
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile, generic capture and on-fiber differentiation of exosomes via confocal immunofluorescence microscopy using a capillary-channeled polymer fiber solid-phase extraction tip†
Kaylan K.
Jackson
 a,
Rhonda R.
Powell
b,
Terri F.
Bruce
c and
R. Kenneth
Marcus
*a
a,
Rhonda R.
Powell
b,
Terri F.
Bruce
c and
R. Kenneth
Marcus
*a
aDepartment of Chemistry, Clemson University, Clemson, SC 29634, USA. E-mail: marcusr@clemson.edu
bClemson Light Imaging Facility, Clemson University, Clemson, SC 29634, USA
cDepartment of Bioengineering, Clemson University, Clemson, SC 29634, USA
First published on 28th March 2022
Abstract
There is great interest in advancing methodologies for the isolation and characterization of exosomes (30–150 nm, extracellular vesicles (EVs)) for fundamental biochemical research and liquid biopsy applications. This is due to the accessibility of exosomal surface biomarkers, providing relevant biochemical information from their cells of origin. Exosome-based techniques hold potential for diagnostic applications through less invasive sampling (versus the physical extraction methods of pathology). This study demonstrates a simple spin-down tip methodology for generic exosome capture, followed by immunoaffinity-based fluorescent labeling to classify EVs captured on a polyester capillary-channeled polymer (C-CP) fiber stationary phase. An antibody to the generic EV tetraspanin protein (CD81) is employed to confirm the presence of biologically active EVs on the fiber surface. An antibody to the CA125 protein, upregulated in the case of ovarian cell stress, is included as a cancer marker protein. Scanning electron microscopy and confocal fluorescence microscopy were performed directly on the capture fibers to visualize the morphology and assess the bioactivity/identity of captured vesicles. This report provides a proof-of-concept for an efficient means of isolating, purifying, immunolabeling, and fluorescent imaging for the biomarker assessment of extracellular vesicles on a single platform. Herein lies the novelty of the overall approach. The ability to affect the entire isolation, immunolabeling, and imaging process in <5 hours is demonstrated. The C-CP fiber spin-down tip is an efficient exosome isolation methodology for microliter samples from diverse media (human urine and cell culture media here) towards diverse means of characterization and identification.
1. Introduction
The concept of the liquid biopsy, wherein a readily accessible biofluid is used to harvest relevant biomarkers versus the excision of tissue specimens, is moving towards realization.1,2 Crucial treatment time is often lost in early instances of infection, tumor growth, and disease progression simply because of the lack of efficient, early detection, diagnostic tools. Particularly in the case of aggressive cancers like ovarian cancer (OC),3,4 much time is lost due to the inability to identify malignant ailments non-invasively, as many cancers are asymptomatic during early stages of disease.1,5 The introduction of accessible, non-invasive methods for disease detection would open the door for a head-on approach to clinical diagnostics. Taken a step further, this would allow routine screenings for diseases such as OC to become commonplace. This would drastically increase patient survival rates by identifying cancerous instances prior to the presentation of noticeable symptoms.Exosomes are 30–150 nm-sized, cell-derived extracellular vesicles (EVs) released from most cell types by multivesicular bodies (MVBs) uniquely created through the endosomal pathway,6,7 allowing them to consist of origin-representative genetic and molecular cargoes. Exosomes are essential elements in cell-to-cell communication, disease progression, cancer metastasis, tumor growth, and other physiological processes.6,7 Because exosomes contain molecular constituents of their host cells, including DNA, microRNA, and biomarker surface proteins, they are a treasure trove of biomarkers. Likewise, as they are present in many bodily fluids, including urine, blood serum and plasma, saliva, and in cell culture media, they can be harvested to assess the status of the biosystem. Importantly, mother cell-derived exosome surface proteins allow for the identification of the originating environment without direct contact to the primary area of concern (i.e., tumor or infection site).8,9 Profiling of genetic material from the interior of disease-derived exosomes allows for monitoring of disease progression and perhaps treatment effectiveness. To that end, liquid biopsies based on the biomarker analysis of exosome cargoes from readily available biofluids have been investigated.2,10,11
An issue limiting exosome-based biochemical research and their implementation in diagnostics is the lack of efficient tools to isolate the vesicles. Exosome isolations have proven to be a challenge due to their relative size, protein affinity, and innate heterogeneity in size and surface protein makeup.1 Though many techniques are widely used, i.e., ultracentrifugation (UC), density gradient centrifugation, size exclusion chromatography, microfiltration, affinity isolation, and polymer precipitation,12,13 all available isolation methods present concerns regarding purity and recovery efficiency. Because these techniques rely on the density, size, or affinity for antibodies to specific proteins, purification of exosomes from free protein or lipoprotein aggregates is particularly challenging.4,14 Many of these isolation techniques require expensive, time-consuming, and tedious processes, and still produce impure, low yield recoveries, limiting the ultimate progress of the study and use of exosomes. Another limiting factor in the clinical arena is the required primary sample volume, often a few milliliters of the original biofluid. To reduce these sample volume requirements, several microfluidic approaches for EV isolation have been recently explored, using immunoaffinity capture,15 nanoporous membrane sieving,16 and physical EV trapping17 (via nanowires/micropillars) approaches. Despite reductions in applied sample/reagent volume, concerns of purity and yield remain, especially since pre-concentration steps using the common EV isolation methods (e.g., UC) are still required.18
In the realm of EV biochemical assessment, few methods are available for simultaneous imaging and EV biomarker characterization; all fall short in providing both on the same platform. The ExoView R100 (NanoView Biosciences, Boston, MA, USA) is a recently-introduced EV analysis method, consisting of an immunoaffinity capture array chip and interferometry imaging module – designed to assess EV size and surface biomarker content. In theory, this technique would allow for simultaneous insight into the size and tetraspanin compositions of an EV population. However, with the relatively high limits of detection (3.94 × 109 particles per mL) and low specificity of the immunoaffinity capture chip,19 EV pre-purification steps are required. Hence, the challenges faced by impure EV recoveries obtained using the above-mentioned isolation methods remain. Ultimately, while small volumes of material are subjected to these sorts of assays, the volume of the primary sample is not reduced. The introduction of an isolation method to provide highly concentrated, biologically-active, and pure EV populations fit for immunological assessment methods, from minute (μL) initial sample volumes, is of much interest. The future of exosome-based clinical and diagnostic applications relies on the implementation of timely, yet efficient exosome isolation and screening methods, able to be tailored to a diversity of relevant biomarkers.
Demonstrated here is a method for rapid EV isolations using a hydrophobic interaction chromatography (HIC) immobilization/purification process and the subsequent immunofluorescence (IF) labeling of exosomes based on their surface marker proteins; on a singular platform. This is done using a capillary-channeled polymer (C-CP) fiber stationary phase and a solid phase extraction (SPE) spin-down tip process. The generic capture of EVs is driven by the HIC solvent system, wherein ionic species and proteins are sequentially removed from the host matrix, allowing exosome isolations from diverse biofluids and culture media.20–22 The capture and sequential release of free proteins and EVs are induced by a solvent change from high (2 M (NH4)2SO4) – to – zero salt content in either continuous or step gradient workflows. The method's efficiency has been demonstrated in protein and exosome separations on C-CP fiber columns via HPLC20–22 and by SPE on spin-down tips.23,24 Overall, the process has proven to provide high EV yields (up to 1 × 1012 EVs per mL) and extremely low concomitant protein content (<0.5% lipoprotein carryover in serum),25,26 on time scales of less than 10 min, without any other form of pre-isolation. While no direct comparisons have been made, the SPE tips have yielded total exosome binding capacities of ∼8 × 1011 EVs from commercial standards,23 while the 30 cm columns more fitting for downstream processing have shown capacities ~5 × 1012 for the far more complex, direct cell milieu purification.22
In this demonstration, EVs from different sources are captured and retained on the fiber surfaces, contaminating host cell proteins (HCPs) are eluted, and the exosome isolates are subjected to immunolabeling and on-fiber identification via confocal fluorescence microscopy. Specifically, exosomes are visualized by fluorescent antibody tagging of the ubiquitous CD81 tetraspanin surface protein and the CA125 protein, a biomarker previously identified as being common to OC.27,28 Other tetraspanins such as CD9 and CD63 have been identified in isolated EVs; however, their contents are highly variable. CD81 is of most prevalence and interest in the populations under study here, as CD81 is also reported to be overexpressed in cancerous EV populations.29,30 EVs originating from known cancer cell lines and patients are readily distinguished from those where OC (or other gynecologic ailments presenting CA125) are not anticipated. In the first demonstration, fiber-immobilized exosomes are processed using standard IF incubation protocols prior to imaging analysis. In this instance, the primary benefits lie in the ability to remove potentially-interfering HCPs and performance of in situ imaging, albeit on much shorter time scales than other methods. Confocal microscopy provides for IF screening on the single-vesicle level. In the subsequent demonstration, a more rapid IF processing concept is broached, wherein EVs are efficiently captured, immunolabeled, and imaged in under 5 hours (a 3× reduction in processing time). The novelty of this work lies in the ability to isolate, purify, immunolabel, and image EVs on a single substrate. While confocal microscopy may not be practical for clinical screening, the rapid, high purity isolation, followed by IF analysis on a single platform, points to significant advances in exosome processing, regardless of the subsequent mode of characterization, biomarker identification, and quantification.
2. Experimental
2.1 Chemicals and reagents
In order to demonstrate the concept of exosome capture and differentiation via immunofluorescence labeling and imaging techniques, EVs were isolated from distinct sources. One example of a generic, non-cancerous source was a cell culture of Dictyostelium discoideum (D. discoideum, a soil-dwelling amoeba), AX2 cells, which were obtained from The Dicty Stock Center (Northwestern University, Chicago, IL, USA). The D. discoideum cells were grown and maintained axenically in HL5 medium supplemented with 100 μg mL−1 ampicillin at room temperature in 25 mL culture flasks. Likewise, HEK293 (human embryonic kidney) cells obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA) were cultured in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% exosome-depleted fetal bovine serum (FBS), and 1% penicillin/streptomycin in a standard humidified incubator at 37 °C with 5% CO2. As an example of an exosome sample derived from a representative human biofluid source, a commercial exosome stock was obtained (HansaBioMed, Tallinn, Estonia), consisting of a urine-derived exosome isolate from supposedly healthy subjects was employed. The example sources that should definitively present biomarkers for OC were the IHOE (immortalized human ovarian epithelial), SKOV-3 (human ovarian adenocarcinoma), and CaOv-3 (human ovarian adenocarcinoma) cell lines. The IHOE cell line was obtained from Applied Biological Materials (Richmond, BC, Canada) and the SKOV-3 and CaOv-3 cell lines were obtained from the ATCC (Manassas, VA, USA). The IHOE and CaOv-3 cells were grown in DMEM, and the SKOV3 cells were grown in McCoy's 5a media. All of the aforementioned ovarian cell lines were supplemented with 10% exosome-depleted FBS and 1% penicillin/streptomycin and cultured in a standard humidified incubator at 37 °C with 5% CO2. Finally, the practical (clinical) efficacy of the method was demonstrated in the extraction and immunofluorescent assay of exosomes derived from a urine sample obtained from an unidentified patient currently under treatment for OC.Ultra-pure ammonium sulfate and biotechnology-grade glycerol were purchased from VWR (Radnor, PA, USA). Bovine serum albumin (BSA) was purchased from Sigma-Aldrich (St. Louis, MO USA). Phosphate-buffered saline (PBS, 0.067 M (PO4), pH 7.4) was obtained from ThermoFisher Scientific (Waltham, MA, USA). The VWR® Symphony™ 4417/R table-top centrifuge (Radnor, PA, USA) was used for SPE spin-down tip processing. A mouse monoclonal antibody to CD81 (1 mg mL−1) was obtained from Santa Cruz Biotechnology (Dallas, TX, USA). A rabbit monoclonal antibody to CA125/MUC16 (1 mg mL−1) was obtained from ThermoFisher (Waltham, MA, USA). A goat anti-rabbit IgG (H + L) cross-adsorbed secondary antibody (Alexa Fluor 488, 2 mg mL−1) and a goat anti-mouse IgG (H + L) cross-adsorbed secondary antibody (Alexa Fluor 647, 2 mg mL−1) were obtained from ThermoFisher Scientific (Waltham, MA).
2.2 C-CP fiber spin-down tip SPE procedure
The exosome isolation and on-fiber immunofluorescence labeling steps are depicted diagrammatically in Fig. 1. The isolation method, to the point of presenting purified EVs on the fiber surface, was executed as described previously,23,24 with the initial labeling and imaging procedures following what is normal in the fluorescence immunoimaging field.31 Following the basic demonstration of the methodology, efforts towards more rapid on-fiber processing were initiated.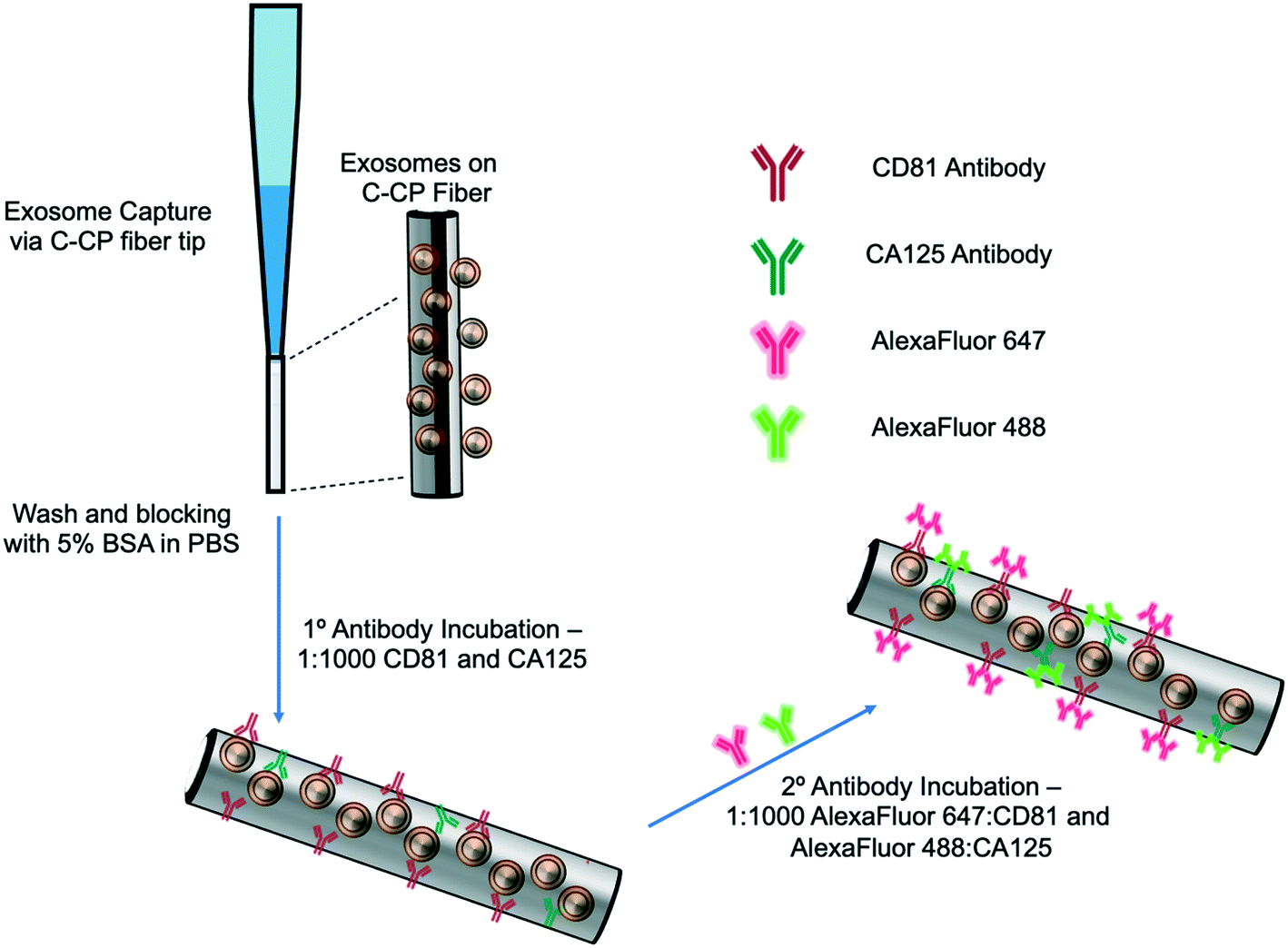 | ||
| Fig. 1 Graphic depiction of the on-fiber exosome extraction and immunolabeling process. | ||
The polyester (PET) SPE tips were prepared as previously published23 using an eight-rotation loop of PET fibers (a total of 448 fibers) collinearly pulled through 30 cm of 0.8 mm ID fluorinated ethylene polypropylene (FEP) polymer tubing (Cole Parmer, Vernon Hills, IL, USA). The fiber bundle hanging from the end of the 30 cm piece of FEP tubing was pulled and repeatedly cut to create 1 cm fiber-packed tips with a 0.5 cm open space for attachment to a 200 μL micropipette tip (Fisherbrand, Waltham, MA). During spin-down processing, the fiber stationary phase was attached to a 200 μL micropipette tip using super glue and placed inside a 1000 μL micropipette tip (Fisherbrand, Waltham, MA) to provide structural support. The spin-down tip set-up was held in place within a 1.5 mL microfuge tube cut laterally, then placed inside of a hollowed center cap of a 15 mL centrifuge tube. The C-CP fiber tips were conditioned by washing with water, acetonitrile, and the starting mobile phase solution before application.
2.3 Exosome SPE immobilization
The isolation of EVs from the test matrices was accomplished by first mixing 100 μL of the sample supernatants with 100 μL of 4 M ammonium sulfate to achieve a 2 M final concentration before being applied to the tip. The total volume (200 μL) of the sample loading mixture was applied to the PET C-CP fiber SPE tip and centrifuged at 300 × g for 60 s. Under these conditions, salts and low molecular weight polar species pass through the tip, while proteinaceous materials and vesicles were retained on the fiber surface. The elution of free proteins and protein aggregates was induced using a mobile phase of 1 M (NH4)2SO4 in 25% glycerol (200 μL, 300 × g, 60 s). Before beginning the immunolabeling process, the fiber surface was further washed five times with 200 μL aliquots of PBS (300 × g, 60 s each, 15 min incubation, 3 buffer changes). For the STEM imaging, the captured exosomes were fixed and imaged directly on the fiber surface after the protein elution step and after release from the fiber surface using 50% glycerol (200 μL, 300 × g, 60 s) upon fixation to a silica wafer, as described in ESI.†2.4 Immunolabeling and imaging
The initial demonstration of the exosome differentiation via immunofluorescence microscopy employed what would be termed a standard labeling protocol.31 In brief, after the sample HIC capture step, the fiber-captured vesicles were washed with PBS and incubated with 5% BSA before and after primary antibody incubation overnight, and incubation of the secondary antibody for 2 hours. That procedure, based on a limited amount of optimization of the antibody concentration and incubation times, is detailed in ESI.† An alternative, higher throughput method was investigated as a means of expediting the labeling process. After the tip capture process, the immobilized and washed exosomes were exposed to a 5% BSA blocking solution in PBS (5 times, 200 μL ea. 300 × g, 60 s ea.) and allowed to incubate in 1 mL of blocking solution for 15 min. to decrease potential non-specific binding between the antibodies to target the exosomal surface biomarkers as well as the PET C-CP fiber surface. Following the blocking step, the fiber surface was washed three times using 200 μl aliquots of PBS and allowed to wash in 1 mL of PBS on a shaker for 15 min (3 buffer changes). Next, antibodies (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000 in PBS) to the CD81 EV marker protein (mouse) and CA125 (rabbit) biomarker protein were applied to the fiber-captured vesicles (200 μL), allowed to wick down the fiber tip for 5 minutes, then placed in the centrifuge for a spin-down at 150 × g for 3 min before incubation in 1 mL of antibody (2 hours, RT). The blocking step was repeated (5 times, 200 μL, 300 × g, 1 min each, 15 min, RT), followed by another PBS wash (5 times, 50 μL, 300 × g, 1 min each, 15 min, RT) to reduce non-specific binding. After this blocking step, more centrifugal force was required to allow the solutions to pass through the fiber tip due to the stationary phase surface saturation. The primary antibody-labeled exosomes on the tip surface were then exposed to Alexa Fluor 488 (anti-rabbit) and Alexa Fluor 647 (anti-mouse) secondary antibodies (1:1000, 200 μL, 500 × g, 3 min) and allowed to incubate in 1 mL of antibody for 1 hour at room temperature. With the addition of the secondary antibody, the solution was allowed to wick down the fiber surface, like previously done with the addition of the primary antibody (200 μL, 5 min), then spun down using the table-top centrifuge (500 × g, 5 min), and allowed to incubate in 1 mL of solution (1 h, RT). Finally, a PBS wash (5 times, 200 μL, 1500 × g, 1 min ea, 15 min) was used to remove unbound secondary antibodies from the fiber tip before confocal microscopy imaging. This entire process is completed in less than 5 hours (with multiple samples processed in parallel) but is by no means rigorously optimized at this point. To be clear, the presented confocal fluorescence approach is not directed at direct antigen quantification. However, the presence of disease states are qualitatively assessed (screened) based on the comparison of the fluorescent images from “healthy” (HEK293, Dictyostelium discoideum) and CA125 protein-upregulating EV sources (SKOV3, CaOv-3). Among other limitations discussed subsequently, without the ability to apply specimen-specific reference material controls in parallel during this experimental workflow, a quantitative microscopic approach remains a challenge. That said, multiple previous efforts have demonstrated the ability to employ simple optical absorbance detection to quantify the total EV content from very diverse biological and culture media.20–24,26 Those methods have been validated through use of diverse quantification modes (e.g. direct response functions and standard addition) as well as the common standard, nanoparticle tracking analysis (NTA).
:1000 in PBS) to the CD81 EV marker protein (mouse) and CA125 (rabbit) biomarker protein were applied to the fiber-captured vesicles (200 μL), allowed to wick down the fiber tip for 5 minutes, then placed in the centrifuge for a spin-down at 150 × g for 3 min before incubation in 1 mL of antibody (2 hours, RT). The blocking step was repeated (5 times, 200 μL, 300 × g, 1 min each, 15 min, RT), followed by another PBS wash (5 times, 50 μL, 300 × g, 1 min each, 15 min, RT) to reduce non-specific binding. After this blocking step, more centrifugal force was required to allow the solutions to pass through the fiber tip due to the stationary phase surface saturation. The primary antibody-labeled exosomes on the tip surface were then exposed to Alexa Fluor 488 (anti-rabbit) and Alexa Fluor 647 (anti-mouse) secondary antibodies (1:1000, 200 μL, 500 × g, 3 min) and allowed to incubate in 1 mL of antibody for 1 hour at room temperature. With the addition of the secondary antibody, the solution was allowed to wick down the fiber surface, like previously done with the addition of the primary antibody (200 μL, 5 min), then spun down using the table-top centrifuge (500 × g, 5 min), and allowed to incubate in 1 mL of solution (1 h, RT). Finally, a PBS wash (5 times, 200 μL, 1500 × g, 1 min ea, 15 min) was used to remove unbound secondary antibodies from the fiber tip before confocal microscopy imaging. This entire process is completed in less than 5 hours (with multiple samples processed in parallel) but is by no means rigorously optimized at this point. To be clear, the presented confocal fluorescence approach is not directed at direct antigen quantification. However, the presence of disease states are qualitatively assessed (screened) based on the comparison of the fluorescent images from “healthy” (HEK293, Dictyostelium discoideum) and CA125 protein-upregulating EV sources (SKOV3, CaOv-3). Among other limitations discussed subsequently, without the ability to apply specimen-specific reference material controls in parallel during this experimental workflow, a quantitative microscopic approach remains a challenge. That said, multiple previous efforts have demonstrated the ability to employ simple optical absorbance detection to quantify the total EV content from very diverse biological and culture media.20–24,26 Those methods have been validated through use of diverse quantification modes (e.g. direct response functions and standard addition) as well as the common standard, nanoparticle tracking analysis (NTA).
To perform the confocal imaging, the PET C-CP fibers (with the immunolabeled exosomes on the surface) were removed from the FEP tubing and placed in one chamber of a 2-well Nunc Lab-Tek Chambered cover glass with a no. 1 borosilicate glass bottom (Thermo Fisher Scientific, Waltham, MA). Optimized excitation wavelengths of 499 and 653 nm were chosen using the Leica Dye Assistant and used to visualize the CA125 and CD81 antigens, respectively, during the confocal imaging using the Leica SP8 confocal microscope. All microscope settings, including the white light laser intensity and power, gain, offset, pinhole, and frame count, remained consistent for the confocal imaging. With this, the localized fluorescence was used to identify specific biomarker protein antigens on the captured EVs. It is important to note that due to the optical resolution limitations of the confocal microscope, it is possible that the vesicles observed as the CD81 and CA125-positive species captured on the C-CP tip may not be individual EVs, but instead, a collection of vesicles in very close proximity, creating the collective fluorescent immune response.
Clean C-CP fiber tips (without the exposure to the EV solution) were run in parallel during the two isolation and immunolabeling protocols to serve as relevant negative controls and are presented with the relevant diagnostic images, herein. Additionally, negative control imaging experiments were carried out using the standard antibody labeling protocol, inclusive of fluorescent imaging of blank fibers, purified CD81 protein exposed to the native fiber surfaces, and a CD81 protein-antibody complex exposed to exosome-immobilized fibers. The latter two situations illustrate the freedom from non-specific binding of the proteins (and subsequently their antibodies) to the native fiber surfaces as well those subjected to the exosome immobilization step. These essential results are included as ESI† Fig. S1a–c.
3. Results and discussion
3.1 Confirmation of exosome physical integrity
To confirm the capture and release of intact vesicles on the C-CP tip surface during the course of the HIC workflow, SEM imaging was performed directly on the surface of the fiber tips as well as on eluted particles. The electron microscopy techniques were employed to investigate the morphological nature and structural integrity of the exosomes captured on the C-CP tip surface. EVs were imaged after fixation, dehydration, and negative staining (as described in ESI†). The SEM images revealed the vesicles' intact morphology on the stationary phase and highlight the channeled structure of the C-CP fibers, affecting highly efficient EV binding. ESI† Fig. S2a presents an SEM micrograph of the commercial-sourced, human urine-derived exosomes bound to the fiber surface after the HIC workflow to the point of the protein wash using 25% glycerol and PBS. As seen, the vast majority of the EVs captured on the fiber surface fall under the 150 nm size cut-off for exosomes and display their expected spherical shape. The visually smooth fiber surface is reflective of the efficient removal of media components and host cell proteins, as needed for high fidelity imaging or chemical characterization. To observe the integrity of the EVs after they are fully processed, the complete spin-down tip method was performed, with the eluted vesicles deposited onto a silica wafer for STEM imaging. Just as an efficient capture of EVs is essential, the release of pure and biologically relevant populations of EVs is vital for further downstream processing, whether for further characterization or biological testing. The STEM micrograph presented in ESI† Fig. S2b provides a magnified view of the exosomes, wherein it is clear that the vesicular structures (as evidenced by the “rings” on the periphery) are indeed retained following complete C-CP fiber tip processing. Because the aim was to capture, retain, and characterize the EVs directly on the fiber tip surface, the vesicles were not subsequently eluted from the stationary phase as demonstrated in previous works.23,24 However, the ability to capture and release of cell-derived EVs (in the exosome size range) using this C-CP tip method was confirmed using nanoparticle tracking analysis (NTA). As shown in ESI† Fig. S3, the NTA size determinations revealed that >93% of the EVs recovered from 100 μL of HEK293 cell culture milieu, fell within the 30–150 nm size range (100.1 nm average diameter, 5.6 × 1010 particles per mL). This confirms the ability to capture and release highly concentrated, structurally-preserved exosomes using the C-CP tip method.3.2 Differentiation of exosome types using a standard immunolabeling protocol
The ability to differentiate exosome populations via on-fiber immunofluorescence imaging is demonstrated in Fig. 2. The fluorescence micrographs encompass the process negative control (blank) and the four EV sources, with the two distinct surface biomarkers probed in situ. It is important to point out that the scales of these images equate to only a couple of channels of a single fiber, making up a very small fraction of the full SPE tip. In this example, the tetraspanin CD81, which is fairly ubiquitous among all EV populations, is used as the benchmark exosome identifier and thus is a positive indication of the vesicles. The target for this analysis is the presence of the CA125 protein, which is one of the primary cancer antigens associated with OC.32,33 To be clear, CA125 is not solely related to OC, as it is also known to be associated with other cancers.34,35 The images presented in Fig. 2 provide confirmation of the presence/location of EVs based on the CD81 (red) responses and those which have CA125 (green) present on the vesicle surfaces (taken simultaneously), following the overnight immunolabeling protocol. The third column is an overlay of the respective responses, providing a correlative verification of the presence of these antigens in close proximity, presumably associated with a given exosomal particle.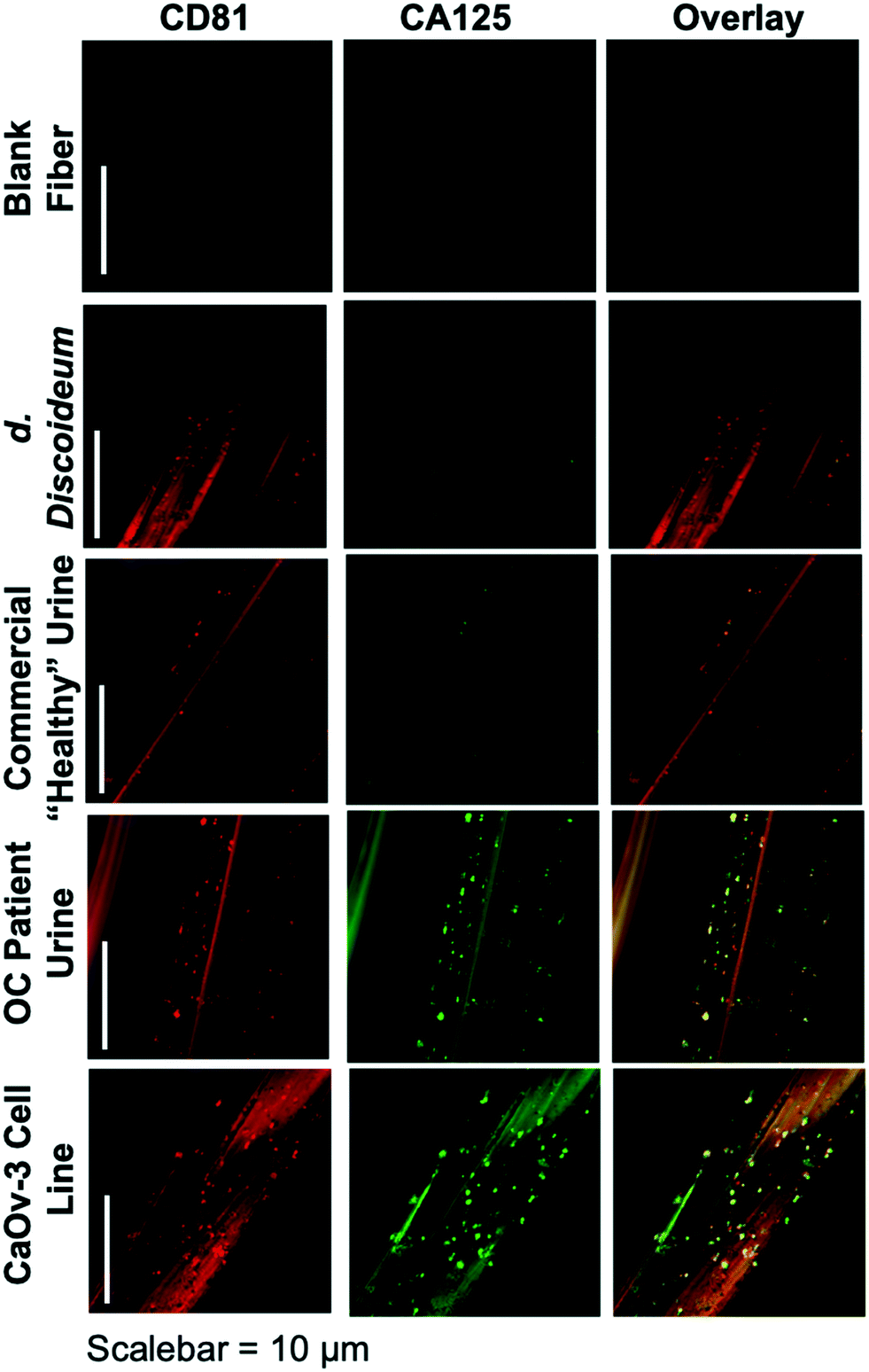 | ||
| Fig. 2 Confocal fluorescence images of C-CP fibers from process blank and test specimens following standard immunolabeling procedure. Red color depicts presence of the CD81 tetraspanin protein on exosome membranes, with green color representing the presence of CA125. | ||
Going through the exosome sources, the immunofluorescence method yields the anticipated results in each case, along with some unanticipated ones. In the case of the non-mammalian D. discoideum amoeba, the imaging only yields positive results towards the CD81 tetraspanin moiety. D. discoideum have previously been reported to contain active homologs to the CD81 EV tetraspanin protein,36 this is further confirmed here. The commercially-sourced sample derived from the urine of supposedly healthy individuals provides positive responses for CD81, but perhaps not anticipated is the fact that some positive responses for CA125 are seen as well. Indeed, the overlay reflects the coexistence of the two proteins on many particles. High levels of CA125 are often released in the body during states of ovarian-related ailments (i.e., endometriosis or ovarian cancers), but healthy women also express low levels of CA125.37 Thus, while CA125 is clearly present on the isolated exosomes, this is not a direct indication of OC because of the comparatively low fluorescent response. As would be expected, the isolates from the CaOv-3 cell line are replete with exosomes presenting both the CD81 and CA125 markers. Again the overlay of the fluorescent images verifies the presence of the same-source exosomes. Finally, the images of the exosomes isolated from the urine of the anonymous OC patient likewise show a high density of particles whose surfaces are populated with both CD81 and CA125. In this way, while CA125 is not solely attributed to OC, it is fairly convincing evidence that a CA125-upregulating source of malignancy (presumably cancer) is present. As a final comment, it is no coincidence that the isolated particle densities (as reflected in the CD81 responses) from the cancerous subject sources appear far higher than the corresponding non-cancerous sources; i.e., the CaOv-3 culture distributions appear far more dense than the D. discoiduem culture, and the patient urine yields far more than the “healthy-source” urine. Certainly, no direct quantitative conclusion can be made here, but it is well known that systems under the stress of disease exhibit far higher EV production and excretion rates.38,39 Ultimately, the method presented suggests a route to a facile, multi-biomarker EV screening and source differentiation.
It is important to note that, as an extension of the SEM images of ESI† Fig. S2, the presence of discrete sources of fluorescence on the EV size scale, provides evidence that the EVs remain intact through the course of the isolation and immunolabeling processes. Should the vesicles have ruptured in the processing, the corresponding fluorescence images would be very diffuse across the fiber surface. Additionally, one might also wish to extend the imaging methodology to the quantitative determination of CD81 and CA125, but that would come with many future challenges, including the detailed characterization of potential non-specific binding, autofluorescence of the fiber substrate, and indeed the ability to generate analytical EV blanks.
3.3 Differentiation of exosome type using a higher-throughput immunolabeling protocol
As presented, the method demonstrated above holds particular promise in terms of the use of minute (100 μL) sample volumes, providing high purity isolates on an inert substrate and ready imaging capabilities, and the possibility to recover exosomes for further characterization or biochemical study. The advantage of sample processing time versus other isolation methods is somewhat negated by the typical overnight antibody/labeling incubation times. As an initial demonstration of the potential to affect higher throughput in the immunolabeling step, Fig. 3 depicts the blank and product images obtained for various-sourced samples following the 5 hour immunolabeling procedure detailed above. In this instance, the commercial, human urine-derived exosomes and a HEK293 culture supernatant are used as presumably CA125-free samples, with the IHOE and SKOV3 cell lines used as likely CA125-positive sources.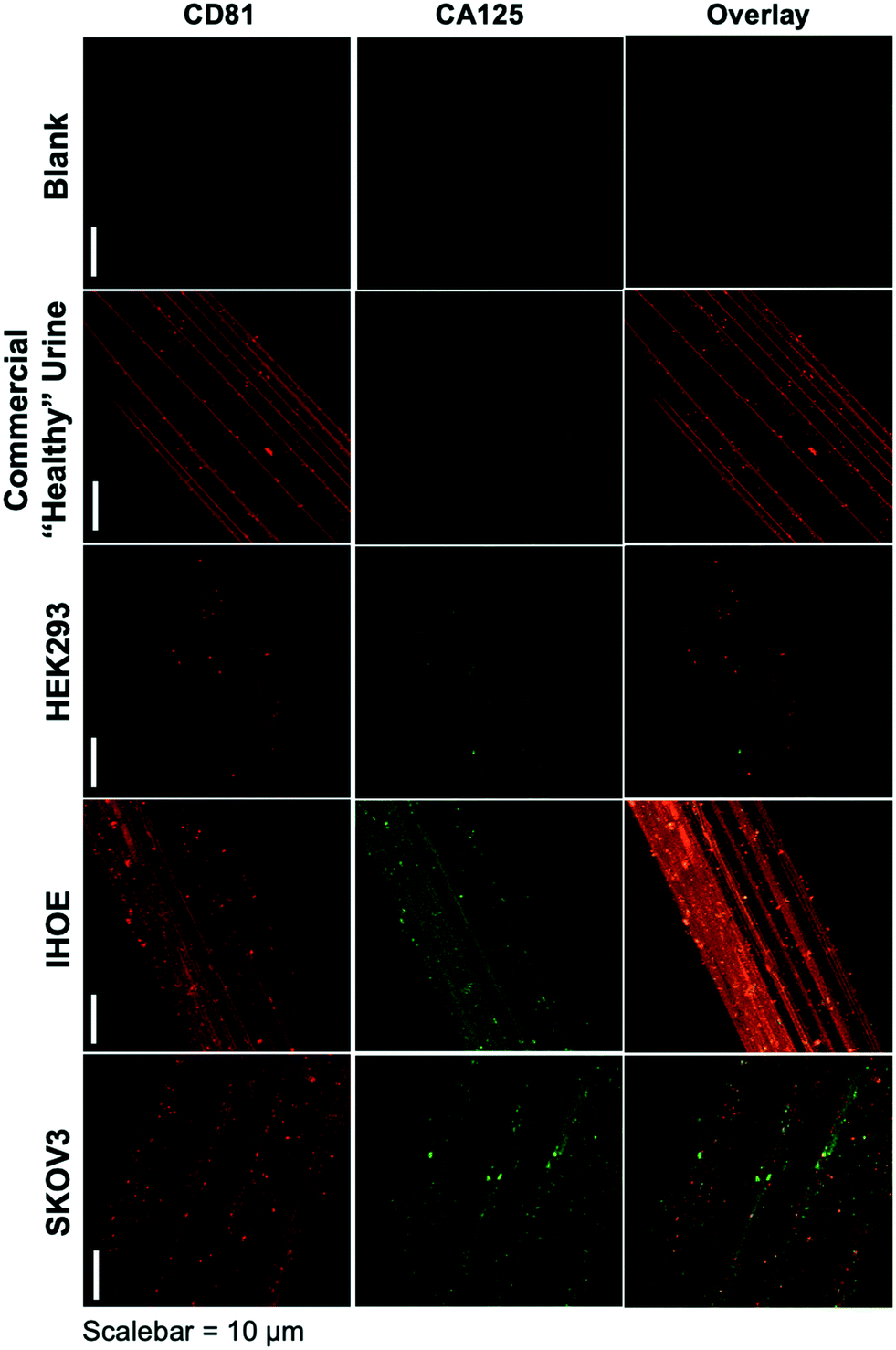 | ||
| Fig. 3 Confocal fluorescence images of C-CP fibers from process blank and test specimens following higher-throughput immunolabeling procedure. Red color depicts presence of the CD81 tetraspanin protein on exosome membranes, with green color representing the presence of CA125. | ||
As seen in the case of the standard immunolabeling protocol, the higher-throughput approach does indeed yield the anticipated results regarding the presence of the expected biomarkers on the exosome surfaces. Here, the commercially sourced exosomes (derived from human urine) and the HEK293 cell line supernatant yield positive results towards the CD81 tetraspanin but little-to-no response towards CA125. The particles providing response to CA125 show little correlation with CD81 in the overlays. Admittedly, the CA125 response seen for the commercial specimen here is far less than the extended incubation time method, which may reflect some sacrifice in sensitivity towards low-abundance proteins (presumably due to the shorter incubation time). As depicted in Fig. 2, the responses towards both immunolabels (CD81 and CA125) are dramatically enhanced in density and correlation for the case of the SKOV3 and IHOE cell lines. While not quantified at this point, this result is surely reflective of the presence of a cancer, with results obtained in less than 5 hours on primary sample volumes of 100 μL. Both metrics lie well in line with what would be desired for a liquid biopsy. Certainly, an extension of the method to urine specimens and a liquid biopsy, as in the case of Fig. 2, is the target following optimization of the high-throughput labeling methodology.
4. Conclusions
This work has demonstrated a novel platform for the isolation and immunolabeling of extracellular vesicles, allowing imaging to be performed directly on the capture fiber. The HIC capture and immunolabeling of EVs isolated on the C-CP fibers opens the door for many research, clinical, and diagnostic applications to be developed. The rapid concentration and classification of exosomal materials based on vesicle-associated biomarker proteins can be tailored to a variety of diagnostic challenges. Advantages in the method are first realized in terms of the sample sizes and the speed and purity of affecting the immobilization. Once isolated, the exosomes' quantity and identity can be determined by multiple on-fiber and post-elution methods. In the example presented here, confocal fluorescence immunoassays provide exquisite sensitivity and specificity, with as few as 10s of EVs being present in the illumination volume. While this method of determination is not suitable for portable point-of-care applications, based on instrument cost and complexity, it is certainly appropriate for many research and clinical laboratories. Further, the biochemical mechanisms of the work demonstrated here can surely be extended to other, more clinically practical, detection approaches (i.e., a C-CP lateral flow immunoassay) for the batch processing of large clinical cohorts on necessary scales of time and cost. Even in the research and clinical laboratory environments, the potential for further optimization of the high-throughput immunolabeling method would be advantageous. For example, Cappi et al. have described an automated 4-plex multistaining technique with 15 min immunolabel incubation times using horseradish peroxidase (HRP)-labeled primary antibodies with tyramide signal amplification-Alexa Fluor secondary antibodies.40 A similar abbreviated EV immunolabeling process is an obvious avenue for future exploration.Beyond the on-fiber single particle detection, one can readily imagine less expensive fluorometric methods applied to the fiber bundles in-mass. Alternatively, on-fiber labeling could be followed by exosome release into microliter volumes for solution-phase immunofluorescence assay, again on very short time scales. Rapid isolation and purification could also be followed by use of conventional dot blot assays, targeting specific biomarkers. Eluted exosomes could also be subsequently lysed for genetic profiling of the vesicle contents. Finally, as has been demonstrated using the C-CP fibers in a column format, the spin-down approach could be readily applied as an isolation stage prior to mass spectrometric exosome proteomics analysis. Ultimately, this method's main benefit may lie in its ease of tailoring to capture nanovesicles from various origins, including viruses, bacteria-derived outer membrane vesicles, lipoproteins, liposomes, and synthetic nanoparticles.
Author contributions
Kaylan K. Jackson: methodology, data curation, visualization, writing – original draft preparation; Rhonda R. Powell: methodology; Terri F. Bruce: conceptualization, supervision; R. Kenneth Marcus: conceptualization, supervision, writing – reviewing and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Science Foundation under grant no. CHE-1608663 and 2107882 is gratefully acknowledged.References
- J. C. Contreras-Naranjo, H. J. Wu and V. M. Ugaz, Lab Chip, 2017, 17, 3558–3577 RSC.
- S. Halvaei, S. Daryani, S. Z. Eslami, T. Samadi, N. Jafarbeik-Iravani, T. O. Bakhshayesh, A. K. Majidzadeh and R. Esmaeili, Mol. Ther.–Nucleic Acids, 2018, 10, 131–141 CrossRef CAS PubMed.
- M. A. Richard, J. J. Grob, M. F. Avril, M. Delaunay, X. Thirion, P. Wolkenstein, P. Souteyrand, B. Dreno, J. J. Bonerandi, S. Dalac, L. Machet, J. C. Guillaume, J. Chevrant-Breton, C. Vilmer, F. Aubin, B. Guillot, M. Beylot-Barry, C. Lok, N. Raison-Peyron and P. Chemaly, Arch. Dermatol., 1999, 135, 269–274 CAS.
- K. Unger-Saldana, World J. Clin. Oncol., 2014, 5, 465–477 CrossRef PubMed.
- W. Li, H. Wang, Z. Zhao, H. Gao, C. Liu, L. Zhu, C. Wang and Y. Yang, Adv. Mater., 2019, 31, e1805344 CrossRef PubMed.
- S. Pant, H. Hilton and M. E. Burczynski, Biochem. Pharmacol., 2012, 83, 1484–1494 CrossRef CAS PubMed.
- N. P. Hessvik and A. Llorente, Cell. Mol. Life Sci., 2018, 75, 193–208 CrossRef CAS PubMed.
- J. Lin, J. Li, B. Huang, J. Liu, X. Chen, X. M. Chen, Y. M. Xu, L. F. Huang and X. Z. Wang, Sci. World J., 2015, 2015, 657086 Search PubMed.
- F. Properzi, M. Logozzi and S. Fais, Biomarkers Med., 2013, 7, 769–778 CrossRef CAS PubMed.
- M. Tomasetti, W. Lee, L. Santarelli and J. Neuzil, Exp. Mol. Med., 2017, 49, e285 CrossRef CAS PubMed.
- C. Sheridan, Nat. Biotechnol., 2016, 34, 359–360 CrossRef CAS PubMed.
- M. L. Alvarez, M. Khosroheidari, R. Kanchi Ravi and J. K. DiStefano, Kidney Int., 2012, 82, 1024–1032 CrossRef CAS PubMed.
- G. K. Patel, M. A. Khan, H. Zubair, S. K. Srivastava, M. Khushman, S. Singh and A. P. Singh, Sci. Rep., 2019, 9, 5335 CrossRef PubMed.
- J. L. Welton, J. P. Webber, L. A. Botos, M. Jones and A. Clayton, J. Extracell. Vesicles, 2015, 4, 27269 CrossRef PubMed.
- C. Chen, J. Skog, C.-H. Hsu, R. T. Lessard, L. Balaj, T. Wurdinger, B. S. Carter, X. O. Breakefield, M. Toner and D. Irimia, Lab Chip, 2010, 10, 505–511 RSC.
- R. T. Davies, J. Kim, S. C. Jang, E.-J. Choi, Y. S. Gho and J. Park, Lab Chip, 2012, 12, 5202–5210 RSC.
- Z. Wang, H.-J. Wu, D. Fine, J. Schmulen, Y. Hu, B. Godin, J. X. J. Zhang and X. Liu, Lab Chip, 2013, 13, 2879–2882 RSC.
- A. Liga, A. D. B. Vliegenthart, W. Oosthuyzen, J. W. Dear and M. Kersaudy-Kerhoas, Lab Chip, 2015, 15, 2388–2394 RSC.
- G. G. Daaboul, P. Gagni, L. Benussi, P. Bettotti, M. Ciani, M. Cretich, D. S. Freedman, R. Ghidoni, A. Y. Ozkumur, C. Piotto, D. Prosperi, B. Santini, M. S. Ünlü and M. Chiari, Sci. Rep., 2016, 6, 37246 CrossRef CAS PubMed.
- T. F. Bruce, T. J. Slonecki, L. Wang, S. Huang, R. R. Powell and R. K. Marcus, Electrophoresis, 2019, 40, 571–581 CrossRef CAS PubMed.
- L. Wang, T. F. Bruce, S. Huang and R. K. Marcus, Anal. Chim. Acta, 2019, 1082, 186–193 CrossRef CAS PubMed.
- S. Huang, L. Wang, T. F. Bruce and R. K. Marcus, Biotechnol. Prog., 2020, 36, e2998 CrossRef CAS PubMed.
- K. K. Jackson, R. R. Powell, T. F. Bruce and R. K. Marcus, Anal. Bioanal. Chem., 2020, 412, 4713–4724 CrossRef CAS PubMed.
- K. K. Jackson, R. R. Powell, T. F. Bruce and R. K. Marcus, Analyst, 2021, 146, 4314–4325 RSC.
- X. Ji, S. Huang, J. Zhang, T. F. Bruce, Z. Tan, D. Wang, J. Zhu, R. K. Marcus and D. M. Lubman, Electrophoresis, 2021, 42, 245–256 CrossRef CAS PubMed.
- S. Huang, X. Ji, K. K. Jackson, D. M. Lubman, M. B. Ard, T. F. Bruce and R. K. Marcus, Anal. Chim. Acta, 2021, 1167, 338578 CrossRef CAS PubMed.
- P. A. Canney, M. Moore, P. M. Wilkinson and R. D. James, Br. J. Cancer, 1984, 50, 765–769 CrossRef CAS PubMed.
- M. J. Duffy, J. M. Bonfrer, J. Kulpa, G. J. Rustin, G. Soletormos, G. C. Torre, M. K. Tuxen and M. Zwirner, Int. J. Gynecol. Cancer, 2005, 15, 679–691 CrossRef CAS PubMed.
- Y. Zhang, H. Qian, A. Xu and G. Yang, Exp. Ther. Med., 2020, 19, 755–761 CAS.
- N. Mizoshiri, T. Shirai, R. Terauchi, S. Tsuchida, Y. Mori, D. Hayashi, T. Kishida, Y. Arai, O. Mazda, T. Nakanishi and T. Kubo, Cell. Oncol., 2019, 42, 861–871 CrossRef CAS PubMed.
- J. G. Donaldson, Curr. Protoc. Cell Biol., 2015, 69, 431–437 Search PubMed.
- V. R. Zurawski Jr., H. Orjaseter, A. Andersen and E. Jellum, Int. J. Cancer, 1988, 42, 677–680 CrossRef CAS PubMed.
- N. Einhorn, K. Sjovall, R. C. Knapp, P. Hall, R. E. Scully, R. C. Bast Jr. and V. R. Zurawski Jr., Obstet. Gynecol., 1992, 80, 14–18 CAS.
- Y. Kimura, T. Fujii, K. Hamamoto, N. Miyagawa, M. Kataoka and A. Iio, Br. J. Cancer, 1990, 62, 676–678 CrossRef CAS PubMed.
- C. Fang, Y. Cao, X. Liu, X.-T. Zeng and Y. Li, Oncotarget, 2017, 8, 63963–63970 CrossRef PubMed.
- B. Zimmerman, B. Kelly, B. J. McMillan, T. C. M. Seegar, R. O. Dror, A. C. Kruse and S. C. Blacklow, Cell, 2016, 167, 1041–1051 CrossRef CAS PubMed , e1011.
- I. Simon, D. Katsaros, I. Rigault de la Longrais, M. Massobrio, A. Scorilas, N. W. Kim, M. J. Sarno, R. L. Wolfert and E. P. Diamandis, Gynecol. Oncol., 2007, 106, 334–341 CrossRef CAS PubMed.
- D. Hoshino, K. C. Kirkbride, K. Costello, E. S. Clark, S. Sinha, N. Grega-Larson, M. J. Tyska and A. M. Weaver, Cell Rep., 2013, 5, 1159–1168 CrossRef CAS PubMed.
- C. Shao, F. Yang, S. Miao, W. Liu, C. Wang, Y. Shu and H. Shen, Mol. Cancer, 2018, 17, 120 CrossRef PubMed.
- G. Cappi, D. G. Dupouy, M. A. Comino and A. T. Ciftlik, Sci. Rep., 2019, 9, 4489 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details for “standard” exosome immunolabeling procedure and methods employed, images resulting from negative control experiments, and images for SEM/STEM confirmation of captured exosomes. See DOI: 10.1039/d2sd00007e |
| This journal is © The Royal Society of Chemistry 2022 |