
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFe2(MoO4)3 assembled by cross-stacking of porous nanosheets enables a high-performance aluminum-ion battery†
Huanyu
Liang‡
a,
Yongshuai
Liu‡
a,
Fengkai
Zuo
a,
Cunliang
Zhang
 *b,
Li
Yang
a,
Linyi
Zhao
a,
Yuhao
Li
a,
Yifei
Xu
a,
Tiansheng
Wang
a,
Xia
Hua
a,
Yue
Zhu
*c and
Hongsen
Li
*a
*b,
Li
Yang
a,
Linyi
Zhao
a,
Yuhao
Li
a,
Yifei
Xu
a,
Tiansheng
Wang
a,
Xia
Hua
a,
Yue
Zhu
*c and
Hongsen
Li
*a
aCollege of Physics, Center for Marine Observation and Communications, Qingdao University, Qingdao 266071, P. R. China. E-mail: hsli@qdu.edu.cn
bSchool of Chemistry and Chemical Engineering, Henan Engineering Center of New Energy Battery Materials, Henan Key Laboratory of Bimolecular Recognition and Sensing, Shangqiu Normal University, Shangqiu, Henan 476000, P. R. China
cMax Planck Institute for Solid State Research, Heisenbergstraße 1, 70569 Stuttgart, Germany
First published on 12th November 2022
Abstract
Rechargeable aluminum-ion batteries have attracted increasing attention owing to the advantageous multivalent ion storage mechanism thus high theoretical capacity as well as inherent safety and low cost of using aluminum. However, their development has been largely impeded by the lack of suitable positive electrodes to provide both sufficient energy density and satisfactory rate capability. Here we report a candidate positive electrode based on ternary metal oxides, Fe2(MoO4)3, which was assembled by cross-stacking of porous nanosheets, featuring superior rate performance and cycle stability, and most importantly a well-defined discharge voltage plateau near 1.9 V. Specifically, the positive electrode is able to deliver reversible capacities of 239.3 mA h g−1 at 0.2 A g−1 and 73.4 mA h g−1 at 8.0 A g−1, and retains 126.5 mA h g−1 at 1.0 A g−1 impressively, after 2000 cycles. Furthermore, the aluminum-storage mechanism operating on Al3+ intercalation in this positive electrode is demonstrated for the first time via combined in situ and ex situ characterization studies and density functional theory calculations. This work not only explores potential positive electrodes for aluminum-based batteries but also sheds light on the fundamental charge storage mechanism within the electrode.
Introduction
The rapid consumption of fossil fuels, together with severe threats posed by global warming, has boosted the demand for new rechargeable battery technologies.1 As the current prevailing energy storage device, lithium-ion batteries (LIBs) have been extensively studied mainly as they enable high density energy storage.2 Despite a high energy density value of 250 W h kg−1, the large-scale application of LIBs to meet the ever increasing demand is still challenging.3 The typical issues include the scarcity of lithium sources, safety problems as well as high cost.4 To address these issues, considerable attention has been paid to aluminum-ion batteries (AIBs) that use aluminum (Al) metal as the negative electrode, which can theoretically deliver an extremely high gravimetric capacity by taking advantage of the trivalent charge carrier.5 These favorable features promise AIBs to be one of the most outstanding next-generation electrochemical storage technologies.5a,6Despite the afore-mentioned benefits of an Al negative electrode, AIBs have major obstacles on the positive electrode side, such as electrode material disintegration and slow charge transport kinetics inside electrode material.3,7 The performance of AIBs thus critically depends on the rational design and construction of a positive electrode structure to solve the problems mentioned above. Previous efforts in searching for suitable AIB positive electrode have included carbon materials,8 metal sulfides,9 and tellurium.10 Research has shown that although carbon-related materials can achieve high voltage plateaus of about 2 V, they usually suffer from a low specific capacity.11 On the other hand, metal sulfides and tellurium are host materials with better charge transport dynamics, but the low discharge plateaus near 1 V hinder their practical applications.10b,12 So far, these challenges have not been fully addressed and the development of these materials in AIBs has been limited. Thereupon, there is an urgent need for continuously exploring positive electrode materials that can simultaneously realize large specific capacity and high voltage plateaus.5c,13
Recently, ternary metal oxide Fe2(MoO4)3 has emerged as a potential candidate in alkali-ion batteries as it was found to possess high electrochemical reactivity and robust structural stability.14 For example, Im et al. reported that the as-synthesized Fe2(MoO4)3 nanosheets achieve an ultrahigh lithium storage capacity of 1983.6 mA h g−1 at a current density of 100 mA g−1 after cycling 400 times.14a Mai et al. developed a facile one-step water-bath method to synthesize graphene oxide-decorated Fe2(MoO4)3 microflowers, which exhibit a reversible discharge capacity of 107.0 mA h g−1 at 10 A g−1 as a negative electrode for sodium ion batteries.15 Our group also previously reported a uniform assembly of layered Fe2(MoO4)3 which achieves an excellent Na storage capability of 504 mA h g−1 at 0.1 A g−1.14b Hitherto, there have still been no efforts to investigate the electrochemical properties of this ternary metal oxide as the electrode material in AIBs. In particular, insights into regulating microscopic morphology and uncovering the aluminum storage mechanism to achieve high performance AIBs are lacking in the literature.
Herein, for the first time, Fe2(MoO4)3 (hereafter expressed as FMO) assembled by cross-stacking of porous nanosheets is being presented as a positive electrode material for AIBs, highlighting a high operating potential (∼1.9 V) along with many excellent electrochemical properties. Remarkably, FMO assembled by cross-stacking of porous nanosheets (denoted as P-FMO) provides abundant active sites for Al3+ storage, hence achieving large discharge capacity (239.3 mA h g−1 at 0.2 A g−1). In the meantime, the intrinsically holey architecture enables highly reversible cycling (78.8% capacity retention after 2000 cycles at 1.0 A g−1). Not only that, a deep understanding of the Al storage mechanism was acquired by means of ex situ X-ray photoelectron spectroscopy (XPS), X-ray diffraction (XRD), high-resolution transmission electron microscopy (HRTEM), and in situ Raman spectroscopy techniques, and density functional theory (DFT), revealing that the positive electrode undergoes intriguing Al3+ intercalation/extraction processes during cycling. This work manifests that multicomponent material, for instance the ternary metal oxide shown here, represents a promising platform on which AIB positive electrodes overcoming current limitations can be widely explored.
Results and discussion
As depicted in Fig. 1a, the synthesis was carried out by mixing ferric nitrate (Fe(NO3)3·9H2O) and sodium molybdate (Na2MoO4·2H2O) in ethylene glycol and subsequently, heating at 200 °C for 15 h in an autoclave. The resulting thin-sheet stacked P-FMO precursor was then annealed to induce pyrolysis and formation of the final crystalline FMO nanoparticles, which interconnected chemically to retain the original two-dimensional morphology but with substantial porosity (for details, see the Experimental section). | ||
| Fig. 1 (a) Schematic illustration of the P-FMO fabrication process. (b) FESEM image of the P-FMO precursor. (c) FESEM image, (d and e) TEM image, (f) HRTEM image and (g) SAED pattern of the P-FMO product. | ||
According to X-ray diffraction (XRD) analysis (Fig. S1†), a highly crystalized pure FMO phase was obtained in accordance with the reported JCPDS data (PDF#72-0935). All diffraction peaks could be well indexed on the basis of monoclinic FMO with a space group P21 (4) and the lattice parameters a = 15.693(3) Å, b = 9.235(1) Å, and c = 18.218(4) Å. The monoclinic phase of Fe2(MoO4)3 has an open three-dimensional framework, which has many interstitial empty sites and diffusion channels for guest ions and allows for minimized volume expansion when redox reaction occurs.14a In terms of morphology, the low magnification field scanning electron microscopy (FESEM) image (Fig. S2†) shows that the FMO precursors are assembled by cross-stacking of nanosheets. A zoomed-in FESEM image (Fig. 1b) signifies that numerous nanosheets, with a typical size of about 1 μm in width and 2 μm in length, are stacked to form the precursor. The typical FESEM images (Fig. S3a, b† and 1c) of the final product after high-temperature calcination indicate that the morphology stayed almost intact. Nevertheless, the stacked nanosheets have been transformed from a dense structure with smooth surfaces (Fig. 1b) into porous nanosheets (Fig. 1c). The as-obtained porous nanosheets made of numerous nanoparticles show good uniformity in the lateral size of about 2 μm. As depicted in Fig. S3c, d† and 1d, the transmission electron microscopy (TEM) image confirms that as-prepared P-FMO possesses a porous structure and is constructed by the agglomeration of numerous nanoparticles. The TEM image at a higher magnification (Fig. 1e) reveals that the nanoparticles are uniform in size of about ∼12 nm, consistent with the FESEM results (Fig. 1c). This rational design of a hierarchical architecture allows Fe2(MoO4)3 to tolerate the dimensional strains and to provide more active sites, which will benefit the stability and performance gains.
The high-resolution TEM (HRTEM) image of the product P-FMO is shown in Fig. 1f. Lattice fringe distances of 0.25 and 0.31 nm in accordance with the (![[6 with combining macron]](https://www.rsc.org/images/entities/char_0036_0304.gif) 02) and (
02) and (![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) 24) planes of FMO can be identified based on the monoclinic P1211 structure. The selected area electron diffraction (SAED) pattern shows a series of diffraction rings (Fig. 1g), which can be assigned to the (24), (204), and (412) planes of FMO, and also reflects the polycrystalline nature of the product. The XPS spectrum of P-FMO (Fig. S4†) displays elemental signals of Fe, Mo and O. In addition, the energy dispersion spectroscopy (EDS) mapping reveals that Fe, Mo and O elements are evenly distributed in the obtained P-FMO (Fig. S5†), confirming the successful preparation of the target material. The nitrogen adsorption–desorption isotherms were obtained to analyze the specific surface area and porous characteristics of P-FMO. As shown in Fig. S6,† a specific surface area of 40.7 m2 g−1 can be calculated as per the Brunauer–Emmett–Teller method. Moreover, according to the nonlocal density functional theory analyses from the inset of Fig. S6,† it can be concluded that P-FMO is indeed highly porous in nature, and the abundant pores are a result of the interspaces among numerous nanoparticles. As has been proven by previous research, this type of hierarchical porous structure is usually beneficial for the storage of metal ions, meanwhile accelerating their migration rate.16
24) planes of FMO can be identified based on the monoclinic P1211 structure. The selected area electron diffraction (SAED) pattern shows a series of diffraction rings (Fig. 1g), which can be assigned to the (24), (204), and (412) planes of FMO, and also reflects the polycrystalline nature of the product. The XPS spectrum of P-FMO (Fig. S4†) displays elemental signals of Fe, Mo and O. In addition, the energy dispersion spectroscopy (EDS) mapping reveals that Fe, Mo and O elements are evenly distributed in the obtained P-FMO (Fig. S5†), confirming the successful preparation of the target material. The nitrogen adsorption–desorption isotherms were obtained to analyze the specific surface area and porous characteristics of P-FMO. As shown in Fig. S6,† a specific surface area of 40.7 m2 g−1 can be calculated as per the Brunauer–Emmett–Teller method. Moreover, according to the nonlocal density functional theory analyses from the inset of Fig. S6,† it can be concluded that P-FMO is indeed highly porous in nature, and the abundant pores are a result of the interspaces among numerous nanoparticles. As has been proven by previous research, this type of hierarchical porous structure is usually beneficial for the storage of metal ions, meanwhile accelerating their migration rate.16
