
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConstruction and application of an efficient dual-base editing platform for Bacillus subtilis evolution employing programmable base conversion†
Wenliang
Hao‡
,
Wenjing
Cui‡
,
Feiya
Suo
,
Laichuang
Han
,
Zhongyi
Cheng
 * and
Zhemin
Zhou
*
* and
Zhemin
Zhou
*
The Key Laboratory of Industrial Biotechnology (Ministry of Education), School of Biotechnology, Jiangnan University, 1800 Lihu Avenue, Wuxi 214122, Jiangsu, China. E-mail: zhmzhou@jiangnan.edu.cn; zyCheng@jiangnan.edu.cn
First published on 1st December 2022
Abstract
The functionally evolved bacterial chassis is of great importance to manufacture a group of assorted high value-added chemicals, from small molecules to biologically active macromolecules. However, the current evolution frameworks are less efficienct in generating in vivo genomic diversification because of insufficient tunability, rendering limited evolution spacing for chassis. Here, an engineered genomic diversification platform (CRISPR-ABE8e-CDA-nCas9) leveraging a programmable dual-deaminases base editor was fabricated for rapidly evolving bacterial chassis. The dual-base editor was constructed by reprogramming the CRISPR array, nCas9, and cytidine and adenosine deaminase, enabling single or multiple base conversion at the genomic scale by simultaneous C-to-T and A-to-G conversion in vivo. Employing titration of the Cas-deaminase fusion protein, the platform enabled editing any pre-defined genomic loci with tunable conversion efficiency and editable window, generating a repertoire of mutants with highly diversified genomic sequences. Leveraging the genomic diversification platform, we successfully evolved the nisin-resistant capability of Bacillus subtilis through directed evolution of the subunit of lantibiotic ATP-binding cassette. Therefore, our work provides a portable and programmable genomic diversification platform, which is promising to expedite the fabrication of high-performance and robust bacterial chassis used in the development of biomanufacturing and biopharmaceuticals.
Introduction
Construction of an evolved bacterial chassis is usually dependent on the directed evolution of functional proteins.1 The evolved proteins substitute the natural counterparts in the hosts, resulting in an evolved bacterial chassis with specific phenotypes,2 such as the evolved RpsE in Escherichia coli and PfDHFR in yeast, which endow spectinomycin resistance3 and pyrimethamine resistance,4 respectively. However, the substitution of an exogenous DNA would affect the safety of hosts, which limits the host application in some fields, especially in the food industry. Therefore, the evolved protein of the host itself is expected. The technical framework for protein-directed evolution has been developed from in vitro to in vivo.5–7 Typical strategies used for directed evolution are random mutagenesis, semi-rational design, and rational design. All of them rely heavily on the process, including several iterative steps from gene cloning, in vitro mutagenesis, heterologous or integrated expression, and characterization.5 These steps are time-consuming and labor-intensive, especially when iterative cycles of mutagenesis and selection are needed. Moreover, a large number of variants generated by in vitro mutagenesis are mostly ineffective and the integration efficiency of target genes is limited and are the main reasons for the time-consuming and laborious process. Importantly, this process is difficult to be used for the evolution of some special proteins, such as membrane proteins, labile proteins, and multi-subunit protein complexes, due to the difficulties of heterologous expression and purification and functional determination.8In recent years, several state-of-the-art techniques have been developed to produce genetic diversification, which could be used for protein evolution in vivo. As a powerful in vivo mutagenesis platform, multiple automated genome editing (MAGE) was created to diversify the genomic sequence across the whole genome. However, iterative cycles of transformation with oligonucleotide libraries followed by high-throughput screenings are required.9,10 The OrthoRep platform was established using a highly error-prone orthogonal DNA polymerase and an orthogonal plasmid to enable more specific and precise genome editing;11 accordingly, the EvolvR system was designed employing CRISPR-guided DNA polymerases, enabling multiplexed and continuous diversification of the coding sequence within a tunable window length at user-defined loci.3
Different from these editing tools that introduce mutations by substituting native sequences or bases, base conversion platforms, such as the base editors (BEs), that employ base deaminase to produce genetic diversification have also been developed.4,12–14 These tools allow the diversification of the target gene based on combining deaminase with other guiding systems, such as CRISPR-Cas systems and T7 transcription systems. One type of base editor is the cytosine base editor (CBE), which is composed of a catalytically inactive Cas9 (dCas9 or nCas9) bearing the H840A and D10A or D10A mutation fused to a cytidine deaminase. Usually, the fusion is followed by an uracil DNA glycosylase (UDG) inhibitor (UGI). CBE introduces C:G > T:A base conversion directly into DNA sites targeted by single guide RNA (sgRNA). The deaminase deaminates cytidines (Cs) to uridines (Us) in the non-targeted strand, which is the single-strand DNA (ssDNA) part of the R-loop generated by the dCas9 (D10A and H840A)/nCas9 (D10A)-sgRNA complex. Meanwhile, the UGI prevents UDG from deaminating cytidines to apyrimidinic (AP) sites. When nCas9 (D10A) induces a nick on the targeted strand, the DNA mismatch repair pathway (MR, or other DNA repair pathways) is activated. It preferentially resolves the U:G mismatch into the desired U:A and a T:A product following DNA replication, thereby generating a C:G > T:A base conversion. Another type of base editor is the adenine base editor (ABE), which expands base editing to include A:T > G:C substitutions using adenosine deaminase fused to nCas9 (D10A). Adenosine deaminase deaminates adenosines (As) to inosines (Is), which are recognized as guanosines (Gs) by DNA polymerase during DNA repair and replication. In addition, all of these base editors contain a suitable editable window, where the conversion efficiency of C:G > T:A or A:T > G:C is different. The editing window for target-AID was from approximately protospacer positions 1 to 5. In addition, the editing window for ABE7.10 was from approximately protospacer positions 4 to 7, counting the PAM as positions 21–23. Most of these platforms are commonly applied in mammalian and plant cells,13–16 and few are applied in bacteria.12,17–19 Likewise, we developed a platform for protein engineering in vivo by employing a cytosine deaminase/nCas9 fusion in Bacillus subtilis.20 Although these base editing platforms are powerful for genetic diversification in vivo, they primarily induce either C to T or A to G conversion. To further increase genetic diversification, several base editors based on dual deaminases were constructed in plant and mammalian cells, which concurrently introduced A-to-G and C-to-T substitutions.21–24 Considering the generation of more base conversions, the dual-base editor would be an efficient platform for the manufacturing of evolved bacterial chassis through the directed evolution of natural proteins in vivo.
Here, we report an efficient chassis cell evolution platform for B. subtilis based on the dual-deaminases base editor. The platform was constructed by a CRISPR-Cas system with fusion of the mutant TadA from E. coli (ABE8e) and CDA from Petromyzon marinus (PmCDA) in the N-terminal of nCas. The platform enabled both C:G > T:A and A:T > G:C substitutions in the same target sequence with a single sgRNA (Fig. 1). Guiding by an sgRNA array, the platform allowed editing of the defined genomic loci with a tunable conversion efficiency and editable window. The conversion efficiency could be tuned by regulating the inducer concentration and induction time, resulting in a mutant library containing large amounts of mutants. The evolved proteins and the evolved bacterial chassis could be isolated through character screening from the library. As proof-of-concept, we successfully enhanced the resistance against a lantipeptide nisin by evolving PsdB in vivo using the platform. As the construction of the platform does not rely on any host-dependent factors, such a platform can be quickly created and is applicable to a wide range of aspects of microbial cell evolution.
