, and wide availability. Although it is an effective antimicrobial drug, it can cause various harmful side effects, such as gastrointestinal disturbances, hypersensitivity reactions, and a number of hematological abnormalities.1,7 Moreover, SMX has poor biodegradability. A significant percentage (up to 30–70%) of the consumed dose is not completely metabolized and is excreted in unchanged and active form,8,9 which accumulates in environmental waters (surface waters, groundwater, and secondary wastewater effluents) and endangers the environmental safety. The SMX residues in foods, such as milk, meat, egg, and others, may endanger human health and lead to various diseases, even cancer (such as thyroid cancer).10 Therefore, China, the European Union, and the USA have established 0.1 ppm (100 μg kg−1) as the maximum residue limit for the total amount of SMX in edible tissues. Thus, there is a need for accurate, sensitive, and simple approaches to quantify the low levels of antibiotics in environmental samples.A number of analytical techniques have been developed for SMX quantification, including chromatography-based techniques (HPLC, GC, LC-MS/MS),11,12 spectrophotometry (fluorometry, chemiluminescence, photometry),13 biosensors (optical biosensor, antibody-based immunosensor, and surface plasmon resonance-based biosensor),14,15 and enzyme-linked immune assays.16 However, most of these methods require high maintenance costs, tedious sample pre-treatment processes, and skilled operators. In some cases, however, the detection limit is unacceptably low.
Electrochemical techniques have several advantages, including a fast response, high selectivity and sensitivity (comes from the electrochemical reaction depends on the applied potential, which can be regulated as required), relatively low preparation costs, simple operational requirements, and easy miniaturization.17–23 These techniques can be used to overcome the aforementioned obstacles for facile on-site environmental analysis. They can also be used for high-fidelity antibiotic quantification. The electrochemical quantification of SMX is mainly based on the electrochemical oxidation of –NH2 (aromatic amino group) to the hydroxylamine group (–NHOH).8,10,24 It is a complex process that requires a suitable electrocatalyst to catalyse the electrochemical oxidation of –NH2 with high activity. In this respect, nanomaterials have special advantages because of their high specific surface areas and high catalytic activities. Many types of nanomaterials have been studied for the electrochemical oxidation of –NH2 groups in SMX, including carbon materials (carbon nanotubes, graphene, reduced graphene oxide),1,7 boron-doped diamond nanoparticles,12 Fe-doped ZnO nanorods,3 CeO2/chitosan nanocomposites,10 antimony nanoparticles,24 and Prussian blue nanocube-decorated multiwalled carbon nanotubes.9 Carbon materials have received considerable attention because of their good electrical conductivity, low cost, high stability, and high affinity for organic molecules,25–27 and, more importantly, their high catalytic activity for the electrochemical oxidation of –NH2.
The electrocatalytic activities of carbon materials can be further improved by incorporating them with metal nanoparticles, in particular, single-atom metal catalysts (single-atom catalysts, SACs). These anchored metal nanoparticles or SACs can alter the coordination environments and improve the electronic structures of the carbon materials.28–31 Moreover, SACs have the smallest catalytically active sites and can maximize the exposure of active sites and use-efficiency of the metal atoms, affording extraordinarily high catalytic activities.32 SAC-incorporated carbon material catalysts are widely used in many fields, varying from electrocatalysis (catalysing the reduction of CO2 and O2, evolution of O2 and H2, oxidation of CO, and electrochemical synthesis of NH3 from N2)28–30,33 to organic synthesis (unsaturated compound hydrogenation and alkene carboboration)34,35 and biosensing.36,37 These catalysts have a strong adsorption ability for aromatic compounds because of the strong π–π interactions between the carbon framework and aromatic molecules such as SMX antibiotics.
This work reports the synthesis of a single-atom cobalt-anchored nitrogen-doped carbon catalyst (Co/N–C), which exhibits high electrocatalytic activity for the oxidation of the –NH2 group in SMX and can be used as an efficient electrocatalyst to develop an electrochemical sensor for quantifying SMX antibiotics in environmental water samples. The Co/N–C SACs are synthesized by pyrolyzing zeolitic imidazolate frameworks (Co-substituted ZIF-8, denoted as Co-ZIF-8) at 900 °C. The electrochemical oxidation of SMX is used to demonstrate the effect of the single-atom Co-anchoring-dependent electrocatalytic activity of Co/N–C. The anchoring of single-atom Co enhances the catalytic activity of Co/N–C SACs. In addition, a possible mechanism for the enhancement is discussed based on the results of theoretical calculations and experimental measurements. Finally, highly sensitive quantification of SMX in water samples is demonstrated using a Co/N–C-based electrochemical sensor. The study results indicate that Co/N–C SACs are an efficient electrochemical sensing platform with great potential for applications in fields such as biomedical analysis, environmental pollutant detection, and water safety evaluation.
Materials and methods
Chemicals and reagents
Co(NO3)2·6H2O (purity >98%), Zn(NO3)2·6H2O (purity >98%), and 2-methylimidazole (2-mIm, purity >98%) were purchased from Sinopharm Chemical Reagent Co. Ltd. (China). SMX (purity >98%), trimethoprim (purity >98%), ciprofloxacin (purity >98%), and norfloxacin (purity >98%) were purchased from Shanghai Aladdin Biochemical Technology Co. Ltd. Nafion (5 wt%) was obtained from Sigma-Aldrich. Other chemicals used in this work were analytical-grade reagents and obtained from Sinopharm Chemical Reagent Co., Ltd. The aqueous solutions used in all the measurements were prepared using deionized water with a resistivity of approximately 18.2 MΩ cm. Phosphate buffer (PBS, 0.1 M) was prepared using Na2HPO4 and NaH2PO4.Synthesis of ZIF-8
ZIF-8 was synthesized following previously reported procedures.38,39 Briefly, 1.848 g of 2-mIm was dissolved in 45 mL of methanol. Then, 1.674 g of Zn(NO3)2·6H2O (in 45 mL of methanol) was added under continuous vigorous stirring for 8 h at room temperature (23 ± 2 °C). The ZIF-8 product was collected by centrifugation, washed with methanol three times, and dried under vacuum at ∼55 °C overnight.Synthesis of Co-ZIF-8
First, 0.819 g of Co(NO3)2·6H2O and 1.674 g of Zn(NO3)2·6H2O were dissolved in 45 mL of methanol (Co![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Zn = 0.5:1 (molar ratio)) and then 1.848 g of 2-mIm (in 45 mL of methanol) was injected under continuous vigorous stirring for 8 h at room temperature. The product (denoted as Co0.5Zn1ZIF-8) was collected by centrifugation, washed three times with methanol, and dried under vacuum at ∼55 °C overnight.
:Zn = 0.5:1 (molar ratio)) and then 1.848 g of 2-mIm (in 45 mL of methanol) was injected under continuous vigorous stirring for 8 h at room temperature. The product (denoted as Co0.5Zn1ZIF-8) was collected by centrifugation, washed three times with methanol, and dried under vacuum at ∼55 °C overnight.
To study the effects of the amount of Co single atoms on the electrocatalytic activity of Co/N–C catalysts on SMX oxidation, Co-substituted ZIF-8 with different Co:Zn molar ratios (0.25:1 and 0.75:1) was also synthesized using the same procedures as those used for Co0.5Zn1ZIF-8 synthesis by altering the molar ratio of Co2+ and Zn2+ in the synthesis mixtures. They were denoted as Co0.25Zn1ZIF-8 and Co0.75Zn1ZIF-8, respectively.
Synthesis of Co/N−C SACs
Powders of ZIF-8 or Co-ZIF-8 (Co0.25Zn1ZIF-8, Co0.5Zn1ZIF-8, and Co0.75Zn1ZIF-8) were transferred to a corundum crucible placed in the centre of a tube furnace for pyrolysis under an atmosphere of high-purity Ar gas. The mixture was first heated by ramping the temperature to 900 °C at 5 °C min−1. This temperature was maintained for 3 h to complete the pyrolysis. Metal Zn in ZIF-8 evaporated at this temperature, while Co remained and generated Co/N–C SACs. The furnace was turned off, and the temperature was allowed to cool naturally to the ambient temperature.The as-synthesized catalysts were purified by overnight refluxing in 100 mL of H2SO4 solution (2 M) in air at 110 °C to remove any metallic Co and cobalt oxide particles produced during pyrolysis. The catalysts were then heated in forming gas (Ar/10% H2) at 900 °C for 2 h and refluxed in a 2 M H2SO4 solution in a similar manner to the first refluxing step. After each refluxing step, the suspensions were filtered and washed with distilled water until the pH of the filtrate reached ∼7. Finally, the catalyst was obtained after drying the filter cake in air at 80 °C.
Characterization
The instruments employed to characterize the synthesized materials are described in the ESI.†Electrochemical measurements
A glassy carbon electrode (GC, 3 mm diameter, CH Instruments), treated and cleaned using procedures similar to those reported in our previous study before use,40–42 was modified with Co/N–C SACs (Co/N–C/GC) and used as the working electrode. To fabricate the electrode, 1 mg of Co/N–C SACs was dispersed in 1 mL of a mixture of water, ethanol, and Nafion (at a volume ratio of 14:5:1) by ultrasonication to form a suspension with a concentration of 1 mg mL−1. Next, 5 μL of the suspension was deposited onto the surface of the cleaned GC electrode. Then, the electrode was stored in a desiccator at ambient temperature to allow evaporation of the solvent. The amount of Co/N–C SACs on the electrode surface was estimated to be 71 μg cm−2.
The electrochemical oxidation of SMX on the electrodes (bare GC, Co/N–C/GC, and N–C/GC electrodes) was studied by cyclic voltammetry (CV), which was performed on an Autolab PGSTAT 302 N electrochemical station (Metrohm) in a two-compartment three-electrode cell with a sample volume of approximately 10 mL. KCl-saturated Ag/AgCl and Pt sheet (0.5 × 0.5 cm) electrodes were used as the reference and counter electrodes, respectively. To eliminate the effect of oxygen on the electrochemical responses, the dissolved oxygen in the solution was removed by purging the buffer with Ar gas for more than 30 min before the measurements. Ar atmosphere was maintained over the solution to exclude oxygen. All electrochemical measurements were conducted in 0.1 M PBS (pH 7.0) at room temperature.
Quantitative analysis of SMX using the electrodes was performed by amperometry, which showed better sensitivity than potential sweep techniques for quantifying low-concentration targets. The measurements were conducted at a constant potential of +0.90 V in a continuously stirred solution. After the background current of the electrode reached a steady state, a drop of SMX solution at different concentrations was injected and the current–time (i–t) curve was recorded. The background-subtracted response currents were used as the analysis signals.
The selectivity and anti-interference ability of the electrode were evaluated using differential pulse voltammetry (DPV), which was conducted using a potential step of 5 mV and pulse width, period, and amplitude of 50 ms, 100 ms, and 50 mV, respectively. Electrochemical impedance spectroscopy (EIS) measurements were also performed at room temperature in 0.1 M KCl solution in the presence of 5 mM Fe(CN)63−/4– with a voltage amplitude of 10 mV and a frequency range of 0.1–1 × 105 Hz. The data were presented as Nyquist plots.
Theoretical calculations
All calculations were performed by using the density functional theory (DFT) method implemented in CASTEP code. The generalized gradient approximation combined with the Perdew, Burke, and Ernzerhof (PBE) functional was employed to describe the exchange–correlation interactions.43 A DFT-D semiempirical correction with Grimme method (DFT-D2) is applied to account for the dispersion interaction.44 The double numerical plus polarization (DNP) was chosen as the basis set for other elements (C and H atoms). To sample the Brillouin zone, Monkhorst–Pack (MP) mesh of 3 × 3 × 1 k-point grid in reciprocal space were used for free energy calculations and 3 × 3 × 1 for density of states (DOS) calculations. The real-space global cutoff radius is set to be 5.2 Å. Fermi occupation is applied to achieve electronic convergence. In all of the calculations, the convergence criteria for energy, force, and displacement were set to 10−5 Ha, 0.002 Ha Å−1, and 0.005 Å, respectively.For simulating SMX adsorption, a (2 × 2) Co/N–C monolayer supercell was employed. The SMX molecule was placed near the Co/N–C or N–C plane at a distance of 3 Å and the optimized geometric structures of the adsorbed SMX at the surface of Co/N–C or N–C. The adsorption energy (Ead), defined as the zero-point energy (ZPE) difference between the total system including the adsorbed molecules and the isolated system, was derived from frequency analysis after the initial structure optimization.45 The energy of the isolated system is the sum of the energies of Co/N–C or N–C and the isolated adsorbed molecule. A negative Ead indicates the adsorption is energetically favorable.
Results and discussion
Synthesis and characterization of Co/N–C SACs
Co/N–C SACs were synthesized by pyrolyzing Co-ZIF-8 to study the effects of a single Co atom on the electrocatalytic activity of the catalyst toward the electrochemical oxidation of aromatic –NH2 in SMX and to achieve electrochemical quantification of antibiotics in environmental waters. Co-ZIF-8, synthesized using Co2+, Zn2+, and 2-mIm as precursors (Fig. 1a), has a well-defined rhombododecahedral shape of ∼195 nm (SEM images in Fig. 1b and c). Theoretically, Co-ZIF-8 is a sodalite structure with a pore diameter of ∼11.6 Å, containing ZnN4 and CoN4 tetrahedra.46 The inactive Zn in Co-ZIF-8 is a sacrificial element and is easily evaporated at elevated temperatures (such as 900 °C) during pyrolysis, generating free vacancies at the surface of nitrogen-doped carbon (N–C). This feature is beneficial for adjusting the final structure of the catalyst and is conducive to the formation of active catalytic sites with single Co atoms (in the form of CoN4).47 Moreover, the specific structure of M–mIm–M (M: Co and Zn) of Co-ZIF-8 can effectively prevent the agglomeration of single Co atoms and produce a uniform spatial dispersion of Co-based (CoN4) active sites in the generated catalyst. | ||
| Fig. 1 Synthesis and characterization of Co/N–C SACs. (a) Schematic illustration of Co/N–C synthesis. (b and c) SEM images of Co-ZIF-8. Co-ZIF-8 was synthesized under a Co:Zn molar ratio of 0.5:1. (d and e) SEM images of Co/N–C SACs. (f–h) TEM images of Co/N–C SACs under different magnifications. (i) HAADF-STEM image of the catalyst and the corresponding elemental mapping for C, N, and Co. (j) Atomic-resolution HAADF-STEM image of Co/N–C SACs. The inset shows histogram analysis of the size distribution of Co atoms; 150 Co atoms were counted from HAADF-STEM images. | ||
After pyrolysis at 900 °C, the Co/N–C catalyst was generated. The catalyst retains its rhombododecahedral shape (Fig. 1d and e). The morphology of the catalyst was uniform. Its size was ∼130 nm, smaller than that of the catalyst before pyrolysis, probably owing to the shrinkage caused by pyrolysis. The transmission electron microscopy (TEM) images in Fig. 1f–h also show the uniform dispersion of the catalyst. No metallic particles were observed in the high-resolution TEM (HRTEM) images, implying that no metal particles were produced during pyrolysis (because of the specific structure of ZIF-8), or that the metal particles were completely removed during purification. The size of the Co particles became less than the resolution of HRTEM owing to purification. Only a typical graphene-like structure (Fig. 1h) can be observed in the HRTEM image, indicating a high degree of graphitization. This characteristic can enhance the electrical conductivity of the catalyst and improve its activity.31
The high degree of graphitization can be further verified from the X-ray diffraction (XRD) patterns (Fig. S1†), which show well-defined peaks corresponding to the (002) and (101) planes of graphitic carbon at ∼25.1° and 44.2°,29 respectively. No XRD patterns of metal (such as Co) particles were observed, agreeing with that observed from the HRTEM images. Elemental distribution mapping by high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) also indicated uniform distribution of C, N, and Co on the catalyst surface (Fig. 1i). No Zn element was detected because of the evaporation during pyrolysis. The atomically dispersed Co species on the catalyst surface are likely to be coordinated to N atoms in the form of CoN4,46,47 producing a single-atom Co active site. The aberration-corrected HAADF-STEM image shown in Fig. 1j confirms this assumption. Many bright spots corresponding to individual heavy atoms (∼0.1 nm) were observed, which were attributed to the single-atom-sized Co species in the synthesized catalyst. The formation of metal aggregates was not observed. The histogram analysis shows very narrow size distributions of 1.32 ± 0.55 Å (Fig. 1j inset). This size agrees well with the characteristic diameter of the single Co atom, further indicating that the Co species in the catalyst exist as individual Co atoms without metal Co aggregation.
Further information on the surface nature and chemical components of the catalyst was obtained using X-ray photoelectron spectroscopy (XPS), which can provide insightful information about chemical bonds. The XPS spectrum shows that the catalyst surface mainly contains carbon, nitrogen, oxygen, and cobalt elements, which generate peaks at approximately 284.8, 398.6, 532.6, and 781.0 eV (Fig. 2a), respectively. The carbon element undergoes easy oxidation because of the oxygen present in the catalyst, a typical phenomenon of carbon materials when handled under ambient conditions.29 The catalyst has ∼84.8%, 6.7%, 7.6%, and 0.9% of C, N, O, and Co, respectively. The high-resolution spectrum of the C1s peak is asymmetrical and shows a broad satellite at high binding energy owing to the delocalized π-electron system (Fig. 2b), a typical feature of carbon materials. The C1s spectrum shows the presence of four peaks, one each at approximately 289.6, 287.2, 285.5, and 284.7 eV, corresponding to carbon in C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, C–O, CN, and CC (sp2) groups, respectively.
O, C–O, CN, and CC (sp2) groups, respectively.
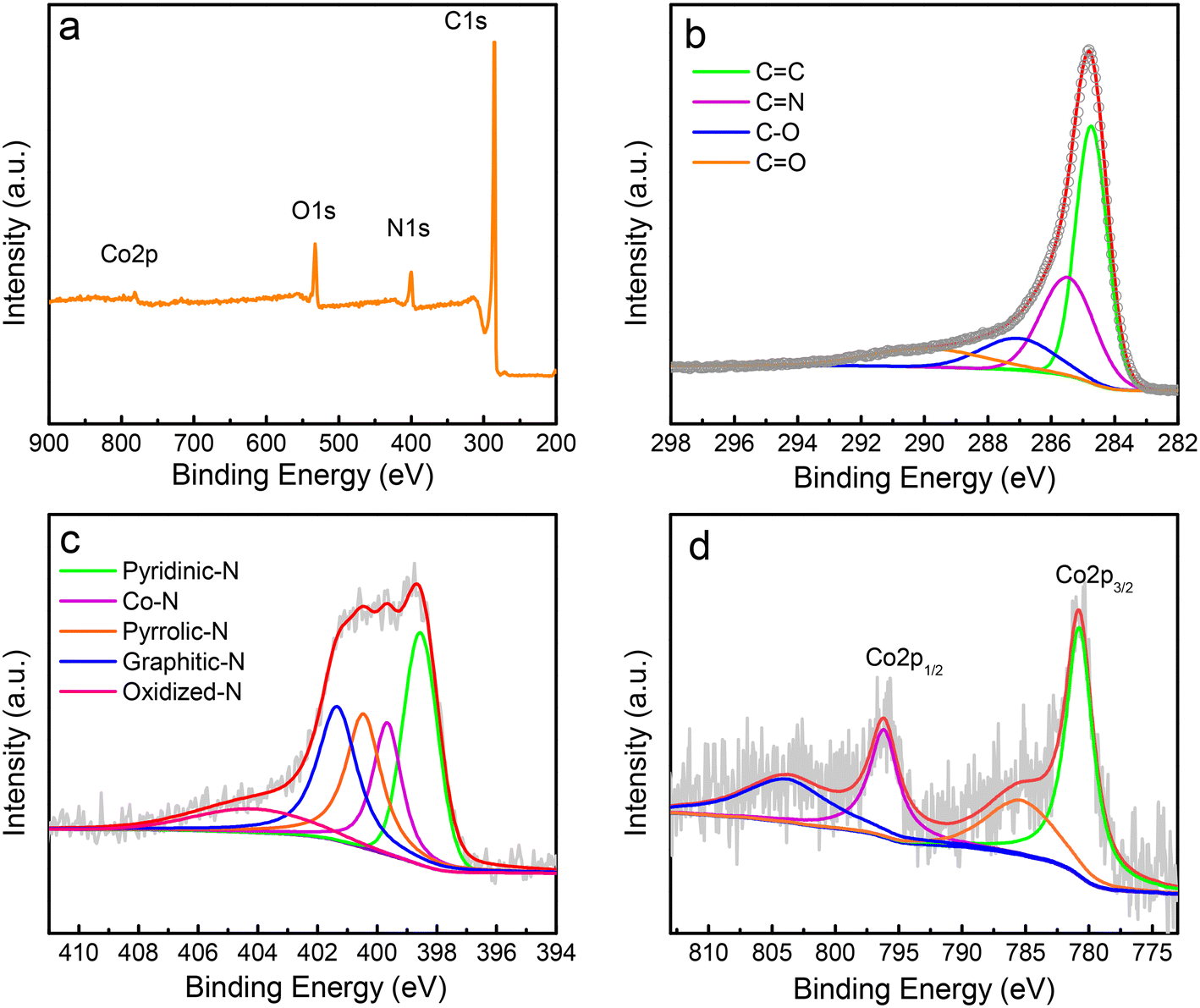 | ||
| Fig. 2 XPS characterization of Co/N–C SACs. (a) Survey XPS spectrum the catalyst. (b−d) High-resolution XPS spectra of C1s (b), N1s (c), and Co2p (d), and their related curve-fitted components. The circles in panel (b) represent the experimental data. | ||
The deconvolution of the XPS of the N1s main peak exhibits four peaks (Fig. 2c), three of which have binding energies of ∼398.6, 400.5, and 401.4 eV that correspond to pyridinic, pyrrolic, and graphitic N, respectively. The peak at the binding energy of ∼399.7 eV is commonly ascribed to the energy of the N atom bonded to the metal48 (the Co–N bond here). The Co atom in the Co–N bond should be bound with the pyridinic N, not pyrrolic N, because the XPS peak for the Co bound to pyrrolic N, such as in porphyrins, should appear at a relatively lower binding energy (∼398.5 eV), as confirmed by the theoretical simulation results of Artyushkova.49 The broad peak at ∼404.3 eV is caused by the oxidized N. The amounts of different types of N were calculated to be (in atomic ratio) ∼30.8%, 20.5%, 23.3%, 17.4%, and 8.0% for pyridinic, pyrrolic, graphitic N, Co–N, and oxidized N, respectively.
The high-resolution XPS of Co2p indicates two predominant peaks caused by the Co–N bond at approximately 780.8 and 796.1 eV (Fig. 2d), respectively. These binding energies are higher than those of metallic Co, which is most likely caused by the transfer of the electron to the N atom from the Co atom because of the higher electronegativity of nitrogen (∼3.0) than that of Co (∼1.9), implying the formation of the Co–N bond in the synthesized Co/N–C SACs.
Electrocatalytic oxidation of SMX
After the synthesis of Co/N–C SACs, their catalytic characteristics toward the electrochemical oxidation of SMX were investigated by CV. The catalytic activity of Co/N–C (synthesized by pyrolyzing Co0.5Zn1ZIF-8) was compared with that of a bare GC electrode, which showed negligible activity for the oxidation of SMX because almost no anodic current could be observed even when the electrode potential was scanned to +1.2 V (Fig. 3a, curve i). However, for the Co/N–C/GC electrode, a well-defined anodic peak is observed at approximately +0.92 V in PBS solution at a pH of 7.0 (Fig. 3a, curve ii), suggesting that the Co/N–C SACs exhibit catalytic activity for the electrochemical oxidation of SMX. Upon reverse scanning, no cathodic peak was observed, indicating an irreversible electron transfer in the electrochemical oxidation of SMX on the Co/N–C/GC electrode. Moreover, the anodic peak position depended on the solution pH (Fig. S2†), the anodic peak potential at pH of 5, 6, 7, 8, and 9 is about +1.03, 0.98, 0.92, 0.86, and 0.81 V, respectively, implying that the electrochemical oxidation is a proton-coupled electron-transfer process. This oxidation process starts with the one-electron oxidation of the –N2H group to generate a cation radical at the N atom (see the electron-transfer process depicted in Fig. S3†), followed by the rapid loss of a second electron and a proton to produce the iminium ion. This ion is then attacked by a solvent molecule (H2O) to form a hydroxylamine group (–NHOH).50,51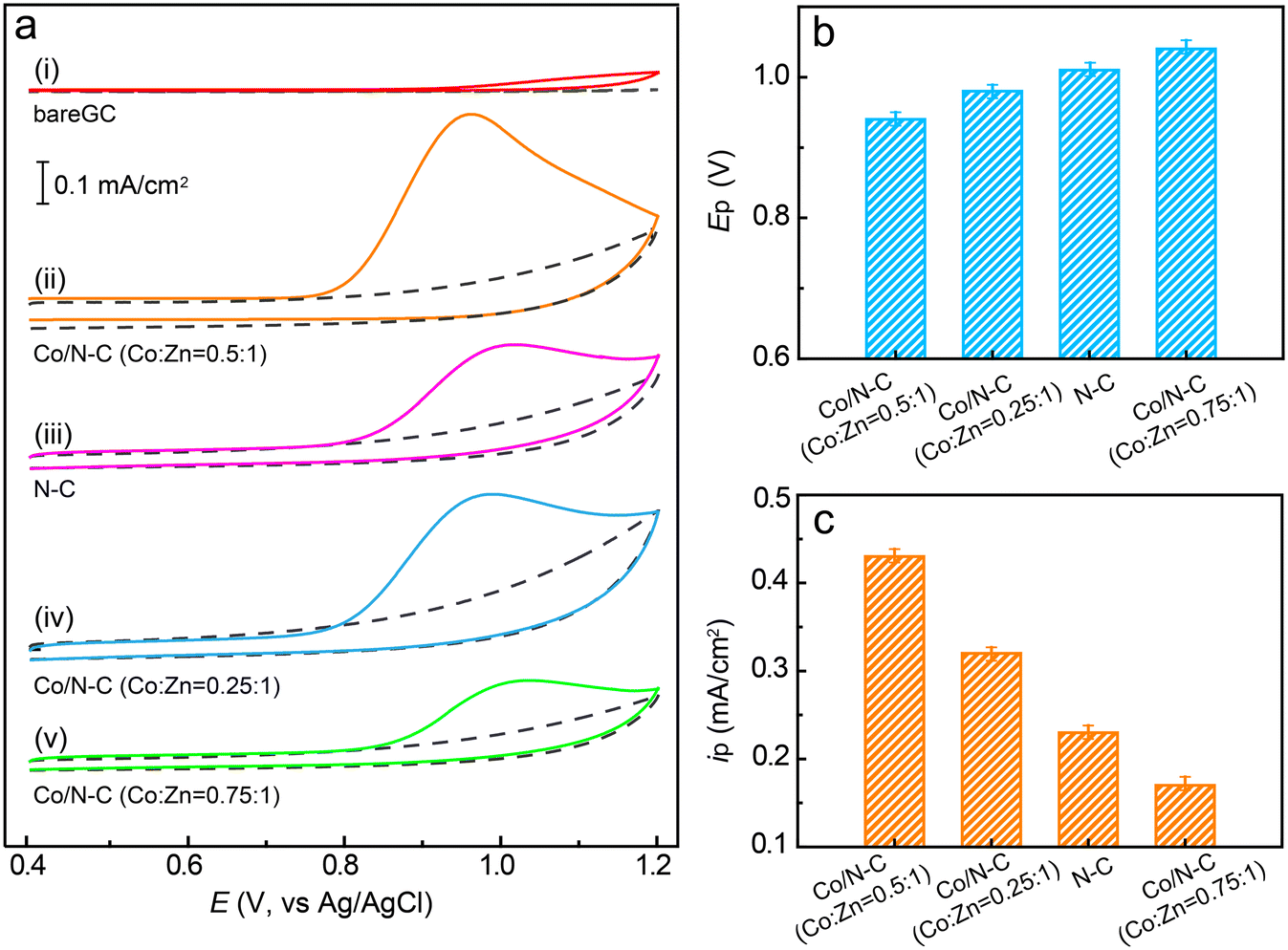 | ||
| Fig. 3 Electrocatalytic oxidation of SMX. (a) CV responses of SMX (50 μM) in 0.1 M PBS (pH 7.0) at the bare GC (i), Co/N–C/GC (ii, synthesized with Co:Zn ratio of 0.5:1), N–C/GC (iii), and Co/N–C/GC (iv, synthesized with Co:Zn ratio of 0.25:1), and Co/N–C/GC (v, synthesized with Co:Zn ratio of 0.75:1) electrodes. The doted curves show the CV responses of each electrode in PBS in the absence of SMX. The scan rate was 50 mV s−1. (b and c) Comparison of anodic peak potentials (b) and peak currents (c) for the electrochemical oxidation of SMX on different electrodes. The data were obtained by averaging the results of three measurements. | ||
The anodic peak potential of the electrochemical oxidation of SMX catalysed by the Co/N–C SACs (+0.92, at pH 7.0) is similar to that catalysed by antimony nanoparticles (+0.88 V, pH 7.0)21 and multiwalled carbon nanotubes (+0.90 V, pH 6.0).1 In addition, it is more negative than those obtained on graphene oxides (1.05–1.13 V, pH 7.0)7 and boron-doped diamond nanoparticle-modified (1.1 V, pH 3.0)12 electrodes. Hence, the Co/N–C SACs exhibit high electrocatalytic activity for SMX oxidation, may be owing to the incorporation of a single Co atom into N–C materials.
To study the effects of anchoring a single Co atom on the electrocatalytic activity of the catalyst, the CV of SMX on the N–C/GC electrode (without Co atom anchoring) was recorded under the same conditions as that of the Co/N–C/GC electrode. The position of the anodic peak at approximately +1.03 V (Fig. 3a, curve iii) shows a ∼110 mV positive shift in comparison with that observed on the Co/N–C/GC electrode (Fig. 3b). Moreover, the height of the anodic peak current is lower than that recorded on the Co/N–C/GC electrode (approximately 0.23 vs. 0.43 mA cm−2, Fig. 3c). Hence, the catalytic activity of Co/N–C SACs for SMX electrochemical oxidation is higher than that of N–C. These results highlight the role of incorporation of a single atom of Co on the electrocatalytic activity of Co/N–C SACs, probably by altering the electronic structure (discussed later) and increasing the number of active catalytic sites, as evidenced by the higher ID/IG value of Co/N–C (∼1.18) than that of N–C (∼1.05) in the Raman spectra (Fig. S4†).
The higher electrocatalytic activity exhibited by the Co/N–C SACs can be ascribed to the enhancement of their electrical conductivity compared to that of N–C. This enhanced conductivity was verified by EIS measurements (Fig. S5†). The electron-transfer resistance of the Co/N–C/GC electrode is approximately 11 Ω, which is one-third of that of the N–C/GC electrode (approximately 30 Ω) and much smaller than that of the bare GC electrode (approximately 170 Ω). The increased conductivity should improve the electron-transfer ability of Co/N–C SACs and enhance their catalytic activity for SMX oxidation.
The higher electrocatalytic activity of the Co/N–C SACs could also be a result of improved electronic characteristics. Theoretical calculations show that the energy gap, defined as the energy difference between the lowest unoccupied molecular orbital (LUMO) and highest occupied molecular orbital (HOMO), that is, the L–H gap, of Co/N–C, is ∼0.54 eV, which is lower than that of N–C (∼0.86 eV). The small L–H gap of Co/N–C implies a decrease in the energy barrier of electron transfer, which improves the catalytic activity of Co/N–C SACs. The alteration of the electronic characteristics of Co/N–C SACs can also be experimentally evidenced from the CV measurements of the redox reaction of the Fe(CN)63− mode molecule, which exhibited higher redox currents on the Co/N–C/GC electrode than on the N–C/GC and bare GC electrodes (Fig. S6†). The potential separation between the cathodic and anodic peaks observed for the Co/N–C/GC electrode is ∼61 mV, which is lower than that observed for the N–C/GC (∼68 mV) and bare GC electrodes (∼79 mV), indicating the greater redox reversibility of Fe(CN)63− on the Co/N–C/GC electrode and the higher electrocatalytic reactivity of this electrode.
The higher electrocatalytic activity of Co/N–C SACs can also be attributed to their high adsorption ability. Theoretical simulation indicated that the Co atom in the catalyst acted as an adsorption site to adsorb the SMX molecule. The calculated adsorption energy for SMX on the Co/N–C surface is approximately −1.49 eV, much lower than that for SMX adsorption on the N–C surface (−0.81 eV). This result indicates the high adsorption ability of the Co/N–C SACs for SMX. Moreover, the adsorption configurations of SMX on the Co/N–C surface were inclined (Fig. S7†), which is different from the lying-down mode on the N–C surface, suggesting that Co/N–C SCAs adsorb more SMX molecules; therefore, they exhibit higher electrocatalytic activity than that of N–C.
The catalytic activity of the Co/N–C SACs can also be affected by the loading of Co atoms on the catalyst. To evaluate this suggestion, the catalyst was synthesized with different ratios of Co to Zn by altering the concentrations of Co2+ and Zn2+ ions in the synthesis mixtures (see the Experimental section). When the Co/N–C catalyst was synthesized with a Co:Zn ratio of 0.25:1, the anodic peak potential for SMX oxidation was observed at ∼+0.98 V (Fig. 3a, curve iv), which is a 60 mV positive shift compared to that obtained with the catalyst synthesized with a Co:Zn ratio of 0.5:1. The peak current also significantly decreased (0.32 vs. 0.43 mA cm−2, Fig. 3c), probably because of the decrease in the number of catalytic active sites caused by the lower loading of Co atoms. When the Co/N–C catalyst was synthesized by increasing the Co:Zn ratio to 0.75:1, the anodic peak potential appears at an even more positive value of ∼1.04 V (Fig. 3a, curve v) and the anodic peak current decreases to ∼0.17 mA cm−2 (Fig. 3), implying a significant decrease in the catalytic activity. The SEM image shows that the catalyst has many Co nanoparticles of size ∼70 nm when its Co:Zn ratio was increased to 0.75:1 (Fig. S8a† for comparison, the TEM image of the catalyst synthesized under Co:Zn ratio of 0.25:1 is depicted in Fig. S8b,† which shows a well-defined rhombododecahedral shape with a size of ∼95 nm, smaller than that synthesized under the Co:Zn ratio of 0.50:1), even after the catalyst was purified by reflux in a 2 M H2SO4 solution. These metallic Co nanoparticles can block the SMX molecules from accessing the active sites.29,31 Thus, they obstruct the catalytic activity of the catalyst.
A detailed comparison of the anodic peak potentials and currents obtained for the different electrodes is shown in Fig. 3b and c. These results clearly demonstrate that the incorporation of a single Co atom into the Co/N–C catalyst can significantly facilitate electron transfer for SMX oxidation and enhance the electrocatalytic activity of the catalyst. Furthermore, the highest electrocatalytic activity was achieved at a Co:Zn ratio of 0.5:1, demonstrating the Co atom-loading-dependent catalytic activity of the catalyst. Hence, Co/N–C SACs synthesized with a Co:Zn ratio of 0.5:1 were used for SMX quantification.
Electrochemical quantification of SMX using the Co/N–C/GC electrode
Before electrochemically qualifying SMX, several parameters potentially affecting the performance of the electrode, such as the detection potential, solution pH, and loading of Co/N–C on the electrode surface, were optimized (Fig. 4). The anodic potential was first selected because a suitable detection potential can improve the detection limit and eliminate the interference effects of other electroactive substances. The anodic potentials, in a constant potential mode, from +0.80 to +1.10 V with an interval of 50 mV were optimized. The SMX concentration was maintained at 50 μM. The anodic current first increased with an increase in the potential from +0.80 to +0.95 V, followed by a slow decrease as the potential was further increased in the positive direction (Fig. 4a). Moreover, the use of a more positive potential usually causes an increase in noise.41,52 Therefore, +0.95 V was employed as the detection potential in the following measurements.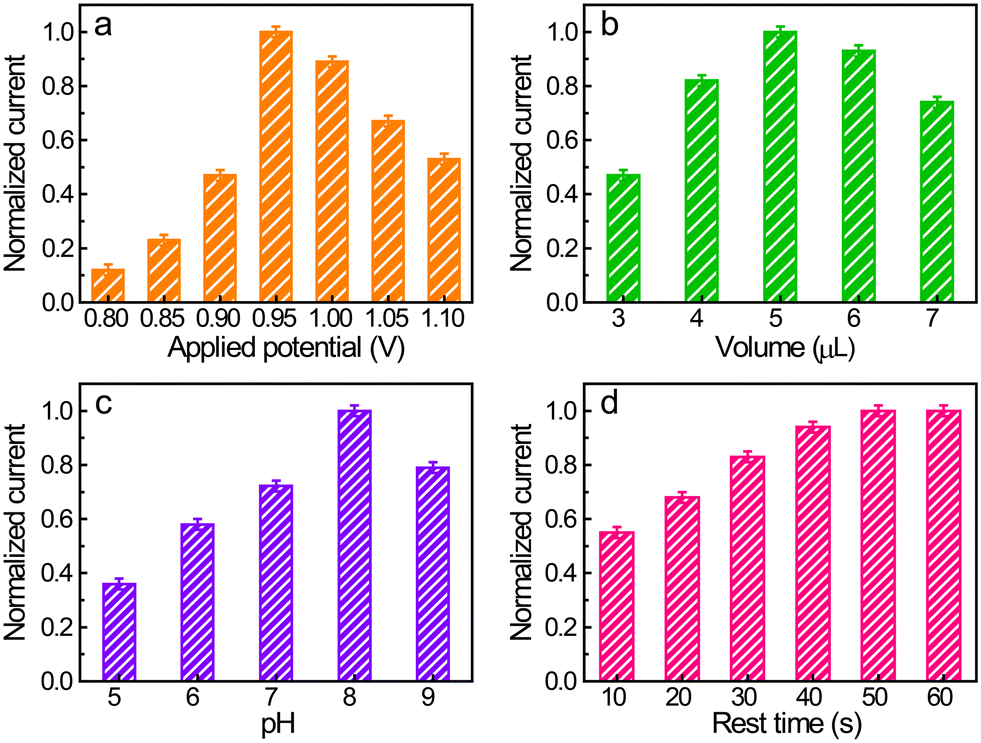 | ||
| Fig. 4 Optimization of electrochemical quantification of SMX. (a)–(d) Dependence of the response of SMX (50 μM) on the detection potential (a), catalyst loading on the electrode surface (b), solution pH (c), and rest time of the electrode in solution before electrochemical measurements (d). Loading of catalyst on the electrode surface is expressed by the deposited volume of the Co/N–C SAC suspension (1 mg mL−1) on the electrode surface. To enable a comparison under each condition, the response currents are displayed in the form of the normalized current with the highest response under each condition. The data presented are the average of three independent measurements. | ||
The Co/N C SAC loading on the electrode surface was selected because it also has a significant effect on the electrode response. The amount of catalyst on the electrode surface was controlled by the volume of Co/N–C suspension (1 mg mL−1) cast on the electrode surface. The response current under an anodic potential of +0.95 V and an SMX concentration of 50 μM increased on increasing the casted suspension volume to up to 5 μL (Fig. 4b), probably owing to the increase in the active sites and the enhancement in the conductivity of the electrode with increasing catalyst loading. However, a further increase in the amount of Co/N–C SACs on the electrode surface decreased the anodic current, probably because the thick film of the catalyst prohibits SMX from accessing these active sites embedded inside the film and hinders effective electron transfer. Therefore, 5 μL of the Co/N–C suspension (1 mg mL−1) was selected as the most suitable catalyst loading on the electrode for subsequent measurements.
Solution pH is another important parameter affecting the response of SMX because the electrochemical oxidation of SMX is a proton-coupled electron-transfer process. In the studied solution pH range (5–9), the response first increased with an increase in pH from 5 to 8 (Fig. 4c) and then decreased when the pH is further increased. Accordingly, 0.1 M PBS with a pH of 8 was selected for the electrochemical quantification of SMX.
In the constant potential analysis, the electrochemical response is usually affected by the rest time of the modified electrode placed in the detection solution before electrochemical measurements. This is because the modified dry film of the catalyst needs time to become infiltrated by the electrolyte and the redox molecule (SMX here) also needs to diffuse into the catalyst film and be adsorbed on the active sites, thereby establishing the equilibrium between molecules in the bulk and the electrode. Fig. 4d shows that the anodic responses of SMX reached a plateau after 50 s of rest time (the electrode was under the open-circuit potential), implying that this time is sufficient for electrode infiltration and equilibrium establishment.
Under these optimized conditions, the amperometric response of the electrochemical oxidation of SMX on the Co/N–C/GC electrode under a potential of +0.9 V was recorded (Fig. 5a). The anodic current increased with the increasing addition of SMX (Fig. 5a, curve i). After each addition, the current increased rapidly, implying that the Co/N–C/GC electrode can sensitively respond to changes in the concentration of SMX in the solution. Moreover, the current increased linearly when the SMX concentrations ranged from 0.08 to 66.5 μM with a correlation coefficient of 0.997. In addition, the current reached a relative saturation level when the SMX concentration was higher than 115.5 μM (Fig. 5b, curve i). The limit of detection (LOD) was estimated to be approximately 15 nM at a signal-to-noise (S/N) ratio of 3. This linear range and LOD are better than those obtained on the N–C/GC electrode, which displays a linear range and LOD of approximately 0.3–43.5 μM and 60 nM (Fig. 5a, and b, curve ii), respectively. Hence, the catalytic activity of the Co/N–C catalyst for the electrochemical oxidation of SMX is higher than that of the N–C catalyst. Furthermore, the linear range and LOD achieved on the Co/N–C/GC electrode are superior to those obtained on other nanomaterial catalyst-modified electrodes. For example, the linear range and LOD are 1.0–10.0 μM and 38 nM, respectively, on the Prussian blue nanocube-decorated multiwalled carbon nanotube-modified screen-printed electrode;9 0.5–50 μM and 40 nM, respectively, on the graphene oxide modified screen-printed electrode;7 and 0.1–0.7 μM and 0.024 μM, respectively, on multiwalled carbon nanotubes modified with antimony nanoparticle-based paraffin composite electrode.24 Hence, the Co/N–C-based SMX electrochemical sensor is better than the previously reported ones, thereby highlighting the high electrocatalytic activity of the Co/N–C SACs toward SMX oxidation.
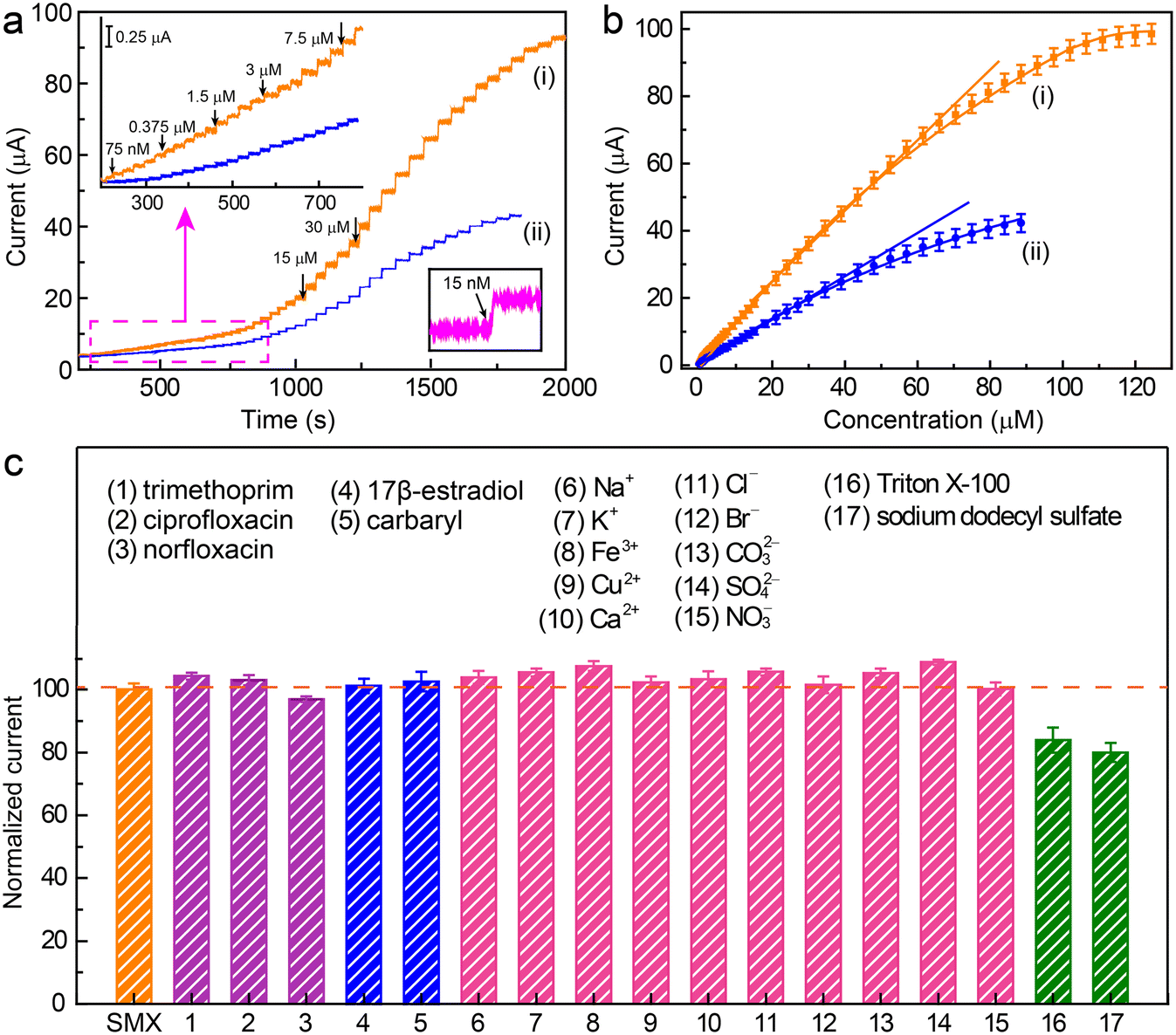 | ||
Fig. 5 Electrochemical quantification of SMX using the Co/N–C/GC electrode. (a) Amperometric responses of SMX oxidation at the Co/N–C/GC (curve i) and N–C/GC electrodes (curve ii) to the successive addition of SMX in 0.1 M PBS (pH 7.0) at a constant potential of +0.90 V. The left upper inset shows the response of the electrodes at low concentrations. The right lower inset shows the response having an S/N of 3 at the Co/N–C/GC electrode. (b) Dependence of the response current of SMX at the Co/N–C/GC (curve i) and N–C/GC electrodes (curve ii) on the SMX concentrations. (c) Evaluation of the selectivity and anti-interference ability of the Co/N–C/GC electrode for SMX quantification in the presence of various interferents. To enable a comparison in the presence of each interferent, the current is displayed in the form of the normalized current, which was calculated as: 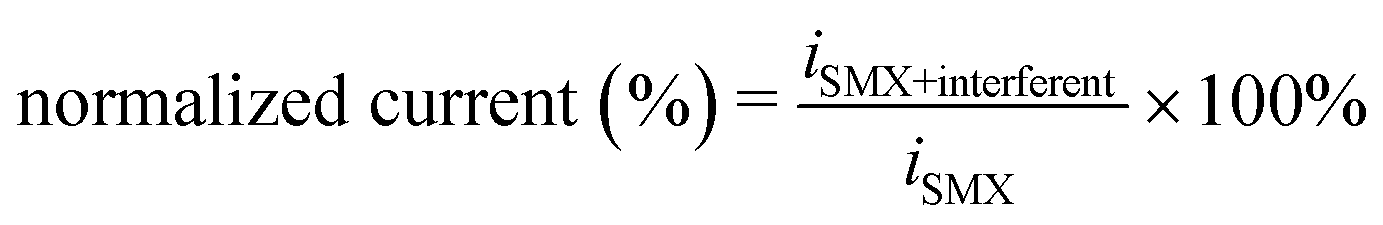 , where iSMX+interferent, and iSMX is the DPV currents of SMX with and without the interferents, respectively. , where iSMX+interferent, and iSMX is the DPV currents of SMX with and without the interferents, respectively. | ||
Repeatability, reproducibility, and stability of the sensor for qualifying SMX were also evaluated. A relative standard deviation (RSD) of ∼2.3% was achieved over ten successive measurements of 50 μM SMX at the Co/N–C/GC electrode, revealing the high repeatability and good precision of the constructed sensor. Furthermore, five different Co/N–C/GC electrodes were used to quantify SMX (50 μM), resulting in an RSD of ∼3.1%, showing an acceptable reproducibility for sensor fabrication. The stability of the sensor was evaluated from the recorded response of an electrode to 50 μM SMX at 2 day intervals over 30 days. The electrode was stored in blank buffer at ambient temperature when not in use. The results showed that the response slightly decreased in the first three measurements but then remained almost stable, retaining more than ∼93% of the initial current value after 30 days. These features demonstrate that the constructed Co/N–C/GC sensor displays good repeatability, reproducibility, and stability.
The selectivity and anti-interference ability of the sensor for SMX quantification were evaluated by recording the DPV signal of the sensor in response to SMX (50 μM) in the presence of other components. Three antibiotics (trimethoprim, ciprofloxacin, and norfloxacin), one endocrine disruptor (17β-estradiol), and one pesticide (carbaryl) were randomly selected to examine their interference with SMX quantification. The presence of trimethoprim, ciprofloxacin, norfloxacin, 17β-estradiol, and carbaryl (50 μM each) did not significantly affect the DPV signal of SMX with a signal change of less than ∼10% (Fig. 5c). In addition, common ions, including Na+, K+, Fe3+, Cu2+, Ca2+, Cl−, Br−, CO32−, SO42−, and NO3−, whose concentrations were 50 times greater than that of SMX, had almost no impact on the DPV signals of SMX. However, the presence of high concentrations of common surfactants, such as Triton X-100 (10 μM) and sodium dodecyl sulfate (10 μM), reduced the response current by >15%, probably because of the strong adsorption of the surfactants on the electrode surface and impeding the SMX oxidation. However, most water samples do not contain such high concentrations of these surfactants, suggesting that these surfactants should not be problematic for SMX quantification. Thus, the Co/N–C-based electrochemical sensor is highly resistant to interference, showing a high potential for use in the quantification of SMX in real water samples.
Quantification of SMX in environmental water samples
Three environmental water samples (lake water from Xuanwu Lake of Nanjing, ground water from Xianlin, and secondary wastewater from the Baixiang wastewater treatment plant) were assayed for SMX using the sensor. Direct measurements showed negligible responses in the three water samples, indicating the absence of SMX in these water samples or the SMX content in these samples was too low to be detected by the sensor, that is, the content was lower than the LOD of the sensor. These results suggest that the sensor can be used for water safety evaluation. The recoveries were determined using a standard addition method. Known concentrations of SMX (5.0, 10.0, and 20.0 μM) were first added to water samples, and then the recoveries were determined. As indicated by the results displayed in Table 1, the average recoveries were ∼101.2%, 100.1%, and 101.9% for the water samples from lake, ground, and wastewater treatment plants, respectively, with the average RSDs of ∼3.47%, 3.83%, and 2.97%, respectively. Furthermore, to validate the accuracy of the sensor, the added SMX in these water samples was assayed by HPLC (the detailed procedures are depicted in the ESI†); the results are also listed in Table 1 for comparison. The results obtained from the electrochemical sensor and HPLC were consistent, demonstrating that the developed sensor has high accuracy for SMX quantification in environmental water samples and suggests its potential use as a cost-effective sensor for SMX in water safety evaluation.| Samples | Spiked amounts (μM) | Measured amountsa (μM) | Recovery (%) | RSD (%) | Measured amountsb (μM) |
|---|---|---|---|---|---|
| a Obtained using the Co/N–C/GC electrode; average of three measurements. b Measured by HPLC; average of three measurements. | |||||
| Lake water | 5.0 | 5.24 | 104.8 | 3.6 | 5.31 |
| 10.0 | 9.95 | 99.5 | 4.2 | 10.33 | |
| 20.0 | 19.88 | 99.4 | 2.6 | 19.94 | |
| Ground water | 5.0 | 4.96 | 99.2 | 3.1 | 5.26 |
| 10.0 | 9.89 | 98.9 | 3.8 | 9.86 | |
| 20.0 | 20.45 | 102.3 | 4.6 | 19.88 | |
| Secondary wastewater | 5.0 | 5.12 | 102.4 | 2.1 | 4.95 |
| 10.0 | 10.36 | 103.6 | 2.9 | 10.54 | |
| 20.0 | 19.91 | 99.6 | 3.9 | 19.85 | |
Conclusions
An electrochemical sensor based on Co/N–C SACs was developed to quantify SMX, a typical aromatic –NH2-containing antibiotic, in environmental water samples. The Co/N–C SACs were synthesized by pyrolyzing Co-ZIF-8 at 900 °C in an inert atmosphere. The Co/N–C/GC sensor was fabricated by casting a Co/N–C SAC suspension onto the GC electrode surface. The quantification was based on the electrochemical oxidation of the –NH2 group of SMX to the –NHOH group, catalysed by Co/N–C SACs with high activity. The effects of single-atom Co anchoring on the electrocatalytic activity of Co/N–C SACs were studied experimentally and theoretically. The electrochemical response of the sensor showed a linear relationship with the concentration of SMX in a wide range of 0.08–66.5 μM with a low LOD (∼15 nM at an S/N of 3), high repeatability, reproducibility, and stability. Moreover, the sensor exhibits high selectivity and resistance to interference in the presence of other antibiotics, endocrine disruptors, pesticides, common aqueous ions, and common surfactants. Furthermore, the sensor was used to quantify SMX in environmental water samples with high accuracy. The use of the sensor can be extended to quantify antibiotics in biological samples such as serum and urine samples. Therefore, Co/N–C SACs are efficient electrochemical sensing platforms with great potential for various applications, including biomedical analysis, environmental pollutant detection, and water safety evaluation.Author contributions
Qingyu Gu: methodology, investigation, writing – original draft. Zhongxue Wang: investigation, resources, data curation, writing – review & editing. Qianwen Ding: formal analysis, software, investigation. Huiling Li: formal analysis, validation. Ping Wu: conceptualization, writing – review & editing. Chenxin Cai: supervision, funding acquisition, project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
C. C. acknowledges the financial support from the Natural Science Foundation of China under Grant 21675088, and P. W. acknowledges the financial support from the Natural Science Foundation of the Jiangsu Province under Grant BK20181383.References
- M. Arvand, R. Ansari and L. Heydari, Mater. Sci. Eng., C, 2011, 31, 1819–1825 CrossRef CAS.
- M. J. Ryeder, S. M. King and S. Read, Pediatr. Infect. Dis. J., 1997, 16, 1028–1031 CrossRef PubMed.
- M. Meshki, M. Behpour and S. Masoum, J. Electroanal. Chem., 2015, 740, 1–7 CrossRef CAS.
- H. da Silva, J. G. Pacheco, J. M. C. S. Magalhães, S. Viswanathan and C. Delerue-Matos, Biosens. Bioelectron., 2014, 52, 56–61 CrossRef CAS PubMed.
- A. Achari, D. O. Somers, J. N. Champness, P. K. Bryant, J. Rosemond and D. K. Stammers, Nat. Struct. Biol., 1997, 4, 490–497 CrossRef CAS PubMed.
- W. Haasnoot, M. Bienenmann-Ploum, U. Lamminmaki, M. Swanenburg and H. Rhijn, Anal. Chim. Acta, 2005, 52, 87–95 CrossRef.
- C. Chen, Y.-C. Chen, Y.-T. Hong, T.-W. Lee and J.-F. Huang, Chem. Eng. J., 2018, 352, 188–197 CrossRef CAS.
- L. Rajith, A. K. Jissy, K. G. Kumar and A. Datta, J. Phys. Chem. C, 2011, 115, 21858–21864 CrossRef CAS.
- L. F. Sgobbi, C. A. Razzino and S. A. S. Machado, Electrochim. Acta, 2016, 191, 1010–1017 CrossRef CAS.
- M. Cai, L. Zhu, Y. Ding, J. Wang, J. Li and X. Du, Mater. Sci. Eng., C, 2012, 32, 2623–2627 CrossRef CAS.
- R. Fernandez-Torres, M. A. Lopez, M. O. Consentino, M. C. Mochon and M. R. Payan, J. Pharm. Biomed. Anal., 2011, 54, 1146–1156 CrossRef CAS PubMed.
- A. Preechaworapun, S. Chuanuwatanakul, Y. Einaga, K. Grudpan, S. Motomizu and O. Chailapakul, Talanta, 2006, 68, 1726–1731 CrossRef CAS PubMed.
- D. Teshima, B. Hino, Y. Itoh and R. Oishi, J. Clin. Pharm. Ther., 2002, 27, 403–408 CrossRef CAS PubMed.
- Y. Chen, R. S. Zheng, D. G. Zhang, Y. H. Lu, P. Wang, H. Ming, Z. F. Luo and Q. Kan, Appl. Opt., 2011, 50, 387–391 CrossRef CAS PubMed.
- W. Haasnoot, M. Bienenmann-Ploum and F. Kohen, Anal. Chim. Acta, 2003, 483, 171–180 CrossRef CAS.
- W. L. Shelver, N. W. Shappell, M. Franek and F. R. Rubio, J. Agric. Food Chem., 2008, 56, 6609–6615 CrossRef CAS PubMed.
- C. Xu, Y. Jiang, P. Yu and L.-Q. Mao, J. Electrochem., 2022, 28, 2108551 Search PubMed.
- J. Li, Z.-P. Li, Y.-F. Bai, S.-X. Luo, Y. Guo, Y.-Y. Bao, R. Li, H.-Y. Liu and F. Feng, J. Electrochem., 2022, 28, 2104211 Search PubMed.
- F. Ying, S.-S. Xu, Y.-B. Xu, M.-M. Liang and J.-F. Li, J. Electrochem., 2022, 28, 2107141 Search PubMed.
- L. Mao, D.-F. Niu, S.-Z. Hu and X.-S. Zhang, J. Electrochem., 2022, 28, 2107061 Search PubMed.
- R. D. Crapnell and C. E. Banks, Sens. Diagn., 2022, 1, 71–86 RSC.
- G. E. Fenoy, W. A. Marmisollé, W. Knoll and O. Azzaroni, Sens. Diagn., 2022, 1, 139–148 RSC.
- W. Cho, H. Rafi, S. Cho, A. Balijepalli and A. G. Zestos, Sens. Diagn., 2022, 1, 460–464 RSC.
- I. Cesarino, V. Cesarino and M. R. V. Lanz, Sens. Actuators, B, 2013, 188, 1293–1299 CrossRef CAS.
- G.-X. Zhao, W. H. Z. Ahmed and F.-L. Zhu, J. Electrochem., 2021, 27, 614–623 Search PubMed.
- Y. Zhao, J. Wan, H. Yao, L. Zhang, K. Lin, L. Wang, N. Yang, D. Liu, L. Song, J. Zhu, L. Gu, L. Liu, H. Zhao, Y. Li and D. Wang, Nat. Chem., 2018, 10, 924–931 CrossRef CAS PubMed.
- Y. Zhao, N. Yang, H. Yao, D. Liu, L. Song, J. Zhu, S. Li, L. Gu, K. Lin and D. Wang, J. Am. Chem. Soc., 2019, 141, 7240–7244 CrossRef CAS PubMed.
- Y. Xu, Z. Cai, P. Du, J. Zhou, Y. Pan, P. Wu and C. Cai, J. Mater. Chem. A, 2021, 9, 8489–8500 RSC.
- Z. Cai, P. Du, W. Liang, H. Zhang, P. Wu, C. Cai and Z. Yan, J. Mater. Chem. A, 2020, 8, 15012–15022 RSC.
- Y. Gao, Z. Cai, X. Wu, Z. Lv, P. Wu and C. Cai, ACS Catal., 2018, 8, 10364–10374 CrossRef CAS.
- Y. Qian, P. Du, P. Wu, C. Cai and D. F. Gervasio, J. Phys. Chem. C, 2016, 120, 9884–9896 CrossRef CAS.
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem., 2018, 2, 65–81 CrossRef CAS.
- K. Jiang, S. Siahrostami, T. Zheng, Y. Hu, S. Hwang, E. Stavitski, Y. Peng, J. Dynes, M. Gangisetty, D. Su, K. Attenkofer and H. Wang, Energy Environ. Sci., 2018, 11, 893–903 RSC.
- H.-J. Qiu, Y. Ito, W. Cong, Y. Tan, P. Liu, A. Hirata, T. Fujita, Z. Tang and M. Chen, Angew. Chem., Int. Ed., 2015, 54, 14031–14035 CrossRef CAS PubMed.
- S. Joung, A. M. Bergmann and M. K. Brown, Chem. Sci., 2019, 10, 10944–10947 RSC.
- X. Xie, D. P. Wang, C. Guo, Y. Liu, Q. Rao, F. Lou, Q. Li, Y. Dong, Q. Li, H. B. Yang and F. X. Hu, Anal. Chem., 2021, 93, 4916–4923 CrossRef CAS PubMed.
- B. Long, Y. Zhao, P. Cao, W. Wei, Y. Mo, J. Liu, C.-J. Sun, X. Guo, C. Shan and M.-H. Zeng, Anal. Chem., 2022, 94, 1919–1924 CrossRef CAS PubMed.
- J. Wang, R. You, C. Zhao, W. Zhang, W. Liu, X.-P. Fu, Y. Li, F. Zhou, X. Zheng, Q. Xu, T. Yao, C.-J. Jia, Y.-G. Wang, W. Huang and Y. Wu, ACS Catal., 2020, 10, 2754–2761 CrossRef CAS.
- N. Zafar, S. Yun, M. Sun, J. Shi, A. Arshad, Y. Zhang and Z. Wu, ACS Catal., 2021, 11, 13680–13695 CrossRef CAS.
- Z. Cai, F. Li, P. Wu, L. Ji, H. Zhang, C. Cai and D. F. Gervasio, Anal. Chem., 2015, 87, 11803–11811 CrossRef CAS PubMed.
- Q. Gu, Z. Wang, L. Qiao, J. Fan, Y. Pan, P. Wu and C. Cai, ACS Appl. Nano Mater., 2021, 4, 12755–12765 CrossRef CAS.
- C. Wu, W. Liang, J. Fan, Y. Cao, P. Wu and C. Cai, Acta Phys.-Chim. Sin., 2021, 37, 1912050 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- A. B. Anderson, Phys. Chem. Chem. Phys., 2012, 14, 1330–1338 RSC.
- Y. He, S. Hwang, D. A. Cullen, M. A. Uddin, L. Langhorst, B. Li, S. Karakalos, A. J. Kropf, E. C. Wegener, J. Sokolowski, M. Chen, D. Myers, D. Su, K. L. More, G. Wang, S. Litster and G. Wu, Energy Environ. Sci., 2019, 12, 250–260 RSC.
- D. Zou, D. Liu and J. Zhang, Energy Environ. Sci., 2018, 1, 209–220 CAS.
- Y. Qian, Z. Liu, H. Zhang, P. Wu and C. Cai, ACS Appl. Nano Mater., 2016, 8, 32875–32886 CrossRef CAS PubMed.
- K. Artyushkova, B. Kiefer, B. Halevi, A. Knop-Gericke, R. Schlogl and P. Atanassov, Chem. Commun., 2013, 49, 2539–2541 RSC.
- O. C. Braga, I. Campestrini, I. C. Vieira and A. Spinelli, J. Braz. Chem. Soc., 2010, 21, 813–820 CrossRef CAS.
- J. D. Voorhies and R. N. Adams, Anal. Chem., 1958, 30, 346–350 CrossRef CAS.
- D. Chen, Q. Wang, J. Jin, P. Wu, H. Wang, S. Yu, H. Zhang and C. Cai, Anal. Chem., 2010, 82, 2448–2455 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The instruments for characterization, details of the theoretical calculations, and HPLC analysis of SMX in water samples; figures. See DOI: https://doi.org/10.1039/d2sd00117a |
| This journal is © The Royal Society of Chemistry 2022 |