To evaluate the feasibility of the P-FMO electrode for aluminum ion storage, a Swagelok cell was assembled with metallic aluminum foil as the counter electrode and the cell was tested within a potential window of 0.01–2.2 V. As shown in Fig. 2a, the electrochemical processes could be unveiled by using the typical cyclic voltametric (CV) curves at a scan rate of 0.5 mV s−1, showing a multistep intercalation process. In the first cathodic scan, three reduction peaks located at 1.9, 1.14, and 0.35 V can be plainly observed (Fig. 2a). The two well-defined peaks situated at 1.9 and 1.14 V can be ascribed to the formation of Al3+-intercalated compounds, while the broad reduction peaks centered at ≈0.01–0.7 V can be attributed to the formation of metallic Fe and a stable SEI layer on the electrode surface. During the following anodic scan, multiple anodic peaks between 0.5 and 2.15 V can be found, which is attributed to the extraction of Al3+ in the sample. The sequent CV curves of P-FMO are distinct from the initial one, which is ascribed to the enhanced reaction dynamics and the efficient utilization of active materials resulting from the microstructure change during the first cycle. Meanwhile, the CV profiles except for the first scan are overlapped, demonstrating an excellent reversibility of the P-FMO electrode. It is worth noting that there are a pair of obvious redox peaks located at around 1.9/2.14 V vs. Al3+/Al, which is in conformity with the working plateaus of the charge–discharge profiles shown in Fig. 2b. In an ionic liquid electrolyte, the P-FMO positive electrode delivers a large discharge capacity of ∼239.3 mA h g−1 at a small current density of 0.2 A g−1, and displays a relatively high discharge voltage plateau of 1.9 V (vs. Al3+/Al) (Fig. 2b). Besides, the P-FMO electrode exhibits superior rate capability, delivering capacities as high as 239.3, 186.4, 166.4, 129.9, 106.1, 93.1, 82.0 and 73.4 mA h g−1 at current densities of 0.2, 0.3, 0.5, 1.0, 2.0, 3.0, 5.0 and 8.0 A g−1, respectively (Fig. S7†). Moreover, the long cycling performance of P-FMO is demonstrated at 1.0 A g−1 for 2000 cycles (pre-conditioned for five cycles at 0.2 A g−1), as plotted and shown in Fig. 2c. A highly reversible capacity of 126.5 mA h g−1 with an extremely low 0.0106% capacity loss per cycle has been achieved. In addition, ex situ electrochemical impedance spectroscopy (EIS) analyses of the P-FMO electrode were carried out after various cycles to assess the transport kinetics within the electrode (Fig. S8†). The resulting curves comprise of a semicircle at a medium high frequency related to charge-transfer resistance and an inclined line at a low frequency related to diffusion of aluminum ions.17 As the cycle number increases, the charge-transfer resistance is nearly at the same level, while the sloped line gradually steepens, indicating a lowering resistance for the diffusion of aluminum ions. It should be pointed out that P-FMO exhibits ultralow charge transfer resistances (≈6 Ω) within 100 cycles, manifesting its outstanding electrochemical kinetics and possibly contributing to its ultra-high cycling stability. To highlight the latter point, the cycling stability of the P-FMO positive electrode is compared with those reported for metal selenide,18 sulfide,6a,9,12a,19 phosphide20 and oxide positive electrodes,21 as plotted and shown in Fig. 2d and Table S1.† More importantly, a relatively high operating voltage has been achieved in P-FMO (Fig. S9†), which is definitely beneficial to improve the overall energy density of AIBs.13,22
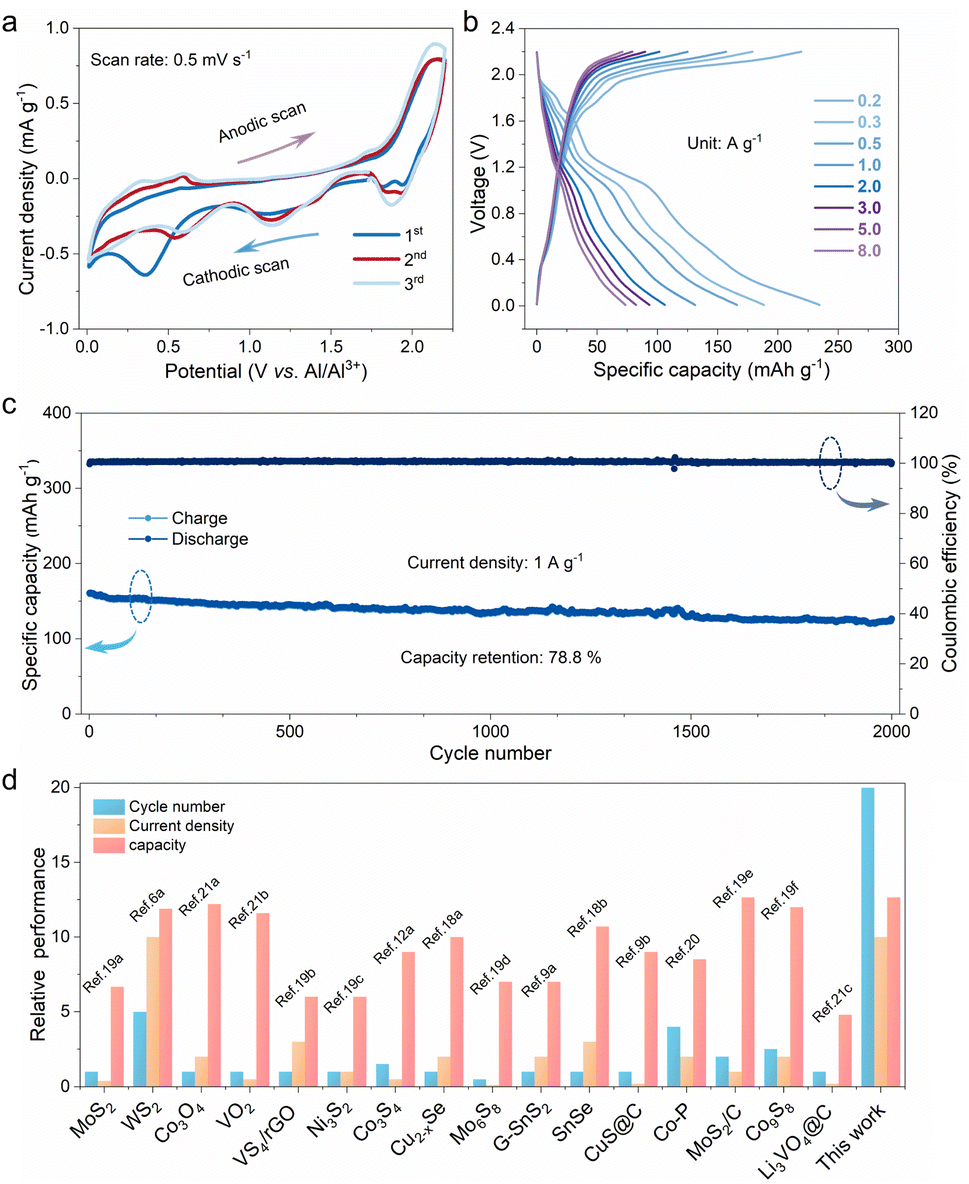 | ||
| Fig. 2 (a) CV profiles for the P-FMO electrode at a scan rate of 0.5 mV s−1. (b) Charge–discharge curves at different current densities. (c) Long cycling stability at a current density of 1.0 A g−1. (d) Comparison of cycling performance of P-FMO with previously reported electrode materials for AIBs. | ||
EIS of the electrode after 50/100 cycles was also analyzed quantitively to further understand the electrochemical kinetics during cycling. Generally, Warburg impedance is directly associated with diffusion resistance. The Warburg factor (σ) can be obtained as the slope of Z′ ∝ w−1/2 plots as shown in Fig. S10† and is determined to be 699.1, 373.2 and 283.1 for the initial electrode, and after 50 cycles and 100 cycles, respectively. Subsequently, the diffusion coefficient DAl3+ can be calculated by using:23