 | ||
| Fig. 1 Schematic diagram of the base editing strategy for the dual-base editor in B. subtilis. The target nucleotides within the editing window (As and Cs) are framed in a red rectangle (with 1 being the most proximal base relative to the protospacer adjacent motif (PAM)). First, sgRNA (blue) binds to D10A nCas9 (cyan), ending up with the nCas9 (D10A):sgRNA complex. Second, the nCas9 (D10A):sgRNA complex finds and binds its target DNA, which mediates the separation of the double-stranded DNA to form the R-loop structure. Third, for A-to-G and C-to-T editing, two tethered B. subtilis-optimized deaminases (orange: adenosine deaminase-ABE8e; pink: cytidine deaminase-CDA) convert the target As and Cs in the nontargeted strand to Is and Us by adenosine deamination and cytidine deamination, respectively. For C-to-T editing, the cellular mismatch repair (MMR) pathway preferentially repairs the mismatch in a nicked strand. Therefore, the Gs in the targeted strand, which are nicked by nCas9 (D10A), are going to be replaced by As, and in the next replication cycle, repaired to a T:A base pair. For A-to-G editing, as Is are read as Gs by DNA polymerase, the resulting I:T heteroduplex is permanently converted to a G:C base pair during DNA replication. | ||
Results
Design and construction of the dual-base editor in B. subtilis
To construct a dual-base editor in a microbial cell, an nCas9-fused dual deaminase module and an sgRNA targeting module were designed in B. subtilis (Fig. 1). Five dual-base editors were designed to isolate a dual-base editor with high conversion efficiency. As shown in Fig. 2a, two of them were fusions of CDA from Petromyzon marinus (PmCDA)25 and evolved ADA from E. coli (ABE7.10)26 in the N-terminus of nCas9 in different orders. One of them was the two deaminases fused in either hand of nCas9. Another two were similar to those of the first two dual-base editors, except that ABE7.10 was substituted by ABE8e (a mutant ecTadA exhibited higher activity).22 The five different combinations were integrated into the lacA locus of the B. subtilis genome, yielding strains BS1, BS2, BS3, BS4, and BS5. One of the sigma factors, sigE in B. subtilis, was selected as the target protein for editing (the protospacer sequence for sigE editing was shown in Fig. 2b). The plasmid (pHY-ECBE) used for sgRNA expression targeting sigE was constructed using a strong promoter, P43, of B. subtilis. The conversion efficiency of the five dual-base editors was tested after incubation. As shown in Fig. 2b, the five dual-base editors displayed varied ranges of editable windows and distinct editing efficiencies. Among them, the dual-base editor harboring the ABE8e-CDA-nCas9 fusion exhibited the highest conversion efficiency (most of them were higher than 83%) and the widest editable window (∼9 nt). Therefore, the dual-base editor harboring the ABE8e-CDA-nCas9 fusion was chosen for the following study.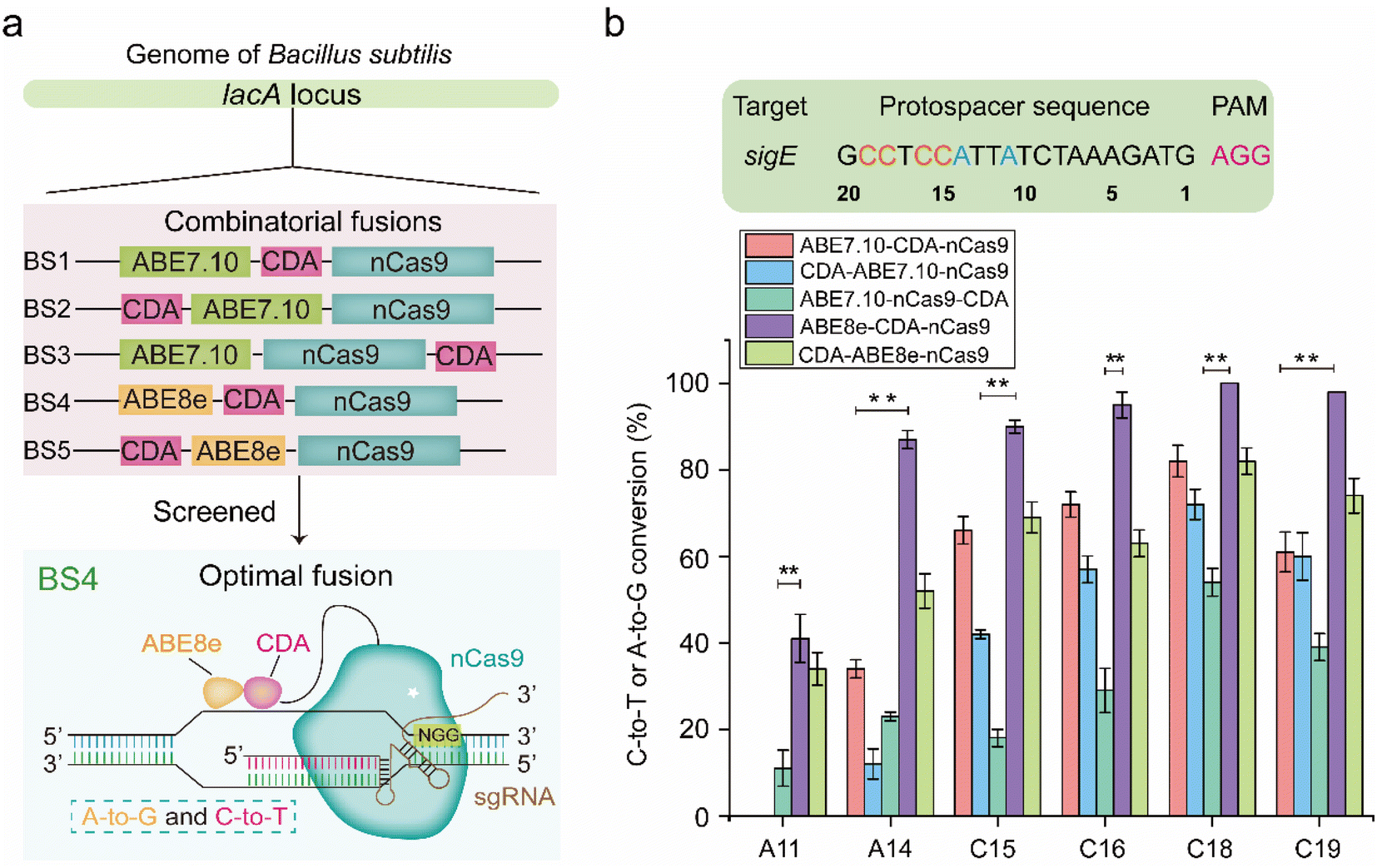 | ||
| Fig. 2 Construction and characterization of the dual-base editor in B. subtilis. (a) Architectures of five dual-deaminase base editors and schematic model for the dual-base editor. Five dual-base editor constructs were separately integrated into the lacA locus of B. subtilis, generating BS1, BS2, BS3, BS4, and BS5. (b) The conversion efficiencies of A-to-G and C-to-T of five dual-deaminase base editors targeting sigE. The protospacer sequence for sigE editing is shown. Bars represent the average conversion efficiency, and error bars represent the S.D. of three independent biological replicates. The asterisks indicate significant editing based on a comparison between the experimental groups (**, p < 0.01, Student's t-test). | ||
The dual-base editing platform is robust in genomic editing
To identify the editing robustness (excluding the influence of the genetic context on editing performance) mediated by the dual-base editor, another protein, BceB, in B. subtilis was chosen to evaluate the conversion efficiency and editing window. Seven sgRNAs (termed B1–B7, the protospacer sequences are shown in Fig. S1a†) targeting different sites in bceB were designed and constructed into expression plasmids, yielding the corresponding recombinant strain after transformation into BS4 (Tables S3 and S4†). The editing performance was determined by population sequencing after induction by 1% xylose. As shown in Fig. S1b† and 3a, the dual-base editor exhibited high editing efficiency at different edited positions and the editing window of approximately 8 nt in length. Within the editing window, most of the site showed an editing efficiency of more than 60% (Fig. S1b†). To further clarify the frontiers of the editing window of the platform, we have selected another large gene cluster in B. subtilis, pks operon (pksABCDEFGHIJLMNRS) to determine the editing window in diverse genetic loci with different genetic context. Ten genomic loci were then selected from 7 genes (pksABCDEFG) within the gene cluster (Fig. S1c† and 3b). Ten sgRNAs (the protospacer sequences were shown in Fig. S1c† and 3b) were designed and constructed into expression plasmids to target the predesigned genomic loci in pks operon. These plasmids were then transformed into BS4, yielding the corresponding recombinant strains. The evaluation of the editing performance of the pks operon was performed similarly to that of bceB. The population sequencing showed that the dual-base editor exhibited high efficiency for different editable positions in most genes (Fig. S1d†). Moreover, we noted that the editing of the pks operon generated a ∼11 nt editing window (Fig. 3b). These results demonstrate that the dual-base editor is robust to edit different sites in a single gene (bceB) or multiple genes (pks operon) at the genomic scale. In addition, combining the editing results of the bceB gene and pks operon, we can roughly determine that the editing window of the dual-base editor is 8–11 nt in length (Fig. 3a and b).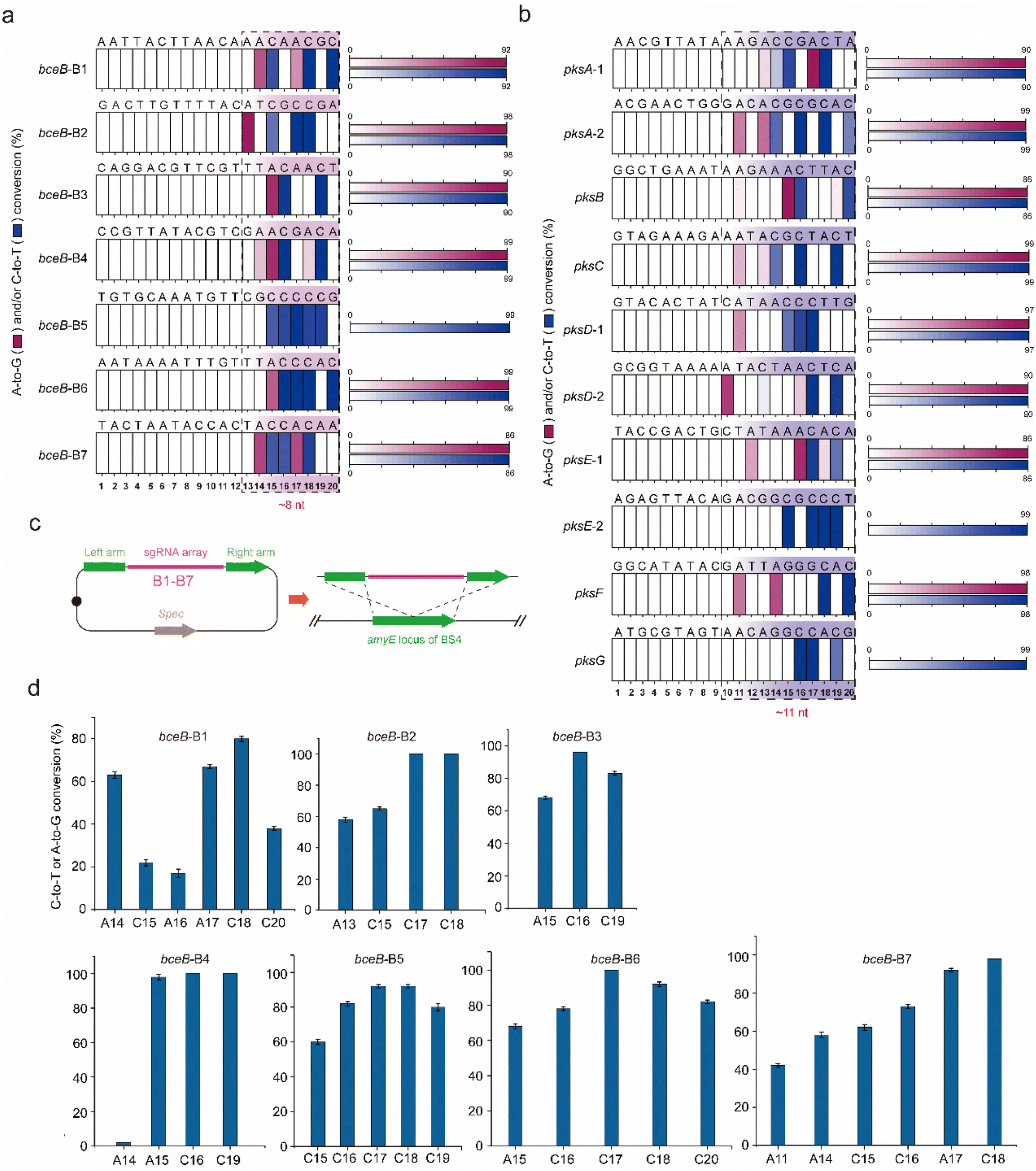 | ||
| Fig. 3 Evaluation of the editing performance for single editing and multiplex editing. (a) Evaluation of the editing window for the dual-base editor with the corresponding single sgRNAs (B1–B7) targeting bceB. (b) The evaluation of the editing window for the dual-base editor with the corresponding single sgRNAs targeting pksABCDEFG. (c) Multiplex editing of the dual-base editor with an sgRNA array created by assembly of the seven sgRNAs. A schematic illustration was applied. (d) Base conversion efficiencies of the dual-base editor with an sgRNA array (B1–B7) targeting bceB. Bars represent the average conversion efficiency, and error bars represent the S.D. of three independent biological replicates. | ||
A key property of a targeted mutagenesis system is the specificity towards the on-target sequence, keeping off-target mutagenesis as low as possible.12 To further investigate the off-target effect of the platform, we separately transformed plasmids pHY-B1, pHY-B2, pHY-B3, pHY-B5, and pHY-B7 into the sBS4 strain. In addition, the BS4 strain was used as the control. The strains BS4, BS4-pHY-B1, BS4-pHY-B2, BS4-pHY-B3, BS4-pHY-B5, and BS4-pHY-B7 on the corresponding plates were randomly selected for incubation and induction. The genome of the induced culture was extracted for whole genome sequencing. Compared with the reference genome of BS4, we observed no mutations at other sites having a prevalence of ≥20% within the population (Fig. S2† only shows the on-target efficiencies of targeted sites). The raw data of the whole genome sequencing can be accessed under BioProject ID PRJNA808834 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA808834).
The performance of the base editing platform in multiplex editing differs from single editing
Multiplex genome editing is very important for producing diverse mutant libraries. To characterize the editing performance of the multiplex genome editing, an sgRNA array was created by assembly of the seven sgRNAs (B1–B7) used for bceB and integrated into the amyE locus in the genome of BS4, yielding recombinant strain BS6 (Fig. 3c). Population sequencing was performed after induction by 1% xylose for 10 hours. As shown in Fig. 3d, the editing window and the editing efficiency were quite similar to those of single editing. The conversion efficiency was relatively high for most positions at each site, while there were some prominent differences between the two editing systems. The editable A16 position at the B1-targeting site was converted by multiplex editing with 17% efficiency, but not by the single editing (Fig. 3d and S1b†). In contrast, A18 at the B4-targeting site was converted by single editing, but not by multiplex editing (Fig. 3d and S1b†). Compared to adenosine deaminase, cytidine deaminase was able to maintain the stable edited features in the fusion, even when editing several consecutive cytidines (Fig. 3d and S1b†). These results demonstrate that the dual-base editor exhibited a multiplex genome editing function and that CDA exhibited a more stable edited feature than ADA.The genomic diversity generated by the platform was fine-tuned by titrating the fusion protein and editing time
Although the dual-base editor converted the base (A or C) at each editable position in the window, it is unknown whether the system could be tuned. The property is of great importance to mutagenetic diversity, which determines the capacity of the mutant library. To test the conversion efficiency tunability of the dual-base editor mediated by the level of the ABE8e-CDA-nCas9 fusion, BS6 was incubated with different concentrations of inducer (xylose in 0%, 0.01%, 0.02%, 0.04%, 0.08%, 0.2%, and 0.5%). Population sequencing was carried out to determine the conversion efficiency after induction for 10 h. For the negative control (0% of xylose), no significant editing was detected in five of the seven sites (B1, B2, B4, B5, and B7, Fig. S3†). Only two sites (B3 and B6) showed observed editing with relatively low efficiency (Fig. S3†), indicating the stringency of the xylose-induced genomic editing. Afterwards, different concentrations of xylose (0.01%, 0.02%, 0.04%, 0.08%, 0.2%, and 0.5%) were employed to induce the platform, by which the editing spectra were profiled. We found that the editing efficiency for each position within most of the targeted loci was increased along with the elevated xylose concentration, especially over the low concentration range (0.01–0.2%) (Fig. S4,† the red asterisks). Only a few sites showed editing efficiencies that were less dependent on the dose of xylose, both at low and high levels (Fig. S4,† the black asterisks). In particular, there were also some positions that were able to reach the highest level at a very low xylose concentration (0.01% xylose concentration), and then kept a constant editing efficiency even though the xylose concentration increased (Fig. S4,† the green asterisks). Thus, to evaluate the dependence of the editing efficiency on the inducer concentration, the editing efficiency grouped by xylose concentration was averaged and compared. The results showed that the conversion efficiency of the dual-base editor increased with the increase of xylose concentration (Fig. 4a). | ||
| Fig. 4 Fine-tuned conversion efficiency of the dual-base editor for multiplex editing. (a) Effects of the inducer concentration on the conversion efficiency. Box and dot plots indicating the aggregate distribution of the editing efficiency of all sites grouped by the xylose concentration. In the box plots, the box spans the interquartile range (IQR) (first to third quartiles), the horizontal line shows the median (second quartile) and the whiskers extend to ±(1.5 × IQR). The single dots represent the editing efficiency of all sites under the same inducer concentration. (b) Effects of induction time on conversion efficiency. All sampling times are calculated after induction. Values and error bars reflect the means and S.D. of three independent experiments. | ||
Subsequently, the effect of the induction time on the conversion efficiency was also determined. Firstly, the bacterial cells harboring the editing systems were pre-cultured for 4 h without xylose induction. Then, 0.5% xylose was added to each culture to trigger the editing. The culture was then sampled at different times (1 h, 2 h, 3 h, 4 h, and 6 h post-induction), and detected by population sequencing. All of the editable positions displayed increased conversion efficiency along with the induction time until the achievement of maximum (Fig. 4b). Taken together, these data indicated that the conversion efficiency of the dual-base editor could be fine-tuned by regulating the inducer concentration and induction time, which was beneficial to the construction of a mutant library and kept the diversity of the mutants in the library as high as possible.
Genomic diversification by multiplex editing generates differentially diversified mutants
To identify the diversity in the mutagenesis library resulting from multiplex editing and the effect of different concentrations of inducers on the diversity of mutagenesis library, the diversity of the mutagenesis library was verified at the single-clone level. The strains BS6 harboring the multiplex editing system targeting the 7 loci of bceB (Fig. 5a) were induced by different concentrations of xylose (0.01%, 0.02%, 0.05%, and 0.5%). From each plate, 20 colonies were randomly chosen for sequencing. To clearly demonstrate the editing diversity, we adopted a classification method by calculating the combination of editable sgRNAs (Fig. 5b and c). From the analysis of mutagenesis diversity mediated by the combinatorial sgRNA, we observed that induction by different concentrations of xylose (0.01%, 0.02%, 0.05%, and 0.5%) yielded numbers of diverse mutations. Noticeably, the numbers of different mutations were increased along with the increasing concentration of xylose, such that there were 15 and 17 distinct mutants induced by 0.01% and 0.02% xylose, respectively. Surprisingly, two sets of 19 distinct mutants were obtained either by 0.05% or by 0.5% xylose induction (Fig. 5b, c and S5a†), even including mutations occurring simultaneously at all seven sgRNAs-targeted sites. These results indicate that a high level of xylose was prone to generating more multiple mutations occurring simultaneously at more than 3 sites than a low level of xylose. | ||
| Fig. 5 Identification of genetic diversification resulting from a dual-base editor at the single-clone level. (a) The protospacer sequence of the 7 sgRNAs. PAM motifs are indicated in pink. Adenines (As) and cytosines (Cs) in the editable window are shown in blue and orange, respectively. (b) The diversity of sgRNA combinatorial mutations for the multiplex editing of bceB at 0.01% and 0.02% xylose concentrations. (c) The diversity of sgRNA combinatorial mutations for the multiplex editing of bceB at 0.05% and 0.5% xylose concentrations. The Arabic numerals in the box represent the number of mutants. (d) The heatmap showing the amino acid conversion associated with the editing of bceB induced by the dual-base editor. Missense and silent mutations were all counted. | ||
In addition, the amino acid conversion associated with the editing of bceB induced by the dual-base editor is shown in the form of a heatmap, including missense mutations, silent mutations, and unsubstituted (Fig. 5d). In practical application, different (combinatorial) mutations could be induced by a single sgRNA or multiple sgRNAs through flexible adjustment by xylose concentration. These results suggest that the dual-base editor exhibited the capability to efficiently produce large amounts of mutants.
Genomic diversity generated by multiplex editing is higher than multiple rounds of sequential single editing
According to the differential editing efficiency at different targeting sites, genetic diversity caused by multiplex editing should be higher than that by multiple rounds of single editing. To identify the difference in the genetic diversity caused by multiplex editing and multiple rounds of single editing, 3 sgRNAs targeting bceB (B2, B3, and B4) were selected to perform multiplex and single editing. For multiplex editing, the 3 sgRNAs were created into an array and integrated into the amyE locus in the BS4 genome, and then single-clone sequencing was performed after incubation and induction (Fig. 6a). For the multiple rounds of single editing, 3 temperature-sensitive plasmids harboring sgRNAs B2, B3, and B4 were constructed. The three plasmids were transformed into BS4 successively for base editing under the same incubation conditions as the multiplex editing, during which a plasmid cure process was performed before the later plasmid transformation (Fig. 6a, see the detailed protocol in the Experimental section). The protospacer sequences of B2, B3, and B4 are shown in Fig. 6b. Thirty colonies in each system were randomly selected to perform single colony sequencing. As shown in Fig. 6c and S6a,† multiplex editing produced a total of 16 distinct mutants among the 30 colonies, and the mutant sites were observed in single, double, or triple sgRNA targeted positions. In contrast, sequential editing yielded a total of 9 distinct mutants, most of which contained two identical mutants (Fig. 6c and S6a†).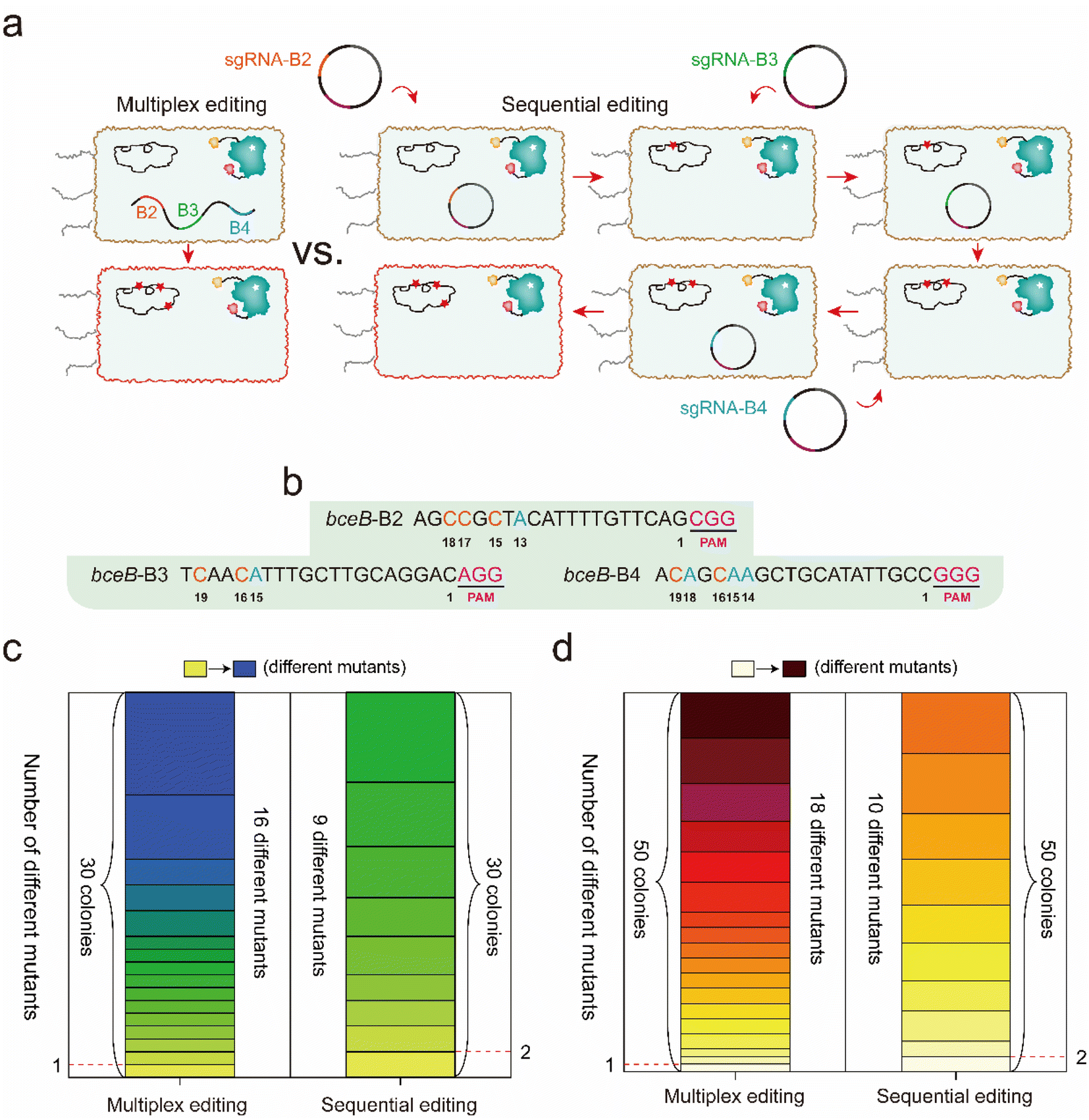 | ||
| Fig. 6 Comparison of the genetic diversification by the corresponding single sgRNAs and an sgRNA array. (a) The rationale and workflow of simultaneous multiplex editing and multiple rounds of single editing. (b) The protospacer sequence of the 3 sgRNAs (B2, B3, and B4). (c) and (d) Comparison of genetic diversification between multiplex editing and multiple rounds of single editing at the single-clone level. (c) Comparison of genetic diversification of 30 colonies from multiplex editing system and multiple rounds of single editing system. (d) The comparison of genetic diversification of 50 colonies from multiplex editing system and multiple rounds of single editing system. | ||
To further demonstrate which system was prone to generate higher mutagenetic diversity, another fifty colonies formed by these two systems were randomly selected to perform single colony sequencing. The sequencing results showed that 18 distinct mutants out of the 50 colonies were obtained by multiplex editing, while there were 10 distinct mutants out of the 50 colonies that were obtained by sequential editing (Fig. 6d). In addition, we observed that 13 new mutants (purple rectangle) among the 50 colonies were different from the previous 16 mutants among the 30 colonies for multiplex editing (Fig. S6b†). However, we only observed that 4 new mutants (purple rectangle) among the 50 colonies were different from the previous 9 mutants among the 30 colonies for sequential editing (Fig. S6b†). Many more mutants should be further observed from many more colonies, and from the more diverse sgRNA combination. These results clarify that multiplex editing produces higher genomic diversity than that of multiple rounds of sequential single editing.
Altering the cellular resistance to lanthipeptide nisin via evolving ATP-binding cassette transporter PsdB in B. subtilis by the dual-base editing platform
The dual-base editor exhibited the capability to efficiently produce large amounts of mutants, which is beneficial for achieving protein-directed evolution in vivo, and for obtaining evolved bacterial chassis resulting from the evolved proteins. To confirm the performance of the dual-base editor in functional protein-directed evolution, a lantibiotic ABC transporter permease PsdB was selected as the target to construct a mutagenesis library. PsdB is a membrane protein that is involved in nisin uptake and efflux processes in B. subtilis.27–29 An evolved bacterial chassis based on evolved PsdB would enhance the resistance to nisin, which is helpful for the overproduction of nisin.Five critical regions (20 aa–41 aa, 53 aa–76 aa, 112 aa–135 aa, 155 aa–177 a, and 229 aa–252 aa) that potentially affect the topology of PsdB according to UniProt annotation were selected as mutagenesis candidates (Fig. 7a). Five corresponding sgRNAs (P1–P5) were designed for these candidates in an array, and then integrated into the amyE locus of BS4, yielding BS8 (Fig. 7b). After induction by 0.2% xylose for 10 h, BS8 was spread onto the plate, and then 6 colonies were randomly selected to quantitatively measure the antimicrobial rate against nisin by the Alamar blue method. This method measured the increased nisin-resistance through the ratio of fluorescence intensity (FI, this reflects the biomass of cell) between wild-type strain and the mutants. Moreover, we have set the wild-type strain as the control group. If the FI of the experimental group is greater than that of the control group, then the final value is less than 0. This indicates that the mutant has increased activity to nisin. Conversely, nisin activity did not increase if the final value was greater than 0. Among these colonies, 5 exhibited increased resistance to nisin, and one colony (M1) exhibited decreased resistance to nisin (Fig. 7c). The sequences of the 6 PsdB mutants were compared. All of the colonies had corresponding base conversions, resulting in amino acid substitutions (Fig. S7 and Table S5†). Notably, M1, which was the only one exhibiting increased sensitivity to nisin, contained a different mutation of V26T from the others (V26A). Additionally, M3 exhibited the most resistance to nisin, containing a special mutation of L235S. These results indicated that the two sites in PsdB might be related to nisin resistance. Subsequently, mutants PsdB-V26A (M7) and PsdB-V26A-L235S (M8) were constructed and characterized, and M8 exhibited the highest nisin resistance compared with the other mutants (Fig. 7c).
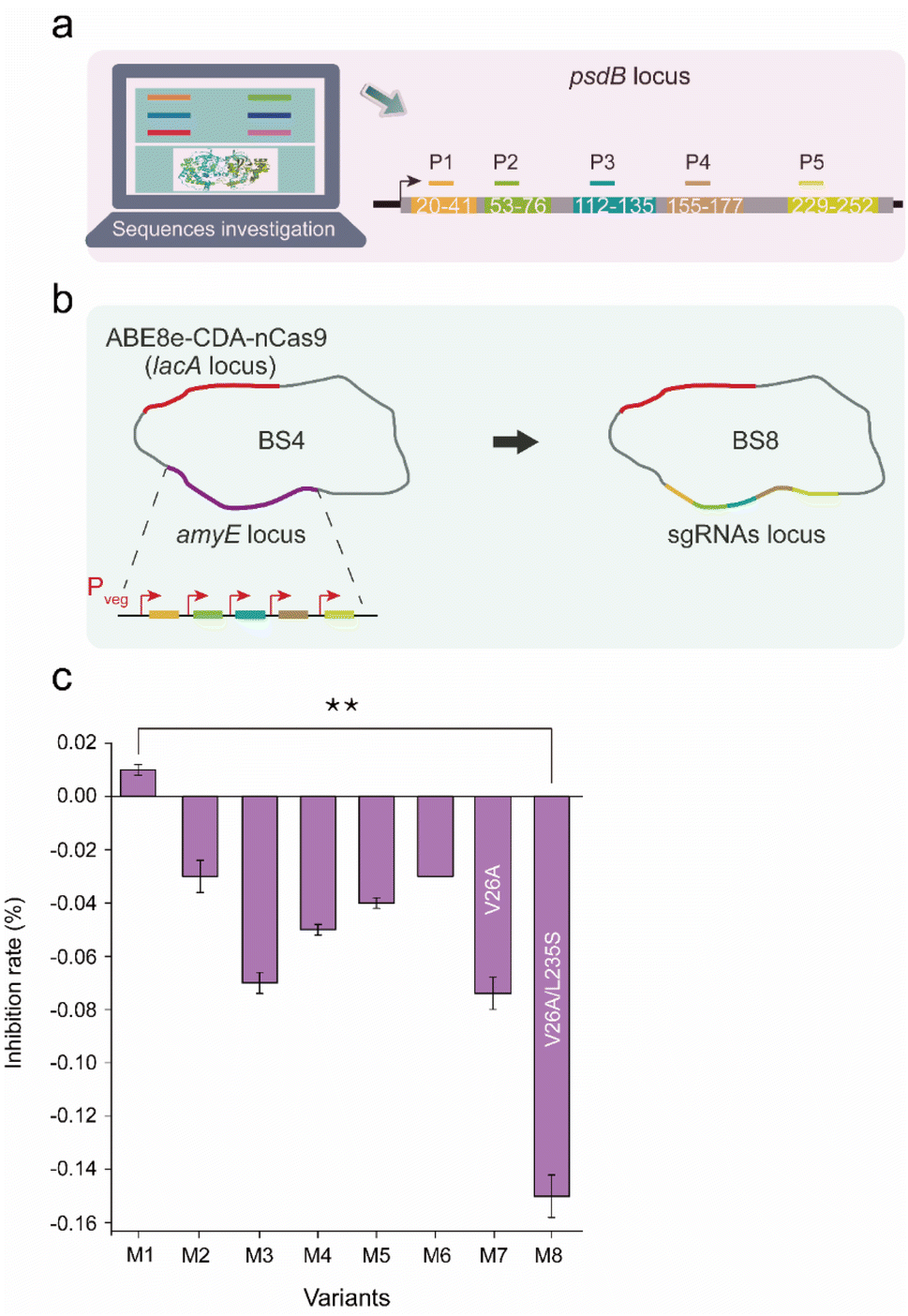 | ||
| Fig. 7 Evolution of nisin resistance mutations based on the dual-base editor. (a) Five sgRNAs (P1–P5) designed for psdB editing. The topological regions targeted by sgRNAs are displayed in different colors. (b) Construction of the strain harboring the dual-base editor and an sgRNA array (P1–P5). The five sgRNAs were sequentially ligated by Golden Gate assembly and integrated into the amyE locus of BS4. (c) The inhibition rate (%) of nisin in the PsdB mutants. The inhibition rate (%) was quantitatively calculated by the ratio of the fluorescence intensity (FI) of the control group to the fluorescence intensity (FI) of the experimental group in the fifth hour by the Alamar blue method. Values and error bars reflect the means and S.D. of three independent experiments. The locations where mutations occurred (including base conversion and amino acid replacement) are shown in Fig. S3 and Table S5.† The asterisks indicate significant nisin-resistant capability based on a comparison between the experimental groups (**, p < 0.01, Student's t-test). | ||
Discussion
Protein evolution is critical to the rapid development of synthetic biology.7 In the past decades, a handful of platforms have been developed to site-directed mutagenesis or random mutagenesis in vitro.5,30In vitro mutagenesis techniques, such as epPCR,31–33 site saturation mutagenesis,34–36 and DNA shuffling,37,38 are able to quickly produce large number of variants of the target gene. However, their selection requires multiple steps in most cases, including cloning and transformation into a host cell or integrating the target gene into the genome of host cell for expression, which are time-consuming and labor-intensive, especially when they require iterative cycles of mutagenesis and selection. Moreover, in these in vitro mutagenesis systems, huge numbers of mutants are usually lost in the transformation process because of the different transformation efficiencies in diverse bacterial chassis. Moreover, these systems are limited to the direct application to some proteins, but difficult to utilize in an array of other proteins, such as membrane-integral proteins and diverse multi-subunit protein complexes because of the challenging soluble expression and inconvenient in vitro characterization of function. In contrast, in vivo mutagenesis platforms mediated by base editors are preferred because the generation of variants, their expression, and selection can be done in a continuous process, which accelerates directed evolution. Due to the specificity of the base editor, it can consciously mutate specific hotspots according to the semi-rational design to form a limited but specific library. The base editor is also suitable for the directed evolution of special proteins because it avoids the heterologous overexpression of these proteins. We recently developed an in vivo mutagenesis system named CRISPR-CDA-nCas9-UGI for the directed evolution of these special proteins.20 However, it only achieves C to T conversion in a 6 bp editable window, which severely limits the genetic diversity in the library.In this work, to further diversify the edited genomic sequences, an in vivo mutagenesis platform, ABE8e-CDA-nCas9, for genomic diversification in bacterial cells was constructed and applied (Fig. 1). ABE8e-CDA-nCas9 exhibited high editing efficiency for C-to-T or A-to-G that was comparable to that of the reported single-deaminase base editors such as dCas-CDA-UL,18 CRISPR-BEST,39 and ABE8e.22 Additionally, it exhibited a more expanded editable window (∼8–11 nt, Fig. 2b; 3a and b). Unlike the dual base editor A&C-BEmax that had highly differential conversion efficiency at diverse alleles in human cells,24 the simultaneous A/C conversion efficiency within the editable window was relatively consistent across different edited sites by both single editing system and multiplex editing system (Fig. S1b† and 3d). Furthermore, even a continuum of several Cs was edited at high efficiency (Fig. S1b† and 3d). Similar to most novel editing systems, ABE8e-CDA-nCas9 edited defined genomic targets in high-fidelity without off-targeting editing (Fig. S2†).
The effects of the fusion position of two deaminases (cytosine deaminase and adenosine deaminase) and nCas9 (D10A) on the editing performance of the dual-base editors have been reported.24,40 Unlike the fusion pattern of Cas protein and deaminase in SPACE,21 Target-ACEmax,23 and A&C-BEmax,24 we reprogramed the fusion order of nCas9 (D10A), cytosine deaminase (CDA),25 and the evolved TadA (ABE8e)22 in a different N-to-C terminal order, ABE8e-CDA-nCas9. Upon the newly constructed system, the editing efficiencies of A-to-G and C-to-T ranged from 6% to 92% and 7% to 100%, respectively (Fig. S1b and d;†3a and b), which is significantly higher than that of the reported dual-base editors.21,23,24,40 We considered that two reasons might explain the better performance of ABE8e-CDA-nCas9. First, different from the systems of Target-ACEmax23 and SPACE,21 in which CDA is fused to the C-terminal of nCas9,18,19,41 the fusion of CDA into the N-terminal of nCas9 could efficiently introduce C-to-T substitutions, which is similar to the CRISPR-CDA-nCas9-UGI reported previously.20 Second, the activity and the adaption of ABE8e in the system are higher than that of ABE7.10 used in other editors.22
The base editing system based on single deaminase exhibited a tunable conversion efficiency by titrating the expression of the editing components.20 In the novel platform, we validated that fine-tuning the expression level of ABE8e-CDA-nCas9 enabled the generation of differential editing spectra, resulting in highly diversified mutants (Fig. 4a, S4, 5, and S5†). Moreover, the simultaneous editing of multiple targets generated more mutagenesis diversity than multiple rounds of sequential single editing, although they were mediated by the same number of sgRNAs (Fig. 6c and S6a†). Thus, both the tunable conversion efficiency and the expanded editable window contribute to the high diversity of genomic mutagenesis, which are beneficial toward evolving the cognate proteins and eventually rendering the evolution of bacterial chassis.
Similar to the most recent state-of-the-art mutagenesis systems that engineer proteins by screening and selection,3,4,12 we successfully isolated an evolved B. subtilis with enhanced resistance against nisin by evolving PsdB in vivo using the platform (Fig. 7c and S7†). An evolved B. subtilis cell without any factor of the ABE8e-CDA-nCas9 would be easily obtained by substitution of the two amino acids based on the information from the platform. It is notable that the gene of PsdB is the endogenous gene of the B. subtilis, but not the inserted exogenous genes. The safety of the evolved B. subtilis would not be affected theoretically, which is important for the hosts being applied in biotechnology industry. In addition, the mutant sites were ensured to be in the appointed positions by the designed sgRNAs for targeting genes. This enabled the avoiding of mutations occurring on other genes in the genome, which are often found in mutants caused by chemical and physical mutagenesis strategies. Therefore, this is a semi-rational strategy for protein and cell evolution, which could significantly increase the evolutionary efficiency.
Compared with the single-deaminase base editor, the dual-base editor can efficiently produce a mutant library with much more diversity in vivo,21 which is promising to protein and cell evolution. In addition, the dual-base editor can balance the content of base types by concurrently introducing C-to-T and A-to-G mutations, which is beneficial to the growth of cells. The construction of ABE8e-CDA-nCas9 does not rely on any additional or host-dependent factors, indicating that such BEs may be readily constructed and applicable to a wide range of bacteria. The protein evolution information resulting from such platform would provide guidelines for powerful host cells and chassis cells obtained through protein engineering. Together with the rapid growth characteristics of microbial cells and the simple operation feature of the dual-base editor construction, this study would give an effective strategy for the construction of evolved bacterial chassis.
Conclusions
We designed a novel dual-base editor in microbial cells by reprogramming the CRISPR-Cas9, cytidine and adenosine deaminase, enabling the performance of simultaneous C-to-T and A-to-G conversion in vivo. The dual-base editor can generate a large number of mutations in the defined editing window by adjusting the editing spectrum. This feature is useful for the construction of protein libraries in vivo. In addition, the simultaneous editing of multiple targets generated more mutant diversity than sequential editing, although they were mediated by the same number of sgRNAs. Using the in vivo gene diversity platform, we evolved PsdB and obtained a series of evolved bacterial chassis resistant to nisin. Overall, this study provided a programmable in vivo genomic diversification platform with tunable editing properties, which is promising to expedite the fabrication of high-performance and robust bacterial chassis used in the field of biomanufacturing and biopharmaceuticals.Experimental section
Bacterial strains, plasmids, primers and targeting sgRNA design
The strains used in this study are described in Table S1.† The E. coli JM109 strains (General Biosystems, China) were used as the host for plasmid propagation. E. coli strains were grown aerobically at 37 °C in Luria–Bertani (LB) liquid medium (10 g per L tryptone, 5 g L−1 yeast extract, 10 g per L NaCl, pH 7.0), or on LB-agar plates supplemented with ampicillin (100 μg mL−1) or spectinomycin (50 μg mL−1) when necessary. B. subtilis 168 strains were cultivated in LB liquid medium or LB solid medium supplemented with spectinomycin (50 μg mL−1), chloramphenicol (5 μg mL−1), kanamycin (50 μg mL−1) or tetracycline (15 μg mL−1). The primers and plasmids used in this study are listed in Tables S2 and S3,† respectively. The sgRNAs were designed based on a previous study via online software (https://chopchop.cbu.uib.no/).42 The relevant protospacer sequences are listed in Table S4.†Plasmids construction
The targeting plasmid pHYT-P43-G10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 43 (kindly provided as a gift from Prof. Guimin Zhang from Hubei University) was used as the backbone to construct the sgRNA-expression vectors. The plasmid pHY-ECBE harboring sigE-targeting sgRNA was used, which was reported in our previous study.20 The plasmid pHY-B1 harboring bceB-targeting sgRNA was constructed by reverse PCR (rPCR) using the primer pHY-B1-F/pHY-B1-R. Similarly, the construction method of the pHY-B2-7 plasmids and pHY-pksA-G plasmids was the same as that of pHY-B1.
43 (kindly provided as a gift from Prof. Guimin Zhang from Hubei University) was used as the backbone to construct the sgRNA-expression vectors. The plasmid pHY-ECBE harboring sigE-targeting sgRNA was used, which was reported in our previous study.20 The plasmid pHY-B1 harboring bceB-targeting sgRNA was constructed by reverse PCR (rPCR) using the primer pHY-B1-F/pHY-B1-R. Similarly, the construction method of the pHY-B2-7 plasmids and pHY-pksA-G plasmids was the same as that of pHY-B1.
To construct dual base editors based on ABE8e, ABE7.10, and CDA, we fused two kinds of deaminases into nCas9 in different order. Here, we selected the plasmid pAX-CDA-nCas9, which was constructed in our previous research, as the template for constructing these different fusions. Specifically, pAX-CDA-nCas9 and ABE7.10 were amplified using the primers TadA-CDA-nCas9-b-F/TadA-CDA-nCas9-b-R (used by both fragments) and TadA1-F/TadA1-R, respectively. The PCR product of the ABE7.10 fragment was then assembled into a linearized plasmid using the Gibson assembly method, yielding plasmid pAX-ABE7.10-CDA-nCas9. Similarly, pAX-CDA-nCas9 and ABE8e were amplified using the primers ABE8e-CDA-nCas9-b-F/ABE8e-CDA-nCas9-b-R and ABE8e1-F/ABE8e1-R, respectively. The resulting PCR products were ligated by Gibson assembly, generating the pAX-ABE8e-CDA-nCas9 plasmid. The ABE8e and ABE7.10 genes were synthesized by GENEWIZ (Inc. SuZhou, China). For the construction of plasmid pAX-CDA- ABE7.10-nCas9, pAX-CDA-nCas9 and ABE7.10 were amplified using primers CDA-TadA-nCas9-b-F/CDA-TadA-nCas9-b-R and TadA2-F/TadA2-R, respectively. The PCR product of the ABE7.10 fragment was then assembled into a linearized plasmid using the Gibson assembly method, yielding plasmid pAX-CDA-TadA-nCas9. Similarly, for the construction of plasmid pAX-CDA-ABE8e-nCas9, pAX-CDA-nCas9 and ABE8e were amplified using primers CDA-ABE8e-nCas9-b-F/CDA-ABE8e-nCas9-b-R and ABE8e2-F/ABE8e2-R. The resulting PCR products were ligated by Gibson assembly, generating the pAX-CDA-ABE8e-nCas9 plasmid. For the construction of plasmid pAX-ABE7.10-nCas9-CDA, pAX-nCas9-CDA and ABE7.10 were amplified using primers TadA-nCas9-CDA-b-F/TadA-nCas9-CDA-b-R and TadA3-F/TadA3-R, respectively. The PCR product of the ABE7.10 fragment was then assembled into a linearized plasmid using the Gibson assembly method, yielding plasmid pAX-ABE7.10-nCas9-CDA.
To construct a multiplex base editing system for the editing of bceB, plasmid pAD123 was used as the donor vector for the expression of sgRNA driven by the strong constitutive promoter Pveg. Using pAD123, the plasmids pAD-B1, pAD-B2, pAD-B3, pAD-B4, pAD-B5, pAD-B6, and pAD-B7, which harbored sgRNAs targeting bceB (B1), bceB (B2), bceB (B3), bceB (B4), bceB (B5), bceB (B6), and bceB (B7), respectively, were generated by reverse PCR using primers bceB-B1-F/bceB-B1-R, bceB-B2-F/bceB-B2-R, bceB-B3-F/bceB-B3-R, bceB-B4-F/bceB-B4-R, bceB-B5-F/bceB-B5-R, bceB-B6-F/bceB-B6-R, and bceB-B7-F/bceB-B7-R, respectively. Similarly, the method of the construction of plasmids targeting psdB for multiplex base editing was the same as that described above. The plasmids pAD-P1, pAD-P2, pAD-P3, pAD-P4, and pAD-P5 that harbored sgRNAs targeting psdB (P1), psdB (P2), psdB (P3), psdB (P4), and psdB (P5), respectively, were generated by reverse PCR using primers psdB-P1-F/psdB-P1-R, psdB-P2-F/psdB-P2-R, psdB-P3-F/psdB-P3-R, psdB-P4-F/psdB-P4-R, and psdB-P5-F/psdB-P5-R, respectively.
pDGT-GFP-Ampm20 was used as the backbone to construct plasmids harboring sgRNAs for multiplex base editing. To generate a multi-editing plasmid targeting bceB, pDGT-B1-7 harboring sgRNAs targeting the bceB (B1), bceB (B2), bceB (B3), bceB (B4), bceB (B5), bceB (B6), and bceB (B7) loci was constructed using primers B1-Go-F/B1-Go-R, B2-Go-F/B2-Go-R, B3-Go-F/B3-Go-R, B4-Go-F/B4-Go-R, B5-Go-F/B5-Go-R, B6-Go-F/B6-Go-R, B7-Go-F/B7-Go-R, and bceB-Go-b-F/R, respectively. Similarly, the plasmid pDGT-P1-5 harboring sgRNAs targeting psdB (P1), psdB (P2), psdB (P3), psdB (P4), and psdB (P5) loci was constructed using primers P1-Go-F/P1-Go-R, P2-Go-F/P2-Go-R, P3-Go-F/P3-Go-R, P4-Go-F/P4-Go-R, P5-Go-F/P5-Go-R, and psdB-Go-b-F/R, respectively. The plasmid pDGT-P1-P5 harboring sgRNAs targeting psdB (P1) and psdB (P5) loci was constructed using primers P1-Go-F/P1-Go-R, P2-Go-F/P5-Go-R, and psdB-Go-b-F/R. To construct the pJOE series plasmids for sequential editing, the pJOE8999 plasmid44 and pAD-B2-4 plasmids were used as templates to amplify the pJOE skeleton and expression cassette of sgRNA targeting bceB (B2, B3, and B4) using primers pJOE-b-F/R and pJOE-F/R, respectively. The PCR products were ligated using the Gibson assembly, yielding plasmid pAX-TadA-nCas9-CDA. The generated sgRNAs targeting bceB (B2, B3, and B4) were connected with the pJOE receptor vector using the Gibson assembly, yielding plasmids pJOE-B2, pJOE-B3, and pJOE-B4, respectively.
Methods for the transformation and genome modification of B. subtilis
For the protocol of B. subtilis transformation, a modified method of Anagnostopoulos and Spizizen was used in this study.Protocol: (a) A volume of 2 ml SPI culture was inoculated overnight from a fresh colony at 37 °C in a 14 mL disposable tube, with 200 rpm shaking. (b) The following morning, 40 μL of the culture was diluted with 2 mL fresh SPI and continued incubation at 37 °C with 200 rpm shaking for 4–5 h. (c) A volume of 200 μL of the culture was mixed with 2 mL fresh SPII with continued incubation at 37 °C for an additional 1.5–2 h. (d) A volume of 20 μL EGTA (10 mM) was added to the culture, and the competent cells was obtained after an additional 10 min shake cultivation. (e) A volume of 500 μL of the competent cells was divided into 1.5 mL sterile tubes, and then incubated at 37 °C for 1.5–2 h after adding 0.5–2 μg DNA into every tube. (f) After centrifugation (4500 g) for 2 min, the cell pellet was gently resuspended in 150–200 μL of the saved supernatant, and then plated onto LB agar plates with selective antibiotic(s). For the preparation of the relevant medias, see the ESI Method.†
A genome engineering method based on a homologous recombination system and PCR was used for overexpression of the target gene in B. subtilis. In brief, the gene (such as ABE8e-CDA-nCas9) to be integrated needs to be cloned into an integration plasmid with a pair of specific homologous arms (such as lacA homologous arms on the pAX01 plasmid). The expression of the ABE8e-CDA-nCas9 fusion gene is regulated by xylose on this plasmid (pAX-ABE8e-CDA-nCas9). Then, a pair of specific primers were used to amplify the upstream and downstream homologous arms (the gene to be expressed and the resistance marker were in the middle of the homologous arms) to prepare the fragments to be transformed. Transformation of the prepared fragment (0.5–2 μg) into competent cells was achieved according to the above transformation method. The target gene can be integrated into a specific locus (such as lacA locus) through its own recombinase system and dual-crossing of homologous arms. The important genetic parts of the related integrated plasmids (pAX and pDGT plasmids) have been shown in the ESI Sequence.†
Protocol for the multiple rounds of sequential single editing
For the first editing generation, pJOE-B2 was transformed into BS4, yielding BS4-pJOE-B2. After induction in the LB liquid medium by xylose, the BS4-pJOE-B2 culture was diluted to the appropriate density and spread onto a LB plate to cure pJOE-B2 at 45–50 °C for overnight. For the second editing generation, the cured-pJOE-B2 strains on the LB plate were completely washed with sterile water. The following culture was then inoculated into the transformation solution and made into competent cells. The pJOE-B3 was transformed into the above competent cells and cultured overnight on the plate. The resulting BS4-pJOE-B3 strains on the plate were completely washed with sterile water, and then the following culture was inoculated into fresh LB liquid medium for induction. The induced BS4-pJOE-B3 was diluted to an appropriate density and spread onto a LB plate to cure pJOE-B3 at 45–50 °C for overnight. For the third editing generation, the cured-pJOE-B3 strains on the LB plate were completely washed with sterile water, and then the following culture was inoculated into the transformation solution and made into competent cells. The pJOE-B4 was transformed into the above competent cells and cultured overnight on the plate. The resulting BS4-pJOE-B4 strains on the plate were completely washed with sterile water, and then the following culture was inoculated into fresh LB liquid medium for induction. The induced BS4-pJOE-B4 was diluted to the appropriate density and spread onto a LB plate to cure pJOE-B4 at 45–50 °C for overnight. The following colonies can be selected for the identification of genetic diversity by sequencing.Plasmid curing
Here, the pJOE8999 plasmid44 was selected for multiple rounds of editing of B. subtilis to accumulate more mutations. The vector pJOE8999 contained the minimal pUC origin for replication in E. coli and the temperature-sensitive replicon repEts for replication in B. subtilis. Three plasmids derived from the pJOE8999 plasmid were generated (pJOE-B2, pJOE-B3, and pJOE-B4), which contained sgRNA targeting different loci of bceB. When B. subtilis containing pJOE series plasmids propagates, it needs to be cultured at 30 °C. The pJOE series plasmids (pJOE-B2, pJOE-B3, and pJOE-B4) were cured when the mutant strains were cultured at 45–50 °C for 12 h.Construction of a single editing and multiplex editing system
To construct a single editing system, the integration vector pAX-ABE8e-CDA-nCas9 harboring the fusion protein ABE8e-CDA-nCas9 was transformed into the genome of B. subtilis at the lacA site. For the editing of sigE, a plasmid harboring a single sgRNA was introduced into the ABE8e-CDA-nCas9-integrated host (BS4). To edit bceB, seven sgRNAs targeting different positions on bceB were designed. These sgRNAs were overexpressed through the P43 promoter on the pHYT-P43-G10 vector. The editing of the pks operon is the same as that of bceB. To construct a multiplex editing system for the BceB mutagenesis experiments, sgRNAs with 20 nt targeting bceB (B1), bceB (B2), bceB (B3), bceB (B4), bceB (B5), bceB (B6), and bceB (B7) were combined and integrated into the amyE site of BS4. The construction of a multiplex editing system targeting PsdB was similar to the BceB mutagenesis experiments. A marker-free genome editing approach was used to perform gene overexpression in B. subtilis as previously reported.45DNA sequencing at the single-clone level and population level
Two methods of sequencing, single-clone sequencing and population-level sequencing, were used to analyze the editing efficiency of the dual-base editor. The colonies were randomly selected from the LB agar plates after induction by xylose for single-clone sequencing. Colony PCR was performed on the selected colonies, and the PCR products were sequenced to verify C-to-T and A-to-G conversions. The colonies were cultured for approximately 10 h in a test tube containing different concentrations of xylose. Then, the induced culture was used as a template to amplify the position of the expected mutation by using customized primer pairs. The PCR products were used for population sequencing, and the data was analyzed quantitatively by online software.46Alamar blue method
The growth inhibition activity of nisin was determined against mutant B. subtilis 168 (M1–M8) by Alamar blue. Wild-type B. subtilis 168 strains were used as controls. The inhibition rate of mutants was performed by culturing 100 μL diluted overnight cells (OD600 = 1.7) into LB medium supplemented with 10% Alamar blue (v/v) and 5% (v/v) 0.1 mg per mL nisin, and incubating at 37 °C for 5 h (the total volume was 2 mL). Two hundred microliters of the culture was used for fluorescence detection (the excitation wavelength and emission wavelength were 570 nm and 590 nm, respectively). The inhibition rate was measured by the above culture fluorescence intensities, and the inhibition rate was calculated using the following equation: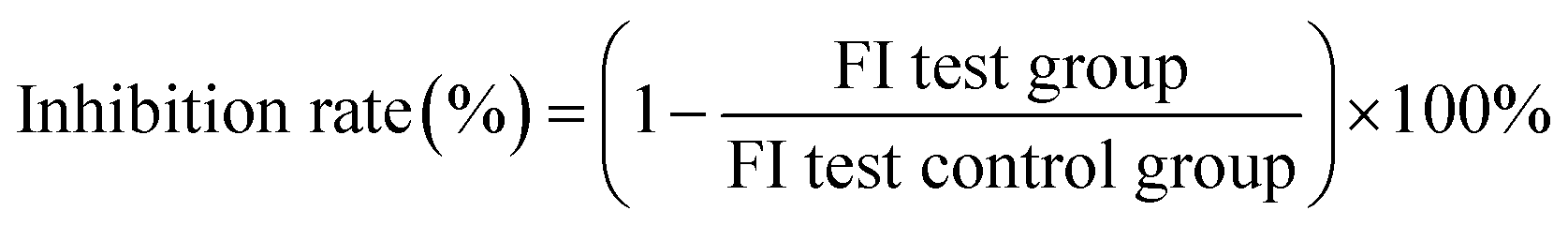
When the value is less than 0, it indicates that the tolerance of the mutants is increased compared with the wild type. When the value is greater than 0, it indicates that the mutants have higher sensitivity to nisin.
Genome-wide off-target identification of a dual-base editor in B. subtilis
Illumina sequencing libraries were constructed from each strain using the VAHTS® Universal Pro DNA Library Prep Kit from Vazyme (China) according to the manufacturer's protocol. Input DNA from each strain was adjusted to 1 ng μL−1, and 11 PCR cycles were run with multiplex indexing primers. VAHTS® DNA Clean Beads (Vazyme, China) were used to size select approximately 200 nt fragments prior to PCR amplification. Libraries were quantified with a Fragment Analyser™ (Agilent Technologies, USA) and Qubit (Thermo Fisher Scientific, USA). A single Illumina MiSeq 2 × 150 nt run yielded between 14418952 and 18735478 reads per sample, and the corresponding coverage was 99.98%. Raw sequencing reads were adapter and quality trimmed using AdapterRemoval with the switches-trimns-trimqualities. Breseq (v0.33.2)47 was used for SNP calling with the parameters -p and -polymorphism-frequency-cut-off 0.2 to allow for variants existing in 20% to 100% of the reads. All raw data and the sequenced genome sequence of B. subtilis have been deposited at NCBI under BioProject accession PRJNA808834.
Statistical analysis
The statistical significance of editing efficiency was assessed by comparing the mean values (±S.D.) using Student's t-test. p < 0.01 was considered significant (**, p < 0.01).Data availability
Deep-sequencing data are available under BioProject ID PRJNA808834 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA808834).Author contributions
H. W. and C. W. performed all of the experiments with help from all authors. H. W., C. W., C. Z. and Z. Z. wrote the manuscript with input from all authors. S. F. and H. L. provided technical advice. H. W., C. W. and Z. Z. conceived the project.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was financially supported by a project funded by the International S&T Innovation Cooperation Key Project (2017YFE0129600), the National Natural Science Foundation of China (21878125, 32171420), the Natural Science Foundation of Jiangsu Province (BK20181206), the Priority Academic Program Development of Jiangsu Higher Education Institutions, the 111 Project (No. 111-2-06), the Jiangsu Province “Collaborative Innovation Centre for Advanced Industrial Fermentation” Industry Development Program, and First-Class Discipline Program of Light Industry Technology and Engineering (LITE2018-04). We greatly appreciate Prof. Guimin Zhang for providing plasmids (pHY-G10-sgRNA and pDG-P43-GFP) for our research.Notes and references
- D. Tavares and J. J. A. van der Meer, Subcellular localization defects characterize ribose-binding mutant proteins with new ligand properties in Escherichia coli, Appl. Environ. Microbiol., 2022, 88, e0211721, DOI:10.1128/aem.02117-21.
- J. D. Keasling, Synthetic biology for synthetic chemistry, ACS Chem. Biol., 2008, 3, 64–76 CrossRef CAS PubMed.
- S. O. Halperin, et al., CRISPR-guided DNA polymerases enable diversification of all nucleotides in a tunable window, Nature, 2018, 560, 248–252, DOI:10.1038/s41586-018-0384-8.
- A. Cravens, O. K. Jamil, D. Kong, J. T. Sockolosky and C. D. Smolke, Polymerase-guided base editing enables in vivo mutagenesis and rapid protein engineering, Nat. Commun., 2021, 12, 1579, DOI:10.1038/s41467-021-21876-z.
- Y. Wang, et al., Directed evolution: methodologies and applications, Chem. Rev., 2021, 121, 12384–12444, DOI:10.1021/acs.chemrev.1c00260.
- A. Currin, N. Swainston, P. J. Day and D. B. Kell, Synthetic biology for the directed evolution of protein biocatalysts: navigating sequence space intelligently, Chem. Soc. Rev., 2015, 44, 1172–1239, 10.1039/c4cs00351a.
- A. Currin, et al., The evolving art of creating genetic diversity: from directed evolution to synthetic biology, Biotechnol. Adv., 2021, 50, 107762, DOI:10.1016/j.biotechadv.2021.107762.
- F. Katzen, T. C. Peterson and W. Kudlicki, Membrane protein expression: no cells required, Trends Biotechnol., 2009, 27, 455–460, DOI:10.1016/j.tibtech.2009.05.005.
- H. H. Wang, et al., Programming cells by multiplex genome engineering and accelerated evolution, Nature, 2009, 460, 894–898, DOI:10.1038/nature08187.
- A. Nyerges, et al., A highly precise and portable genome engineering method allows comparison of mutational effects across bacterial species, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 2502–2507, DOI:10.1073/pnas.1520040113.
- A. Ravikumar, G. A. Arzumanyan, M. K. A. Obadi, A. A. Javanpour and C. C. Liu, Scalable, continuous evolution of genes at mutation rates above genomic error thresholds, Cell, 2018, 175, 1946–1957, DOI:10.1016/j.cell.2018.10.021.
- B. Alvarez, M. Mencia, V. de Lorenzo and L. A. Fernandez, In vivo diversification of target genomic sites using processive base deaminase fusions blocked by dCas9, Nat. Commun., 2020, 11, 6436, DOI:10.1038/s41467-020-20230-z.
- G. T. Hess, et al., Directed evolution using dCas9-targeted somatic hypermutation in mammalian cells, Nat. Methods, 2016, 13, 1036–1042, DOI:10.1038/nmeth.4038.
- Y. Ma, et al., Targeted AID-mediated mutagenesis (TAM) enables efficient genomic diversification in mammalian cells, Nat. Methods, 2016, 13, 1029–1035, DOI:10.1038/nmeth.4027.
- C. Li, et al., Expanded base editing in rice and wheat using a Cas9-adenosine deaminase fusion, Genome Biol., 2018, 19, 59, DOI:10.1186/s13059-018-1443-z.
- Y. Zong, et al., Efficient C-to-T base editing in plants using a fusion of nCas9 and human APOBEC3A, Nat. Biotechnol., 2018, 36, 950–953, DOI:10.1038/nbt.4261.
- C. L. Moore, L. J. Papa III and M. D. Shoulders, A processive protein chimera introduces mutations across defined DNA regions in vivo, J. Am. Chem. Soc., 2018, 140, 11560–11564, DOI:10.1021/jacs.8b04001.
- S. Banno, K. Nishida, T. Arazoe, H. Mitsunobu and A. Kondo, Deaminase-mediated multiplex genome editing in Escherichia coli, Nat. Microbiol., 2018, 3, 423–429, DOI:10.1038/s41564-017-0102-6.
- Y. Wang, et al., MACBETH: Multiplex automated Corynebacterium glutamicum base editing method, Metab. Eng., 2018, 47, 200–210, DOI:10.1016/j.ymben.2018.02.016.
- W. Hao, et al., Development of a base editor for protein evolution via in situ mutation in vivo, Nucleic Acids Res., 2021, 49, 9594–9605, DOI:10.1093/nar/gkab673.
- J. Grunewald, et al., A dual-deaminase CRISPR base editor enables concurrent adenine and cytosine editing, Nat. Biotechnol., 2020, 38, 861–864, DOI:10.1038/s41587-020-0535-y.
- M. F. Richter, et al., Phage-assisted evolution of an adenine base editor with improved Cas domain compatibility and activity, Nat. Biotechnol., 2020, 38, 883–891, DOI:10.1038/s41587-020-0453-z.
- R. C. Sakata, et al., Base editors for simultaneous introduction of C-to-T and A-to-G mutations, Nat. Biotechnol., 2020, 38, 865–869, DOI:10.1038/s41587-020-0509-0.
- X. Zhang, et al., Dual base editor catalyzes both cytosine and adenine base conversions in human cells, Nat. Biotechnol., 2020, 38, 856–860, DOI:10.1038/s41587-020-0527-y.
- K. Nishida, et al., Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems, Science, 2016, 353, aaf8729 CrossRef PubMed.
- N. M. Gaudelli, et al., Programmable base editing of A·T to G·C in genomic DNA without DNA cleavage, Nature, 2017, 551, 464–471, DOI:10.1038/nature24644.
- A. Staron, D. E. Finkeisen and T. Mascher, Peptide antibiotic sensing and detoxification modules of Bacillus subtilis, Antimicrob. Agents Chemother., 2011, 55, 515–525, DOI:10.1128/AAC.00352-10.
- Y. Quentin, G. Fichant and F. Denizot, Inventory, assembly and analysis of Bacillus subtilis ABC transport systems, J. Mol. Biol., 1999, 287, 467, DOI:10.1006/jmbi.1999.2624.
- E. Rietkotter, D. Hoyer and T. Mascher, Bacitracin sensing in Bacillus subtilis, Mol. Microbiol., 2008, 68, 768–785, DOI:10.1111/j.1365-2958.2008.06194.x.
- M. S. Packer and D. R. Liu, Methods for the directed evolution of proteins, Nat. Rev. Genet., 2015, 16, 379–394, DOI:10.1038/nrg3927.
- T. Vanhercke, C. Ampe, L. Tirry and P. Denolf, Reducing mutational bias in random protein libraries, Anal. Biochem., 2005, 339, 9–14, DOI:10.1016/j.ab.2004.11.032.
- R. Fujii, M. Kitaoka and K. Hayashi, One-step random mutagenesis by error-prone rolling circle amplification, Nucleic Acids Res., 2004, 32, e145, DOI:10.1093/nar/gnh147.
- R. Fujii, M. Kitaoka and K. Hayashi, Error-prone rolling circle amplification: the simplest random mutagenesis protocol, Nat. Protoc., 2006, 1, 2493–2497, DOI:10.1038/nprot.2006.403.
- M. D. Matteucci and H. L. Heyneker, Targeted random mutagenesis: The use of ambiguously synthesized oligonucleotides to mutagenize sequences immediately 5′ of an ATG initiation codon, Nucleic Acids Res., 1983, 11, 3113–3121 CrossRef CAS PubMed.
- J. F. Reidhaar-Olson and R. T. Sauer, Combinatorial cassette mutagenesis as a probe of the informational content of protein sequences, Science, 1988, 241, 53–57 CrossRef CAS PubMed.
- S. Byrappa, D. K. Gavin and K. C. Gupta, A highly efficient procedure for site specific mutagenes of full-length plasmids using Vent DNA polymerase, Genome Res., 1995, 5, 404–407 CrossRef CAS PubMed.
- W. P. Stemmer, DNA shuffling by random fragmentation and reassembly: in vitro recombination for molecular evolution, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 10747–10751 CrossRef CAS PubMed.
- W. P. Stemmer, A. Crameri, K. D. Ha, T. M. Brennan and H. L. Heyneker, Single-step assembly of a gene and entire plasmid from large numbers of oligodeoxyribonucleotides, Gene, 1995, 164, 49–53 CrossRef CAS PubMed.
- Y. Tong, et al., Highly efficient DSB-free base editing for streptomycetes with CRISPR-BEST, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 20366–20375, DOI:10.1073/pnas.1913493116.
- C. Li, et al., Targeted, random mutagenesis of plant genes with dual cytosine and adenine base editors, Nat. Biotechnol., 2020, 38, 875–882, DOI:10.1038/s41587-019-0393-7.
- S. Yu, et al., CRISPR-dCas9 mediated cytosine deaminase base editing in Bacillus subtilis, ACS Synth. Biol., 2020, 9, 1781–1789, DOI:10.1021/acssynbio.0c00151.
- K. Labun, et al., CHOPCHOP v3: expanding the CRISPR web toolbox beyond genome editing, Nucleic Acids Res., 2019, 47, W171–W174, DOI:10.1093/nar/gkz365.
- Z. Lu, et al., CRISPR-assisted multi-dimensional regulation for fine-tuning gene expression in Bacillus subtilis, Nucleic Acids Res., 2019, 47, e40, DOI:10.1093/nar/gkz072.
- J. Altenbuchner, Editing of the Bacillus subtilis genome by the CRISPR-Cas9 system, Appl. Environ. Microbiol., 2016, 82, 5421–5427, DOI:10.1128/AEM.01453-16.
- X. Yan, H. J. Yu, Q. Hong and S. P. Li, Cre/lox system and PCR-based genome engineering in Bacillus subtilis, Appl. Environ. Microbiol., 2008, 74, 5556–5562, DOI:10.1128/AEM.01156-08.
- M. G. Kluesner, et al., EditR: a method to quantify base editing from sanger sequencing, CRISPR J., 2018, 1, 239–250, DOI:10.1089/crispr.2018.0014.
- D. E. Deatherage and J. E. Barrick, Identification of mutations in laboratory-evolved microbes from next-generation sequencing data using breseq, Methods Mol. Biol., 2014, 1151, 165–188, DOI:10.1007/978-1-4939-0554-6_12.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc05824c |
| ‡ Wenliang Hao and Wenjing Cui contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |