
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChirality-controlled polymerization-induced self-assembly†
Haolan
Li
 a,
Erik Jan
Cornel
*a,
Zhen
Fan
a and
Jianzhong
Du
*ab
a,
Erik Jan
Cornel
*a,
Zhen
Fan
a and
Jianzhong
Du
*ab
aDepartment of Polymeric Materials, School of Materials Science and Engineering, Tongji University, 4800 Caoan Road, Shanghai 201804, China. E-mail: 20310048@tongji.edu.cn; jzdu@tongji.edu.cn
bDepartment of Gynaecology and Obstetrics, Shanghai Fourth People's Hospital, School of Medicine, Tongji University, Shanghai 200434, China
First published on 17th November 2022
Abstract
Recent studies have shown that biodegradable nanoparticles can be efficiently prepared with polymerization of N-carboxyanhydrides-induced self-assembly (NCA-PISA). However, thus far, the effect of chiral monomer ratio on such NCA-PISA formulations and the resulting nanoparticles has not yet been fully explored. Herein, we show, for the first time, that the morphology, secondary structure, and biodegradation rate of PISA nanoparticles can be controlled by altering the chiral ratio of the core-forming monomers. This chirality-controlled PISA (CC-PISA) method allowed the preparation of nanoparticles that are more adjustable and applicable for future biomedical applications. Additionally, the complex secondary peptide structure (ratio of α-helix to β-sheet) and π–π stacking affect the polymer self-assembly process. More specifically, a PEG45 macro-initiator was chain-extended with L- and D-phenylalanine (L- and D-Phe-NCA) in various molar ratios in dry THF at 15 wt%. This ring-opening polymerization (ROP) allowed the preparation of homo- and hetero-chiral Phe-peptide block copolymers that self-assembled in situ into nanoparticles. For homo-chiral formulations, polymers self-assembled into vesicles once a sufficiently high phenylalanine degree of polymerization (DP) was obtained. Hetero-chiral formulations formed larger nanoparticles with various morphologies and, much to our surprise, using an equal enantiomer ratio inhibited PISA and led to a polymer solution instead. Finally, it was shown that the enzymatic biodegradation rate of such PISA particles is greatly affected by the polymer chirality. This PISA approach could be of great value to fabricate nanoparticles that exploit chirality in disease treatment.
Introduction
Polymer nanoparticles have interesting properties in biomedical applications, as they can improve the stability and solubility of drugs, and facilitate transmembrane transport effectively.1 Such nanoparticles are, therefore, suitable for drug loading and delivery,2–4 vaccine adjuvants,5 various disease treatments,6–10 and antimicrobial applications.11 An emerging method to prepare polymer nanoparticles is Polymerization-Induced Self-Assembly (PISA).12–14 This is an efficient nanoparticle synthesis method that combines polymer synthesis and self-assembly in a single step. Moreover, nanoparticles with various morphologies (spheres, worms, and vesicles) and chemical compositions can be prepared at high solids concentration in polar15,16 and non-polar media.17–19 Generally, PISA comprises the chain extension of a soluble polymer block (stabilizer block) with a second polymer block (core-forming block) that becomes progressively insoluble as the polymerization proceeds. These growing block copolymers self-assemble during the polymerization reaction once a critical degree of polymerization (DP) of the core-forming block is achieved. Other known PISA driving forces include hydrogen bonding, electrostatic interactions, and crystallization.20–25 Various nanoparticle morphologies can be targeted reproducibly by adjustment of the stabilizer/core-forming block DP, and the polymer concentration (solids content).16,26,27PISA is generally conducted at a relatively high solids content (10–50 wt%), which makes PISA more efficient compared to post-polymerization self-assembly methods such as the solvent switch28,29 or the rehydration approach,30 which are generally conducted at approximately 0.1–3.0 wt%. PISA is well-established in combination with reversible addition–fragmentation chain-transfer (RAFT) polymerization.31–33 However, other polymerization methods have shown potential, including photo-PISA.34–37 Despite the efficiency and versatility of PISA, it remains challenging to prepare biodegradable nanoparticles. A promising PISA method that solves this problem utilizes the ring-opening polymerization (ROP) of N-carboxyanhydrides (NCA-monomers); this method is also known as NCA-PISA or ROPISA.38–41 The resulting core-forming polypeptide block is biodegradable since it can be hydrolyzed with acids or enzymes.38,42–44
The first NCA-PISA example was reported by our group in 2019.38 Here, PEG45-NH2 was chain-extended with phenylalanine-NCA (L-Phe-NCA) in THF to form biodegradable PEG45-b-L-PPhex diblock copolymer nanoparticles. It was shown that specific nanoparticle morphologies could be prepared by varying the core-forming block DP and the copolymer concentration: spheres formed at 10 wt% and vesicles were observed when a longer core-forming block DP was used at 20 wt%. Interestingly, NCA-PISA can also be directly performed in aqueous media, as shown by Lecommandoux and co-workers.39 Spheres, worms, and needles could be prepared by chain extending a PEG110-NH2 with γ-benzyl-L-glutamate-NCA (BLG-NCA) in water. The versatility of this PISA method was demonstrated in a subsequent study where L-leucine-NCA (Leu-NCA) was used instead.40 Additionally, a methionine (Met)-based one-pot NCA-PISA procedure in DMSO was reported by Battaglia and co-workers.41 These PEG-b-PMet formulations allowed the preparation of nanoparticles with various morphologies (spheres, worms, and vesicles) and allowed the preparation of a pseudo-phase diagram.
A so far scarcely explored factor on NCA-PISA is the role of polymer chirality. This is of considerable interest since there are various literature reports in which chirality is used in RAFT-PISA to prepare chiral nanoparticles with (meth)acrylate monomers.45,46 Here, the chirality of a (meth)acrylate monomer is transferred to the polymer and is, afterwards, transferred during PISA to the nanoparticles, a common example of a chiral nanostructure is the twisted fiber morphology. Additionally, it is known that the adjustment of chirality can lead to various secondary peptide structures and that varying secondary structures can affect the morphology of peptide nanoparticles.47–49 Lecommandoux and co-workers showed that changes in secondary structure can result in more stable nanoparticles.50 Therefore, chirality and the secondary structure might play an important morphology directing role in NCA-PISA. Additionally, nanoparticles that consist of hetero-chiral block copolymers should have an adjustable biodegradation rate. This is important for future biomedical applications, for example, to adjust the drug release rate of drug nanocarriers.2–4
Herein, it is shown that monomer chirality can be used to control the nanoparticle morphology during CC-PISA, the resulting nanoparticles have a differing enzymatic biodegradability profile which depends on the degree of chirality. More specifically, PEG45-b-P(D/L-Phe)x block copolymer nanoparticles were prepared in THF at 15 wt% (Scheme 1). Homo-chiral formulations (where either L-Phe-NCA or D-Phe-NCA is used) resulted in vesicles with a well-defined membrane. Hetero-chiral compositions led to various other kinds of nanoparticles. Polymerizing equal amounts of L- and D-NCA-monomers is also possible; however, this prevents in situ polymer self-assembly. This aspect allowed us to gain insight into the unusual self-assembly mechanism during this PISA reaction. In summary, this work provides a new way to introduce morphological control during NCA-PISA and gives novel insights into the self-assembly mechanism during NCA-PISA. This work is different from the RAFT-PISA formulations where monomer chirality is used to prepare chiral nanoparticles. Here, the chiral monomer ratio was used as a handle to control a peptide-based PISA process, no chiral nanoparticles were obtained. Furthermore, the enzymatic degradability of such nanoparticles can be adjusted. Both factors make the herein discussed nanoparticles suitable for potential future chiral pharmaceutical applications.51
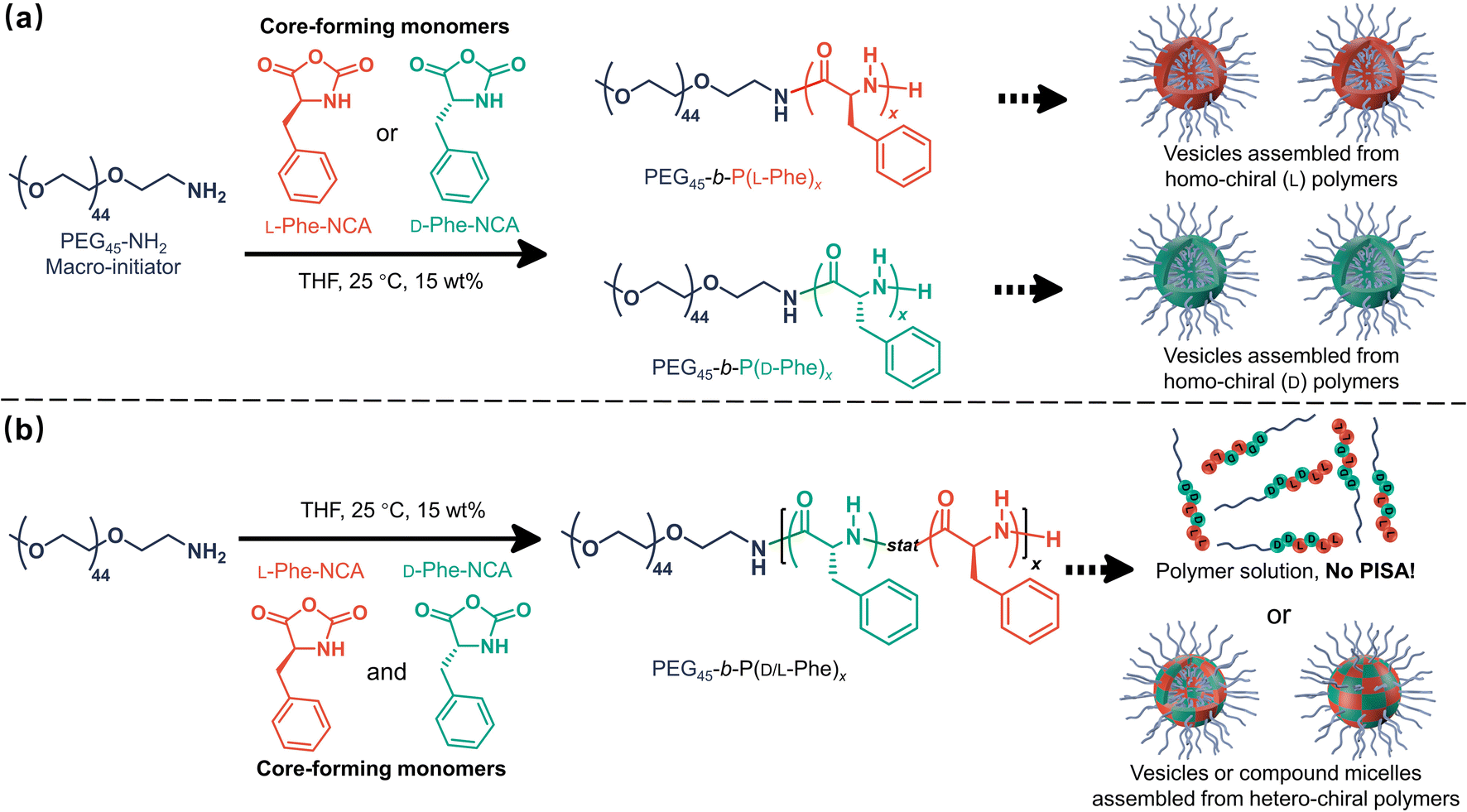 | ||
| Scheme 1 Chirality-controlled polymerization-induced self-assembly (CC-PISA) for preparing nanoparticles with adjustable morphology and biodegradation rate. The effect of core-forming monomer chirality on NCA-PISA was investigated by adjusting the initial ratio of L-Phe-NCA to D-Phe-NCA in THF (15 wt%) at 25 °C. (a) Vesicles self-assembled from homo-chiral polymers were prepared from either L-Phe-NCA or D-Phe-NCA. (b) Vesicles or compound micelles self-assembled from hetero-chiral polymers were prepared from a mixture of these two monomers. PISA was inhibited when equal amounts of L-Phe-NCA and D-Phe-NCA were used, leading to a block copolymer solution. NB. The polymer chains in section ‘b’ are a schematic representation, in reality the length and the sequence of the polymer chains vary. | ||
Results and discussion
Effect of chirality in CC-PISA
The effect of core-forming monomer chirality on the PEG45-b-P(Phe)x PISA formulation in THF was explored. This was achieved by performing this PISA protocol with L-Phe-NCA, D-Phe-NCA, and mixtures thereof (Scheme 1). A range of block copolymers with a PPhe target core-forming block DP ranging from 5 to 20 were prepared at 15 wt% in THF. The effect of monomer chirality (and chiral ratio) was systematically investigated by adjusting the initial ratio between L-Phe-NCA and D-Phe-NCA. Varying this parameter drastically affects nanoparticle self-assembly behavior (Fig. 1). Using equal amounts of chiral monomers led to a free-flowing polymer solution for all assessed core-forming block DPs. Homo-chiral formulations formed dispersed nanoparticles in the form of a free-flowing dispersion when a DP of 5 was targeted; a gel was formed when longer DPs were targeted. Interestingly, formulations with intermediate chiral ratios (with an L-Phe-NCA content of around 10–30%, and 70–90%) did not form a gel and appeared less turbid than the homo-chiral formulations, indicating changes in nanoparticle morphology. The nanoparticle-polymer chain boundary seemed to be affected by the target DP. A transparent solution was observed when an L-Phe-NCA content between 30–70% was used for the DP 5 samples. This range decreased to 40–60% when higher core-forming block DPs were targeted. This indicates that both the targeted DP and the L-Phe-NCA content play an important role in this CC-PISA formulation. These data show that the self-assembly process of the DP 5 species is more affected by the introduction of hetero-chirality compared to the DP 10 species. This can be expected as the ability of the shorter chain to form a PISA-favoring secondary structure is more hindered by the amount of hetero-chirality, compared to the formulations with higher DPs (3 to 4 amino acid residues are required to form an α-helix, a more detailed discussion on the effect of the secondary peptide structure is given at a later stage in this article). | ||
| Fig. 1 Digital images of the final dispersions and solutions obtained from the polymerization of L-Phe-NCA and/or D-Phe-NCA in various ratios in THF at 15 wt%. The L-Phe-NCA content ranged from 0 to 100% (from left to right) and the target core-forming block DPs were 5, 10, 15, and 20 (from top to bottom). The red border identifies the experiments where PISA occurred (turbid dispersions). PISA did not occur in the other experiments and transparent solutions were formed instead. Homo-chiral formulations with a target core-forming block DP > 5 formed a free-standing gel. Here, D-V = D-Vesicles, D/L-V = D/L-Vesicles, CM = Compound Micelles, and L-V = L-Vesicles. Please note that ‘D-, L- and D/L-’ only refer to the type of chiral monomer, and hence the chiral composition of the polymer, not the nanoparticle chirality. | ||
1H nuclear magnetic resonance (NMR) and size exclusion chromatography (THF-SEC) were used to confirm that the targeted DPs were obtained during the polymerization (Fig. S1–S3†). Here, only the block copolymers with an L-Phe content of 50% were evaluated, this is because an equivalent chiral ratio ensures the highest polymer solubility (as in our previous work, vesicles self-assembled from homo-chiral polymers remained insoluble in a range of organic solvents38). Prior to analysis, the samples were purified by dialysis against water and pure copolymer powders were obtained after lyophilization. A small amount of purified polymer was dissolved in deuterated trifluoroacetic acid (d-TFA) prior to 1H NMR analysis. This deuterated solvent was selected to minimize the effect of peptide folding on this analysis method. As expected, the 1H NMR determined DPs were consistent with the targeted DP. This was further supported by THF-SEC (Table S1†). Plotting the calculated 1H NMR DPs versus the obtained SEC Mn gave a linear correlation, indicating a good pseudo-living character for this NCA polymerization (Fig. 2a). Nevertheless, relatively large THF-SEC dispersities were observed for the samples with target DPs of 15 and 20. However, it is known that the dispersities can be artificially enlarged owing to undesired peptide-column interactions that become more prominent when longer DPs are used, as our data confirms. Nevertheless, well-defined vesicles and compound micelles were obtained in this DP range.
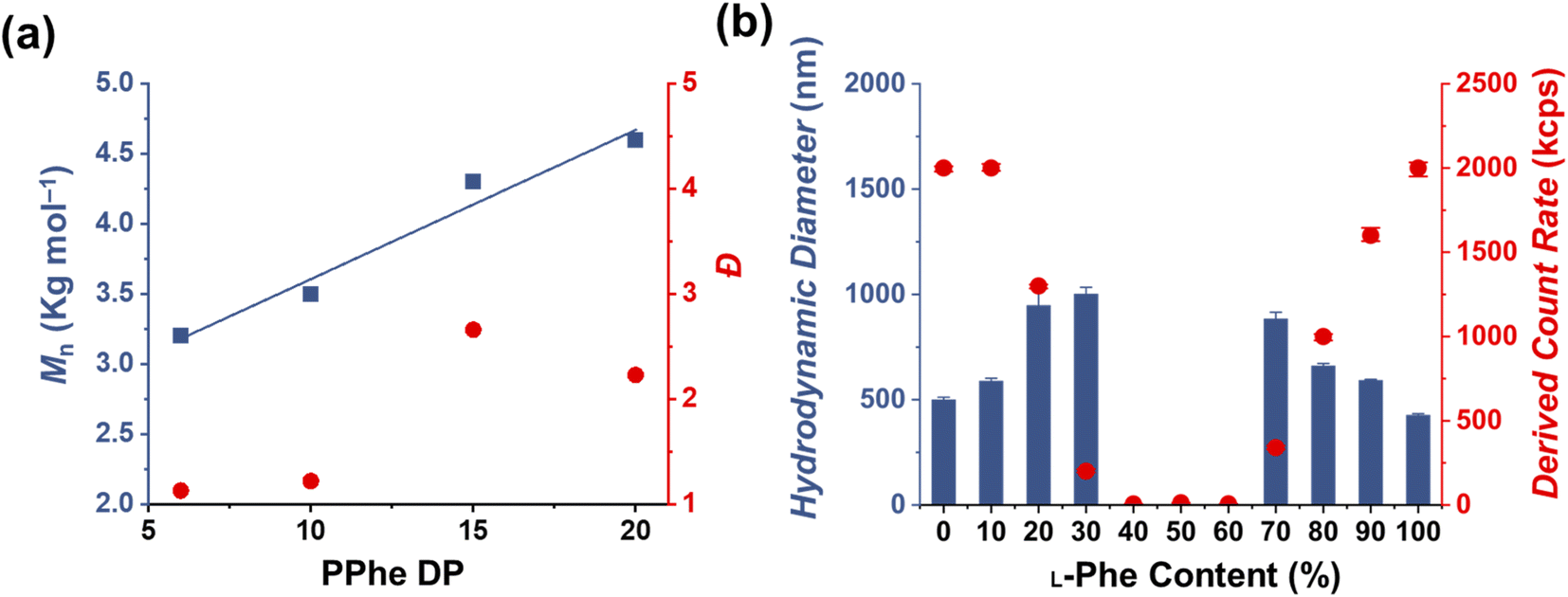 | ||
| Fig. 2 (a) A linear increase in Mn (determined by SEC) was observed with an increased core-forming DP (blue squares), indicating good pseudo-living character. The obtained PPhe DPs were determined by 1H NMR for a series of PEG45-b-P(DL-Phe)x (here, the L-Phe content is 50%) prepared at 15 wt%. SEC dispersities (Đ) (red circles) tend to reach larger values as the target DP increases. (b) DLS study of PEG45-b-P(D/L-Phe)10 copolymer formulations obtained from CC-PISA in THF at 15 wt%. Here, the L-Phe-NCA content is increased from 0–100%. L-Phe contents between 40% and 60% do not show evidence of self-assembled nanoparticles obtained from PISA. | ||
Dynamic light scattering (DLS) studies were performed to assess the change in turbidity upon altering the L-Phe-NCA content for the PEG45-b-P(D/L-Phe)10 formulations (Fig. 2b); the hydrodynamic diameter (Dh) and the count rate were determined for all formulations (Fig. S4†). These data support the optical observation of a reduced turbidity in the range of 40–60% L-Phe-NCA. A derived count rate of approximately 2000 kcps was observed for the homo-chiral compositions, this value significantly reduced when mixtures of chiral monomers were used. The formulations with an L-Phe-NCA content between 40–60% exhibited the least light scattering. The Dh of vesicles self-assembled from homo-chiral polymers was around 465 nm (as confirmed later by TEM). Interestingly, the DLS data of the hetero-chiral formulation where an L-Phe content of 30% was used showed a larger Dh of approximately 1000 nm. This is likely related to nanoparticle solvation by solvent, which suggests that these block copolymer nanoparticles become more solvated when a more equal chiral ratio is used: completely dissolved block copolymer chains were observed for the PEG45-b-P(D/L-Phe)10 group with a L-Phe content between 40–60%.
Transmission electron microscopy (TEM) analysis was used to examine the obtained nanoparticle morphologies. Nanoparticle dispersions in THF were directly applied to the TEM grid and analyzed after the solvent evaporated, no TEM stain was required. A range of various nanoparticle morphologies were observed by altering the target core-forming block DP and the chiral monomer ratio, which allowed the preparation of a pseudo-phase diagram (Fig. 3). Vesicles with a well-defined membrane (TEM diameter = 530 ± 65 nm) were observed for the homo-chiral PEG45-b-P(D/L-Phe)10 formulations (DP 10 series, L-Phe content of 0% and 100%). Compound micelles (TEM diameter = 565 ± 82 nm) were detected for nanoparticles with an L-Phe content of either 10% or 90%. Disrupted compounds micelles (TEM diameter = 720 ± 70 nm) were observed for formulations with an L-Phe content of either 20% or 80%. TEM images of the particles obtained from formulations with a L-Phe contents of either 30% or 70% appeared with a dark spot in the nanoparticle core, indicating that the particles are more solvated at the particle exterior and are less solvated by THF within the particle core. This morphology is therefore classified herein as ‘swollen compound micelles’ (TEM diameter = 882 ± 85 nm) (Fig. 3, Table S2†). Here, the appearance of this dark spot in the center of the nanoparticle is likely a result of the initial non-uniform solvated ‘wet’ state. The drying process of such particles, during the preparation of the TEM sample and the vacuum in the TEM, will lead to a dark spot since the polymer chains are more compact in the core, in comparison to the nanoparticle exterior. This more solvated nature is also supported by DLS analysis, larger Dh values were observed for the hetero-chiral nanoparticle compositions (L-Phe content: 10–30%) and compositions with an L-Phe content of 40–60% appeared as dissolved chains.
 | ||
| Fig. 3 (Top) Pseudo-phase diagram of PEG45-b-P(D/L-Phe)x assemblies obtained via CC-PISA at 15 wt% in THF. Various morphologies were targeted by adjusting the DP (y-axis) and the L-Phe-NCA content (x-axis). Here, D-V = D-Vesicles, D/L-V = D/L-Vesicles, CM = Compound Micelles, and L-V = L-Vesicles. Please note that ‘D-, L- and D/L-’ only refer to the used type of chiral monomer, and hence the chiral composition of the polymer, not the nanoparticle chirality. (Bottom) Representative TEM images obtained for PEG45-b-P(D/L-Phe)10 copolymer nano-objects with different L-Phe content ranging from 0–100%; the letters correspond to the samples in the pseudo-phase diagram (a, h) vesicles, (b, g) compound micelles, (c, f) disrupted compound micelles, (d, e) swollen compound micelles. | ||
Regarding the other compositions with other target DPs: homo-chiral formulation with a DP of 5 resulted in polymer vesicles with TEM diameters of 500 ± 45 nm and homo-chiral formulations with core-forming block DPs of 15 and 20 produced vesicles with a TEM diameter of about 542 ± 85 nm and 560 ± 100 nm, respectively. Similar to the DP 10 series, the introduction of hetero-chirality led to larger DLS and TEM diameters (Table S2, Fig. S5–S7†). A mixed vesicle/sphere phase was observed for the DP 5 formulation with L-Phe contents of 10% and 90%. Surprisingly, but consistent with our previous study,38 the samples that formed a gel did not consist of worm-like nanoparticles, which is usually the case in PISA. Instead, vesicular nanoparticles with a well-defined membrane were detected. This observation deserves to be further examined in a subsequent study; this characteristic could possibly be explained with the percolation theory.52
More insight into the PISA reaction was obtained by determining the reaction kinetics by FTIR (Fig. 4a–c), additionally this process was monitored by DLS (Fig. 4d and e). Three representative formulations were evaluated: PEG45-b-P(L-Phe)10, PEG45-b-P(D-Phe)10, and PEG45-b-P(DL-Phe)10 at 15 wt%. Samples were withdrawn from the reaction solution and directly analyzed by FTIR (Fig. S8–S10, Tables S3–S5†). The distinctive NCA-monomer bands at 1779 cm−1 and 1848 cm−1 progressively disappeared as the reaction proceeded, the PEG band at 1108 cm−1 was used as an internal standard. The FTIR data shows that full conversion was reached after 120 min. DLS analysis during CC-PISA (Fig. 4d and e) gave insight into the self-assembly behavior. The reaction mixture of the two homo-chiral formulations turned into a turbid dispersion after approximately 5–10 min. These data show that nucleation occurred during the polymerization reaction, this was further confirmed optically as the dispersion turned gradually more turbid over time (Fig. S11†). DLS data show that both the Dh and PD approach a stable value at around 120 min and the derived count rate (DCR) became constant at this time, indicating the end of the CC-PISA reaction. These data are consistent with the FTIR reaction kinetics (Fig. 4a and b). The final Dh of the prepared vesicles is ∼500 nm.
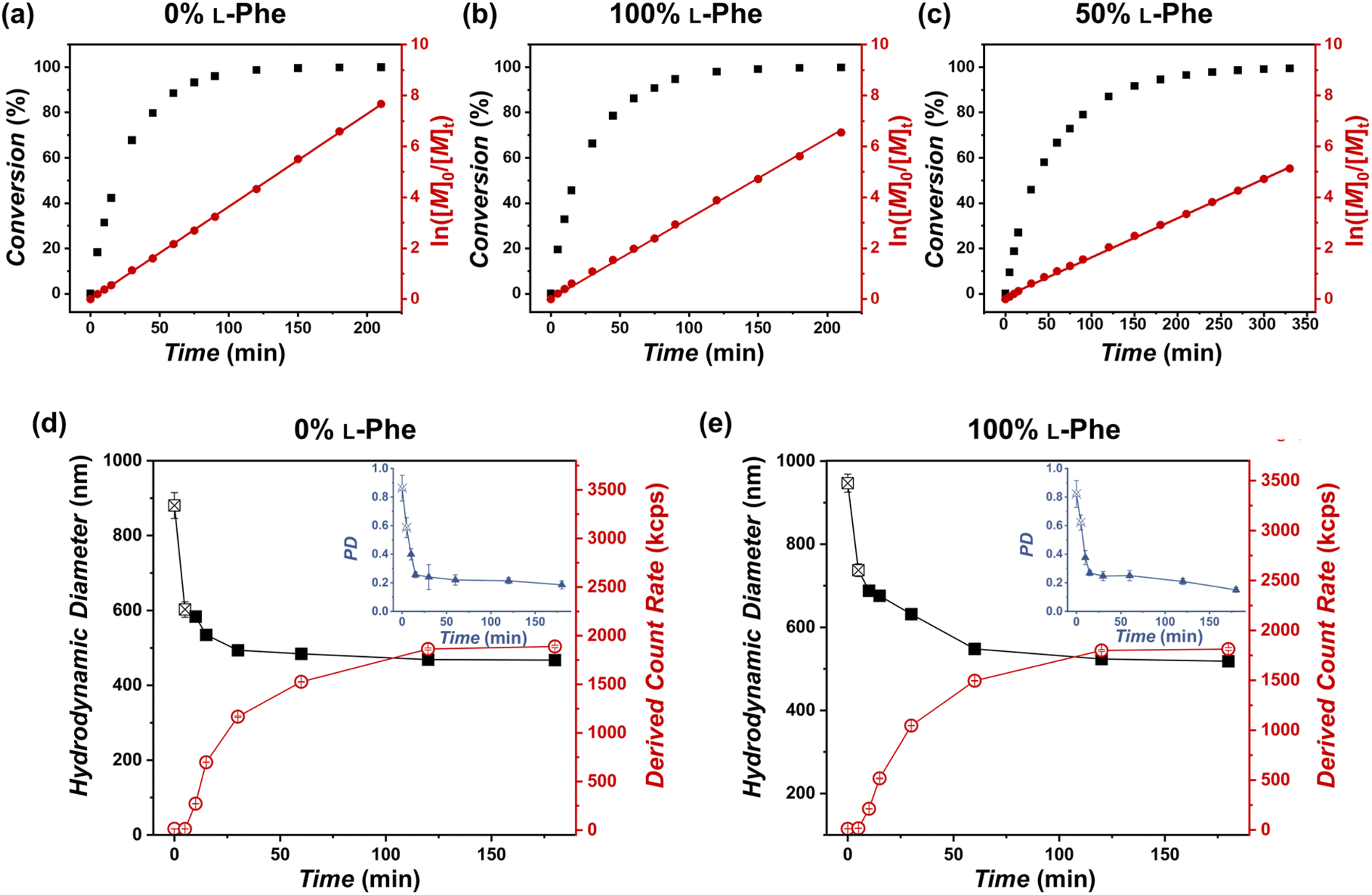 | ||
| Fig. 4 FTIR reaction kinetics of the PEG45-b-P(D/L-Phe)10 formulations with a varying L-Phe content at 15 wt% and DLS analysis during these reactions. (a, b) For the homo-chiral CC-PISA synthesis, where L-Phe content is either 0% or 100%, full conversion was achieved after approximately 120 min. (c) The hetero-chiral formulation exhibited a slower polymerization rate; full conversion was achieved after 270 min. All data were fitted to first order reaction kinetics (red line). (d, e) DLS analysis during homo-chiral CC-PISA, where L-Phe content is either 0% or 100%. Here, the Dh values are reported from the point when the count rate started to increase (i.e., after nucleation at ∼5–10 min). A stable derived count rate (DCR) was observed after 120 min and vesicles with a diameter of approximately 500 nm (PD ∼ 0.15) were obtained. | ||
Interestingly, the hetero-chiral formulation, where no PISA occurred, reached full conversion at approximately 270 min. These observations are expected since it is known that nucleation causes an increase in reaction rate during PISA, owing to the monomer solvation of the nascent nanoparticles.53 However, no characteristic increase in first-order reaction kinetics was observed to support nanoparticle swelling by monomer. The increased reaction rate can also be explained by the secondary peptide structure of the core-forming block. It is known that an α-helix structure has the ability to enhance the polymerization rate.54 The secondary peptide structure of the obtained nanoparticles and polymers was investigated next.
It is known that various factors can influence the secondary PPhe structure.47–49,55,56 For example, stable PPhe nanoparticles with a β-sheet secondary structure can be formed in water/DMSO mixtures.57–59 PPhe can also form nanoparticles with α-helix secondary structure in THF or water/HFIP mixtures.60,61 It seems, therefore, likely that the secondary structure is affected in the herein discussed CC-PISA formulation when monomer chirality is altered. This was confirmed by FTIR analysis57–64 for the DP 10 formulations in THF (with an L-Phe content ranging from 0–100%). The FTIR spectra of all the formulations have bands in the amide I region at 1658 cm−1 and 1630 cm−1 (Fig. 5a and S12†). The two homo-chiral formulations have a dominant band at 1658 cm−1, which indicates the presence of an α-helix. As the L-Phe content reaches 50%, a reduction in the band intensity at 1658 cm−1 is observed, while the band intensity at 1630 cm−1 increases; the latter band indicates the presence of a β-sheet secondary structure.57–59,62,63,65
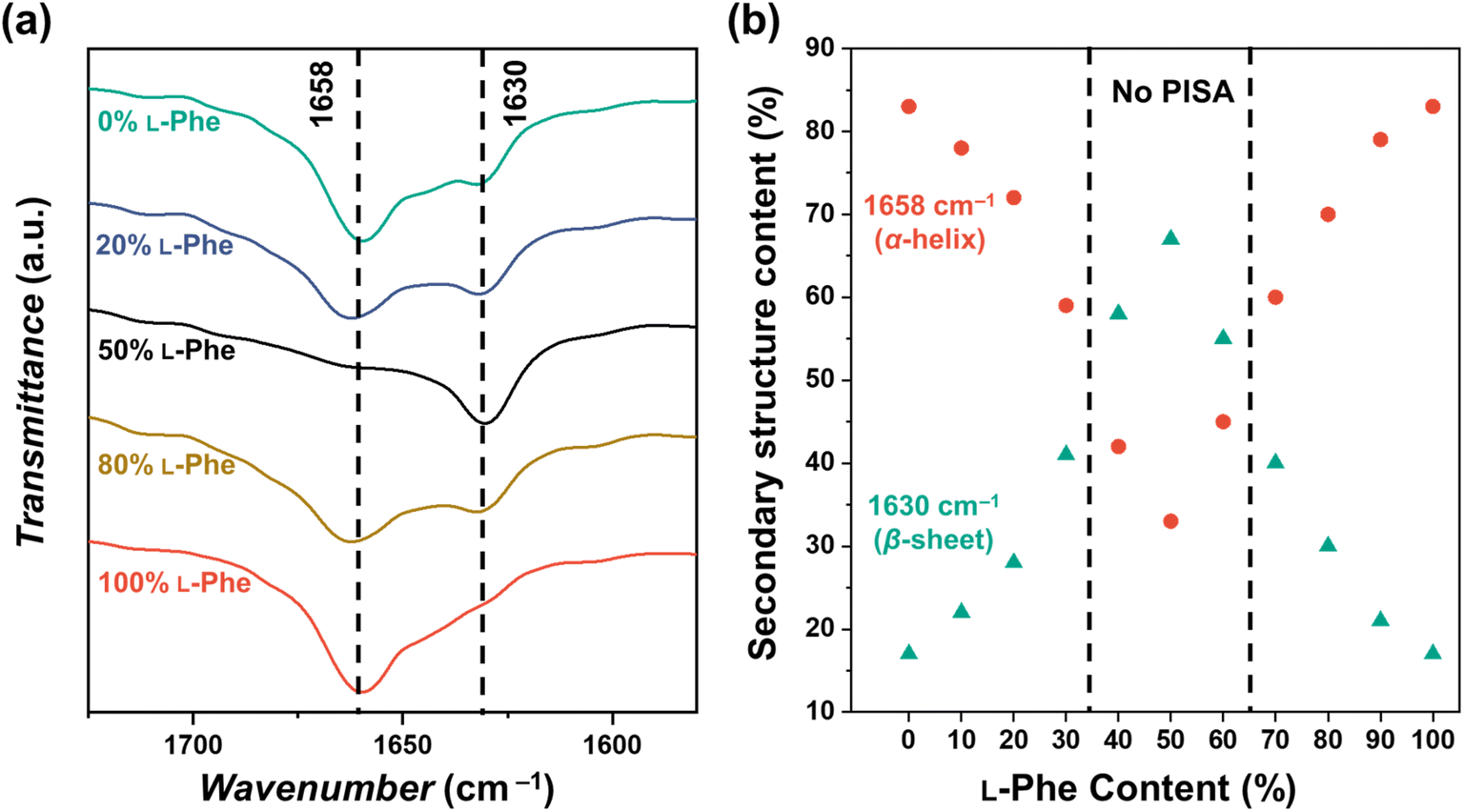 | ||
| Fig. 5 (a) FTIR spectra recorded for PEG45-b-P(D/L-Phe)10 copolymer nanoparticles with a varying L-Phe content (0%, 20%, 50%, 80%, and 100%) in THF. The bands in the amide I region at 1658 cm−1 and 1630 cm−1 correspond to the α-helix and β-sheet secondary structure, respectively. The complete FTIR spectra of these samples are shown in Fig. S12.† (b) Deconvoluted amide I band areas of PEG45-b-P(D/L-Phe)10 copolymer nanoparticles plotted against the L-Phe content. | ||
The bands in the amide I region were deconvoluted and band areas were plotted against the L-Phe content Fig. 5b. The α-helix content in the homo-chiral formulations were both 87%, this amount reduced as a chiral content of 50% L-Phe was approached. A minimal α-helix amount of 33% was observed when equal amount of chiral monomers were used. In contrast, the content of the β-sheet fraction increases from 17% to 67% when approaching an L-Phe content of 50% (Fig. S13, Table S6†). When comparing these observations with the nanoparticle morphology (Fig. 3), no polymer self-assembly occurs when the β-sheet fraction exceeds the α-helix fraction. Indeed, these data indicate that an increasing amount of β-sheets leads to weaker self-assembly and more solvated nanoparticles; PISA does not occur above a β-sheet fraction of approximately 50%. Similar FTIR data were obtained for the DP 5, 15, and 20 formulations (Fig. S14, Table S7†).
Interestingly, the appearance of a β-sheet fraction seems counterintuitive since a β-sheet can connect various peptides via inter-molecular interactions, which, in turn, should lead to aggregation. Nevertheless, these samples did not appear as nanoparticles by DLS and TEM and the fully dissolved state even allowed SEC analysis. Therefore, we suggest that the β-sheet originates from intra-molecular interactions when the core-forming block is sufficiently long, or is present in the form of a single β-strand. We refer to this secondary structure as β-sheet throughout the manuscript.
Another common method to gain insight into the secondary structure is circular dichroism (CD) spectroscopy (Fig. S15†). Unfortunately, this method seemed to be of little value in the herein presented research. This is because of the high solvent cut-off wavelength of THF (212 nm) and, more importantly, because of the nature of the prepared polymer; it is well-known that mixtures of enantiomers have reduced optical activity.49 Nevertheless, the homo-chiral formulations exhibited a strong band around 230 nm, which can be assigned to n–π* transitions resulting from aromatic stacking interactions of the phenylalanine residues, suggesting an ordered secondary structure.59
More insight into the interactions of the secondary peptide structure were obtained by using UV-Vis spectroscopy (Fig. S16†). This method allows insight into the aromatic interactions, such as π–π stacking, within the nanoparticles and dissolved polymer chains. A larger redshift indicates the presence of stronger aromatic interactions. It was found that the maximum absorbance was detected at a higher wavelength for the homo-chiral formulations, compared to the hetero-chiral formulations. Lower wavelengths were detected for formulations with an L-Phe content between 40–60%. Interestingly, this L-Phe content has the largest β-sheet fraction, as shown by FTIR. This result indicates that a larger α-helix fraction, and a larger amount of homo-chirality, promote aromatic stacking and, therefore, favor nanoparticle stability. Indeed, it is known that such aromatic interactions can support the formation of stable nanostructures.50,66
Self-assembly mechanism
The FTIR data of the PEG45-b-P(D/L-Phe)10 with a varying L-Phe content show two main bands in the amide I region. The band at 1658 cm−1 corresponds to an α-helix secondary structure, whereas the band at 1630 cm−1 represents a β-sheet secondary structure.57–59,62,63,65 Clearly, these data indicate that an increasing β-sheet fraction, in addition to hetero-chirality, disfavors polymer self-assembly in THF. Indeed, a greater α-helix fraction, and a higher degree of homo-chirality, seem to work in favor of in situ polymer self-assembly and causes a faster rate of polymerization. In other words, a more ordered secondary structure promotes in situ self-assembly,40,67 this is likely because a more ordered structure has stronger intermolecular interactions.37,69 These observations are consistent with the literature where the formation of an α-helix structure is energetically favorable when the peptide comprises L-amino acids, in contrast to chiral mixtures.68,69 Our data also indicate that self-assembly does not initially promote α-helix formation during CC-PISA. This is because our data suggest that the structure of the polypeptide chain drives the PISA process. If self-assembly promotes α-helix formation, we should observe polymer aggregation before the formation of the secondary α-helix structure within the nanoparticles. If this was the case, we should observe nanoparticles regardless of the secondary peptide structure. Since we do not see any self-assembled structures for the 50% L-Phe composition (where a dominant β-sheet fraction was found), we can discount this possibility.It is known that the α-helix secondary structure can promote homo polypeptide-assembly, as shown by Gimeno and co-workers.61 Here it was shown that the α-helix secondary structure of PPhe aids self-assembly into nanorods in water on the addition of hexafluoro-2-propanol. It was hypothesised that the formation of nanorods is likely triggered by the long-range orientation of peptide bonds along the axis, which creates a hydrophilic region inside the α-helix with outside-oriented hydrophobic aromatic rings. However, the herein reported CC-PISA system is conducted in THF instead of water. The hydrophobicity of the α-helix plays therefore a less important role compared to aqueous formulations.
Consistent with our previous work, the nanoparticles self-assembled from homo-chiral L- and D-PPhe polymers were insoluble in all common solvents (water, DMSO, DCM, CHCl3, etc.). This indicates that these nanoparticles with a large fraction of α-helix secondary structure form robust intra-molecular interactions, which are likely not able to originate from dissolved random coils. Additional forces that can contribute to polymer self-assembly are π–π stacking interactions from the aromatic group on the PPhe core-forming block.66,70 It is possible that the strength of these aromatic interactions is affected by the changing secondary structure and/or that these interactions are governed by the degree of hetero-chirality. Nevertheless, more research is required to further elucidate the driving force for this intriguing PISA formulation that is aided by the structure of the peptide chain.
Additionally, the herein presented data show that the solvent choice is evidently important in the formation of the secondary structure, as other reports showed that this polymer (PPhe) can form stable nanoparticles with a β-sheet secondary structure in water/DMSO mixtures.57,58 To further elaborate on the importance of the choice of solvent during such PISA formulations, the herein described inter-molecular interactions are not necessarily the main driving force for all the so-far reported NCA-PISA formulations.38–41 This is because peptide solubility might vary depending on the solvent type.
Chirality to control biodegradation rate
As shown in this work, polymer chirality can be used to control the final PEG45-b-P(D/L-Phe)x nanoparticle morphology during PISA. In addition, control over polymer chirality should affect the nanoparticle biodegradability, since enzymes are generally designed to cleave amide bonds with only left-handed R-groups.38,71,72 Introduction of polymer hetero-chirality in such nanoparticles should therefore influence the rate of enzymatic degradation. To test this hypothesis, we performed an enzymatic degradation experiment where nanoparticles were transferred to water by extensive dialysis and degraded with the enzyme trypsin at 37 °C. As expected, the vesicle morphology was preserved during this THF-to-water transfer procedure. The degradation process was monitored by in situ DLS, the DCR and Dh were monitored for 192 h (8 days) (Fig. 6a, b, and S17†). Additionally, samples were taken at intermediate time points and analyzed by TEM (Fig. 7, S18, and S19†). The DLS data of the nanoparticles with an L-Phe content of 100%, 80%, and 20% showed 71%, 36%, and 12% reduction in DCR after 192 h (8 days), respectively (Fig. S17†). This indicates that the biodegradability of these polymer nanoparticles decreased when an increased fraction of D-Phe was used for their preparation. TEM was used to gain insight into the nanoparticle morphology during the degradation experiment. Final degraded nanoparticles appeared as less defined heterogeneous aggregates, for the nanoparticles with an L-Phe content of 80% and 20%. In contrast, vesicles that were solely prepared from D-Phe-NCA monomer did not show any signs of degradation. A constant count rate and hydrodynamic diameter were observed for 192 h and TEM images show that the vesicles remained stable during the experiment. Decreasing the trypsin concentration from 1.0 mg mL−1 to 0.8 mg mL−1 led to slower and less complete nanoparticle degradation, but gave us valuable insights into the nanoparticle degradation mechanism for the 100% L-Phe formulation. Nevertheless, a similar degradation profile was observed for both evaluated enzyme concentrations (Fig. S17c and d†). The 100% L-Phe formulation was chosen for this purpose owing to its well-defined initial nanoparticle structure and good degradability. Here, an initial abrupt drop in DCR was observed, which becomes constant after 18 h (remaining DCR ∼ 75%) (Fig. 6a, blue arrow). Subsequently, a very modest decrease in DCR was observed until 42 h (remaining DCR ∼ 70%), after which a second rapid reduction was observed (Fig. 6a, brown arrow). No further reduction in DCR was observed after 96 h (remaining DCR ∼ 40%). Interestingly, the Dh of the 100% L-Phe formulation also undergoes a two-stage degradation profile (Fig. 6b). In the first 30 h, the Dh increases due to a progressively more solvated membrane upon enzymatic degradation. A subsequent decrease in Dh was observed afterwards. TEM images further supported the degradation profile obtained from the in situ DLS study (Fig. 7 and S18†). Initially, vesicles with a TEM diameter of 570 ± 60 nm were observed. The TEM image after 12 h shows vesicles with a similar diameter, but with membrane disruptions. After 24 h, the vesicle membrane appeared more disturbed and smaller vesicles started to form. Only smaller vesicles with a disrupted membrane were observed by TEM analysis after 48 h (TEM diameter of 350 ± 60 nm). Finally, fragmented heterogeneous aggregates were observed after 72 h and 96 h. These TEM observations suggest a two-step mechanism: first, the vesicle membrane is disturbed; secondly, vesicle fragmentation occurs once the membrane is sufficiently degraded. For the 80% L-Phe formulation (using a trypsin concentration of 1.0 mg mL−1), a similar degradation profile was observed where the DLS nanoparticle diameter seemed to increase at an initial stage. This result indicates that the nanoparticles become more solvated as the degradation proceeds (Fig. S17 and S19†). As expected, no full degradation was observed within 8 days as the degradation products suffer from inferior solubility compared to that of the initial nanoparticles. Others have reported that this poor solubility significantly hinders the degradation rate.72 However, the degradation rate and extent of degradation can be enhanced by increasing the trypsin concentration and degradation time (Fig. S17†); eventually full degradation can be expected owing to the degradable nature of the polypeptide block.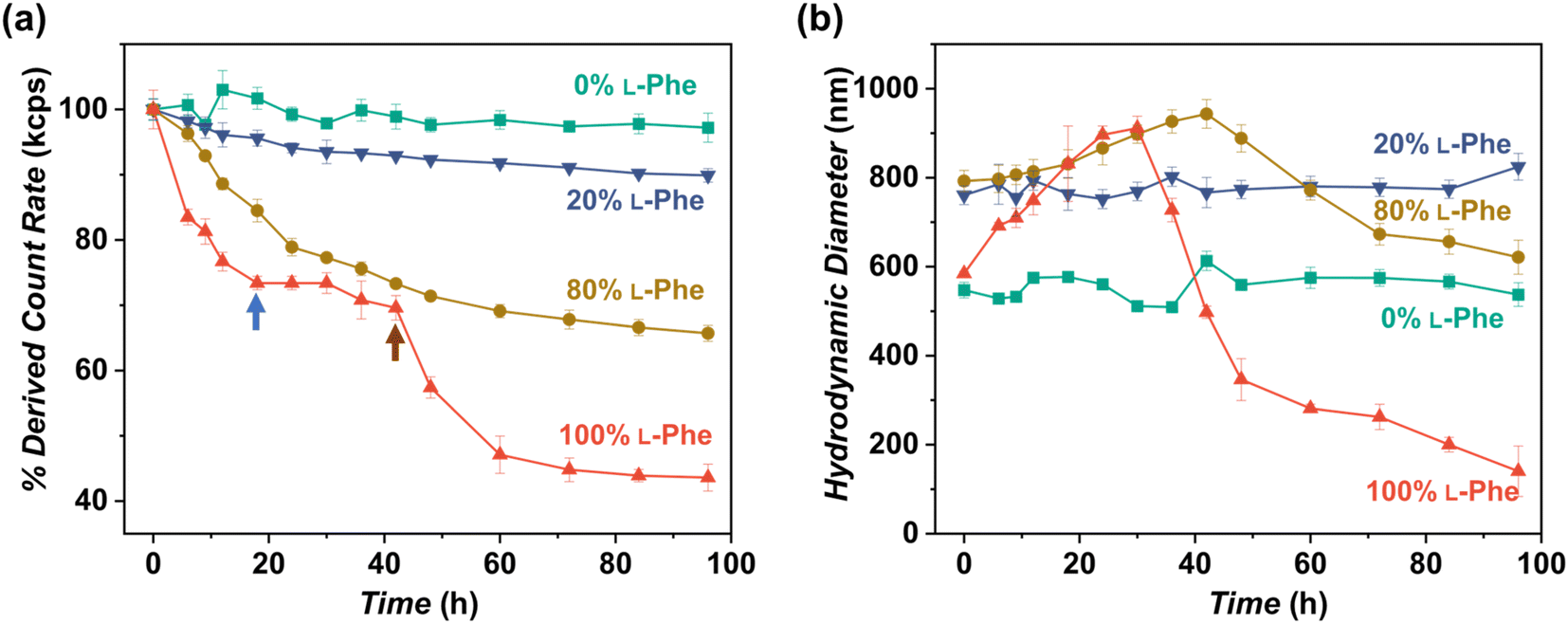 | ||
| Fig. 6 In vitro enzymatic degradation studies of PEG45-b-P(Phe)10 polymer nanoparticles prepared with varying L-Phe-NCA content. The enzyme trypsin was used at 37 °C at 1.0 mg mL−1 (0.8 mg mL−1 for the red data) and samples were monitored over time by DLS for 192 h (see complete data set in Fig. S17†). Copolymer nanoparticles were obtained via NCA-PISA in THF and transferred to water by dialysis prior to this study. (a) Percentage of the initial derived count rate (DCR) plotted over time. No significant reduction in PEG45-b-P(D-Phe)10 copolymer nanoparticles was observed over 96 h. Two separate degradation stages were observed for the PEG45-b-P(L-Phe)10 copolymer nanoparticles: membrane degradation (blue arrow), followed by fragmentation (brown arrow). (b) Hydrodynamic diameter (Dh) plotted over time. No significant change in the size of PEG45-b-P(D-Phe)10 copolymer nanoparticles was observed over 96 h. In contrast, an initial increase in Dh up until 30 h was observed for PEG45-b-P(L-Phe)10; this might be caused by a progressively more solvated membrane upon enzymatic degradation. Error bars correspond to the observed average DCR and Dh range. | ||
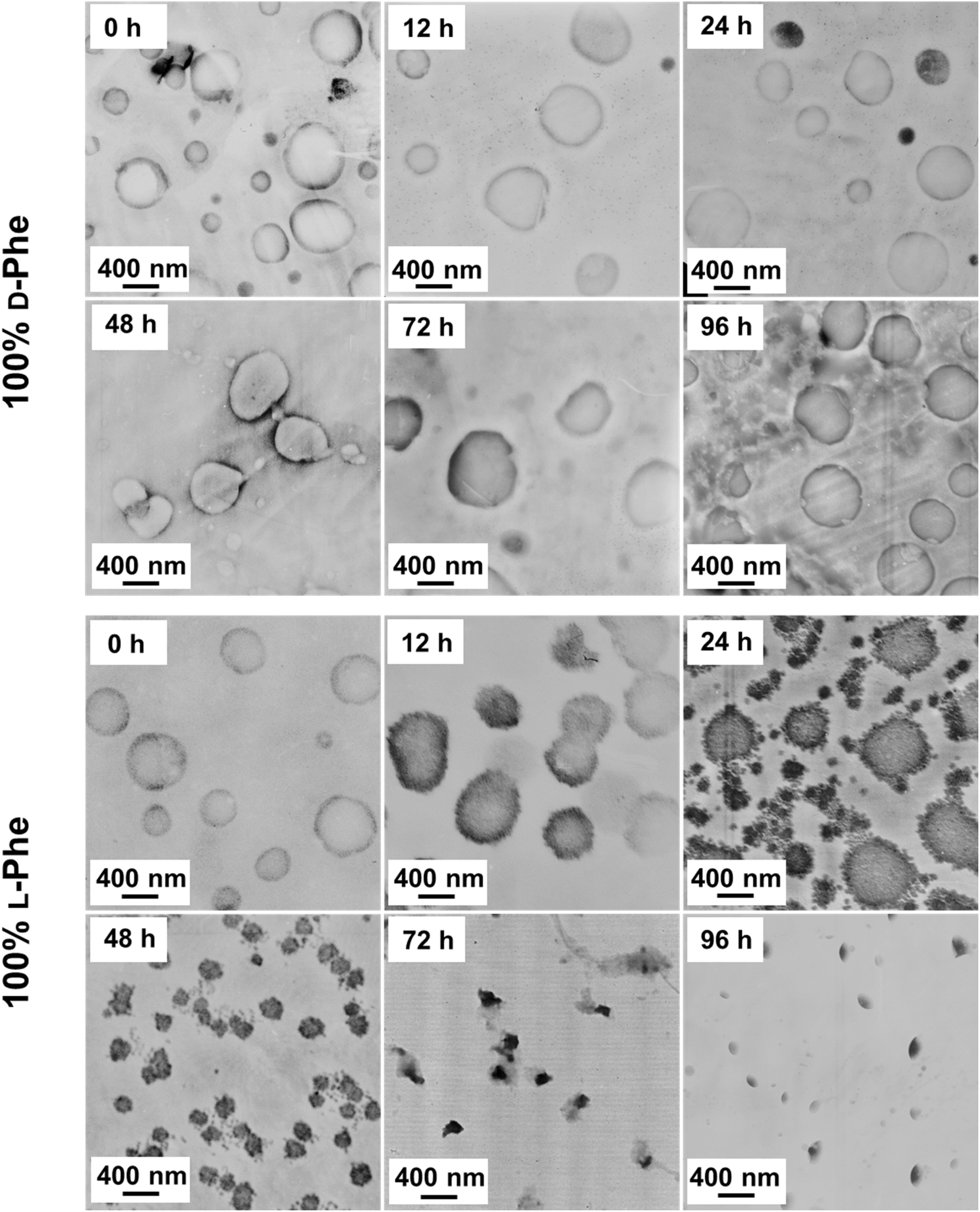 | ||
| Fig. 7 TEM images obtained during the enzymatic degradation process for both PEG45-b-P(D-Phe)10 and PEG45-b-P(L-Phe)10 polymer nanoparticles. These data show minor membrane disruption after 12 h, major membrane disturbance after 24 h, nanoparticle fragmentation into smaller vesicles after 48 h, and dissolving nanoparticles after 72 h. These TEM observations correspond to the two-stage DLS degradation patterns. | ||
In summary, these enzymatic degradation experiments show that nanoparticle chirality has a significant effect on the biodegradation rate. These data show that the adjustment of the chiral ratio is an efficient strategy to control the biodegradability of such PISA nanoparticles. Such insights are important in terms of biological applications and could, for example, be a potential handle to control the drug release rate.
Conclusion
PEG45-b-P(D/L-Phe)x block copolymer nanoparticles were prepared via CC-PISA in THF at 15 wt%. This NCA-PISA formulation can be controlled by altering the chirality of the core-forming peptide block simply by using various ratios of L-Phe-NCA and D-Phe-NCA monomers during PISA. 1H NMR and SEC analyses showed that the polymerization proceeds regardless of the monomer chirality and that good control over such polymerizations was achieved, as the desired DPs were obtained; however, relatively high SEC dispersities (∼2.3–2.7) were observed when the targeted DPs are above 15. These are likely artificially enlarged during the SEC analysis due to undesired polymer–column interactions that become more pronounced when targeting longer DPs. Nevertheless, well-defined vesicles and compound micelles were obtained in this DP range.For the first time, it was shown that the enantiomeric ratio greatly affected the final nanoparticle morphology and the secondary structure during NCA-PISA. Homo-chiral formulations self-assembled into vesicular nanoparticles, regardless of the targeted core-forming DP. In contrast, compositions with an L-Phe content between 40–60% did not self-assemble into nanoparticles, dissolved block copolymer chains were observed instead. Hetero-chiral formulations outside this range self-assembled into nanoparticles with various morphologies: vesicles, compound micelles, disrupted compound micelles, and swollen compound micelles. Interestingly, the DLS diameter of such nanoparticles self-assembled from hetero-chiral polymers increased when using a more equal chiral ratio as such nanoparticles become more solvated under such conditions.
FTIR was used to obtain reaction kinetics. A higher rate of reaction was observed for the homo-chiral CC-PISA formulations compared to the non-self-assembling hetero-chiral solution polymerization (with an L-Phe content of 50%). This increased rate of reaction likely originates from the forming α-helix secondary structure, but unlikely from monomer solvation which is usually the case in PISA, since no characteristic increase in first-order reaction kinetics was observed.
The presence of this secondary structure was confirmed with FTIR and CD spectroscopy; it was shown that the nanoparticles of these CC-PISA formulations contained both an α-helix and β-sheet secondary structure. Additionally, for the first time, it was shown that the ratio of these secondary structures can be controlled during PISA by using judicious amounts of L- and D-Phe-NCA. An increased amount of hetero-chirality leads to a larger β-sheet fraction and more solvated nanoparticles with various morphologies. An L-Phe content in the range of 40–60% inhibited PISA and gave rise to polymer chains where the β-sheet fraction exceeds that of the α-helix fraction. Our data indicate that the formulations with a larger degree of hetero-chirality have weaker π–π stacking interactions, and that the stronger π–π stacking interactions that are present in the homo-chiral formulations and the secondary structure play an important role in this PISA process. In other words, herein, in situ self-assembly is likely driven by the inter-molecular attraction between growing polypeptide blocks that adopt a secondary structure once a critical DP is obtained, instead of polymer–solvent interactions (polymer solubility).
Furthermore, adjustment of the degree of polymer chirality can also be used to control the enzymatic nanoparticle degradation rate. Nanoparticles that were solely prepared from L-Phe-NCA degraded via a two-step mechanism: first, the vesicle membrane degrades, afterwards this nanoparticle fragments into smaller vesicles that further degrade and finally dissolve. In contrast to the nanoparticles self-assembled from homo-chiral L-Phe-based polymers, vesicles self-assembled from homo-chiral polymers with a D-Phe core-forming block appeared to be stable in the presence of trypsin. Increasing the D-Phe content leads to a slower degradation reaction and more robust nanoparticles that suffer from more incomplete degradation.
In summary, we show that: (1) the secondary peptide structure of the CC-PISA nanoparticles can be controlled by adjustment of the chiral molar ratio of the core-forming monomers. (2) The monomer chirality affects the polymer self-assembly behavior during the CC-PISA process. (3) The driving force for this CC-PISA formulation is related to inter-molecular peptide–peptide interactions, instead of polymer–solvent interactions. (4) In addition to morphological control, monomer chirality can be used to prepare PISA nanoparticles with different enzymatic degradation rates.
These observations are of great interest for further PISA studies since it suggests a new handle to control the nanoparticle morphology and secondary structure during PISA, and offers a new way to control the biodegradability of nanoparticles that are prepared via PISA. These aspects make such PISA nanoparticles more applicable for future biomedical applications. Additionally, this PISA approach could be of great value for preparing pharmaceutical nanoparticles that use chirality in disease treatment.73–75
Nevertheless, more work is required to precisely establish the peptide interactions that drive this PISA process and the effect of the peptide sequence. This work is, therefore, of considerable interest to scientists that work in the fields of polymer nanoparticle synthesis and applicable biomedical nanoparticles (such as drug carriers).
Data availability
All necessary information is included in the ESI.†Author contributions
Haolan Li – investigation, writing; Erik Jan Cornel – investigation, writing, supervision, funding acquisition; Zhen Fan – supervision, funding acquisition; Jianzhong Du – supervision, funding acquisition, writing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Natural Science Foundation of China (Nos. 21925505, 22101207, 22075212, and 52222306), Shanghai International Scientific Collaboration Fund (No. 21520710100) and Shanghai Municipal Education Commission, E. J. C. is supported by the China Postdoctoral Science Foundation (2020M671197), and the International Postdoctoral Exchange Fellowship Program. J. D. is the recipient of National Science Fund for Distinguished Young Scholars.References
- L. G. Xu, X. X. Wang, W. W. Wang, M. Z. Sun, W. J. Choi, J. Y. Kim, C. L. Hao, S. Li, A. H. Qu, M. R. Lu, X. L. Wu, F. M. Colombari, W. R. Gomes, A. L. Blanco, A. F. de Moura, X. Guo, H. Kuang, N. A. Kotov and C. L. Xu, Nature, 2022, 601, 366–373 CrossRef CAS PubMed.
- J. Yeom, P. P. G. Guimaraes, H. M. Ahn, B. K. Jung, Q. Hu, K. McHugh, M. J. Mitchell, C. O. Yun, R. Langer and A. Jaklenec, Adv. Mater., 2020, 32, 1903878 CrossRef CAS PubMed.
- M. Ying, Q. Shen, Y. Liu, Z. Q. Yan, X. L. Wei, C. Y. Zhan, J. Gao, C. Xie, B. X. Yao and W. Y. Lu, ACS Appl. Mater. Interfaces, 2016, 8, 13232–13241 CrossRef CAS PubMed.
- X. L. Wei, C. Y. Zhan, Q. Shen, W. Fu, C. Xie, J. Gao, C. M. Peng, P. Zheng and W. Y. Lu, Angew. Chem., Int. Ed., 2015, 54, 3023–3027 CrossRef CAS PubMed.
- H. M. Wang, Z. C. Luo, Y. Z. Wang, T. He, C. B. Yang, C. H. Ren, L. S. Ma, C. Y. Gong, X. Y. Li and Z. M. Yang, Adv. Funct. Mater., 2016, 26, 1822–1829 CrossRef CAS.
- Y. H. Shen, Y. F. Wang, I. W. Hamley, W. Qi, R. X. Su and Z. M. He, Prog. Polym. Sci., 2021, 123, 101469 CrossRef CAS.
- Z. L. Luo, S. K. Wang and S. G. Zhang, Biomaterials, 2011, 32, 2013–2020 CrossRef CAS PubMed.
- J. F. Liu, J. J. Liu, L. P. Chu, Y. M. Zhang, H. Y. Xu, D. L. Kong, Z. M. Yang, C. H. Yang and D. Ding, ACS Appl. Mater. Interfaces, 2014, 6, 5558–5565 CrossRef CAS PubMed.
- Z. L. Luo, Y. Y. Yue, Y. F. Zhang, X. Yuan, J. P. Gong, L. L. Wang, B. He, Z. Liu, Y. L. Sun, J. Liu, M. F. Hu and J. Zheng, Biomaterials, 2013, 34, 4902–4913 CrossRef CAS PubMed.
- L. D. Blackman, S. Varlas, M. C. Arno, Z. H. Houston, N. L. Fletcher, K. J. Thurecht, M. Hasan, M. I. Gibson and R. K. O’Reilly, ACS Cent. Sci., 2018, 4, 718–723 CrossRef CAS PubMed.
- Z. Ye, X. Zhu, S. Acosta, D. Kumar, T. Sang and C. Aparicio, Nanoscale, 2019, 11, 266–275 RSC.
- D. Le, D. Keller and G. Delaittre, Macromol. Rapid Commun., 2019, 40, e1800551 CrossRef PubMed.
- L. D. Blackman, S. Varlas, M. C. Arno, A. Fayter, M. I. Gibson and R. K. O’Reilly, ACS Macro Lett., 2017, 6, 1263–1267 CrossRef CAS PubMed.
- S. Varlas, P. G. Georgiou, P. Bilalis, J. R. Jones, N. Hadjichristidis and R. K. O’Reilly, Biomacromolecules, 2018, 19, 4453–4462 CrossRef CAS PubMed.
- R. R. Gibson, S. P. Armes, O. M. Musa and A. Fernyhough, Polym. Chem., 2019, 10, 1312–1323 RSC.
- D. Zehm, L. P. D. Ratcliffe and S. P. Armes, Macromolecules, 2012, 46, 128–139 CrossRef.
- M. J. Derry, L. A. Fielding and S. P. Armes, Polym. Chem., 2015, 6, 3054–3062 RSC.
- L. P. D. Ratcliffe, B. E. McKenzie, G. M. D. Le Bouëdec, C. N. Williams, S. L. Brown and S. P. Armes, Macromolecules, 2015, 48, 8594–8607 CrossRef CAS.
- L. A. Fielding, M. J. Derry, V. Ladmiral, J. Rosselgong, A. M. Rodrigues, L. P. D. Ratcliffe, S. Sugihara and S. P. Armes, Chem. Sci., 2013, 4, 2081–2087 RSC.
- Q. Z. Liu, X. Y. Wang, L. Ma, K. W. Yu, W. X. Xiong, X. H. Lu and Y. L. Cai, ACS Macro Lett., 2020, 9, 454–458 CrossRef CAS PubMed.
- K. W. Yu, X. Y. Wang, C. H. Luo, Y. Cao, J. Cai, X. H. Lu and Y. L. Cai, Chem. Commun., 2022, 58, 6793–6796 RSC.
- W. X. Xiong, X. Y. Wang, Y. Y. Liu, C. H. Luo, X. H. Lu and Y. L. Cai, Macromolecules, 2022, 55, 7003–7012 CrossRef CAS.
- P. Gao, H. Cao, Y. Ding, M. Cai, Z. G. Cui, X. H. Lu and Y. L. Cai, ACS Macro Lett., 2016, 5, 1327–1331 CrossRef CAS PubMed.
- H. Ren, Z. M. Wei, H. C. Wei, D. S. Yu, H. Y. Li, F. H. Bi, B. B. Xu, H. Zhang, Z. Hua and G. Yang, Polym. Chem., 2022, 13, 3800–3805 RSC.
- P. J. Hurst, A. M. Rakowski and J. P. Patterson, Nat. Commun., 2020, 11, 4690 CrossRef CAS PubMed.
- A. Blanazs, A. J. Ryan and S. P. Armes, Macromolecules, 2012, 45, 5099–5107 CrossRef CAS.
- A. Blanazs, J. Madsen, G. Battaglia, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2011, 133, 16581–16587 CrossRef CAS PubMed.
- H. Khan, M. J. Cao, W. F. Duan, T. Y. Ying and W. Q. Zhang, Polymer, 2018, 150, 204–213 CrossRef CAS.
- G. Riess, Prog. Polym. Sci., 2003, 28, 1107–1170 CrossRef CAS.
- S. Chen, E. J. Cornel and J. Z. Du, Chin. J. Polym. Sci., 2022, 40, 1–10 CrossRef.
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS.
- E. J. Cornel, J. H. Jiang, S. Chen and J. Z. Du, CCS Chem., 2021, 3, 2104–2125 CrossRef CAS.
- F. D’Agosto, J. Rieger and M. Lansalot, Angew. Chem., Int. Ed., 2020, 59, 8368–8392 CrossRef PubMed.
- J. Yeow and C. Boyer, Adv. Sci., 2017, 4, 1700137 CrossRef PubMed.
- S. Varlas, J. C. Foster and R. K. O’Reilly, Chem. Commun., 2019, 55, 9066–9071 RSC.
- C. Zhu and J. Nicolas, Polym. Chem., 2021, 12, 594–607 RSC.
- N. J. W. Penfold, J. Yeow, C. Boyer and S. P. Armes, ACS Macro Lett., 2019, 8, 1029–1054 CrossRef CAS PubMed.
- J. H. Jiang, X. Y. Zhang, Z. Fan and J. Z. Du, ACS Macro Lett., 2019, 8, 1216–1221 CrossRef CAS PubMed.
- C. Grazon, P. Salas Ambrosio, E. Ibarboure, A. Buol, E. Garanger, M. W. Grinstaff, S. Lecommandoux and C. Bonduelle, Angew. Chem., Int. Ed., 2020, 59, 622–626 CrossRef CAS PubMed.
- C. Grazon, P. Salas Ambrosio, S. Antoine, E. Ibarboure, O. Sandre, A. J. Clulow, B. J. Boyd, M. W. Grinstaff, S. Lecommandoux and C. Bonduelle, Polym. Chem., 2021, 12, 6242–6251 RSC.
- A. Duro Castano, L. Rodríguez Arco, L. Ruiz Pérez, C. De Pace, G. Marchello, C. Noble Jesus and G. Battaglia, Biomacromolecules, 2021, 22, 5052–5064 CrossRef CAS PubMed.
- C. C. Zhou, M. Z. Wang, K. D. Zou, J. Chen, Y. Q. Zhu and J. Z. Du, ACS Macro Lett., 2013, 2, 1021–1025 CrossRef CAS PubMed.
- R. Luxenhofer, C. Fetsch and A. Grossmann, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2731–2752 CrossRef CAS.
- Y. J. Xi, Y. Wang, J. Y. Gao, Y. F. Xiao and J. Z. Du, ACS Nano, 2019, 13, 13645–13657 CrossRef CAS PubMed.
- X. X. Cheng, T. F. Miao, L. Yin, Y. J. Ji, Y. Y. Li, Z. B. Zhang, W. Zhang and X. L. Zhu, Angew. Chem., Int. Ed., 2020, 59, 9669–9677 CrossRef CAS PubMed.
- K. Bauri, A. Narayanan, U. Haldar and P. De, Polym. Chem., 2015, 6, 6152–6162 RSC.
- Y. L. Liu, C. Z. Zhao and C. Y. Chen, Macromolecules, 2022, 55, 3801–3810 CrossRef.
- K. Hu, H. Geng, Q. Z. Zhang, Q. S. Liu, M. S. Xie, C. J. Sun, W. J. Li, H. C. Lin, F. Jiang, T. Wang, Y. D. Wu and Z. G. Li, Angew. Chem., Int. Ed., 2016, 55, 8013–8017 CrossRef CAS PubMed.
- J. Skey, C. F. Hansell and R. K. O’Reilly, Macromolecules, 2010, 43, 1309–1318 CrossRef CAS.
- J. Rodríguez Hernández and S. Lecommandoux, J. Am. Chem. Soc., 2005, 127, 2026–2027 CrossRef PubMed.
- Z. H. Chen, Z. G. Chi, Y. F. Sun and Z. Y. Lv, Chirality, 2021, 33, 618–642 CrossRef CAS PubMed.
- J. R. Lovett, M. J. Derry, P. Yang, F. L. Hatton, N. J. Warren, P. W. Fowler and S. P. Armes, Chem. Sci., 2018, 9, 7138–7144 RSC.
- E. J. Cornel, S. van Meurs, T. Smith, P. S. O’Hora and S. P. Armes, J. Am. Chem. Soc., 2018, 140, 12980–12988 CrossRef CAS PubMed.
- S. X. Lv, H. J. Kim, Z. Y. Song, L. Feng, Y. F. Yang, R. Baumgartner, K. Y. Tseng, S. J. Dillon, C. Leal, L. C. Yin and J. J. Cheng, J. Am. Chem. Soc., 2020, 142, 8570–8574 CrossRef CAS PubMed.
- E. Peggion, M. Palumbo, G. M. Bonora and C. Toniolo, Bioorg. Chem., 1974, 3, 125–132 CrossRef CAS.
- C. Noble Jesus, R. Evans, J. Forth, C. Estarellas, F. L. Gervasio and G. Battaglia, ACS Macro Lett., 2021, 10, 984–989 CrossRef CAS PubMed.
- M. Matsusaki, M. Matsumoto, T. Waku and M. Akashi, J. Biomater. Sci., Polym. Ed., 2011, 22, 1035–1048 CrossRef CAS PubMed.
- M. Matsusaki, T. Waku, T. Kaneko, T. Kida and M. Akashi, Langmuir, 2006, 22, 1396–1399 CrossRef CAS PubMed.
- V. Castelletto and I. W. Hamley, Biophys. Chem., 2009, 141, 169–174 CrossRef CAS PubMed.
- E. Peggion, A. S. Verdini, A. Cosani and E. Scoffone, Macromolecules, 1969, 2, 170–174 CrossRef CAS.
- A. Romero Montero, I. S. Aguirre Díaz, J. Puiggalí, L. J. del Valle and M. Gimeno, Polym. Chem., 2021, 12, 1199–1209 RSC.
- T. Waku, M. Matsusaki, S. Chirachanchai and M. Akashi, Chem. Lett., 2008, 37, 1262–1263 CrossRef CAS.
- T. Waku, M. Matsusaki, T. Kaneko and M. Akashi, Macromolecules, 2007, 40, 6385–6392 CrossRef CAS.
- C. Bonduelle, Polym. Chem., 2018, 9, 1517–1529 RSC.
- E. Goormaghtigh, V. Cabiaux and J. M. Ruysschaert, in Physicochemical Methods in the Study of Biomembranes, Springer US, Boston, MA, 1994, pp. 329–441 Search PubMed.
- J. K. Wychowaniec, R. Patel, J. Leach, R. Mathomes, V. Chhabria, Y. Patil-Sen, A. Hidalgo-Bastida, R. T. Forbes, J. M. Hayes and M. A. Elsawy, Biomacromolecules, 2020, 21, 2670–2680 CrossRef CAS PubMed.
- Y. Liu, H. Tang, M. Zhu, H. Y. Zhu and J. Y. Hao, Polymer, 2022, 254, 125093 CrossRef CAS.
- P. Y. Chou, M. Wells and G. D. Fasman, Biochemistry, 1972, 11, 3028–3043 CrossRef CAS PubMed.
- P. Y. Chou and G. D. Fasman, J. Mol. Biol., 1973, 74, 263–281 CrossRef CAS PubMed.
- J. H. Jiang, H. Sun, P. Wei, M. Sun, S. Lin, M. C. Lv, Z. Fan, Y. Q. Zhu and J. Z. Du, Chem. Mater., 2022, 34, 4937–4945 CrossRef CAS.
- A. Rodríguez Galán, M. Pelfort, J. E. Aceituno and J. Puiggalí, J. Appl. Polym. Sci., 1999, 74, 2312–2320 CrossRef.
- Y. J. Fan, M. Kobayashi and H. Kise, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 385–392 CrossRef CAS.
- X. L. Wu, L. G. Xu, L. Q. Liu, W. Ma, H. H. Yin, H. Kuang, L. B. Wang, C. L. Xu and N. A. Kotov, J. Am. Chem. Soc., 2013, 135, 18629–18636 CrossRef CAS PubMed.
- H. Y. Zhang, S. Li, A. H. Qu, C. L. Hao, M. Z. Sun, L. G. Xu, C. L. Xu and H. Kuang, Chem. Sci., 2020, 11, 12937–12954 RSC.
- X. L. Zhao, S. Q. Zang and X. Y. Chen, Chem. Soc. Rev., 2020, 49, 2481–2503 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Materials, synthesis, and analysis section. NMR spectra and analyses, SEC chromatograms, additional DLS data and TEM images, in situ FTIR, digital images of samples, FTIR spectra of monomers and all assessed formulations, CD spectra, UV-Vis analyses, and additional TEM images during enzymatic degradation. See DOI: https://doi.org/10.1039/d2sc05695j |
| This journal is © The Royal Society of Chemistry 2022 |