| DAl3+ = R2T2/(2A2n4CAl3+2σ2) |
To evaluate the charge storage behavior for the P-FMO electrode, detailed kinetic analyses based on a current/sweep rate correlation were carried out.24 Fig. S11† shows CV profiles with an increasing scan rate from 0.1 to 2.0 mV s−1. The equation (i = avb)25 was applied to identify the storage process of Al3+ in the P-FMO positive electrode material, where a and b are constants.26 The b values of the cathodic and anodic peaks were obtained as the slope of log(i) vs. log(v). It would be 0.5 for a diffusion-controlled insertion process, and 1 for a surface capacitive behavior. As a general rule, a b value between 0.5 and 1.0 signifies a mixed kinetics process.27 As displayed in Fig. S12,† by plotting the log(i)–log(v) curves, the corresponding b values of the cathodic and anodic peaks were calculated to be 0.69 and 0.55, respectively, reflecting that the electrochemical reaction in the FMO positive electrode is dominated by ion diffusion and capacitance concomitantly.28
To gain mechanistic insights into the Al storage mechanisms in FMO, a series of ex situ and in situ measurements were performed at the selected states of the first discharge/charge cycles. The collected ex situ X-ray photoelectron spectroscopy (XPS) spectra are presented in Fig. 3a, b, S12a and b.† As shown in Fig. 3a, the Fe 2p core level peak of the pristine electrode can be deconvoluted into two characteristic peaks centered at ≈724.3 and ≈711.1 eV, corresponding to the 2p1/2 and 2p3/2 peaks of Fe3+. In contrast, the appearance of Fe2+ (Fe 2p3/2 at 709.8 eV and Fe 2p1/2 at 722.9 eV) and Fe0 (Fe 2p3/2 at 707.4 eV and Fe 2p1/2 at 720.6 eV) upon discharge indicates the reduction of Fe3+ to Fe2+ and Fe0, on account of the incorporation of Al3+ into the FMO phase.29 Nevertheless, the high-resolution Fe 2p spectrum of P-FMO in the fully charged state shows paired deconvoluted peaks corresponding to both Fe2+ and Fe3+. The presence of a small fraction of Fe2+ can be attributed to part of the Fe0 being oxidized to Fe2+. Fig. 3b exhibits the high-resolution Mo 3d core level XPS spectra. It shows that the Mo peaks of the pristine electrode can be assigned to Mo6+ (Mo 3d3/2 at 235.4 eV and Mo 3d5/2 at 232.2 eV).14b,30 Upon being fully discharged, the spectrum in the Mo 3d region shows evidence of Mo4+ (at 228.5 and 229.6 eV). When fully charged, the peaks of Mo4+ decrease significantly, indicating partially reversible oxidation of Mo4+ during the redox reaction process. It is well known that Al3+, AlCl4− and AlCl2+ can all be used as charge carriers for AIBs.31 As far as we know, however, the charge storage of AlCl2+ intercalation prefers to take place in the organic electrode.32 The XPS spectra of Al 2p and Cl 2p at different electrochemical states are often used to determine the types of charge carriers in an Al//inorganic electrode system.3,29a It is worth mentioning that the intensity of Al 2p (Fig. S13a†) at the fully discharged state (0.01 V) is significantly stronger than that in the fully charged state (2.2 V), whereas the intensity of Cl 2p (Fig. S13b†) remains relatively unchanged, suggesting that the carriers are most likely to be Al3+.10b,23a
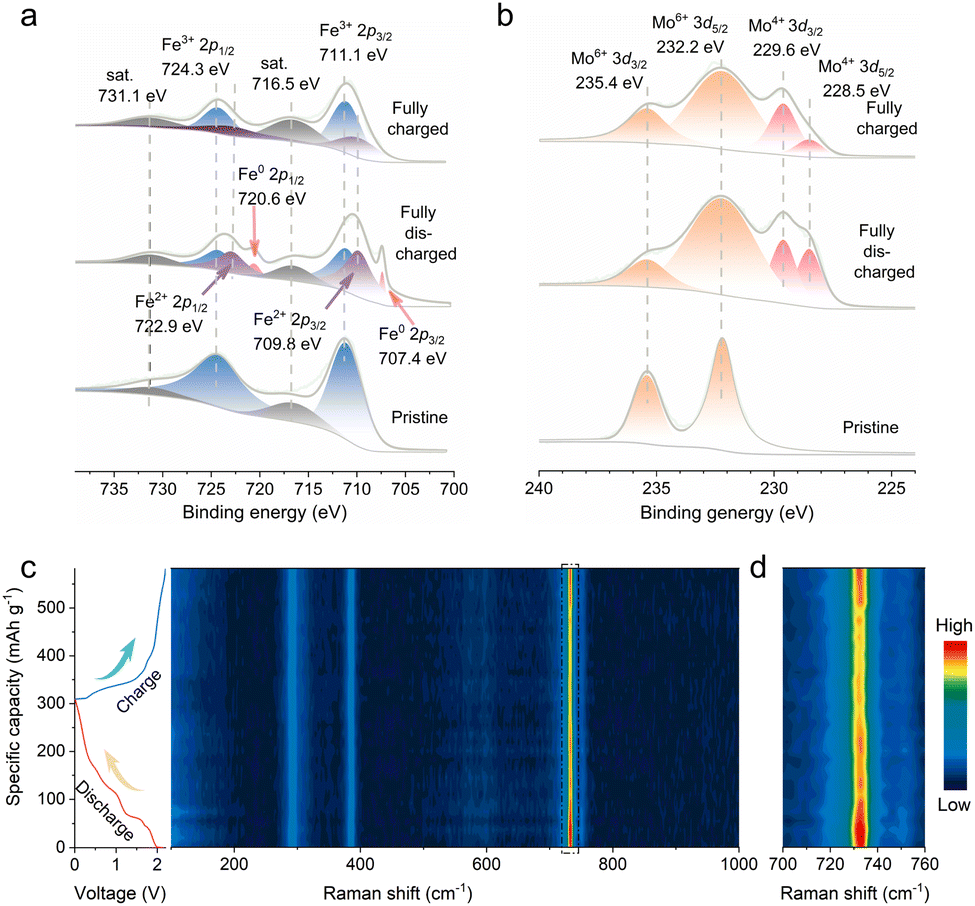 | ||
| Fig. 3 (a and b) Ex situ XPS Fe 2p and Mo 3d at different discharge/charge states. (c and d) In situ Raman spectra of a P-FMO positive electrode in an AIB. | ||
To probe the electrochemical storage mechanism in the FMO material, ex situ HRTEM analyses were carried out at its discharged and charged states. The HRTEM image details the information about the products generated by the incorporation of aluminum ions. As depicted in Fig. S14a,† a lattice spacing of 0.21 nm is recognizable after discharging to 0.01 V, which can be assigned to the (100) plane of Fe0. Also, a few expanded lattice fringes of 0.51 nm are detectable owing to Al3+ insertion, indicating that the conversion reaction is incomplete.3,33 The collected results signify that Fe3+ in FMO can transfer to zero-valent Fe during the insertion of Al3+, which is in accordance with the XPS results. When the electrode is charged back to 2.2 V, the lattice fringes of Fe0 disappear and those of FMO reappear, indicating the highly reversible nature of the conversion reaction (Fig. S14b†). Moreover, although still present in the charge state, the lattice fringes of AlxFe2(MoO4)3 shrink substantially, illustrating the clear existence of an extraction process of Al3+. From the XPS and TEM analyses, it can be concluded that the FMO material undergoes a mixed electrochemical process with synergistic conversion and Al-ion (de)intercalation reactions, positively contributing to its excellent cycle stability without sacrificing its high specific capacity.
To further confirm the Al3+ storage mechanism of the FMO material, an in situ Raman test was conducted during cycling. As shown in Fig. 3c, several noticeable characteristic spectral features which lie in the range of 900–700 cm−1, 500–280 cm−1 and below 280 cm−1 are identified, belonging to the anti-symmetric stretching modes, bending modes and lattice modes, respectively.34 An enlarged view of Fig. 3c shows the evolution of FMO in the black-box range (Fig. 3d), and it can be clearly observed that the intensity becomes weaker during the discharge process, providing strong evidence for the insertion of Al3+.3
When charging back to 2.2 V, the intensity gradually recovers owing to the incomplete extraction of Al3+. Additionally, the insertion mechanism and the high reversibility during the discharge/charge processes were also proved by ex situ XRD (Fig. S15†). Apparently, the (122) and (![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) 24) diffraction peaks of FMO shift towards lower angles during the discharge process, corroborating the Al3+ insertion which causes lattice expansion. During the charging process, Al3+ extraction from FMO restores the (122) and (24) diffraction peaks to their original positions, again illustrating the reversibility of the proposed intercalation mechanism.
24) diffraction peaks of FMO shift towards lower angles during the discharge process, corroborating the Al3+ insertion which causes lattice expansion. During the charging process, Al3+ extraction from FMO restores the (122) and (24) diffraction peaks to their original positions, again illustrating the reversibility of the proposed intercalation mechanism.
Altogether, from the combined ex situ XPS, HRTEM, XRD and in situ Raman analyses of the electrodes at different states, it can be concluded that FMO as an AIB positive electrode undergoes the following stepwise intercalation-conversion process:
For the positive electrode:
Discharge process:
| Fe2(MoO4)3 + xAl3+ + 3xe− → AlxFe2(MoO4)3 |
| AlxFe2(MoO4)3 → AlxFen(MoO4)3 + (2 − n)Fe0(0 < n < 2) |
Charge process:
| AlxFen(MoO4)3 + (2 − n)Fe0 → AlxFe2(MoO4)3 (0 < n < 2) |
| AlxFe2(MoO4)3 → xAl3+ + 3xe− + Fe2(MoO4)3 |
For the negative electrode:
| Al + 7AlCl−4 ↔ 4Al2Cl7− + 3e− |
Based on the above understanding, the phase evolution mechanism of FMO in AIBs is schematically depicted in Fig. 4. At first, Al3+ is incorporated into the FMO phase to initially form AlxFe2(MoO4)3, which is attributed to a pure intercalation reaction. With more Al3+ incorporated, the decomposition of AlxFe2(MoO4)3 starts to take place, leading to the formation of metallic Fe. In the charging process, undecomposed AlxFen(MoO4)3 reacts with metallic Fe to transform into AlxFe2(MoO4)3 and the subsequent Al3+ extraction converts it back into Fe2(MoO4)3.
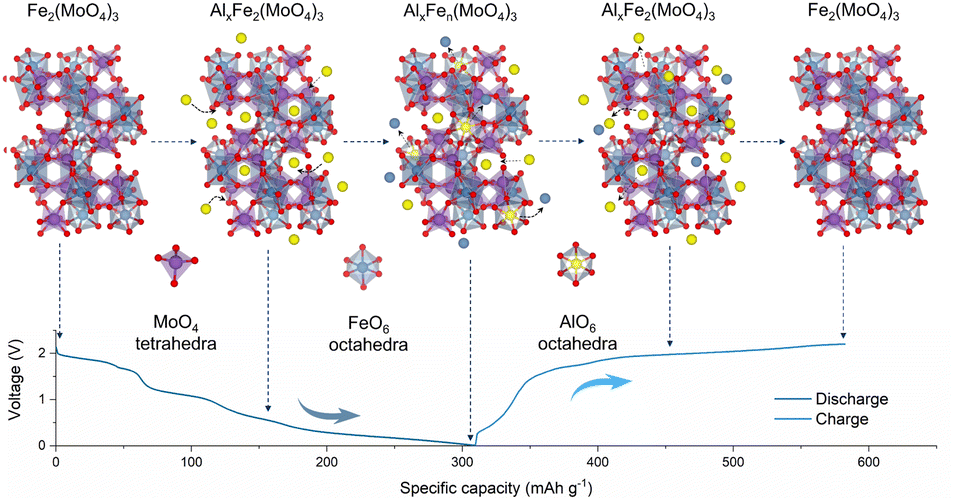 | ||
| Fig. 4 Schematics of the aluminum reaction mechanisms in P-FMO. | ||
To understand the origin of the outstanding Al storage performance of the P-FMO better, DFT calculations were carried out to elucidate the reaction process and electronic transport properties of the FMO material. The high operating voltage and superior capacity of the Al/P-FMO battery possibly originates from the distinctive electronic structure of FMO, which can be inferred from its density of state (DOS). As shown in Fig. 5a, the electronic state near the Femi level composed of Fe-3d, Mo-4d and O-2p orbitals demonstrates that the electrons accompanied by Al3+ insertion in the discharge process of FMO are well accommodated by Fe, V, and O sites. Meanwhile, with merits of high electronic conductivity provided by its metallic properties,35 which is beneficial for the cation host to accommodate electrical field changes during Al3+ insertion, FMO is thus a very suitable positive electrode material for AIBs. To verify thermodynamically the soundness of the proposed storage mechanism, the formation energy (Eb) of the insertion of Al (complex) ions (e.g. Al3+ or AlCl4−) is taken into account according to the equation, E(b) = E(Total) − E{Fe2(MoO4)3} − E(ion), where E(Total) and E{Fe2(MoO4)3} are the total energies of the Fe2(MoO4)3 monoclinic crystals with/without an inserted ion, respectively (Fig. 5b and c). E(ion) is the total energy of the Al3+ ions in the reservoir. According to the computational results of E(b), Al3+ displays a more negative value (−2.859 eV, Fig. 5d and Table S2†), accentuating that the insertion reaction is highly plausible in theory.21b Most notably, because of the positive E(b) of AlCl4− (1.08 eV > 0, Fig. 5d), the anion cannot exist as intercalated ions inside the FMO crystal.6a On the basis of this conclusion, the Al3+ diffusion process within the FMO structure can be readily simulated and displayed, as shown in the illustration of Fig. 5e and f. Finally, Fig. 5g shows an Al3+ diffusion barrier of about 0.285 eV, which is considerably small and favorable for diffusion kinetics, thus contributing to the superior electrochemical capability of FMO.
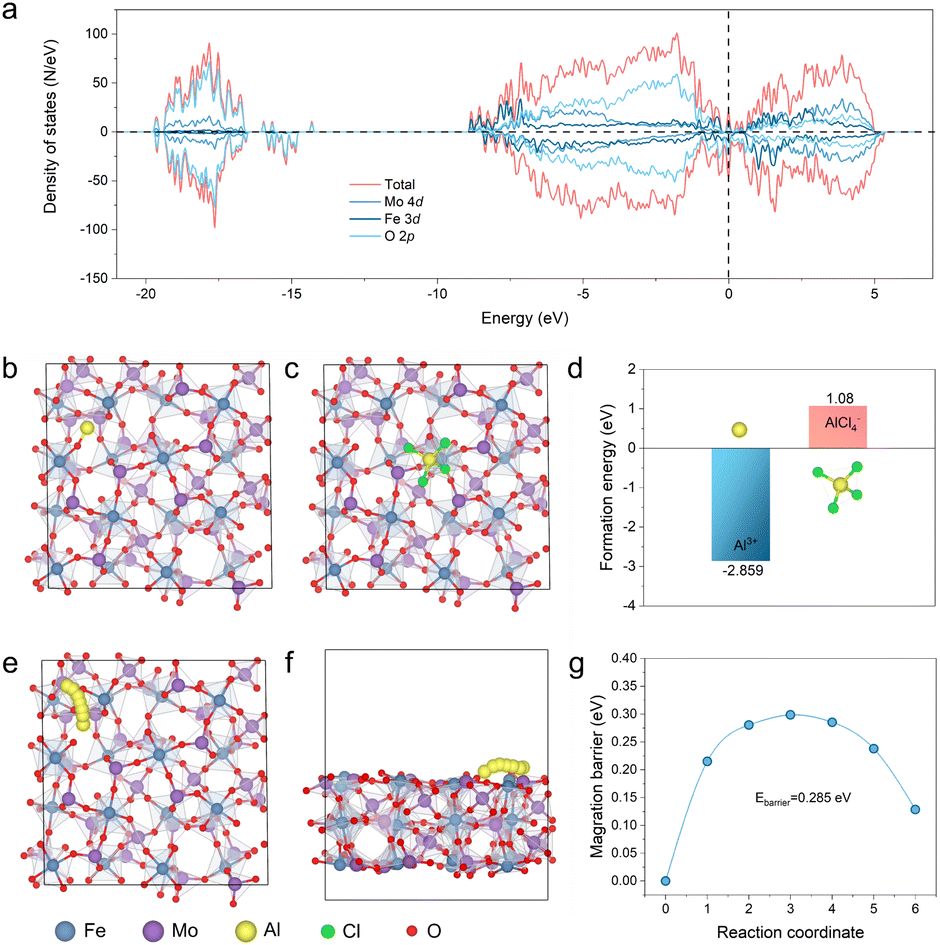 | ||
| Fig. 5 (a) Total DOS and partial DOS of Fe 3d, Mo 4d and O 2p orbitals. The calculated formation energies of (b) Al3+ and (c) AlCl4− in the monoclinic structure of FMO. (d) The formation energy of Al3+ and AlCl4−. Top view (e) and side view (f) of diffusion paths for Al3+. (g) The Al-ion diffusion energy barrier of FMO. | ||
Conclusions
To summarize, we report the fabrication of a P-FMO material by a facile solvothermal method with a post-annealing procedure, and its outstanding performance as a positive electrode for AIBs. Such a uniformly nanosized FMO assembly can provide desired compatibility with AIBs during Al3+ (de)insertion, substantially accelerating diffusion kinetics and achieving excellent electrochemical capability. Specifically, the as-fabricated electrode features a high operating potential (∼1.9 V), superior high-rate discharge capacity (73.4 mA h g−1 at 8.0 A g−1), and excellent cyclability (2000 cycles at 1.0 A g−1). Beyond device performance, the underlying reaction mechanism of Al3+ cation insertion into FMO was first systematically corroborated by in situ Raman spectroscopy, DFT calculations, as well as ex situ XPS, TEM and XRD characterization. Electrochemical kinetic analyses coupled with first principles calculations suggest that the high diffusion coefficients and low diffusion barrier of Al3+ are the key to its superior electrochemical properties. This work demonstrates the potential of utilizing ternary metal oxides as the positive electrode for aluminum storage, and provides mechanistic insights towards the realization of high performance and stable AIBs.Data availability
All data have been provided in the main text and ESI.† We do not have addition data to provide.Author contributions
H. S. L. designed and supervised the project. H. Y. L. and Y. S. L. carried out the experiments. H. Y. L., Y. S. L., F. K. Z., C. L. Z., L. Y., L. Y. Z., Y. F. X., T. S. W., X. H. and Y. Z. analyzed and discussed the data. H. Y. L., Y. S. L. and Y. Z. wrote the manuscript with help from all of the co-authors. All authors revised the manuscript and discussed the results.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors appreciatively acknowledge the financial support of the National Natural Science Foundation of China (51804173), Youth Innovation Technology Project of Higher School in Shandong Province (2021KJ020), Open Fund of Key Laboratory for Intelligent Nano Materials and Devices of the Ministry of Education NJ2022002 (INMD-2022M06), Science and Technology Research Project of Henan Province (222102240025), and Henan New Engineering Research and Practice Project (2020JGLX067).Notes and references
- (a) I. H. Son, J. H. Park, S. Park, K. Park, S. Han, J. Shin, S.-G. Doo, Y. Hwang, H. Chang and J. W. Choi, Nat. Commun., 2017, 8, 1561 CrossRef PubMed; (b) H. Li, Z. Hu, Q. Xia, H. Zhang, Z. Li, H. Wang, X. Li, F. Zuo, F. Zhang, X. Wang, W. Ye, Q. Li, Y. Long, Q. Li, S. Yan, X. Liu, X. Zhang, G. Yu and G.-X. Miao, Adv. Mater., 2021, 33, 2006629 CrossRef CAS; (c) D.-Y. Wang, C.-Y. Wei, M.-C. Lin, C.-J. Pan, H.-L. Chou, H.-A. Chen, M. Gong, Y. Wu, C. Yuan, M. Angell, Y.-J. Hsieh, Y.-H. Chen, C.-Y. Wen, C.-W. Chen, B.-J. Hwang, C.-C. Chen and H. Dai, Nat. Commun., 2017, 8, 14283 CrossRef CAS; (d) Q. Li, H. Li, Q. Xia, Z. Hu, Y. Zhu, S. Yan, C. Ge, Q. Zhang, X. Wang, X. Shang, S. Fan, Y. Long, L. Gu, G.-X. Miao, G. Yu and J. S. Moodera, Nat. Mater., 2021, 20, 76–83 CrossRef CAS PubMed; (e) F. Li, Y. Li, L. Zhao, J. Liu, F. Zuo, F. Gu, H. Liu, R. Liu, Y. Li, J. Zhan, Q. Li and H. Li, Adv. Sci., 2022 DOI:10.1002/advs.202203895.
- (a) L. Liang, W. Zhang, F. Zhao, D. K. Denis, F. u. Zaman, L. Hou and C. Yuan, Adv. Mater. Interfaces, 2020, 7, 1901749 CrossRef CAS; (b) Z. Hao, X. Shi, W. Zhu, X. Zhang, Z. Yang, L. Li, Z. Hu, Q. Zhao and S. Chou, Chem. Sci., 2022, 13, 11376–11381 RSC; (c) L. Ma, Y. Lv, J. Wu, C. Xia, Q. Kang, Y. Zhang, H. Liang and Z. Jin, Nano Res., 2021, 14, 4442–4470 CrossRef CAS.
- L. Xing, K. A. Owusu, X. Liu, J. Meng, K. Wang, Q. An and L. Mai, Nano Energy, 2021, 79, 105384 CrossRef CAS.
- (a) M. T. Ahsan, Z. Ali, M. Usman, Y. Hou, Carbon Energy, 4, 776–819 Search PubMed; (b) L. Guojin, L. Xinliang, W. Yanbo, Y. Shuo, H. Zhaodong, Y. Qi, W. Donghong, D. Binbin, Z. Minshen and Z. Chunyi, Nano Res. Energy, 2022, 1, e9120002 CrossRef; (c) Z. Li, Y. Zhang, X. Li, F. Gu, L. Zhang, H. Liu, Q. Xia, Q. Li, W. Ye, C. Ge, H. Li, H. Hu, S. Li, Y.-Z. Long, S. Yan, G.-X. Miao and Q. Li, J. Am. Chem. Soc., 2021, 143, 12800–12808 CrossRef CAS PubMed; (d) Z. Jin, X. Zhou, Y. Hu, X. Tang, K. Hu, K. M. Reddy, X. Lin and H.-J. Qiu, Chem. Sci., 2022, 13, 12056–12064 RSC.
- (a) Y. Liu, Y. Li, F. Zuo, J. Liu, Y. Xu, L. Yang, H. Zhang, H. Wang, X. Zhang, C. Liu, Q. Li and H. Li, Small, 2022, 18, e2203236 CrossRef PubMed; (b) K. Zhang, X. Han, Z. Hu, X. Zhang, Z. Tao and J. Chen, Chem. Soc. Rev., 2015, 44, 699 RSC; (c) G. A. Elia, K. Marquardt, K. Hoeppner, S. Fantini, R. Lin, E. Knipping, W. Peters, J.-F. Drillet, S. Passerini and R. Hahn, Adv. Mater., 2016, 28, 7564–7579 CrossRef CAS; (d) X. Wang and H. Wang, Adv. Powder Mater., 2022, 1, 100057 CrossRef.
- (a) Z. Zhao, Z. Hu, Q. Li, H. Li, X. Zhang, Y. Zhuang, F. Wang and G. Yu, Nano Today, 2020, 32, 100870 CrossRef CAS; (b) X. Zhang and S. Jiao, Int. J. Miner., Metall. Mater., 2022, 29, 896–904 CrossRef CAS.
- Y. Hu, H. Huang, D. Yu, X. Wang, L. Li, H. Hu, X. Zhu, S. Peng and L. Wang, Nano-Micro Lett., 2021, 13, 159 CrossRef CAS PubMed.
- M.-C. Lin, M. Gong, B. Lu, Y. Wu, D.-Y. Wang, M. Guan, M. Angell, C. Chen, J. Yang, B.-J. Hwang and H. Dai, Nature, 2015, 520, 324–328 CrossRef CAS PubMed.
- (a) Y. Hu, B. Luo, D. Ye, X. Zhu, M. Lyu and L. Wang, Adv. Mater., 2017, 29, 1606132 CrossRef PubMed; (b) S. Wang, S. Jiao, J. Wang, H.-S. Chen, D. Tian, H. Lei and D.-N. Fang, ACS Nano, 2017, 11, 469–477 CrossRef CAS PubMed.
- (a) X. Zhang, S. Jiao, J. Tu, W.-L. Song, X. Xiao, S. Li, M. Wang, H. Lei, D. Tian, H. Chen and D. Fang, Energy Environ. Sci., 2019, 12, 1918–1927 RSC; (b) Z. Yu, S. Jiao, J. Tu, Y. Luo, W.-L. Song, H. Jiao, M. Wang, H. Chen and D. Fang, ACS Nano, 2020, 14, 3469–3476 CrossRef CAS PubMed.
- (a) L. Zhang, L. Chen, H. Luo, X. Zhou and Z. Liu, Adv. Energy Mater., 2017, 7, 1700034 CrossRef; (b) Y. Wu, M. Gong, M.-C. Lin, C. Yuan, M. Angell, L. Huang, D.-Y. Wang, X. Zhang, J. Yang, B.-J. Hwang and H. Dai, Adv. Mater., 2016, 28, 9218–9222 CrossRef CAS PubMed.
- (a) H. Li, H. Yang, Z. Sun, Y. Shi, H.-M. Cheng and F. Li, Nano Energy, 2019, 56, 100–108 CrossRef CAS; (b) Y. Hu, D. Ye, B. Luo, H. Hu, X. Zhu, S. Wang, L. Li, S. Peng and L. Wang, Adv. Mater., 2018, 30, 1703824 CrossRef PubMed.
- H. Zhang, Y. Liu, L. Yang, L. Zhao, X. Dong, H. Wang, Y. Li, T. Sun, Q. Li and H. Li, Energy Storage Mater., 2022, 47, 336–344 CrossRef.
- (a) H. Tran Huu and W. B. Im, ACS Appl. Mater. Interfaces, 2020, 12, 35152–35163 CrossRef CAS PubMed; (b) H. Liang, H. Zhang, L. Zhao, Z. Chen, C. Huang, C. Zhang, Z. Liang, Y. Wang, X. Wang, Q. Li, X. Guo and H. Li, Chem. Eng. J., 2022, 427, 131481 CrossRef CAS; (c) H. T. Huu, N. S. M. Viswanath, N. H. Vu, J.-W. Lee and W. B. Im, Nano Res., 2021, 14, 3977–3987 CrossRef CAS.
- C. Han, X. Ren, Q. Li, W. Luo, L. Huang, L. Zhou and L. Mai, Nano Res., 2018, 11, 1285–1293 CrossRef CAS.
- (a) R. Muruganantham, T.-H. Hsieh, C.-H. Lin and W.-R. Liu, Mater. Today Energy, 2019, 14, 100346 CrossRef; (b) L.-F. Zhao, Z. Hu, W.-H. Lai, Y. Tao, J. Peng, Z.-C. Miao, Y.-X. Wang, S.-L. Chou, H.-K. Liu and S.-X. Dou, Adv. Energy Mater., 2021, 11, 2002704 CrossRef CAS; (c) J. Zhang, H. Huang, J. Bae, S.-H. Chung, W. Zhang, A. Manthiram and G. Yu, Small Methods, 2018, 2, 1700279 CrossRef; (d) L. Peng, Z. Fang, Y. Zhu, C. Yan and G. Yu, Adv. Energy Mater., 2018, 8, 1702179 CrossRef.
- Y. Jiang, Y. Wu, Y. Chen, Z. Qi, J. Shi, L. Gu and Y. Yu, Small, 2018, 14, 1703471 CrossRef PubMed.
- (a) J. Jiang, H. Li, T. Fu, B.-J. Hwang, X. Li and J. Zhao, ACS Appl. Mater. Interfaces, 2018, 10, 17942–17949 CrossRef CAS PubMed; (b) Y. Zhang, B. Zhang, J. Li, J. Liu, X. Huo and F. Kang, Chem. Eng. J., 2021, 403, 126377 CrossRef CAS.
- (a) Z. Li, B. Niu, J. Liu, J. Li and F. Kang, ACS Appl. Mater. Interfaces, 2018, 10, 9451 CrossRef CAS PubMed; (b) X. Zhang, S. Wang, J. Tu, G. Zhang, S. Li, D. Tian and S. Jiao, ChemSusChem, 2018, 11, 709–715 CrossRef CAS PubMed; (c) S. Wang, Z. Yu, J. Tu, J. Wang, D. Tian, Y. Liu and S. Jiao, Adv. Energy Mater., 2016, 6, 1600137 CrossRef; (d) L. Geng, G. Lv, X. Xing and J. Guo, Chem. Mater., 2015, 27, 4926–4929 CrossRef CAS; (e) W. Yang, H. Lu, Y. Cao, B. Xu, Y. Deng and W. Cai, ACS Sustainable Chem. Eng., 2019, 7, 4861–4867 CrossRef CAS; (f) Z. Hu, K. Zhi, Q. Li, Z. Zhao, H. Liang, X. Liu, J. Huang, C. Zhang, H. Li and X. Guo, J. Power Sources, 2019, 440, 227147 CrossRef CAS.
- S. Lu, M. Wang, F. Guo, J. Tu, A. Lv, Y. Chen and S. Jiao, Chem. Eng. J., 2020, 389, 124370 CrossRef CAS.
- (a) J. Liu, Z. Li, X. Huo and J. Li, J. Power Sources, 2019, 422, 49–56 CrossRef CAS; (b) W. Wang, B. Jiang, W. Xiong, H. Sun, Z. Lin, L. Hu, J. Tu, J. Hou, H. Zhu and S. Jiao, Sci. Rep., 2013, 3, 3383 CrossRef PubMed; (c) J. Jiang, H. Li, J. Huang, K. Li, J. Zeng, Y. Yang, J. Li, Y. Wang, J. Wang and J. Zhao, ACS Appl. Mater. Interfaces, 2017, 9, 28486–28494 CrossRef CAS PubMed.
- L. E. Blanc, D. Kundu and L. F. Nazar, Joule, 2020, 4, 771–799 CrossRef CAS.
- (a) C. Li, S. Dong, P. Wang, C. Wang and L. Yin, Adv. Energy Mater., 2019, 9, 1902352 CrossRef CAS; (b) W. Zhang, L. Liang, F. Zhao, Y. Liu, L. Hou and C. Yuan, Electrochim. Acta, 2020, 340, 135871 CrossRef CAS.
- (a) N. Cheng, W. Zhou, J. Liu, Z. Liu and B. Lu, Nano-Micro Lett., 2022, 14, 146 CrossRef CAS PubMed; (b) X. Ma, X. Cao, M. Yao, L. Shan, X. Shi, G. Fang, A. Pan, B. Lu, J. Zhou and S. Liang, Adv. Mater., 2022, 34, 2105452 CrossRef CAS PubMed.
- (a) C. Liu, Y. Li, Y. Feng, S. Zhang, D. Lu, B. Huang, T. Peng and W. Sun, Chin. Chem. Lett., 2021, 32, 3601–3606 CrossRef CAS; (b) B. Wu, S. Qi, X. Wu, H. Wang, Q. Zhuang, H. Yi, P. Xu, Z. Xiong, G. Shi, S. Chen and B. Wang, Chin. Chem. Lett., 2021, 32, 3113–3117 CrossRef CAS.
- Z. Sun, H. Liang, H. Wang, J. Shi, M. Huang, J. Chen, S. Liu, W. Tian, H. Cao and Z. Li, Adv. Funct. Mater., 2022, 32, 2203291 CrossRef CAS.
- (a) Y. Ma, L. Zhang, Z. Yan, B. Cheng, J. Yu and T. Liu, Adv. Energy Mater., 2022, 12, 2103820 CrossRef CAS; (b) W. Zhang, Y. Wu, Z. Xu, H. Li, M. Xu, J. Li, Y. Dai, W. Zong, R. Chen, L. He, Z. Zhang, D. J. L. Brett, G. He, Y. Lai and I. P. Parkin, Adv. Energy Mater., 2022, 12, 2201065 CrossRef CAS; (c) Y. Jiang and J. Liu, Energy Environ. Mater., 2019, 2, 30–37 CrossRef.
- S. Wang, S. Huang, M. Yao, Y. Zhang and Z. Niu, Angew. Chem., Int. Ed., 2020, 59, 11800–11807 CrossRef CAS.
- (a) T. Cai, L. Zhao, H. Hu, T. Li, X. Li, S. Guo, Y. Li, Q. Xue, W. Xing, Z. Yan and L. Wang, Energy Environ. Sci., 2018, 11, 2341–2347 RSC; (b) H. Wang, L. Zhao, H. Zhang, Y. Liu, L. Yang, F. Li, W. Liu, X. Dong, X. Li, Z. Li, X. Qi, L. Wu, Y. Xu, Y. Wang, K. Wang, H. Yang, Q. Li, S. Yan, X. Zhang, F. Li and H. Li, Energy Environ. Sci., 2022, 15, 311–319 RSC.
- Z. Zhang, W. Li, T.-W. Ng, W. Kang, C.-S. Lee and W. Zhang, J. Mater. Chem. A, 2015, 3, 20527–20534 RSC.
- K. L. Ng, B. Amrithraj and G. Azimi, Joule, 2022, 6, 134–170 CrossRef CAS.
- (a) D. J. Kim, D.-J. Yoo, M. T. Otley, A. Prokofjevs, C. Pezzato, M. Owczarek, S. J. Lee, J. W. Choi and J. F. Stoddart, Nat. Energy, 2018, 4, 51–59 CrossRef; (b) X. Han, S. Li, W. L. Song, N. Chen, H. Chen, S. Huang and S. Jiao, Adv. Energy Mater., 2021, 11, 2101446 CrossRef CAS.
- H. Hong, J. Liu, H. Huang, C. Atangana Etogo, X. Yang, B. Guan and L. Zhang, J. Am. Chem. Soc., 2019, 141, 14764–14771 CrossRef CAS PubMed.
- (a) T. S. Freitas, F. S. Oliveira, R. P. Cruz, R. L. S. Pereira, A. R. P. Silva, J. V. B. Moura, C. Luz-Lima, B. C. Viana, J. H. da Silva, P. T. C. Freire and H. D. M. Coutinho, Eur. J. Pharm. Sci., 2018, 123, 295–300 CrossRef CAS PubMed; (b) J. V. B. Moura, G. S. Pinheiro, P. T. C. Freire, J. M. Filho, G. D. Saraiva, B. C. Viana and C. Luz-Lima, Vib. Spectrosc., 2016, 87, 88–93 CrossRef CAS.
- L. Ma, N. Li, C. Long, B. Dong, D. Fang, Z. Liu, Y. Zhao, X. Li, J. Fan, S. Chen, S. Zhang and C. Zhi, Adv. Funct. Mater., 2019, 29, 1906142 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc05479e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |