
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn situ recalibration of ion selective electrodes
Neel
Sisodia
 a,
Kay L.
McGuinness
a,
Jay D.
Wadhawan
b and
Nathan S.
Lawrence
*a
a,
Kay L.
McGuinness
a,
Jay D.
Wadhawan
b and
Nathan S.
Lawrence
*a
aANB Sensors Ltd., 4 Penn Farm, Haslingfield, Cambridge, CB23 1JZ, UK. E-mail: nlawrence@anbsensors.com
bDepartment of Chemical Engineering, The University of Hull, Cottingham Road, HU6 7RX, UK
First published on 27th October 2021
Abstract
Reference electrode drift resulting from the exchange of ions at the solution/reference electrode chamber interface is the number one reason why ion selective electrodes and pH sensors in particular require recalibration, costing companies up to 70% of the total lifetime cost of a pH probe. In this work, a novel and innovative methodology is proposed in which the reference potential is tracked, allowing for in situ recalibration of the sensor without the need for end-user intervention.
Ion selective electrode (ISE) sensors are used to detect ionic concentration in aqueous solutions. Throughout the years a number of ISE's have been produced, the majority focusing on how to achieve selectivity for the particular analyte.1 As our environment changes, and legislation tightens across the globe regarding discharges into the world's waterways and oceans, there is a real need to improve the reliability of today's sensing system.2 Although there has been significant progress over the last decade with the development of the internet of things and networkable sensors, the information provided to these networks is only ever as reliable as the sensors monitoring the environment. Issues around recalibration and maintenance often mean mass deployment is economically unviable. ISE's fall foul of this issue of manual recalibration, making up to 70% of the sensor's lifetime cost.
In the majority of cases the potential of the ion selective sensing component is measured with respect to a standard Ag/AgCl reference electrode housed in a known and defined potassium chloride solution.3 A fundamental issue with ISE sensors is reference electrode drift or poisoning.4 Reference electrode poisoning occurs due to ion exchange at the porous frit used to connect the reference chamber to the external environment, a fundamental component of the sensor to operate. Ion exchange means that the composition of the reference solution is altered. Variations in the chloride concentration will cause a shift in the reference potential, and may give rise to soluble complexes of the form AgCln(n−1)−; ingress of ions such as sulphides, bromides, iodides that form salts with silver that are less soluble than silver chloride (AgCl) will also impact reference stability. Moreover, reducing agents can precipitate silver from silver ions, and complexing agents form metal complexes with silver ions, all of which will adversely affect the reference electrode potential.5
Several researchers and organisations have tried to overcome the drift issue. These have focused on improving the stability of the reference electrolyte to minimize ion mobility and exchange, improving the performance of the porous frit, or introducing junction systems.6
Hach reported a new method known as the differential electrode technique.7 This technique utilises two glass electrodes to make a measurement, which differs from the normal way of using a reference electrode in a potassium chloride solution. The two glass electrodes make the measurement differentially with respect to a third metal electrode. It was reported that a dilution of 100 to 1 only saw a 0.05 pH shift.
More classically, different electrolyte filling solutions have been studied.5 Due to the desire of miniaturising sensors, the idea of developing sensors without internal filling was investigated.8 It was said that removing the filling could also improve the sensors durability and temperature range. This theory is also presented elsewhere and the liquid contact has been described as ‘cumbersome’.9 However, they also do go on to report the several major challenges to the use of all-solid state ISEs. One of these issues was the ion to electron transduction was interrupted at the solid/membrane interface. Even after several improved designs, it was seen that the all solid-state ISEs still didn't match the performance of the conventional sensor.10,11 It seems the effectiveness of solid state ISEs is heavily dependent on the solid contact that is utilised.
Companies have also tried altering the type of frit used inside the sensors. Sentek (amongst others) explore the idea of a double junction probe as well as the common single junction probe.12 The double junction contains an extra reference channel; this is to stop any ‘leaching’ of contaminants to the silver chloride reference. Rather than overcoming the drift issue they merely act to slow the impact of the poisoning, elongating the time between calibrations. This however does not remove the necessity for calibration to ensure accurate and reliable sensing systems.
In this work we propose an innovative method to overcome this drift, rather than trying to stop the drift, we understand that it will always occur. Therefore, we report a novel means of verifying the electrochemical potential/performance of the reference electrode through the introduction of an additional stable voltammetric electrochemical measurement (Scheme 1A). Scheme 1B and C highlight the methodology of the approach. Scheme 1B highlights the voltammetric response of the new calibration electrode whereby the peak potential of the new redox couple is monitored periodically. This merely acts as a guide to monitor any shift in the reference electrode potential with time. Scheme 1C shows the response of the classical electrode when a reference shift occurs (red line). The peak potential shift taken from Scheme 1B is then fed back to the sensor and the outputted response is then amended to provide the correct pH signal (Scheme 1C, blue response). This reference electrode potential drift correction occurs immediately and without any end user intervention.
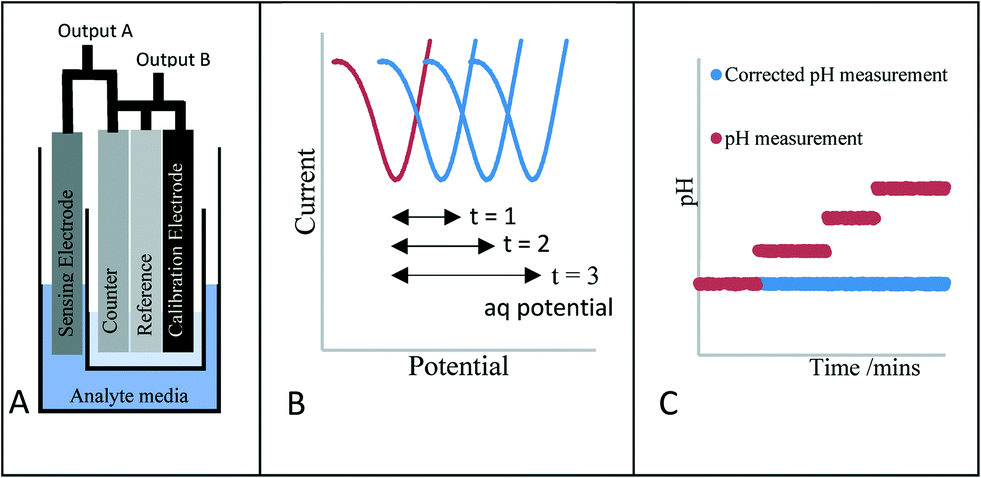 | ||
| Scheme 1 (A) The proposed reference system set-up (B) the reference tracking measurement (aq potential refers to the potential of the anthraquinone species used as the redox indicator) (C) how it is applied in real time. | ||
All reagents were obtained from Sigma-Aldrich; these were of the highest grade available and used without further purification. All solutions and subsequent dilutions were prepared using deionized water. All experiments were conducted in a 20 cm3 glass cell.
Voltammetric measurements were undertaken using an Anapot potentiostat (Zimmer and Peacock, UK) with a three electrode configuration. A modified carbon rod (3 mm length, 0.5 mm diameter) was used as the working electrode. The counter electrode was an unmodified carbon rod (4 mm length, 1 mm diameter). Unless otherwise stated the reference electrode was a saturated Ag/AgCl electrode (BASI, USA). Square wave voltammograms were collected using the following parameters; frequency = 25 Hz, amplitude 80 mV, step potential 0.5 mV. All the experiments were conducted whilst thermostatted at ambient temperature (20 ± 1 °C). Derivatisation of the sensing electrodes was conducted using the electrochemical reduction of Fast Red Al salt (10 mM) in acetonitrile containing 0.1 M tetrabutylammonium perchlorate.13 The classical cyclic voltammetric signal was observed for the modification of the carbon surface, with a small reduction wave at 0.3 V (vs. Ag wire) consistent with the reduction of the diazonium moiety to generate the reactive quinone species. A larger wave at −0.9 V was also seen due to the reduction of the quinone adduct.13 After the modification, the electrode was washed two times each in fresh acetonitrile and deionised water solutions respectively to ensure any absorbed species were removed from the electrode surface. After which the electrode was ready for interrogation.
The approach uses the results of Compton et al. who showed that in media of low buffer, the electrochemical reduction of anthraquinone perturbs its use as a pH sensor, rather it changes the pH of the solution locale to the electrode interface.14 In the following we use this phenomenon to produce a stable reference system in media of low buffer such as that found in the reference chamber of a typical reference electrode system.
To demonstrate the suitability of the approach as a reference system, the peak potential is required to be stable in a number of solutions of varying concentration and composition. Perhaps one of the most common reasons for reference electrode drift when sensors are placed in aquifer and river systems is the loss of chloride ions from the reference electrode chamber. This is due to the reference potential being very sensitive to the chloride ion concentration. Fig. 1A depicts the reductive square wave voltammetric response of the anthraquinone (AQ) – modified carbon electrode to varying chloride concentrations, ranging from 0.1–3.0 M KCl.
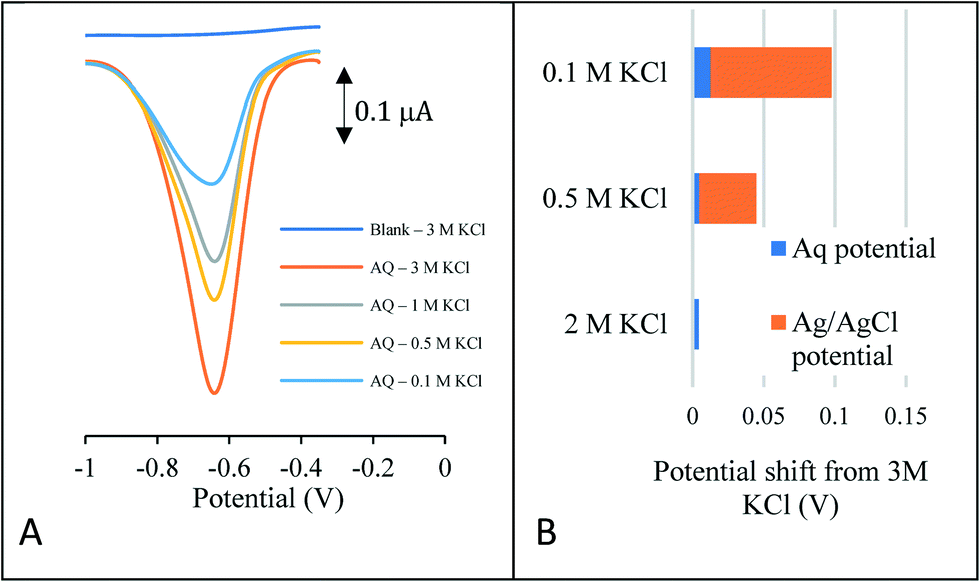 | ||
| Fig. 1 (A, LHS) Square wave voltammetry of the anthraquinone modified carbon electrode in various KCl concentrations (B, RHS) comparison with the unmodified carbon electrode placed in 3 M KCl. | ||
In each case a well-defined redox wave is observed consistent with the anthraquinone redox couple being present on the surface of the electrode. As expected, the peak current decreases with electrolyte concentration. Analysis of the variation of peak potential as a function of chloride concentration is given in Fig. 1B. This compares the peak potential shift of the AQ redox wave with that of redox potential of the Ag/AgCl redox couple for the same concentrations of chloride. This latter shift depicts the error that would occur in a pH measurement should there be a loss of chloride ions to the bulk solution. At the extremity, should the solution concentration change from 0.1–3.0 M KCl, this equates to a 1.42 pH unit error in a classical pH sensor compared to a 0.08 pH error using the novel internal voltammetric reference system.
In industrial applications, ion exchange is not solely the loss of chloride ions from the reference chamber, but rather the uptake ions into the chamber. Fig. 2 illustrates the response of the AQ-modified electrode when directly immersed in a solution covering the range pH 2–13. In Fig. 2A the pH was adjusted by the addition of hydrochloric acid to the 3 M KCl solution, Fig. 2B is the addition of sodium hydroxide and Fig. 2C the addition of acetic acid.
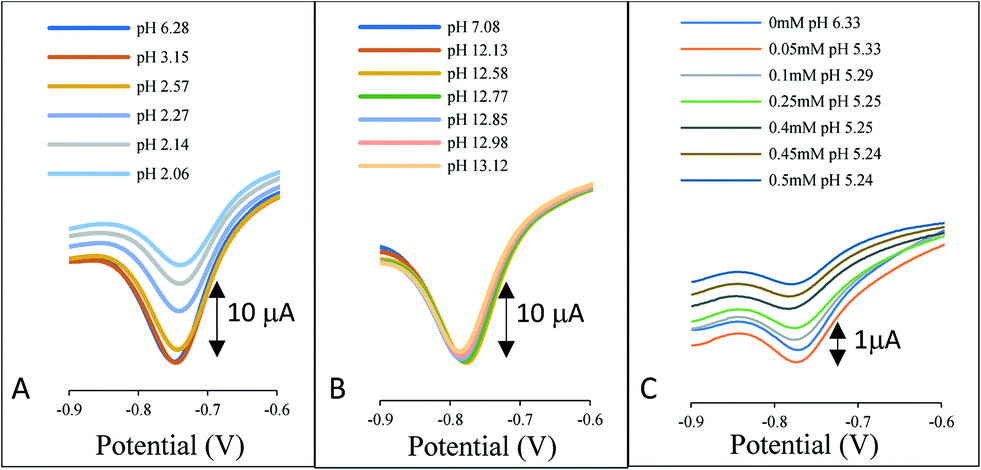 | ||
| Fig. 2 The influence of pH on the reference system signal. A) Addition of HCl to 3 M KCl, B) addition of NaOH to 3 M KCl and C) addition of acetic acid to 3 M KCl. | ||
In each case a voltammetric signal is observed consistent with the AQ redox couple, however, contrary to the belief that the AQ redox potential shifts with pH, no perturbation in the redox potentials is observed. The results are consistent with previously produced data for a species in solution.14 The reduction of the AQ and subsequent uptake of protons from the solution in the immediate vicinity of the electrode perturbs the pH locale to the electrode surface. This phenomenon thus provides a pH independent redox wave even in high and low pH regions and in buffered conditions as highlighted by the addition of acetic acid in Fig. 2C.
Further to demonstrating the independence to protons and hydroxide ions on the redox potential of the AQ-electrode, other potential interferences were studied. Fig. 3 highlights the mean deviation of the AQ-peak potential when in the presence of a number of anions and cations over various concentrations.
 | ||
| Fig. 3 Mean deviation of pH when in the presence of a number of anions and cations over various concentrations. In each case the standard deviation was ca. 0.005 pH unit. | ||
The deviation is measured with respect to the average peak potential measured across all the solutions. A maximum of 0.12 pH units is observed in a solution containing 0.1 M CaCl2 and can be tentatively attributed to the calcium ions chelating to the oxygen moieties of the AQ species. Although this appears large it should be noted should a reference electrode chamber suffer a complete exchange from 3 M KCl to 0.1 M CaCl2 this would equate to a 1.1 pH unit error and shows the stability of the newly proposed reference system. It should also be noted that across 55 separate measurements (five electrodes and eleven solutions of different compositions), a standard deviation of 0.1 pH units was observed, thus showing the reproducibility of this technique. Key poisoning agents of Ag/AgCl reference electrodes are sulfides and organothiols due to the formation of AgS complexes on the surface of the reference electrode. It has been previously shown that the redox potential of anthraquinone is independent of these species.15
The ability of the AQ redox couple to monitor the reference potential change was demonstrated through monitoring the peak potential difference using two different reference electrodes and comparing this to that of a potentiometric response between the two reference electrodes. Table 1 provides the peak potential of the AQ redox wave for three different reference electrodes, a standard Ag/AgCl electrode along with carbon and steel pseudo electrodes. As expected, the peak potential varies with reference electrode. In addition to this, the computed difference in measured peak potential against the potentiometric difference between the two electrodes is detailed.
| Reference electrode | AQ potential (mV) | ΔE wrt Ag/AgCl | ΔE calc-pot (mV) | |
|---|---|---|---|---|
| Calc. (mV) | Potentiometric (mV) | |||
| Ag/AgCl | −739 | N/A | N/A | N/A |
| Carbon pseudo | −736 | 3 | 7 | 4 |
| Steel pseudo | −439 | 300 | 290 | 10 |
It can be seen that in the two cases shown, the calculated difference between the potentiometric and proposed methodology provides a maximum error of 10 mV. This highlights that even in the case where the reference electrode is completely changed to an unstable pseudo reference electrode, the proposed methodology accounts for the reference potential changes.
It is recommended that current pH sensors should be replaced every 12–18 months depending on the environment in which they are deployed. During this lifetime the electrode will be calibrated numerous times, every day, week or month depending on the application. For the proposed voltammetric reference system to be a significant improvement, the recalibration frequency must be greatly decreased; ideally the lifetime of the voltammetric response should exceed the lifetime of the pH probe so it will never be recalibrated. Ideally the new reference electrode system if deployed on a commercial system would be tested at least once day throughout the lifetime of the probe equating to ca. 550 measurements for an 18 month deployment. The lifetime of the electrochemical response is dependent on two parameters; the number of electrochemical sweeps and storage time in the analysis media. These were tested in parallel, where an electrode was continually scanned over a short period of time demonstrating a strong voltammetric signal after 1440 scans, significantly higher than the once a day measurement frequency. In addition, an electrode was stored in 3 M KCl for a period of six months with no degradation in the AQ-response observed after this time, providing a predicted lifetime in excess of the eighteen month time period. These results demonstrate the suitability of the system for long-term deployments within a commercial device.
Commercial optimisation of the final product can be tuned through varying the underlying carbon substrate. The chemistry used in this method is known to modify a variety of carbon based substrates including graphite, C60, glassy carbon and carbon nanotubes.16,17 This will allow the device to be mechanically optimised for a whole range of sensor architectures.
Obviously, for this system to be fully commercialised there needs to be a potentiostat capable of running the voltammetric sweeps designed and implemented within a classical glass pH geometry, which will add cost to the final product. However, this will be low compared to the cost saved of not having to have field engineers routinely calibrate their sensor. Furthermore, any changes of the redox potential associated with temperature will be part of the in-built electronics calibration algorithms as already implemented in commercial ISE's today.
The results highlight an innovative and novel approach to overcome the issues of reference electrode drift within electrochemical devices. Rather than trying to stop drift through modifying aspects of the sensors, it is accepted that drift will always occur due to the design and requirements of the reference electrode system. Thus, the new system demonstrates the suitability to track the reference electrode potential by monitoring the potential of the anthraquinone redox couple. The system has been tested under a range of conditions and in the presence of a number of known interferences to the Ag/AgCl reference system with insensitivity demonstrated throughout. The efficacy of the new voltammetric reference system will provide pH sensor manufacturers the ability to track and recalibrate in situ for reference electrode drift, making these systems suitable for mass deployment in the internet of things.
ANB sensors acknowledges the support from the Royal Society of Chemistry, Innovate UK (133171). Innovate UK is the UK's innovation agency. It works with people, companies and partner organisations to find and drive the science and technology innovations that will grow the UK-economy. This project has received funding from the European Union's Horizon 2020 research and innovation programme under grant agreement No. 82297.
Conflicts of interest
There are no conflicts to declare.Notes and references
- R. Yan, S. Qiu, L. Tong and Y. Qian, Review of progress on clinical applications of ion-selective electrodes for electrolytic ion tests: from conventional ISEs to graphene-based ISEs, Chem. Speciation Bioavailability, 2016, 28, 72 CrossRef CAS.
- P. Namour, M. Lepot and N. Jaffrezic-Renault, Recent Trends in Monitoring of European Water Framework Directive Priority Substances Using Micro-Sensors: A 2007–2009 Review, Sensors, 2010, 10(9), 7947–7978 CrossRef PubMed.
- W. E. Morf, The Principles of Ion-Selective Electrodes and of Membrane Transport, Elsevier, 2012 Search PubMed.
- M. Sophocleous and J. K. Atkinson, A review of screen-printed silver/silver chloride (Ag/AgCl) reference electrodes potentially suitable for environmental potentiometric sensors, Sens. Actuators, A, 2017, 267, 106 CrossRef CAS.
- T. J. Smith and K. J. Stevenson, Handbook of Electrochemistry, ed. C. G. Zoski, Elsevier, 2007 Search PubMed.
- D. T. Sawyer and J. L. Roberts Jr, Experimental Electrochemistry for Chemists, John Wiley, New York, 1974 Search PubMed.
- Hach, https://www.hach.com/cms-portals/hach_com/cms/documents/pdf/Differential-pH-Design-Overcomes-Common-pH-Sensor-Challenges.pdf, Accessed March 2020.
- L. Van de Velde, E. d'Angremont and W. Olthuis, Solid contact potassium selective electrodes for biomedical applications – a review, Talanta, 2016, 160, 56–65 CrossRef CAS PubMed.
- J. Hu, A. Stein and P. Buhlman, Rational design of all-solid-state ion-selective electrodes and reference electrodes, Trends Analyt. Chem., 2016, 76, 102–114 CrossRef CAS.
- K. S. Johnson, J. A. Needoba, S. C. Riser and W. J. Showers, Chemical Sensor Networks for the Aquatic Environment, Chem. Rev., 2007, 107, 623–640 CrossRef CAS PubMed.
- M. Seidel and R. Niessner, Automated analytical microarrays: a critical review, Anal. Bioanal. Chem., 2008, 391, 1521–1544 CrossRef CAS PubMed.
- Sentek, https://www.sentek.co.uk/news/pick-right-ph-testing-equipment/, Accessed March 2020.
- P. Allongue, M. Delamer, B. Desbat, O. Fagebaume, R. Hitmi, J. Pinson and J. M. Saveant, Covalent Modification of Carbon Surfaces by Aryl Radicals Generated from the Electrochemical Reduction of Diazonium Salts, J. Am. Chem. Soc., 1997, 119, 201–207 CrossRef CAS.
- C. Batchelor-McAuley, B. R. Kozub, D. Menshykau and R. G. Compton, Voltammetric Responses of Surface-Bound and Solution-Phase Anthraquinone Moieties in the Presence of Unbuffered Aqueous Media, J. Phys. Chem. C, 2011, 115, 714–718 CrossRef CAS.
- V. G. H. Lafitte, W. Wang, A. S. Yashina and N. S. Lawrence, Athraquinone-ferrocene film electrodes: Utility n pH and oxygen sensing, Electrochem. Commun., 2008, 10, 1831–1834 CrossRef CAS.
- M. Pandurangappa, N. S. Lawrence and R. G. Compton, Homogeneous chemical derivatisation of carbon particles: a novel method for funtionalising carbon surfaces, Analyst, 2002, 127, 1568–1571 RSC.
- M. A. Ghanem, I. Kocak, A. Al-Mayouf, M. AlHoshan and P. N. Bartlett, Covalent modification of carbon nanotubes with anthraquinone by electrochemical grafting and solid phase synthesis, Electrochim. Acta, 2012, 68, 74–80 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |