
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly selective detection of ethanol in biological fluids and alcoholic drinks using indium ethylenediamine functionalized graphene
Ramin
Boroujerdi
 *,
Amor
Abdelkader
and
Richard
Paul
*,
Amor
Abdelkader
and
Richard
Paul
Faculty of Science and Technology, Bournemouth University, Talbot Campus, Fern Barrow, Poole, BH12 5BB, UK. E-mail: rboroujerdi@bournemouth.ac.uk
First published on 5th April 2022
Abstract
A graphene-based electrochemical sensor has been fabricated by attaching indium–ethylenediamine nanoparticles to the surface of reduced graphene oxide nanolayers. The developed screen-printed graphene ink electrode was utilized to detect and measure trace amounts of ethanol in various biological and industrial aqueous authentic samples in a cyclic voltammetry system. The combination of selective interaction between indium nanoparticles with aqueous ethanol and the large surface area provided by two-dimensional graphene layers allows the sensor to detect ethanol concentrations as low as 100 micromolar. The sensor offers a wide linear range and can generate a linear response to the increase of ethanol in the environment up to 3 molar concentrations. The selectivity of the sensor was tested and compared in response to various primary, secondary, and tertiary alcohols, where it proved to be highly selective towards ethanol. After passing standard tests, the sensor has been tested with aqueous authentic samples and was able to successfully detect and measure ethanol in urine, saliva, beer and whisky.
Introduction
In 2004, the discovery of graphene triggered a wave of theoretical and experimental research worldwide due to its unique properties. Graphene-based materials and their composites enjoy the status of being the new super-material on the horizon of condensed matter physics and materials science respectively, owing to their applications in numerous industries. 2D graphene with its honeycomb lattice structure and free π–π electrons not only possesses unique physicochemical properties such as high mechanical strength and flexibility, large surface area, thermal stability, and high conductivity, but also offers wide potential and a variety of applications in different fields.1,2Accurate and rapid measurement of ethanol in aqueous samples is a requirement across many fields, including biological sample testing in medical and pharmaceutical applications as well as forensic applications which may require on-site, portable testing solutions.3–5 Nanolayered graphene, due to its extraordinary characteristics, could be one of the best candidates to develop affordable, durable, and fast response electrochemical sensors. These characteristics have led to the development of various graphene based ethanol sensors, yet almost all of them respond only to the ethanol vapors.1 The mechanism of detection in those sensors relied on measuring changes in resistance at high temperatures.1 There was only one report to date of a graphene–enzyme sensor that could detect ethanol in aqueous samples,6 and a purely chemically functionalized graphene based sensor for measuring ethanol in aqueous samples using cyclic voltammetry is something we report in this paper for the first time. It is worth mentioning that despite the fact that enzyme based sensors are highly selective and can work in aqueous samples, they sometimes suffer from poor detection limit,7,8 short lifetime, poor power density, poor electron transfer rate, and difficulties of enzyme loading problems which all are related to enzyme molecule characteristics and stability.9–11 Whilst the combination of graphene and enzyme fixed some issues such as improving electrochemical behavior of the sensor,12 other issues still need to be addressed before such biosensors can become competitive in practical applications. To address these problems we present here the development of a stable and selective sensor, capable of detecting ethanol in aqueous samples in the compartment of an electrochemical sensor.
Indium based nanoparticles generate a relatively high surface area which is highly preferred for sensors and lets the sensor interact with the environment much more than bulk materials.2,13 The applications and mechanism of ethanol gas sensing (in the form of volatile gases) for indium based compounds, such as indium oxide (In2O3)14 and nitrogen doped indium oxide (N-In2O3)15 has been studied very recently, yet again most of those sensors rely on measuring changes in resistance of the gas, and not measurement of ethanol in aqueous samples.1
While it was expected that different products could have developed from the interaction between indium salts and amines, from possibility of aminolysis and formation of In2O3 (ref. 16) to formation of tris(ethylenediamine) complexes with indium (In(en)3),17 to date no research has reported on the interaction between indium and ethylenediamine. Here we present the synthesis of this complex and the study of its characteristics and applications as an electrochemical sensor. Moreover, the chemical composition of our sensing compartment is novel and as Raman, FTIR and XPS results showed, no sign of In–O bonds has been found in samples. However, we can still expect the general oxidation procedure to be similar to the previously reported sensors.
In this paper we successfully anchored novel nanoparticles of indium–ethylenediamine (In–en) to the surface of graphene by forming chemical bonds and applied this sensor for the detection and measurement of ethanol in various real samples. The selectivity and sensitivity of the sensor was found to come from the In–en nanoparticles, and so the effects of this functional group were also tested separately from bonding with graphene. It was demonstrated that graphene amplifies the signal generated from the selective compartment. The development of a sensing probe made of a combination of 1D and 2D nanoparticles, which offers high surface area, allows us to miniaturize the sensing system, while maintaining the sensitivity and selectivity of the sensor, to achieve portable sensors. Our sensor showed selective response to ethanol in beer, whisky, saliva and urine. After developing a calibration curve in a complex matrix like urine, we were able to measure the alcohol concentration by monitoring the changes in the intensity of the oxidation peak.
Materials and methods
Reagents
Chemicals were purchased from Fisher Scientific, UK, and were analytical reagent grade. Aqueous solutions and dilutions were prepared using distilled water. Beer (5% alcohol; made of barley malt, rice and hops) and American style whisky (35% alcohol; made of corn, barley, rye and natural honey) were used as industrial real samples. Biological samples used in testing was urine donated by an adult male volunteer who was teetotal. Informed consent was obtained from human subject.Apparatus
Cyclic voltammetry measurements were carried out using IviumStat.h potentiostat (Ivium Technologies, the Netherlands) in a 1.5 mL cell with a three-electrode configuration at room temperature (∼25 °C). Ag/AgCl was used as a reference electrode, while a Pt wire (1 mm diameter) worked as the counter electrode in response to the developed graphene-based electrode. Graphene ink was made in U500H Ultrasonic Bath (Ultrawave, UK) and Jeio Tech's OV-11 vacuum oven (South Korea) dried printed electrodes. Platinum ATR – Alpha II FTIR spectrophotometer (Bruker, USA) was applied to collect Fourier transform infrared (FTIR) spectra and XploRA™ PLUS Raman spectrometer was used for Raman spectra. JEOL JSM-6010 tungsten cathode scanning electron microscope was used to carry out both scanning electron microscopy and energy dispersive spectroscopy (SEM and EDX) analysis. High-resolution transmission electron microscopy (HR-TEM) imaging was carried out on the JEOL 2100F FEG-TEM at 80 kV. Flash EA 1112 elemental analyser was used to measure the ratio of carbon and nitrogen in the developed crystals while Vista-PRO Simultaneous Inductively coupled plasma-optical emission spectrometer (ICP-OES) was applied to measure indium ratio in the same crystal. X-ray photoelectron spectroscopy (XPS) data were collected on a Kratos AXIS ULTRA instrument configured with a monochromated Al Kα (1486.6 eV) X-ray source.Graphene oxide (GO) and reduced graphene oxide (rGO)
One of the most widely applied methods which has been used for synthesising graphene oxide in the past decades was developed by Hummers and Offeman, which is known as Hummers method.18,19 For this experiment, we used a slightly modified Hummers' method to obtain graphene oxide. Since the conductivity of reduced graphene oxide is much higher than graphene oxide1 for developing an electrode, and also the removal of oxygen functional groups will minimize unwanted interactions and improves the selectivity. We used aluminum as the reducing metal in hydrochloric acid due to the proven ability to obtain a very high carbon to oxygen ratio.20 Developed reduced graphene oxide washed with centrifuge to neutral pH, and freeze dried to be functionalised later in the next steps.Synthesis of indium–ethylenediamine crystals
1.5 g indium acetate was added to 15 mL ethylenediamine in a 50 mL round bottom flask in a reflux system and stirred at 800 RPM at 130 °C for 24 hours. Brown sediments were separated from the mixture using centrifuge (6000 RPM, 2 h) and washed multiple times with ethanol to form a white solid. Then it is left in vacuum oven at 160 °C to form yellow crystals of indium–ethylenediamine complex.Synthesis of indium–ethylenediamine functionalised graphene
The process of preparing indium–ethylenediamine functionalised graphene (In–en) is similar to that of preparing indium–ethylenediamine crystals, with minor changes in the concentration and addition of rGO. 0.5 grams indium acetate is added to 15 mL ethylenediamine and left for 24 hours reflux, then condenser is removed from the system and heating is reduced (to approximately 50 °C). Then 1.5 grams of rGO was added to the mixture and stirred for 1 hour. The black mixture was then washed with ethanol and left to dry in vacuum oven at 160 °C.Electrode preparation
In order to investigate the mechanism of sensing and determine the role of indium complex and rGO in the process of sensing ethanol, various electrodes were prepared as follows.Results and discussion
Materials characterization
To determine the surface chemistry of the graphene and graphene hybrids one of the widely used spectroscopy methods is Fourier transform infrared spectroscopy (FTIR) due to the non-invasive and non-destructive nature of the method.21Fig. 1a compares the FTIR spectra of reagents (indium acetate and ethylenediamine) and the produced indium complex. The primary amine stretching peaks at 3357, 3286 cm−1 and primary amine N–H bend at 1595 cm−1 belonging to ethylenediamine were not present in the product, but the C–N stretching around 1148 cm−1 and secondary N–H single stretching peak at 3159 cm−1 were found in the product.22 Considering there are no amine groups in the initial indium salt (indium acetate), and conversion of primary amine groups on pure ethylenediamine to secondary amine groups, it is expected that indium formed a bond with ethylenediamine through its amine groups and also eliminates the possibility of aminolysis reaction which could have led to formation of indium oxide.16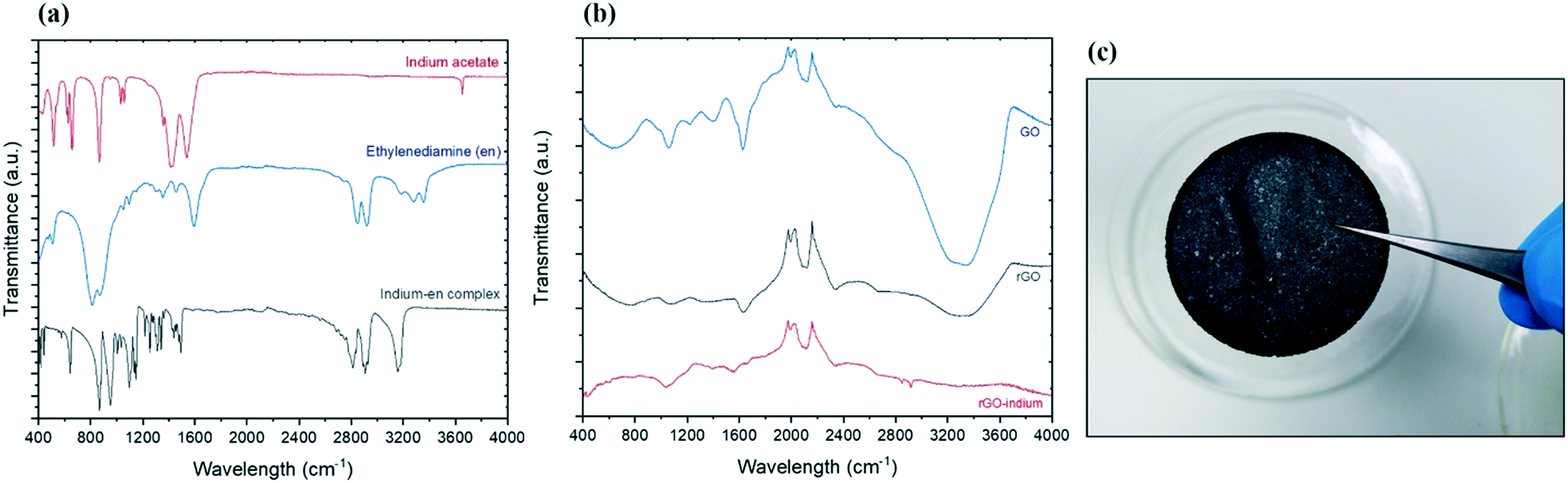 | ||
Fig. 1 (a) FTIR spectra of reagents (indium acetate and ethylenediamine) vs. final product (In–en) confirms the successful dimerization through the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds. (b) FTIR spectra of graphene oxide, reduced graphene oxide and functionalised rGO. (c) Sample obtained after the functionalization of graphene with In–en, in a form of a compact disk. O bonds. (b) FTIR spectra of graphene oxide, reduced graphene oxide and functionalised rGO. (c) Sample obtained after the functionalization of graphene with In–en, in a form of a compact disk. | ||
The formation of graphene oxide confirmed as the FTIR spectrum of GO in Fig. 1b shows the typical characteristics peaks including a wide O–H stretching peak at 3300 cm−1 and sharp C–O stretch peak at 1628 cm−1.22,23 Following the reduction, the intensity of peaks associated with the oxides functional groups between 400–1700 significantly decreased, suggesting the successful removal of the oxidative groups by aluminum and HCl.24 After treatment with indium precursor, the oxides peaks almost disappeared, suggesting more reduction is taking place during the indium functionalization/hybridization. Two new peaks can be observed in the rGO–In samples between 2800 and 3000 cm−1, suggesting nitrogen groups from the ethylenediamine has replaced some of the remaining oxygen functional groups on the rGO.22 These two new nitrogen peaks add more evidence to support our hypothesis that graphene is bonded chemically to indium through a nitrogen containing link.
Further information on the existing phases can be gathered from the Raman spectroscopy analysis.25,26Fig. 2b shows Raman spectra of formed indium–en and indium acetate in the frequency range of 100 to 1600 cm−1 obtained at room temperature. The spectra show clear changes in the molecular bonds after the reaction as evidenced by the appearance of multiple new peaks caused by the ethylenediamine ligands.27,28 Peaks at 890.48, 860.87 and 847.67 cm−1 represent NH2 twist, while the strong peak at 1266.14 cm−1 shows CH2 twist.29 Two small peaks at 431.22 and 443.64 cm−1 caused by metal–nitrogen stretch bond29 while the sharp peak at 556.01 cm−1 could stand for In–N bond, confirms indium linked to the ethylenediamine.30 The Raman spectrum of the developed indium–en complex doesn't show any of the specific indium oxide peaks in the range between 100 and 650 cm−1,31,32 which suggests the homogenous formation of a complex and ruled out the formation of In2O3 as side product in the process. At the same time, the appearance of narrow and sharp bands suggests that the developed organometallic complex has good crystalline structure.33
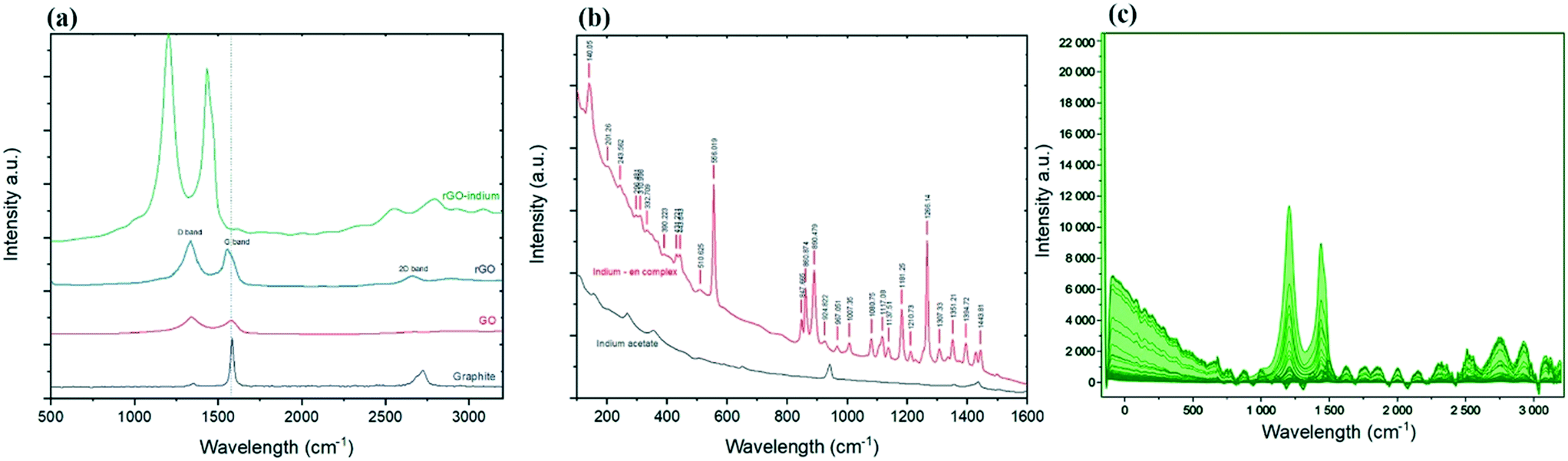 | ||
| Fig. 2 (a) Raman spectra of graphite, graphene oxide, reduced graphene oxide and chemically indium–en functionalised reduced graphene oxide (rGO–indium complex); graph illustrates the evolution of the D, G, and 2D and D′ from graphite to the main product. (b) Raman spectrum of indium acetate (reagent) compared with the indium–ethylenediamine complex crystals (product). (c) Raman mapping of chemically developed rGO–indium complex obtained from studying 1000 data points (10 points in each of X, Y and Z dimensions). Graph shows the entire hyperspectral dataset of spectra collected. | ||
Regarding the Raman spectra for carbon materials (Fig. 2a), graphite shows a G band (at 1584 cm−1) sourced from the scattering caused by carbon atoms with sp2 orbitals, and a weak peak at 2730 cm−1 which is a 2D band caused by the combination of 2 types of phonons (double-resonance process; D phonon + acoustic longitudinal D′ phonon).34 The oxidation process leads to the exfoliation of the staked graphene into monolayer GO, which results in reducing the intensity of the D + D′ band. In addition, the harsh environment during the transformation of graphite into graphene oxide caused significant damage to the graphene plane, evidenced by the appearance of the defects peak (the D peak) at ∼1353 cm−1.35 After chemical reduction no significant changes in the D![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :G ratio can be observed.36 However, the 2D band which once vanished in GO spectra now appears at 2675 cm−1 for rGO. The re-appearance of the 2D band on graphene suggested the ability of the reduction process to recover some of the original graphene structure and properties.37 After chemical functionalization, D and G bands shifted to 1210 and 1436 cm−1 respectively, however 2D band shifted to the opposite side and appeared at 2827 cm−1; while this shift could have been caused partially by the changes on the surface of graphene after functionalization, sample orientation and uniaxial strain could have been another reason for the shift to happen.38–41 Ethylenediamine functionalised graphene oxide has been studied before and it is proven that the location of the G and D peaks of graphene are not affected by ethylenediamine,42 so this shift in the peaks could directly be related to the indium functionalisation. The homogeneity of the functionalized product was confirmed by Raman mapping by recording the Raman spectra over 1000 points on the surface of the functionalized product as illustrated in Fig. 2c. It can be seen that there are no signs of unexpected or irregular peaks, and the graph in Fig. 2c shows that almost all of the studied points present a similar spectrum and peaks.
:G ratio can be observed.36 However, the 2D band which once vanished in GO spectra now appears at 2675 cm−1 for rGO. The re-appearance of the 2D band on graphene suggested the ability of the reduction process to recover some of the original graphene structure and properties.37 After chemical functionalization, D and G bands shifted to 1210 and 1436 cm−1 respectively, however 2D band shifted to the opposite side and appeared at 2827 cm−1; while this shift could have been caused partially by the changes on the surface of graphene after functionalization, sample orientation and uniaxial strain could have been another reason for the shift to happen.38–41 Ethylenediamine functionalised graphene oxide has been studied before and it is proven that the location of the G and D peaks of graphene are not affected by ethylenediamine,42 so this shift in the peaks could directly be related to the indium functionalisation. The homogeneity of the functionalized product was confirmed by Raman mapping by recording the Raman spectra over 1000 points on the surface of the functionalized product as illustrated in Fig. 2c. It can be seen that there are no signs of unexpected or irregular peaks, and the graph in Fig. 2c shows that almost all of the studied points present a similar spectrum and peaks.
More details of the chemical composition of prepared materials were collected by elemental analyser and inductively coupled plasma (ICP) analysis. Results of the measured compositions are listed in Table 1. Nitrogen could be detected by the elemental analyser, despite the low atomic weight compared to that of indium. The EDX elemental analysis (Fig. 3) confirms the homogeneous distribution of the elements in the prepared composite.
| Contents | |||
|---|---|---|---|
| Element | C% | N% | In% |
| Method | EA | EA | ICP |
| Experimental ratios | 6.08 | 2.41 | 75.79 |
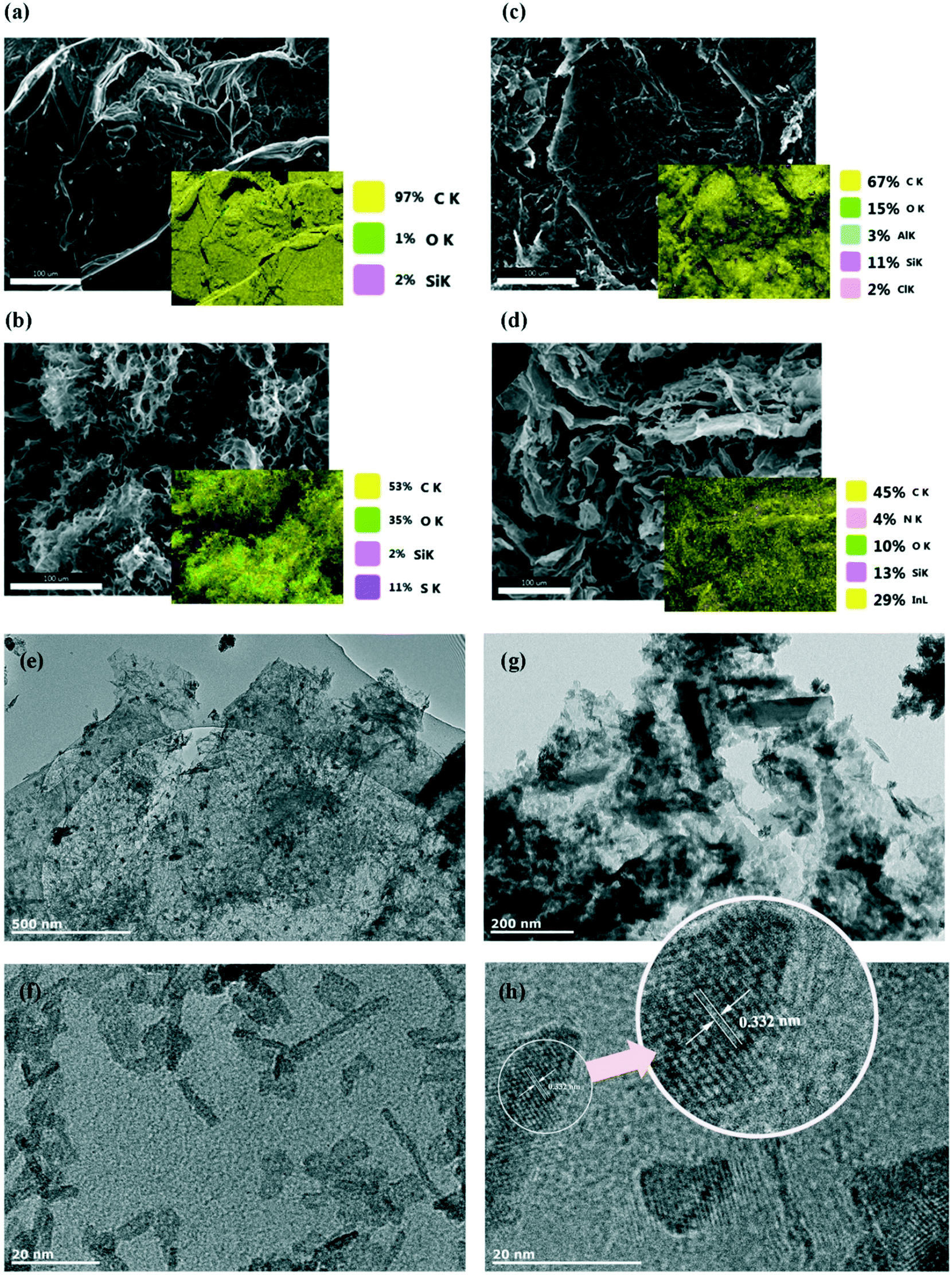 | ||
| Fig. 3 Figures a to d show SEM and EDS analyses of graphite (a), graphene oxide (b) and reduced graphene oxide (c) in comparison with indium functionalised rGO (d). Acceleration voltages, 10 keV. Scale bars are 100 μm for all EDS graphs. Figures e to h shows HR-TEM image of the chemically developed rGO–In composite. Scale bars are (e) 500 nm, (f) 200 nm, (g) 20 nm and (h) 20 nm. | ||
The morphology of the prepared composite was investigated using SEM and TEM techniques. As shown in Fig. 3, the prepared graphene oxide shows a loose, wrinkled and corrugated structure (Fig. 3b), though it can be seen that layers seem to be aggregated after the reduction process (Fig. 3c). After functionalization with indium, the TEM images detected the morphology of several indium-containing nanoparticles; rod-like and almost equiaxial larger particles. The nanoparticles had an average length of about 15.78 nm and average diameter of 2.56 nm (Fig. 3f); however larger nanoparticles with the length of about 217 nm (and thickness of about 53 nm) were also observed (Fig. 3g). Fig. 3h shows an image of the tangled particles accumulated at the interface between graphene layer and indium nanoparticles; the lattice fringe/spacing indicated in the Fig. 3h is 0.332 nm. It can be seen that functionalised graphene shows high crystalline quality due to the presence of the lattice fringes corresponding to the basal plane spacing (0.332).43 The fact that the basal plane distance is slightly smaller, but very close to that of bulk graphite (0.336 nm)44 could be that ethylenediamine linked some of the layers together45 and introduced new functional groups at the same time. Nitrogen doping through the functionalisation process is less possible as that would have enlarged the basal plane spacing of the graphene sample.46
Further information about the chemical composition and bonds in the final functionalized graphene can be collected from the XPS analysis. The carbon spectrum (Fig. 4e) of the chemically functionalized rGO revealed that it consists of four components. The first main components belong to carbon bonds (CC) of the graphene layer which appeared at 284.86 eV. The second predominant peaks belongs to hydroxyl and epoxy C–O bonds at 285.98 eV, which is probably associated with the residual oxygen group from the GO or to the link between graphene and indium.47 The peak at 287.1 eV is for the C–N bond48,49 which might have overlapped with other peaks. The other two minor components are from CN bonds at 283.06 eV (ref. 50) and O–CN bonds at 287.9 eV (ref. 51) which are developed through the functionalisation process. The C–In bond peak is covered by the CC peak of rGO and hence it cannot be pointed out separately in the spectra.52 The presence of carbon–indium bonds can be confirmed by the indium spectrum as illustrated in Fig. 4c. Indium spectrum shows four peaks: two sharp peaks of In 3d5/2 and In 3d3/2 and their satellite. Both sharp peaks at 446.13 and 453.63 eV are related to In+–C bonds,52 while both shoulder peaks at 443.33 and 450.93 eV represent the In–N bonds.53,54
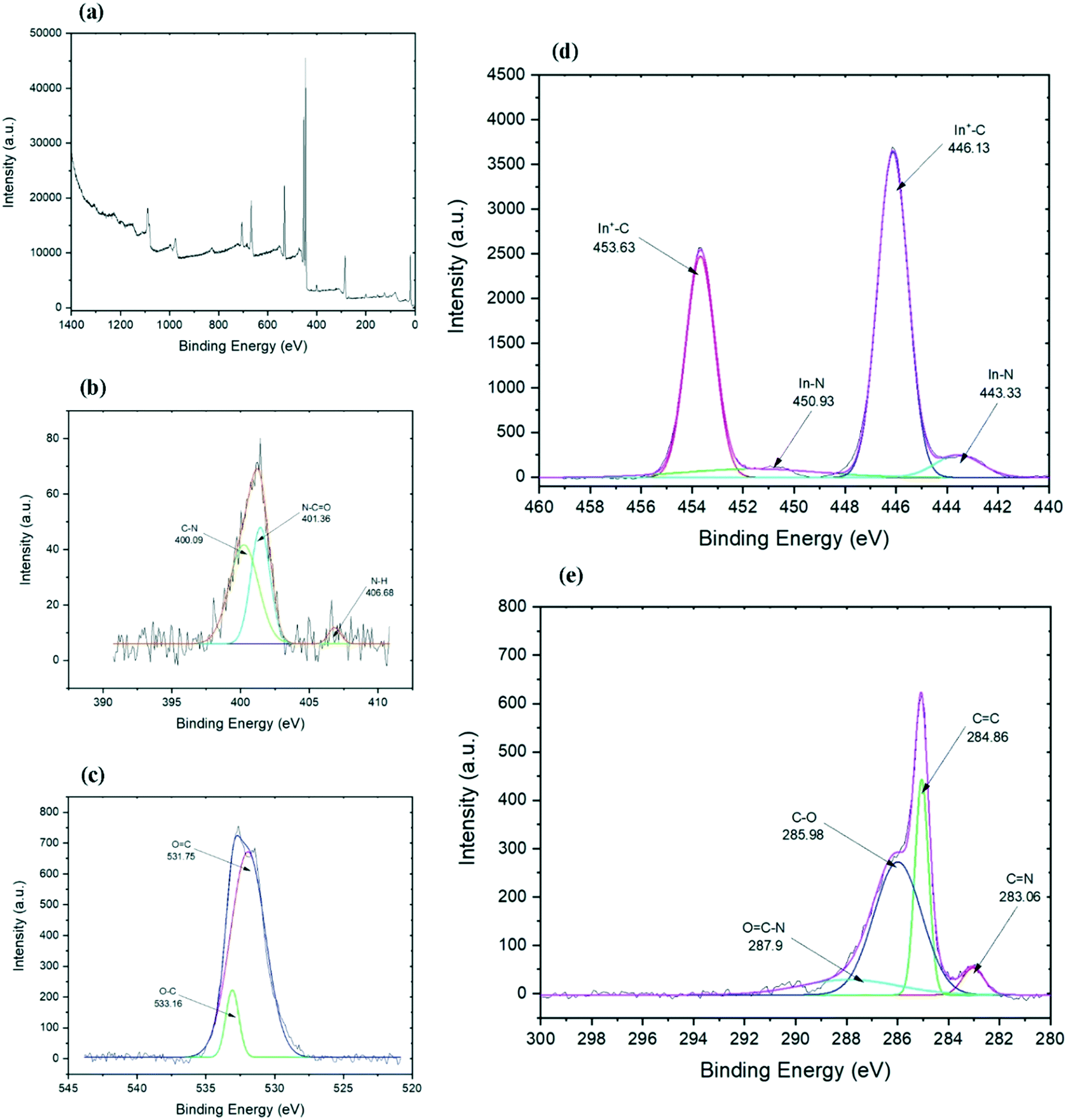 | ||
| Fig. 4 XPS comparison of chemically functionalised indium–rGO. The corresponding deconvoluted peaks are also shown for each case: (a) wide scan XPS spectrum of sample, (b) nitrogen 1s, (c) oxygen 1s, (d) indium 3d, (e) carbon 1s. | ||
The high-resolution nitrogen spectrum shows three peaks (Fig. 4b). The small peak at 406.68 eV could be N–H bonds, while two big peaks could represent sp3 and sp2 C–N bonds.55,56 The peak at 401.36 eV is an sp2 C–N bond and comparing to the carbon spectrum, it could show N–CO bonds,57 while the peak at 400.09 eV relates to C–N bands.49,58,59 Due to the weak intensity of the nitrogen peak, it was hard to investigate and differentiate the weak peak around 396 eV belonging to N–In bond58 from the noise. The oxygen spectrum (Fig. 4c) shows one broad peak which is formed from the overlap of two peaks, one at 531.7 eV and another peak at 533.16 eV which represent OC and O–C bands, respectively.60 Interestingly, as expected from ethylenediamine to further reduce the graphene,61 not only did it eliminate the trace amounts of aluminum and chlorine on the surface (trace amounts of Al and Cl found on rGO based on the EDS results) and almost maintained the carbon to oxygen ratio, but it also helped with the functionalisation with indium. The XPS results suggest that indium–en complex could have connected to the graphene layers both directly through In–C bonds and possibly through nitrogen, by forming In–N–C bonds.
The electrochemical performance of the sensor
To evaluate the selectivity of the developed electrochemical sensor, the change in electrical current, at the oxidation peak potential, was measured in the presence of several types of alcohol, including methanol, ethanol, 1-butanol, 2-butanol, 2-propanol and 1,2-propanediol. The response of the sensor towards 0.1 M aqueous solutions of alcohols is shown in Fig. 5a. Comparing the response of the sensor to diluted alcohol (current at peak potential, I) in comparison to response of the sensor to pure water (current at same potential as peak potential, I′) measured as ΔI = I/I′, and as illustrated in Fig. 5b, the only alcohol that generated higher electrical current in comparison to pure water is ethanol; in other words, the sensor responded to other alcohols in the same manner as it responded to pure water (ΔI ≈ 1) and only shows high selectivity for ethanol. The selectivity toward ethanol could have been caused by the effect of both size and reactivity of different alcohols with indium and also attached amine groups. Smaller molecules can approach and interact with the receptor more easily. At the same time, acidity of alcohols decreases as the size of their conjugated base increases. The mechanism of sensing is explained in the following sections.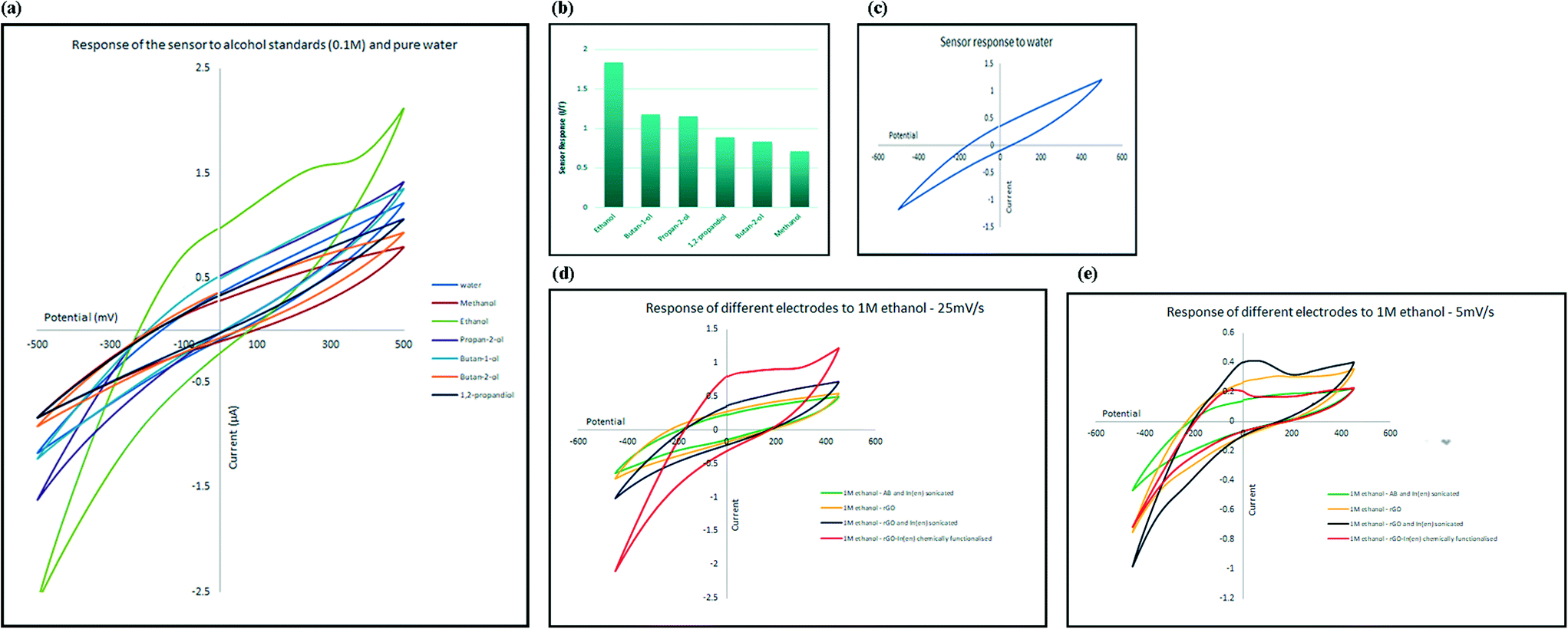 | ||
| Fig. 5 (a) Cyclic voltammetry spectra generated from the response of the sensor toward different alcohol standards and pure water; scan rate was 25 mV s−1. Oxidation peak only appears in the presence of ethanol. (c) and (d) Response of 4 different electrodes (pure rGO, rGO–In–en chemically functionalized composite, rGO–In–en physically mixed composite and AB–In–en physically mixed composite) toward ethanol at different scan rates of 5 mV s−1 (e) and 25 mV s−1 (d). (c) CV of the electrode with [alcohol-free] water, in a separate graph. Graph (b) shows ΔI, which is the response of the sensor to different diluted alcohols at the oxidation peak potential (I) to the response towards pure water at the same potential (I′). | ||
Sensitivity of the sensor towards ethanol
The process of sensing ethanol and obtaining the linear range of the electrochemical sensor was performed at room temperature using aqueous standard solutions. In a typical run, 1.5 mL of standard was added to the micro cell in direct contact with three electrodes system and current changes in a full cycle were recorded. To evaluate the sensitivity towards ethanol, aqueous solutions of different concentrations in a wide range from 10−5 M to 5 M were freshly prepared before use. The CV spectra were recorded in the range between −450 and +450 mV (scan rate: 1 mV s−1) and the current at oxidation peak potential were recorded and used to develop a calibration graph. Two phenomena were recorded while recording the spectra, the first was that slower scan rates caused the peaks to appear at lower potentials and secondly it was observed that by increasing the concentration, the maximum point of the peak slowly shifted towards higher potentials. As illustrated in Fig. 6a, there was a linear correlation between increasing the concentration of ethanol and intensity of the monitored oxidation peak. The sensor showed linear response to the changes in the concentration of ethanol in the range between 10−4 M and 3 M (Fig. 6c).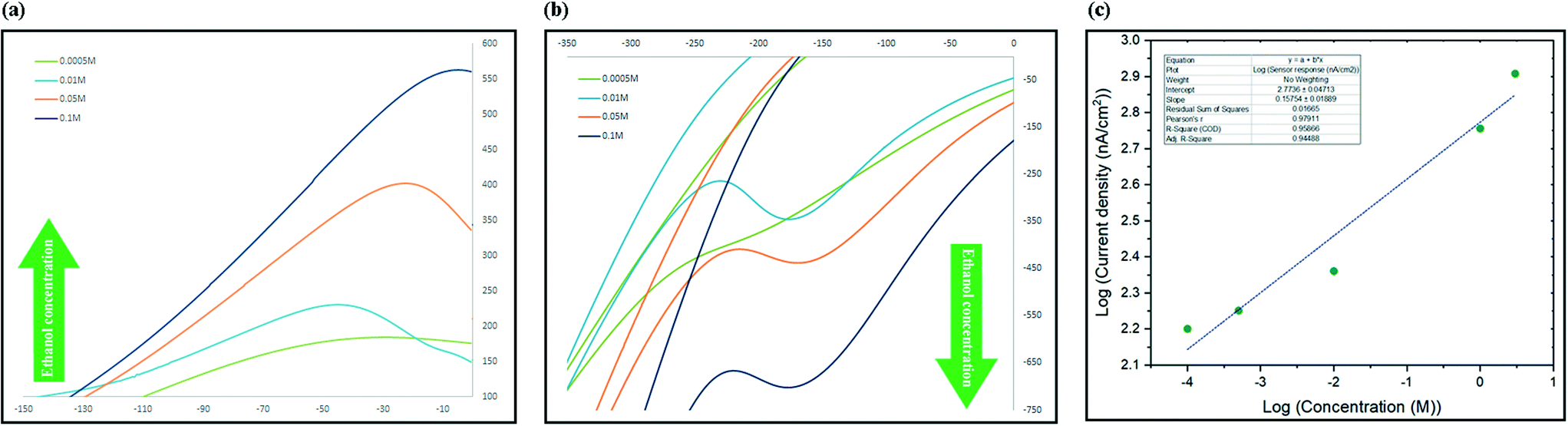 | ||
| Fig. 6 (a) and (b) Response of the sensor to increasing concentrations of ethanol in the environment; scan rate was 1 mV s−1. Graph (c) wide linear range of the sensor between 1 × 10−4 M and 3 M ethanol concentrations; R2 is 0.9587. | ||
The characteristics of the developed sensor has been compared with other graphene-based ethanol sensors in Table 2. As it can be seen, our sensor showed significant advantages comparing to previously developed sensors in different areas such as detection limit, linear range and real samples and working phase (liquid phase) comparing to others. As table shows, graphene-based gas sensor that measure volatile ethanol concentrations, despite their reasonable detection limit, can mostly work at high temperatures and have not been used to study real samples.1 Graphene based sensors that can work in liquid phase and has been tested before not only offer a very limited linear range, they also have not been tested for biological samples such as urine and saliva before.
| Sensing material | Detection limit | Linear range | Real samples | Working phase | Ref. |
|---|---|---|---|---|---|
| VA/CuO/Gr-NPls | 1800 ppm | 1800–4000 ppm | None | Gas | 62 |
| GO/melamine | 70 ppm | 70–1670 ppm | None | Gas | 63 |
| ZnFe2O4NSs/rGO | 0.1 ppm | 0.1–100 ppm | None | Gas | 64 |
| Au–AgNPs/P(Cys)/rGO | 5 μM | 17 μM–1.84 mM | Whisky, soft drink | Liquid | 6 |
| rGO–In2O3 | 68 mM | 0.1–1.2 M | Beer, whisky | Liquid | 2 |
| rGO–In–en | 100 μM | 100 μM–3 M | Urine, saliva, beer, whisky | Liquid | This work |
Mechanism of sensing
In order to understand the role of each of the composite components on the sensing process, four different electrodes with different chemical structure were developed: chemically functionalised rGO–In; physically mixed rGO and In–en; physically mixed AB and In–en; and finally rGO sensor. All materials were tested with the same concentration of ethanol so the responses can be compared with each other.The pristine rGO electrode showed no oxidation peaks in the scanned range, suggesting no interaction between rGO and alcohol. The electrode where rGO or AB was physically mixed with indium–en shows obvious oxidation peaks. The oxidation peak recorded in the presence of rGO has much stronger intensity compared with that of the AB–indium electrode, suggesting rGO helped with amplifying the intensity of the signal. The chemically functionalised rGO–In also shows the same oxidation peak with almost the same intensity. However, careful investigation of the electrochemical processes on the two electrodes shows significant differences. As shown in Fig. 5e at slow scan rate (5 mV s−1) the intensity of the oxidation peak for the physically mixed sensor is higher than the chemically functionalised sensor. At higher scan rates (25 mV s−1; Fig. 5d), only the chemically functionalised sensor still generates a measurable signal, suggesting higher impedance for the physically mixed electrode. As the oxidation peak appears only at the presence of In–en composite no matter what base material has been used (graphene or acetylene black), suggests the selective behavior of the sensor only happens because of the In–en group. However, from the results it can be seen that adding graphene to the synthesis procedure and forming a direct bond between rGO and In–en, not only improved the intensity of the response and makes the sensor more sensitive to lower concentrations, but it also makes the sensor respond at different scan rates and the chemical bond prevents In–en from being washed away from the surface after each test.
When the organometallic nanoparticles of the developed sensor are exposed to ethanol in the sample, the hydrogen atom in the alcoholic (–OH) group of ethanol binds to the nitrogen sites while the oxygen atom binds to the indium site, which could cause the etching of hydrogen (dehydrogenation) or water (dehydration).15 Both routes could be responsible for the appearance of an oxidation peak in cyclic voltammetry measurements.
Authentic samples
The proposed electrochemical sensor has been successfully used for detection and measurement of ethanol in bodily fluids and alcoholic beverages. We have tested the performance of the chemically functionalized rGO electrode in four authentic samples. All of the tests were conducted with a scan rate of 25 mV s−1. The CV of all samples showed some shift to a more positive potential in the location of oxidation peaks as a result of increasing the electrolyte resistance; other factors that can be attributed to the change of the oxidation peak potential such as sluggish charge transfer reaction rates, instrumental delay, and capacitive currents for double layers65 does not apply here as the only difference is the changes in the solution matrix. The sensor still shows an oxidation peak that changes its intensity linear to the concentration of ethanol in the electrolytes. The following section discusses the application of our sensor across a range of authentic samples.Diluted urine samples prepared at ethanol concentrations in the range between 0.0029 M and 0.8 M were tested and the sensor was able to generate an oxidation peak for that range and the intensity of the peak increased in correlation with the increase in the concentration of ethanol. The range covers concentrations of about 10 times below the UK driving limit to much higher amounts and did not show false positive errors for concentrations below the limit (Fig. 7).
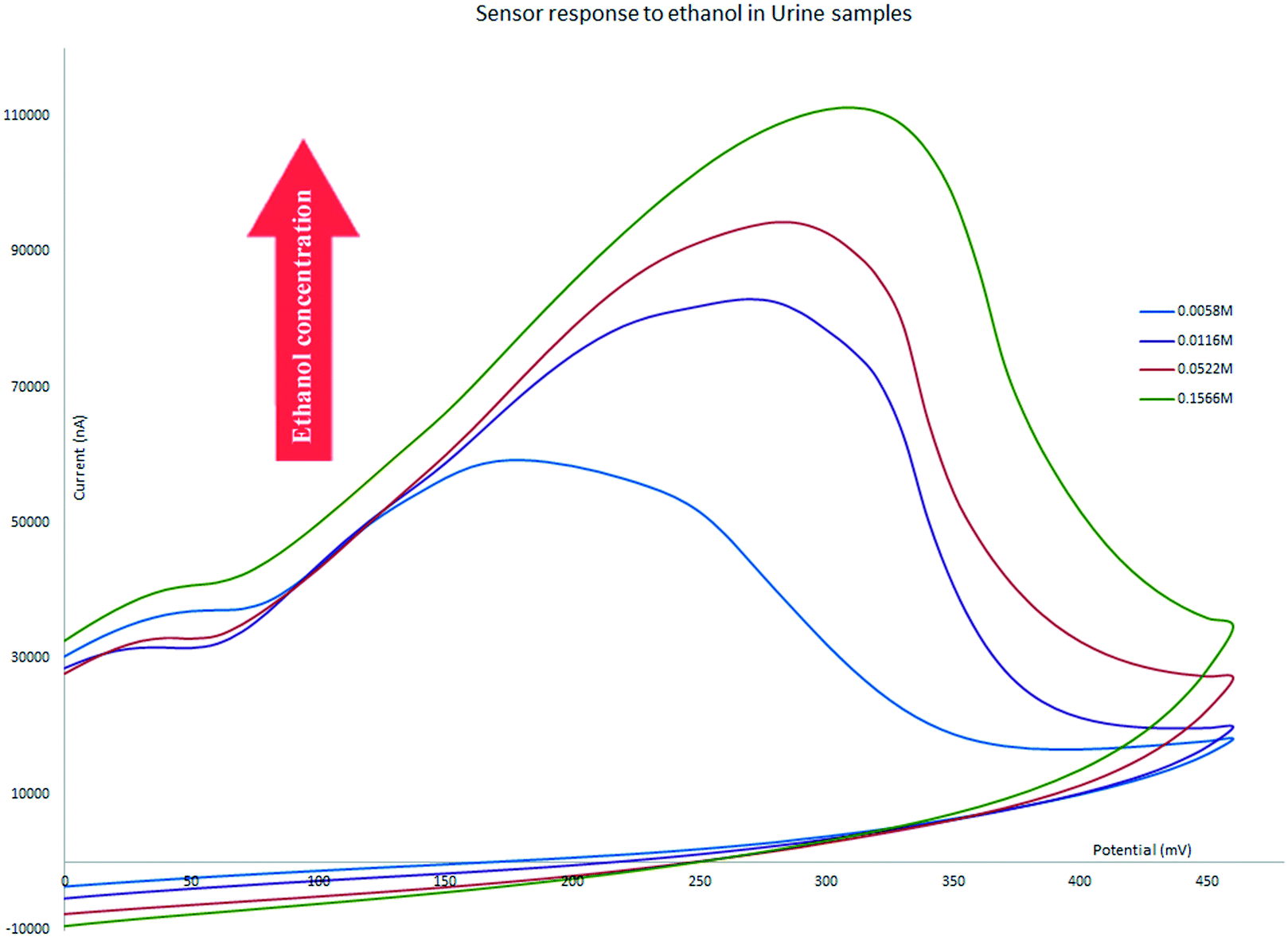 | ||
| Fig. 7 Sensor response to different concentrations of ethanol in urine samples. | ||
Saliva was tested as the second biological sample. While expectoration seems to be a straight forward method for collecting the oral fluid, samples collected using this method often contain food and oral debris which may cover and contaminate the surface of electrodes, even after centrifugation.3 To prevent these issues, an 8 × 8 cm absorbent pad with the thickness of 0.5 mm was chewed for 5 min and then shaken in 5 mL water. Samples were then prepared in the same manner as for urine described above. An oxidation peak only appeared for high concentrations of ethanol and the sensor only responded to relatively high ethanol concentrations in diluted saliva. As it can be seen in Fig. 8 while diluted saliva sample which contains 0.8 M ethanol shows an oxidation peak at around 200 mV; the current intensity of the peak is below the response of the sensor to diluted saliva in the absence of ethanol, which can cause errors.
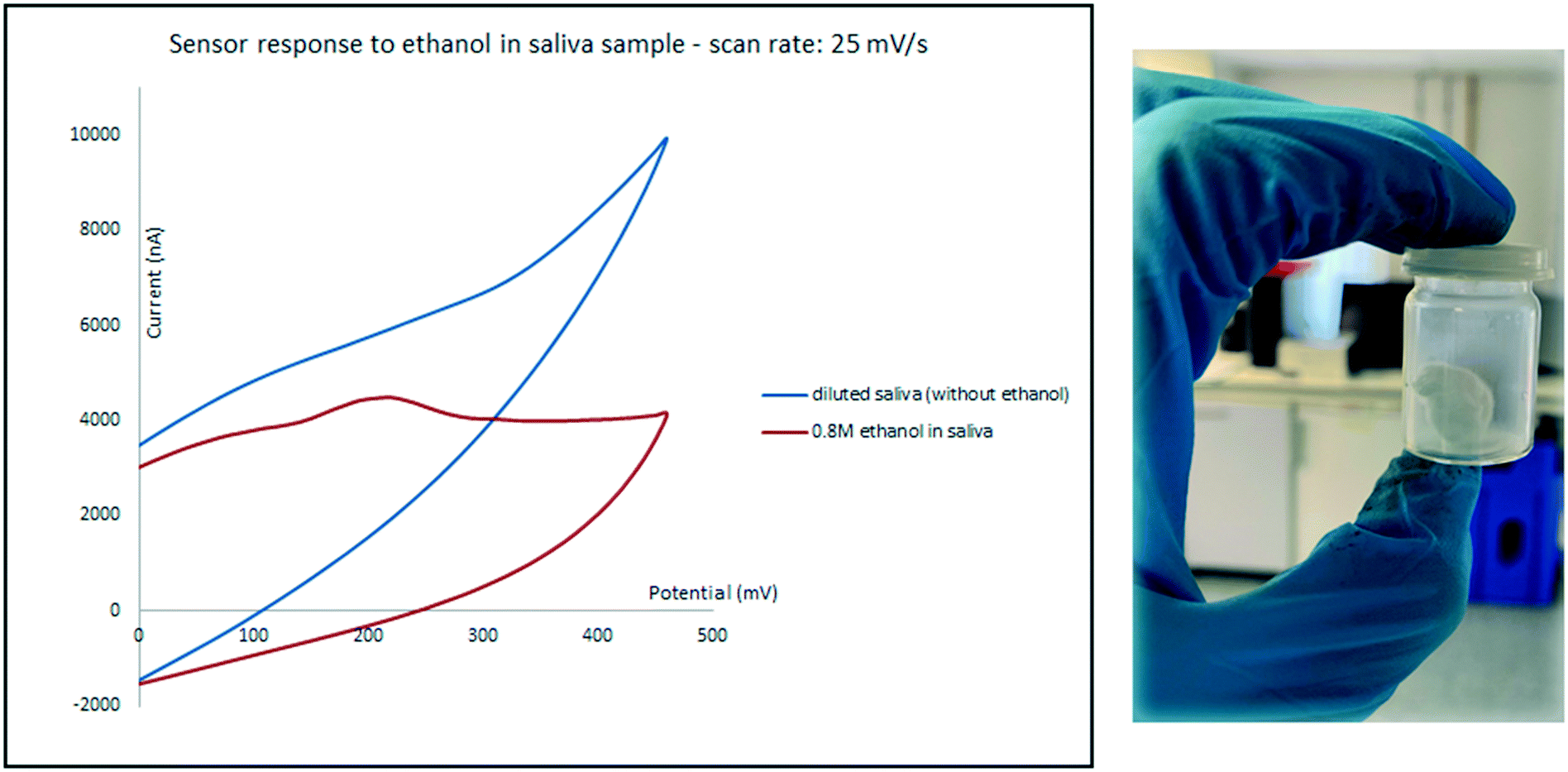 | ||
| Fig. 8 Sensor response to the saliva sample in the presence and absence of ethanol. | ||
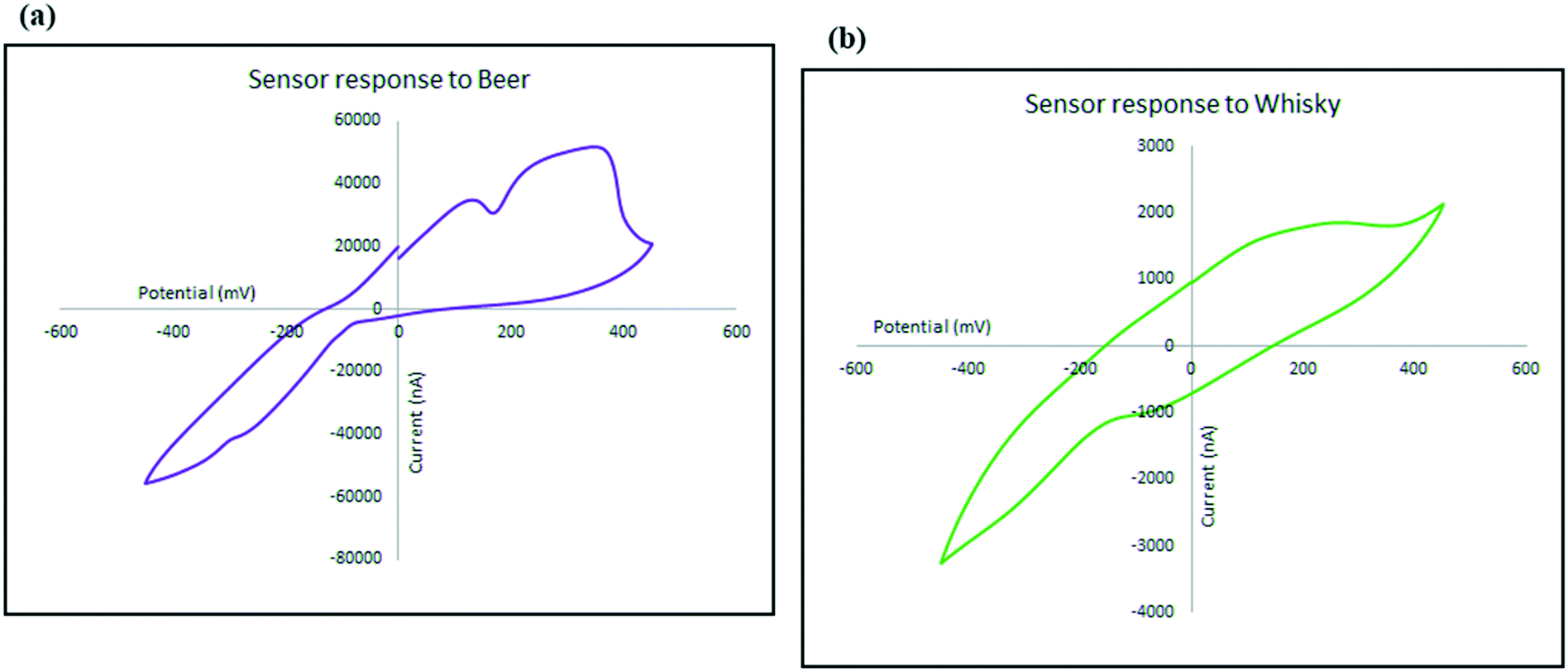 | ||
| Fig. 9 Sensor response to alcoholic drinks, (a) beer and (b) whisky; scan rate was 5 mV s−1. | ||
To investigate the kinetics of the process and the speed of the electrode response, the changes in the intensity (current) and location (potential) of the ethanol oxidation peak has been monitored on the beer sample as explained in Fig. 10. Not only does the oxidation peak potentials slightly shift as the scan rate increases, but also the intensity of the electrical current response of the sensor also increased. The diffusion coefficient describes diffusional transport which can be calculated from voltammetry studies based on theoretical relations such as Randles–Sevcik equation.71–73 The diffusion coefficient for the sensor in the presence of the real sample (beer) was calculated using Randles–Sevcik equation and found to be approximately 4 × 10−4 cm2 s−1 which represents the slow diffusion of sample on the electrode in beer sample which could have been caused by the effect of its complex matrix and the presence of carbon dioxide bubbles74 which may have covered the surface of the electrode.
| Ip = 2.69 × 105AD1/2n3/2y1/2C | (1) |
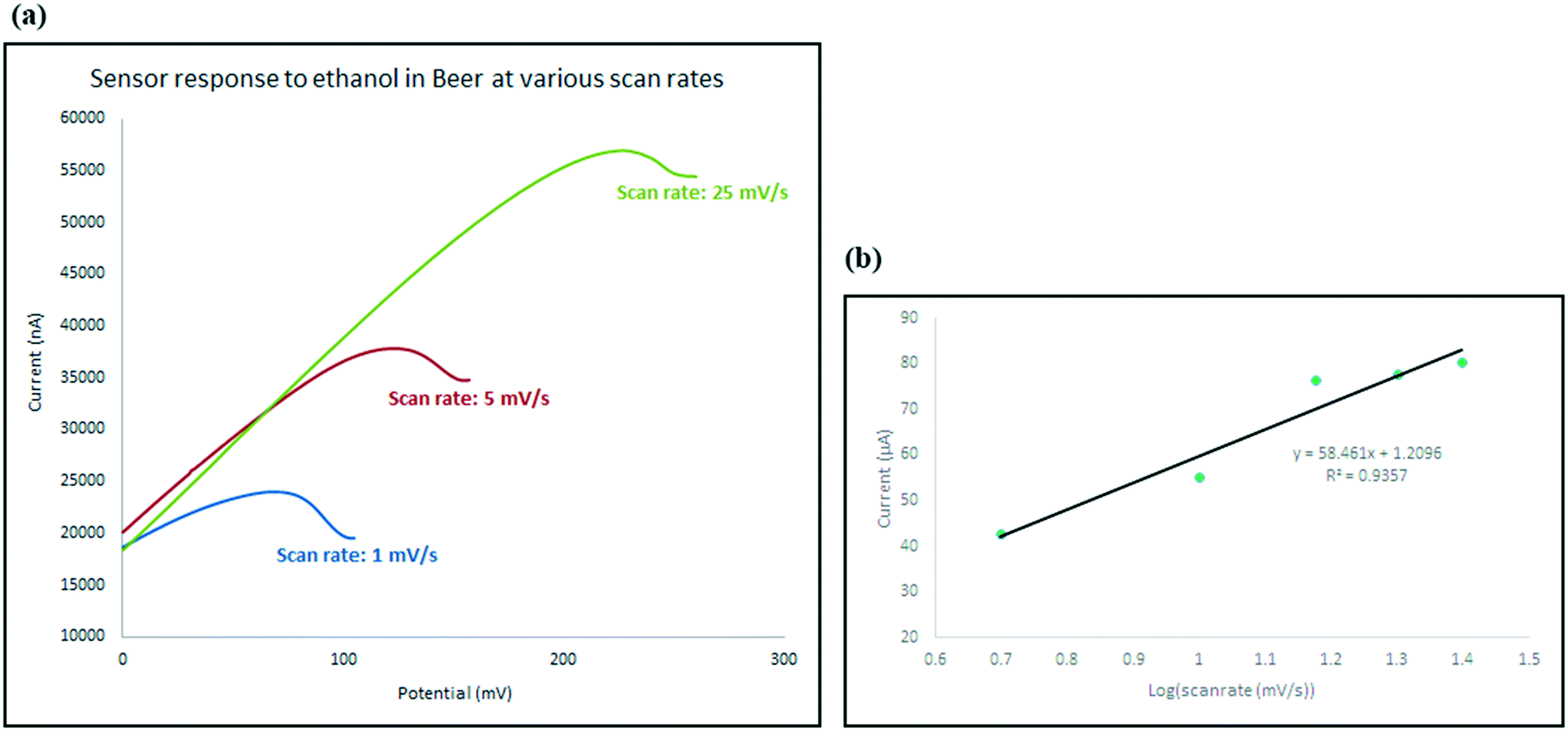 | ||
| Fig. 10 (a) Oxidation peaks obtained from cyclic voltammograms recorded in the presence of beer at scan rates of 1, 5 and 25 mV s−1 at room temperature. (b) Variation of current vs. log (scan rate) at scan rates of 5, 10, 15, 20 and 25 mV s−1. | ||
Conclusion
Indium–ethylenediamine was successfully anchored on rGO to modify the surface of graphene and be used as an electrochemical sensor. Functionalised graphene was then printed on a thin copper substrate to be studied by cyclic voltammetry. The rGO–In–en sensor showed significant selectivity towards ethanol compared to a range of alcohols in aqueous media. Increasing the concentration of the ethanol caused a linear increase in the current response of the electrode in a wide range between 10−4 M and 3 M. The analytical performance of the sensor was also proven by testing biological samples; it was able to detect ethanol concentrations up to 10 times below the UK legal driving limit in urine, with a reliable and reproduceable response. The sensor also showed a good response to industrial authentic samples including beer and whisky. Furthermore, the selectivity and high sensitivity of our electrochemical sensor suggests that while researchers previously have developed and used graphene–metal sensors for measuring small molecules such as ethanol only in the form of volatile gas, our work shows such sensors have potential to be tuned and applied to detect such analytes in aqueous forms as well. There is wide demand for such sensors in a variety of fields, from forensic investigations to pharmaceutical and food industries.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Surescreen Diagnostics for funding this work (Ref: 10757).References
- R. Boroujerdi, A. Abdelkader and R. Paul, Nano-Micro Lett., 2020, 12, 33 CrossRef CAS PubMed.
- R. Boroujerdi and R. Paul, Chemosensors, 2022, 10, 42 CrossRef CAS.
- O. H. Drummer, Clin. Biochem. Rev., 2006, 27, 147 Search PubMed.
- A. W. Jones, Forensic Sci. Int., 2010, 200, 1–20 CrossRef CAS PubMed.
- K. Sutter, CHIMIA International Journal for Chemistry, 2002, 56, 59–62 CrossRef CAS.
- G. Aydoğdu Tığ, Talanta, 2017, 175, 382–389 CrossRef PubMed.
- T. Kuila, S. Bose, P. Khanra, A. K. Mishra, N. H. Kim and J. H. Lee, Biosens. Bioelectron., 2011, 26, 4637–4648 CrossRef CAS PubMed.
- X. Xu, J. Zhou, Y. Xin, G. Lubineau, Q. Ma and L. Jiang, Sci. Rep., 2017, 7(1), 1–10 CrossRef PubMed.
- M. Zhou and S. Dong, Acc. Chem. Res., 2011, 44, 1232–1243 CrossRef CAS PubMed.
- S. Calabrese Barton, J. Gallaway and P. J. C. R. Atanassov, Chem. Rev., 2004, 104, 4867–4886 CrossRef PubMed.
- J. A. Cracknell, K. A. Vincent and F. A. Armstrong, Chem. Rev., 2008, 108, 2439–2461 CrossRef CAS PubMed.
- Y. Shao, J. Wang, H. Wu, J. Liu, I. A. Aksay and Y. Lin, Electroanalysis, 2010, 22, 1027–1036 CrossRef CAS.
- S. Pramanik, Y. Kumar, D. Gupta, V. K. Vashistha, A. Kumar, P. Karmakar and D. K. Das, Mater. Sci. Eng., B, 2021, 272, 115356 CrossRef CAS.
- R. Huang, V. Fung, Z. Wu and D.-e. Jiang, Catal. Today, 2020, 350, 19–24 CrossRef CAS.
- P. K. Shihabudeen and A. Roy Chaudhuri, J. Mater. Res., 2021, 1–12 Search PubMed.
- R. T. Rasheed, S. D. Al-Algawi and S. Z. Tariq, Iraqi Journal of Applied Physics, 2014, 10(4), 15–19 Search PubMed.
- D. N. Pham, M. Roy, J. A. Golen and D. R. Manke, Acta Crystallogr., Sect. C: Struct. Chem., 2017, 73, 442–446 CrossRef CAS PubMed.
- W. S. Hummers, Jr. and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef.
- J. Chen, B. Yao, C. Li and G. Shi, Carbon, 2013, 64, 225–229 CrossRef CAS.
- Z. Fan, K. Wang, T. Wei, J. Yan, L. Song and B. Shao, Carbon, 2010, 48, 1686–1689 CrossRef CAS.
- C. Margariti, Heritage Sci., 2019, 7, 63 CrossRef.
- D. Lin-Vien, N. B. Colthup, W. G. Fateley and J. G. Grasselli, The handbook of infrared and Raman characteristic frequencies of organic molecules, Elsevier, 1991 Search PubMed.
- A. M. Abdelkader, J. Mater. Chem. A, 2015, 3, 8519–8525 RSC.
- T. F. Emiru and D. W. Ayele, Egypt. J. Basic Appl. Sci., 2017, 4, 74–79 Search PubMed.
- P. Vecera, J. C. Chacón-Torres, T. Pichler, S. Reich, H. R. Soni, A. Görling, K. Edelthalhammer, H. Peterlik, F. Hauke and A. Hirsch, Nat. Commun., 2017, 8, 15192 CrossRef CAS PubMed.
- T. M. Barnard and N. E. Leadbeater, Chem. Commun., 2006, 3615–3616 RSC.
- E. Roedern, R.-S. Kühnel, A. Remhof and C. Battaglia, Sci. Rep., 2017, 7, 46189 CrossRef CAS PubMed.
- Y. Suffren, F. G. Rollet and C. Reber, Comments Inorg. Chem., 2011, 32, 246–276 CrossRef CAS.
- K. Krishnan and R. A. Plane, Inorg. Chem., 1966, 5, 852–857 CrossRef CAS.
- E. Alarcón-Lladó, T. Brazzini and J. W. Ager, J. Phys. D: Appl. Phys., 2016, 49, 255102 CrossRef.
- K. Yadav, B. R. Mehta and J. P. Singh, J. Mater. Chem. C, 2014, 2, 6362–6369 RSC.
- D. Liu, W. W. Lei, B. Zou, S. D. Yu, J. Hao, K. Wang, B. B. Liu, Q. L. Cui and G. T. Zou, J. Appl. Phys., 2008, 104, 083506 CrossRef.
- R. Zallen, The physics of amorphous solids, John Wiley & Sons, 2008 Search PubMed.
- S. Roscher, R. Hoffmann and O. J. A. M. Ambacher, Anal. Methods, 2019, 11, 1224–1228 RSC.
- D. López-Díaz, M. Lopez Holgado, J. L. García-Fierro and M. M. Velázquez, J. Phys. Chem. C, 2017, 121, 20489–20497 CrossRef.
- A. M. Abdelkader, C. Vallés, A. J. Cooper, I. A. Kinloch and R. A. W. Dryfe, ACS Nano, 2014, 8, 11225–11233 CrossRef CAS PubMed.
- L. M. Malard, M. A. Pimenta, G. Dresselhaus and M. S. Dresselhaus, Phys. Rep., 2009, 473, 51–87 CrossRef CAS.
- O. Frank, M. Mohr, J. Maultzsch, C. Thomsen, I. Riaz, R. Jalil, K. S. Novoselov, G. Tsoukleri, J. Parthenios and K. J. A. N. Papagelis, ACS Nano, 2011, 5, 2231–2239 CrossRef CAS PubMed.
- G. Tsoukleri, J. Parthenios, C. Galiotis and K. J. D. M. Papagelis, 2D Mater., 2015, 2, 024009 CrossRef.
- T. M. G. Mohiuddin, A. Lombardo, R. R. Nair, A. Bonetti, G. Savini, R. Jalil, N. Bonini, D. M. Basko, C. Galiotis, N. Marzari and K. S. Novoselov, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 205433 CrossRef.
- A. Jorio, International Scholarly Research Notices, 2012, vol. 2012 Search PubMed.
- X. Zhang, L. Hou, F. Richard and P. Samorì, Chem. – Eur. J., 2018, 24(69), 18518–18528 CrossRef CAS PubMed.
- L. Tang, R. Ji, X. Li, G. Bai, C. P. Liu, J. Hao, J. Lin, H. Jiang, K. S. Teng and Z. J. A. N. Yang, ACS Nano, 2014, 8, 6312–6320 CrossRef CAS PubMed.
- J. Wu, P. Wang, F. Wang and Y. J. N. Fang, Nanomaterials, 2018, 8, 864 CrossRef PubMed.
- J. U. Lee, W. Lee, J. W. Yi, S. S. Yoon, S. B. Lee, B. M. Jung, B. S. Kim and J. H. Byun, J. Mater. Chem. A, 2013, 1, 12893–12899 RSC.
- B. Zheng, Y. Chen, P. Li, Z. Wang, B. Cao, F. Qi, J. Liu, Z. Qiu and W. J. N. Zhang, Nanophotonics, 2017, 6, 259–267 CAS.
- F. T. Johra, J.-W. Lee and W.-G. Jung, J. Ind. Eng. Chem., 2014, 20, 2883–2887 CrossRef CAS.
- E. Riedo, F. Comin, J. Chevrier, F. Schmithusen, S. Decossas and M. Sancrotti, Surf. Coat. Technol., 2000, 125, 124–128 CrossRef CAS.
- X. Yan, T. Xu, G. Chen, S. Yang, H. Liu and Q. Xue, J. Phys. D: Appl. Phys., 2004, 37, 907 CrossRef CAS.
- Z. Gao, H. Yang, X. Fu, Q. Jin, Q. Wu, L. Kang and J. Wu, Environ. Sci. Pollut. Res., 2020, 27, 17446–17457 CrossRef CAS PubMed.
- M. Liu, W. Li, J. Rong and C. Zhou, Colloid Polym. Sci., 2012, 290, 895–905 CrossRef CAS.
- I. Y. Jeon, S. W. Kim, S. H. Shin, S. M. Jung and J. B. Baek, Mater. Today Adv., 2020, 6, 100030 CrossRef.
- P. Deminskyi, P. Rouf, I. G. Ivanov and H. Pedersen, J. Vac. Sci. Technol., A, 2019, 37, 020926 CrossRef.
- M. Wang, H.-Z. Chen, J.-L. Shen and S. L. Yang, Sci. China, Ser. A: Math., Phys., Astron., 1994, 37, 497–503 CAS.
- M. Zhao, Y. Cao, X. Liu, J. Deng, D. Li and H. Gu, Nanoscale Res. Lett., 2014, 9, 1–9 CrossRef PubMed.
- Z. Huang, H. Chen, L. Zhao, W. Fang, X. He, W. Li and P. Tian, Environ. Int., 2019, 126, 289–297 CrossRef CAS PubMed.
- H. J. Kim, I.-S. Bae, S.-J. Cho, J.-H. Boo, B.-C. Lee, J. Heo, I. Chung and B. Hong, Nanoscale Res. Lett., 2012, 7, 1–7 CrossRef PubMed.
- K. S. A. Butcher, A. J. Fernandes, P. T. Chen, M. Wintrebert-Fouquet, H. Timmers, S. K. Shrestha, H. Hirshy, R. M. Perks and B. F. Usher, J. Appl. Phys., 2007, 101, 123702 CrossRef.
- A. M. Abdelkader and D. J. Fray, Nanoscale, 2017, 9, 14548–14557 RSC.
- I. Menapace, W. Yiming and E. Masad, Fuel, 2017, 202, 366–379 CrossRef CAS.
- N. H. Kim, T. Kuila and J. H. Lee, J. Mater. Chem. A, 2013, 1, 1349–1358 RSC.
- G. J. Thangamani, K. Deshmukh, K. Chidambaram, M. B. Ahamed, K. K. Sadasivuni, D. Ponnamma, M. Faisal, N. A. Nambiraj and S. K. K. Pasha, J. Mater. Sci.: Mater. Electron., 2018, 29, 5186–5205 CrossRef CAS.
- C. Zhang, Z. L. Hou, B. X. Zhang, H. M. Fang and S. Bi, Carbon, 2018, 137, 467–474 CrossRef CAS.
- C. Zheng, C. Zhang, K. Zhang, J. Zhang, L. Jin, A. M. Asiri, K. A. Alamry, L. He and X. Chu, Sens. Actuators, B, 2021, 330, 129280 CrossRef CAS.
- K. J. Aoki, J. Chen, Y. Liu and B. Jia, J. Electroanal. Chem., 2020, 856, 113609 CrossRef CAS.
- A. W. Jones, Toxicol. Rev., 2006, 25, 15–35 CrossRef CAS PubMed.
- A. W. Jones and P. Holmgren, Forensic Sci. Int., 2003, 135, 206–212 CrossRef CAS PubMed.
- A. W. Jones and P. Holmgren, J. Clin. Pathol., 2001, 54, 699–702 CrossRef CAS PubMed.
- R. Paul, L. Tsanaclis, C. Murray, R. Boroujerdi, L. Facer and A. J. A. Corbin, Alcohol Alcohol., 2019, 54(4), 402–407 CrossRef CAS PubMed.
- J. J. Saady, A. Poklis and H. P. Dalton, J. Forensic Sci., 1993, 38, 1467–1471 CAS.
- M. Bogdan, D. Brugger, W. Rosenstiel and B. Speiser, J. Cheminf., 2014, 6, 1–13 Search PubMed.
- K. Ahmad, P. Kumar and S. M. Mobin, Nanoscale Adv., 2020, 2, 502–511 RSC.
- D. M. MacArthur, J. Electrochem. Soc., 1970, 117, 729 CrossRef.
- G. Liger-Belair and C. Cilindre, ACS Omega, 2021, 6, 9672–9679 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |