
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThree-component carboformylation: α-quaternary aldehyde synthesis via Co(III)-catalysed sequential C–H bond addition to dienes and acetic formic anhydride†
Joseph P.
Tassone
,
Jihyeon
Yeo
and
Jonathan A.
Ellman
 *
*
Department of Chemistry, Yale University, 225 Prospect St., New Haven, CT 06520, USA. E-mail: jonathan.ellman@yale.edu
First published on 21st November 2022
Abstract
All carbon α-quaternary aldehydes are prepared via Co(III)-catalysed sequential C–H bond addition to dienes and acetic formic anhydride, representing a rare example of intermolecular carboformylation. A wide range of internally substituted dienes containing diverse functionality can be employed in this reaction, affording complex α-quaternary aldehydes that would not be accessible via hydroformylation approaches. Mechanistic investigations, including control reactions and deuterium labeling studies, establish a catalytic cycle that accounts for formyl group introduction with an uncommon 1,3-addition selectivity to the conjugated diene. Investigations into the role of the uniquely effective additive Proton Sponge® were also conducted, leading to the observation of a putative, intermediate Co(I) tetramethylfulvene complex at low temperatures via NMR spectroscopy. The synthetic utility of the aldehyde products is demonstrated by various transformations, including proline-catalysed asymmetric aldol addition, reductive amination, and the asymmetric synthesis of amines using tert-butanesulfinamide technology.
Introduction
Aldehydes are highly versatile synthetic intermediates due to the plethora of chemical transformations in which they can participate. Industrially relevant aldehydes are commonly produced on commodity scale using hydroformylation, a metal-catalysed process involving the formal addition of a hydrogen and a formyl group across an unsaturated C–C bond (Scheme 1A).1 Hydroformylation has been studied extensively, with numerous developments for improving the chemoselectivity, regioselectivity and stereoselectivity of the reaction having been reported.1,2 Despite these advances, a longstanding challenge in hydroformylation is the preparation of α-quaternary aldehydes from 1,1-disubstituted alkenes, which are generally considered to be prohibited according to the empirical Keulemans' rule, “Addition of the formyl group to a tertiary C atom does not occur, so that no quaternary C atoms are formed.”3 In keeping with this rule, examples of α-quaternary aldehyde synthesis from 1,1-disubstituted alkenes are limited to the use of highly specialized substrates, namely: alkenes containing electron-withdrawing substituents, including heteroatoms and fluorines (to form α-tetrasubstituted aldehydes);4a,b,d,5a,b,e,g strained exocyclic alkenes;5b and alkenes incorporating a directing group4c,5f or a functionality that reacts with a catalytic directing group.5c,d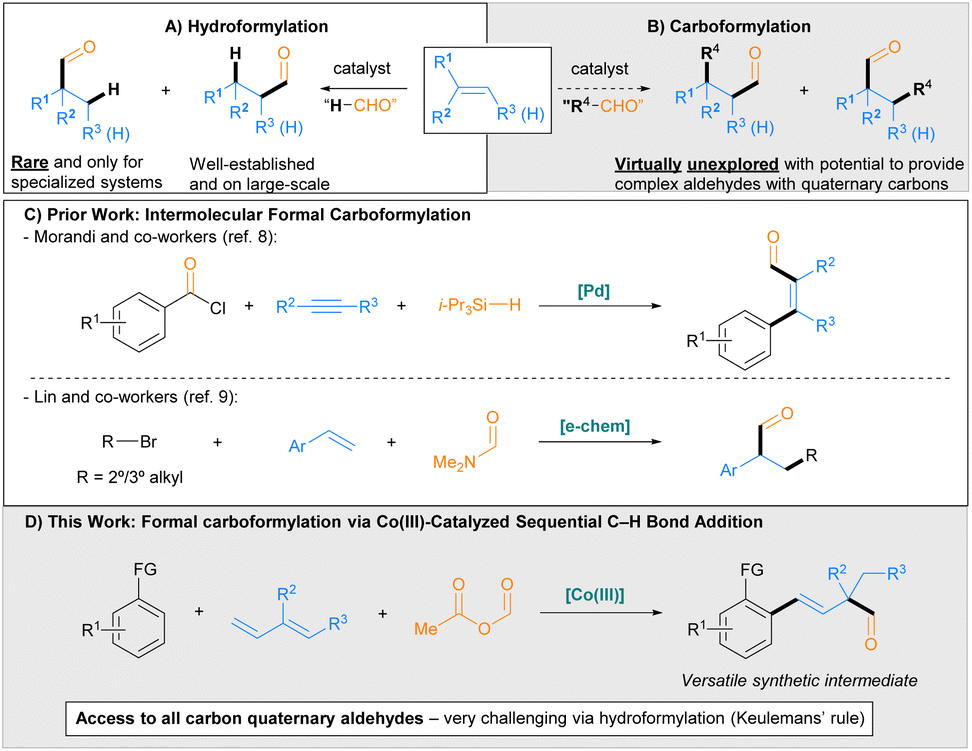 | ||
| Scheme 1 Key issues and challenges of hydroformylation and carboformylation. | ||
Carboformylation whereby an R group and a formyl group are added across an unsaturated C–C bond has the potential to provide more complex value-added aldehydes given that two new C–C bonds are formed (Scheme 1B). Indeed, for 1,1-disubstituted alkenes, either regioisomeric product necessarily incorporates a quaternary carbon, thereby overcoming Keulemans' rule for hydroformylation. However, approaches for achieving carboformylation are virtually unexplored. Initial reports relied on intramolecular transformations via Pd-catalysed cyclization-carboformylation cascades between alkene-tethered aryl iodides, carbon monoxide, and methyldiphenylsilane (as a terminal hydride source) to form indoline and 2,3-dihydrobenzofuran structures.6,7 More recently, two formal intermolecular carboformylation processes have been disclosed. Morandi and co-workers8 developed a procedure for the Pd-catalysed carboformylation of alkynes using aroyl chlorides as the carbon monoxide and ‘R’ source and triisopropylsilane as the terminal hydride source to form α,β-unsaturated aldehydes (Scheme 1C, top). Additionally, Lin and co-workers9 developed a method for the carboformylation of styrenes using alkyl bromides and N,N-dimethylformamide as the formyl source to prepare α-branched aldehydes via an electrochemically mediated, radical-polar crossover mechanism (Scheme 1C, bottom). However, only two examples of α-quaternary aldehydes were synthesized using this method.
Recently, our group10 and Zhou, Chen and co-workers11 have developed Co(III)-catalysed sequential C–H bond additions to dienes and aldehydes, ketones, or an electrophilic cyanating reagent to prepare complex products with two new C–C σ-bonds in a single synthetic step.12,13 Notably, reactions employing internally substituted dienes furnish a quaternary center. With this in mind, we hypothesized that leveraging the sequential C–H bond addition to dienes and an appropriate formylating agent might therefore grant access to complex all carbon α-quaternary aldehydes. Herein, we describe the development of a Co(III)-catalysed sequential C–H bond addition to dienes and acetic formic anhydride to efficiently prepare complex α-quaternary aldehydes via a formal intermolecular carboformylation process. The reaction proceeds with a wide range of internally substituted dienes, affording a broad scope of α-quaternary aldehydes that would not be accessible using existing hydroformylation methods. Mechanistic investigations support a proposed catalytic cycle consistent with the uncommon 1,3-addition to the conjugated diene and the unique effectiveness of Proton Sponge® as an additive. The versatility of the aldehyde products for further synthetic elaboration was illustrated by five distinct transformations, including efficient asymmetric transformations to alcohol and amine products.
Results and discussion
Identification of key reaction parameters towards reaction optimization
After examining a variety of reaction parameters, including catalyst counterion, additive, temperature, solvent, and formylating agent, optimized reaction conditions for the Co(III)-catalysed sequential C–H bond addition of 1-(m-tolyl)-1H-pyrazole (1a) to isoprene (2a) and acetic formic anhydride (3) to form aldehyde 4a are presented in entry 1 of Table 1 (see Tables S1–S5 in the ESI for additional information†). [Cp*Co(C6H6)][B(C6F5)4]2, developed by our lab,14 proved to be a superior catalyst for this transformation because it is completely non-coordinating and provides for high solubility of the catalyst (entry 1). Other pre-formed Co(III) catalysts or catalyst mixtures containing minimally coordinating counterions gave lower yields (entries 2–4 and Table S1 in the ESI†). The addition of Proton Sponge® (1,8-bis(dimethylamino)naphthalene) proved to be uniquely beneficial to the reaction yield in comparison to a reaction run without additive (entry 5). However, the addition of excess Proton Sponge® completely inhibited catalytic activity (entry 6 and Table S2 in the ESI†). Other acidic or basic additives commonly used in two- and three-component C–H bond addition reactions such as HOAc and LiOAc were detrimental (entries 7 and 8). Moreover, in contrast to Proton Sponge®, a variety of other tertiary amines were ineffective (Table S2 in the ESI†).|
|
||
|---|---|---|
| Entry | Variation from standard conditions | Yield of 4ab(%) |
| a Reaction conditions: 1a (0.1 mmol), 2a (0.4 mmol), 3 (0.3 mmol), [Cp*Co(C6H6)][B(C6F5)4]2 (20 mol%), Proton Sponge® (20 mol%) in DCE ([1a] = 1.0 M) at 70 °C. b Yield determined by crude 1H NMR spectroscopic analysis relative to trimethyl (phenyl)silane as standard. Tf = triflyl, DCE = 1,2-dichloroethane, PhCl = chlorobenzene. | ||
| 1 | None | 71 |
| 2 | [Cp*Co(C6H6)](PF6)2 (20 mol%) in place of [Cp*Co(C6H6)][B(C6F5)4]2 | 20 |
| 3 | Cp*Co(CO)I2 (20 mol%) + AgSbF6 (40 mol%) in place of [Cp*Co(C6H6)][B(C6F5)4]2 | 50 |
| 4 | Cp*Co(CO)I2 (20 mol%) + AgNTf2 (40 mol%) in place of [Cp*Co(C6H6)][B(C6F5)4]2 | 45 |
| 5 | No Proton Sponge® | 51 |
| 6 | Proton Sponge® (50 mol%) | 0 |
| 7 | HOAc (20 mol%) in place of Proton Sponge® | 44 |
| 8 | LiOAc (20 mol%) in place of Proton Sponge® | 22 |
| 9 | Performed at 90 °C | 56 |
| 10 | Performed at 50 °C | 75 |
| 11 | In 1,4-dioxane | 62 |
| 12 | In toluene | 75 |
| 13 | In PhCl | 74 |
| 14 | In CH2Cl2 | 76 |
| 15 | No [Cp*Co(C6H6)][B(C6F5)4]2 | 0 |
| 16 | [Cp*RhCl2]2 (10 mol%) and AgSbF6 (40 mol%) in place of [Cp*Co(C6H6)][B(C6F5)4]2 | 0 |

Higher temperatures proved to be deleterious to the yield of 4a (entry 9), although the reaction could be run with isoprene (2a) at 50 °C to afford the product with little change in yield (entry 10). Nevertheless, 70 °C was selected as the optimal reaction temperature because reactions at 50 °C with more sterically hindered dienes (e.g., (E)-(2-methylbuta-1,3-dien-1-yl)benzene) were not as effective (see Table S3 in the ESI†). A modest decrease in the yield of 4a was observed when the reaction was conducted in 1,4-dioxane (entry 11), but yields in toluene, PhCl, and CH2Cl2 were comparable to those in DCE (entries 12–14 and Table S4 in the ESI†). Although DCE was selected as the standard solvent, toluene and PhCl were found to be superior to DCE for certain substrate combinations (vide infra). A control reaction in which the catalyst was omitted demonstrated that it is essential for this transformation (entry 15). Moreover, a closely related cationic Cp*Rh(III) catalyst did not provide any product (entry 16). Finally, acetic formic anhydride was found to be the optimal formylating agent, providing superior yields of aldehyde 4a compared to formic pivalic anhydride, 4-nitrophenyl formate and 2,4,6-trichlorophenyl formate (see Table S5 in the ESI†).
C–H bond substrate and diene scope
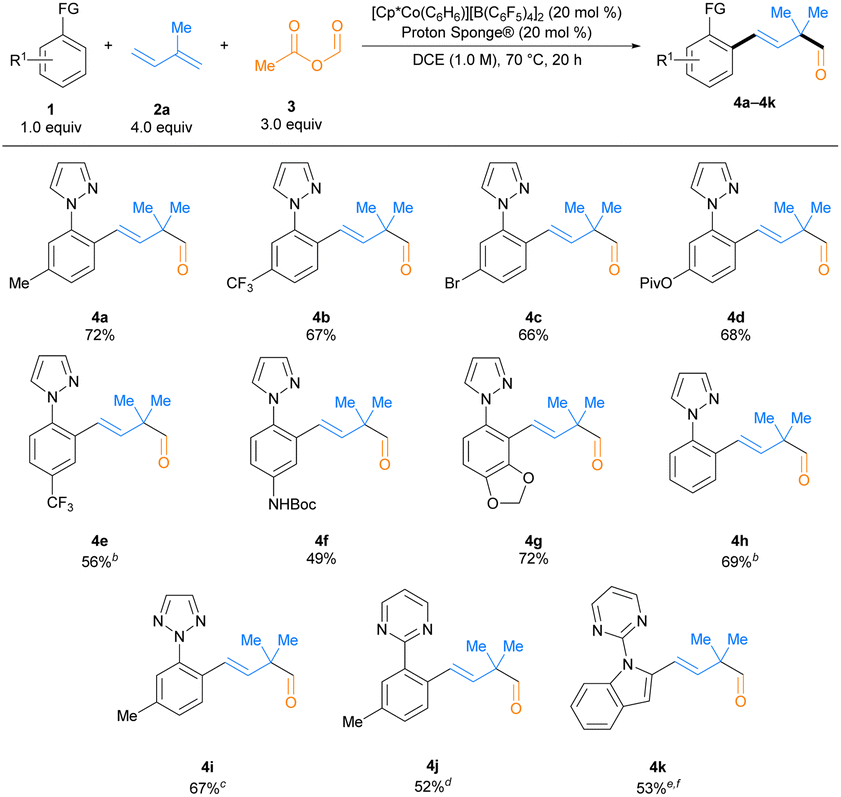
Having identified optimized reaction conditions, we first explored the scope of this transformation with respect to C–H bond substrate (Table 2). C–H bond substrates containing a variety of substituents, including trifluoromethyl (4b and 4e), bromo (4c), pivalate-protected phenol (4d), and Boc-protected amine (4f) groups, were effective reactants. Moreover, a C–H bond substrate without any substituent provided the desired product in a good yield (4h). Heterocyclic C–H bond substrates with benzodioxole (4g) or indole (4k) cores were also successfully employed in this transformation. In addition to substrates containing a pyrazole directing group (4a–4h), those with other N-heterocyclic directing groups such as 1,2,3-triazole (4i) and pyrimidine (4j and 4k) could be used. However, substrates with less basic directing groups such as amides or ketoximes did not afford the corresponding aldehyde product under the standard reaction conditions.| a Reaction conditions: 1(0.2 mmol),2a (0.8 mmol), 3 (0.6 mmol), [Cp*Co(C6H6)][B(C6F5)4]2 (20 mol%), Proton Sponge® (20 mol%) in DCE ([1] = 1.0 M) at 70 °C. Isolated yields reported. b Conducted at 50 °C. c 1,2-dichlorobenzene as solvent. d PhCl as solvent. e Toluene as solvent. f 2a (1.2 mmol, 6.0 equiv.). Piv = pivalate. |
|---|
|
We then surveyed the diene scope of this transformation (Table 3). A wide range of 2-substituted and 1,2-disubstituted dienes were successful inputs. Moderate to good yields were observed for 2-substituted dienes with cyclopentyl (4l) and isobutyl (4m) groups; however, 2-aryl substituted dienes (e.g., 2-phenyl-1,3-butadiene) only provided trace amounts of the desired aldehyde product (<5% NMR spectroscopic yield). While the 1,2-dialkyl substituted diene 1-cyclohexyl-2-methyl diene (4n) coupled in only 40% yield, 1,2-dialkyl substituted dienes with an endocyclic alkene gave much higher yields to provide aldehydes incorporating α-quaternary cyclohexyl (4o), tetrahydropyranyl (4p), and Cbz-protected piperidinyl (4q) ring systems. Various 1-aryl-2-methyl dienes containing diverse functional groups were also effective substrates. Fluoro (4s), bromo (4t), trifluoromethyl (4u), nitro (4v), chloro (4w), methyl ester (4x), methyl (4y), and methoxy (4z) substituents, including at the ortho-, meta-, and para-positions on the aryl ring, could all be incorporated into the carboformylation products. Finally, a 1-naphthyl-2-methyl diene provided aldehyde 4aa in a synthetically useful yield. Butadiene and terminally monosubstituted dienes did not give rise to aldehyde products under these conditions, perhaps because the putative α-tertiary aldehyde products would be readily deprotonated and/or form enol tautomers that could undergo side reactions (see Chart S3 in the ESI for unsuccessful diene substrates†).

Mechanistic studies and proposed mechanism
Several experiments were conducted to gain more insight into the mechanism of this reaction (Scheme 2). Because acetic formic anhydride is known to release carbon monoxide spontaneously and in the presence of base,15,16 we were interested in determining whether carbon monoxide was the source of the formyl carbonyl in this transformation. Reactions employing carbon monoxide gas in place of acetic formic anhydride under optimized reaction conditions did not afford any aldehyde product, and neither omitting Proton Sponge® nor replacing it with equimolar amounts of HOAc or LiOAc had any effect on the reaction outcome (Scheme 2A, top). Furthermore, a reaction where acetic formic anhydride was replaced with acetic anhydride, which cannot serve as a carbon monoxide surrogate, provided the corresponding three-component methyl ketone product 6 (Scheme 2A, bottom). More forcing conditions were likely required for this reaction due to the reduced electrophilicity and increased steric hindrance of acetic anhydride vs. acetic formic anhydride. Taken together, the carbonyl source in this reaction is unlikely to be carbon monoxide.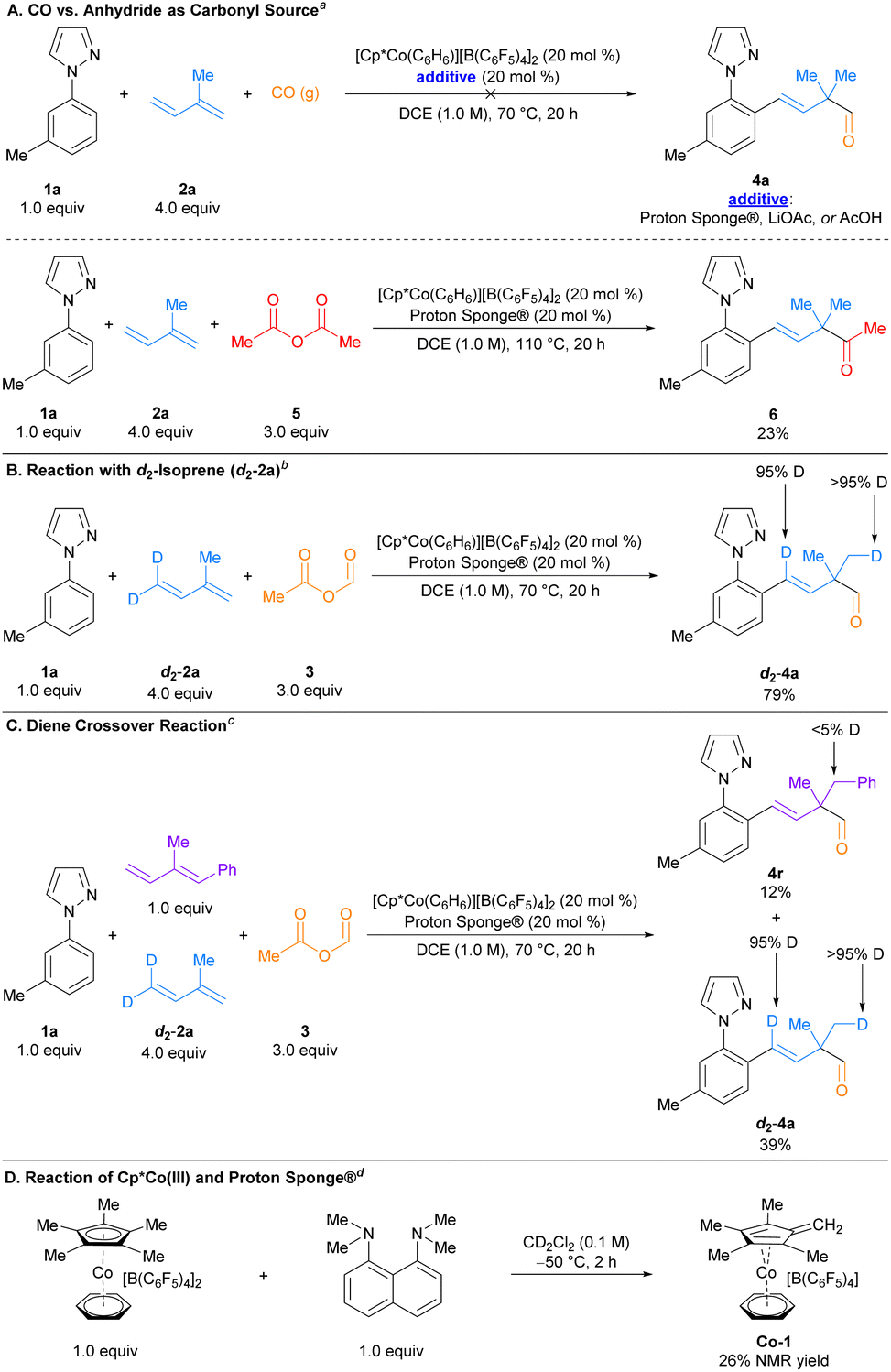 | ||
| Scheme 2 Mechanistic experiments. a(A) Top, reaction conditions: 1a (0.1 mmol), 2a (0.4 mmol), [Cp*Co(C6H5)][B(C6F5)4]2 (20 mol%), additive (20 mol%), in DCE ([1a] = 1.0 M) at 70 °C. (A) Bottom, 1a (0.2 mmol), 2a (0.8 mmol), 5 (0.6 mmol), [Cp*Co(C6H5)][B(C6F5)4]2 (20 mol%), Proton Sponge® (20 mol%), in DCE ([1a] = 1.0 M) at 110 °C. Isolated yield reported. b(B) Reaction conditions: 1a (0.2 mmol), d2-2a (0.8 mmol), 3 (0.6 mmol), [Cp*Co(C6H5)][B(C6F5)4]2 (20 mol%), Proton Sponge® (20 mol%), in DCE ([1a] = 1.0 M) at 70 °C. Isolated yield reported. c(C) 1a (0.2 mmol), d2-2a (0.8 mmol), (E)-(2-methylbuta-1,3-dien-1-yl)benzene (0.2 mmol), 3 (0.6 mmol), [Cp*Co(C6H5)][B(C6F5)4]2 (20 mol%), Proton Sponge® (20 mol%), in DCE ([1a] = 1.0 M) at 70 °C. Isolated yields reported. d(D) Reaction conditions: [Cp*Co(C6H5)][B(C6F5)4]2 (0.04 mmol), Proton Sponge® (0.04 mmol) in CD2Cl2 ([Cp*Co] = 0.1 M) at −50 °C. | ||
We also performed a deuterium labelling experiment in which isoprene (2a) was replaced with terminally deuterated d2-isoprene (d2-2a; Scheme 2B). Under the standard reaction conditions, quantitative deuterium incorporation occurred at the sp2-benzylic position as well as at one of the geminal methyl groups in the resulting aldehyde, d2-4a, consistent with the proposed β-hydride elimination/intramolecular hydride reinsertion pathway that accounts for the observed regioselectivity of the products (vide infra). An alternative mechanism involving intermolecular hydride reinsertion was also considered and probed via a crossover experiment where 1.0 equiv. of (E)-(2-methylbuta-1,3-dien-1-yl)benzene was added to the reaction of 1a, d2-2a, and 3a under standard conditions (Scheme 2C). No significant deuterium incorporation into product 4r was observed, suggesting the intermolecular pathway is not operative.
The uniqueness of Proton Sponge® as an additive in this transformation (and indeed in the realm of C–H functionalisation in general) prompted us to investigate its role in facilitating this reaction. Reacting equimolar amounts of [Cp*Co(C6H6)][B(C6F5)4]2 and Proton Sponge® in CD2Cl2 at −50 °C generates appreciable amounts (26% NMR yield) of a putative Co(I) tetramethylfulvene complex Co-1 of the form [(η4-C5Me4CH2)Co(C6H6)][B(C6F5)4] (Scheme 2D). Proton Sponge® has been shown to deprotonate methyl groups on the Cp* ligand of cationic, piano stool Rh(III) complexes,17 and Co(I) tetramethylfulvene complexes generated from Cp*Co(III) precursors and a suitable base (e.g., KOt-Bu or KHMDS) have been characterized spectroscopically.18 Complex Co-1 displays NMR signals consistent with the proposed structure, as well as related tetramethylfulvene complexes (see Fig. S10–S15 in the ESI†). Attempts to characterize Co-1 crystallographically or by high-resolution mass spectrometry (HRMS) were unsuccessful, consistent with prior efforts to obtain crystallographic or HRMS characterization of Co(I) tetramethylfulvene complexes due to their instability.18 A catalytic amount of complex Co-1 gave comparable yields of product relative to our standard conditions (see Scheme S2 in the ESI†). Additionally, the Co(I) tetramethylfulvene complex Co-1 forms in lower yield when [Cp*Co(C6H6)][B(C6F5)4]2 is reacted with 2.0 equiv. of Proton Sponge® in CD2Cl2 at −50 °C (see Fig. S16 in the ESI†). This result correlates with the lower observed yield when excess Proton Sponge® is added (see entry 6, Table 1).
These experiments suggest the combination of [Cp*Co(C6H6)][B(C6F5)4]2 and Proton Sponge® generates a catalytically competent species via an intermediate Co(I) tetramethylfulvene that might be superior in facilitating the sequential C–H bond addition to dienes and acetic formic anhydride than [Cp*Co(C6H6)][B(C6F5)4]2 alone (see Section 6 in the ESI for additional experiments†). However, further investigations are needed to definitively establish the relevance of Co-1 in the catalytic cycle.
On the basis of these collective mechanistic studies, as well as those performed for other Co(III)-catalysed sequential C–H bond addition reactions with conjugated dienes,10a,c,d,11 a proposed mechanism for this transformation is shown in Scheme 3. Firstly, reversible C–H activation by the cationic Co(III) catalyst via concerted metalation-deprotonation, possibly facilitated by Proton Sponge or another equivalent of 1a,19 gives rise to the cobaltacycle A (see Scheme S1 in the ESI for reversibility experiments†). Next, diene insertion into the Co–C bond affords the Co(III)-allyl species B. This complex then undergoes a reversible β-hydride elimination/hydride reinsertion via Co(III)-hydride species C to give an isomerized Co(III)-allyl complex D, in which one of the diene hydrogens at C4 has been transposed to C1. Evidence for this isomerization pathway is provided by the deuterium incorporation observed in d2-4a when using d2-2a (Scheme 2B). Acetic formic anhydride can then undergo direct nucleophilic attack from complex D, possibly through the six-membered transition state TS1, to afford complex E. Finally, β-OAc elimination from complex E furnishes the aldehyde product 4 and regenerates the active Co(III) catalyst.
 | ||
| Scheme 3 Proposed mechanism for the Co(III)-catalysed sequential C–H bond addition to dienes and acetic formic anhydride. | ||
Synthetic elaboration of aldehyde 4a
Various transformations were conducted on aldehyde 4a to showcase the synthetic versatility of the products generated through Co(III)-catalysed sequential C–H bond addition to dienes and acetic formic anhydride (Scheme 4). A proline-catalysed asymmetric aldol addition of acetone to aldehyde 4a produced β-hydroxy ketone 7 with excellent enantioselectivity (99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 er). A Horner–Wadsworth–Emmons olefination of aldehyde 4a with triethyl phosphonoacetate and sodium hydride gave rise to alkene 8 with complete (E)-selectivity. Additionally, tertiary amine 9 was prepared via reductive amination of aldehyde 4a using 1-Boc-piperazine. Finally, tert-butanesulfinyl imine 10 was synthesized from aldehyde 4a and (R)-tert-butanesulfinamide20 and subsequently reacted with TMSCF3 and TBAT (tetrabutylammonium difluorotriphenylsilicate)21 and allylmagnesium bromide22 to furnish the corresponding α-branched amines 11 and 12 with high diastereoselectivities (>99:1 and 96:4 dr, respectively). These transformations highlight the utility of the aldehyde products 4 for the straightforward and stereoselective introduction of useful functionality.
:1 er). A Horner–Wadsworth–Emmons olefination of aldehyde 4a with triethyl phosphonoacetate and sodium hydride gave rise to alkene 8 with complete (E)-selectivity. Additionally, tertiary amine 9 was prepared via reductive amination of aldehyde 4a using 1-Boc-piperazine. Finally, tert-butanesulfinyl imine 10 was synthesized from aldehyde 4a and (R)-tert-butanesulfinamide20 and subsequently reacted with TMSCF3 and TBAT (tetrabutylammonium difluorotriphenylsilicate)21 and allylmagnesium bromide22 to furnish the corresponding α-branched amines 11 and 12 with high diastereoselectivities (>99:1 and 96:4 dr, respectively). These transformations highlight the utility of the aldehyde products 4 for the straightforward and stereoselective introduction of useful functionality.
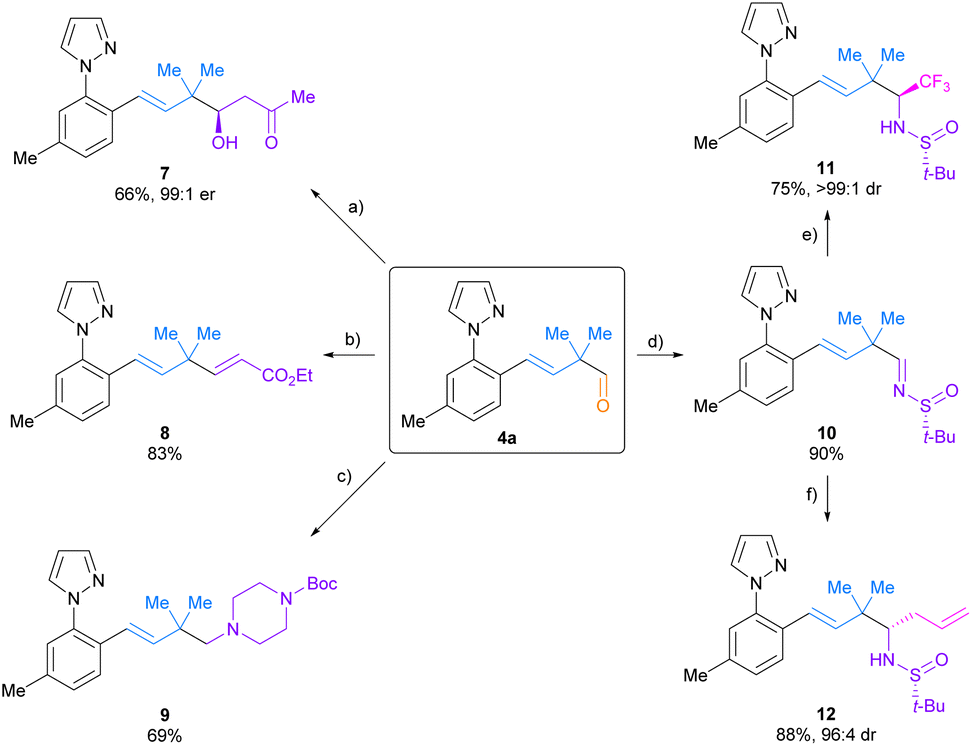 | ||
| Scheme 4 Diversification reactions of aldehyde 4a. (a) (S)-proline, acetone, CHCl3, 30 °C. (b) Triethyl phosphonoacetate, NaH, Et2O, 25 °C. (c) 1-Boc-piperazine, NaBH(OAc)3, MeCN, 25 °C. (d) (R)-tert-butanesulfinamide, Ti(Oi-Pr)4, THF, 50 °C. (e) TMSCF3, TBAT, THF, −55 °C. (f) Allylmagnesium bromide, CH2Cl2, −40 °C. TMS = trimethylsilyl. TBAT = tetrabutylammonium difluorotriphenylsilicate. For complete experimental details, see the ESI.† | ||
Conclusions
We have developed a Co(III)-catalysed sequential C–H bond addition reaction to dienes and acetic formic anhydride for the synthesis of all carbon α-quaternary aldehydes via a three-component process. A wide variety of substituted dienes are successful inputs in this reaction, affording a broad range of complex aldehyde products that are inaccessible via the hydroformylation of specialized 1,1-disubstituted alkenes. Mechanistic studies support that acetic formic anhydride directly provides the formyl group, explain the uncommon 1,3-functionalization of the conjugated diene, and provide insight into the role of the uniquely effective Proton Sponge additive. The versatility of the aldehyde products was also demonstrated through several diversification reactions, including proline-catalysed asymmetric aldol addition, olefination, reductive amination, and nucleophilic additions to the corresponding tert-butanesulfinyl imine derivative. Overall, this procedure highlights the synthetic potential of sequential C–H bond addition reactions for the modular and efficient preparation of complex, value-added products incorporating all carbon α-quaternary aldehyde functionality.Data availability
The data that support the findings of this study are available in the ESI† for this article.Author contributions
J. P. T. and J. A. E. conceptualized the project. J. P. T. and J. Y. performed the experiments and collected the data. J. P. T. wrote the original draft of the manuscript, and all authors contributed to the revision and editing of the manuscript. J. A. E. supervised the project and acquired funding for the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the NIH (R35GM122473). J. P. T. is grateful to the NSERC of Canada for a Postdoctoral Fellowship. We also thank Dr Robert Berry and Dr Fabian Menges of the Yale University Chemical and Biophysical Instrumentation Center for technical assistance in the acquisition of NMR and HRMS data.Notes and references
- R. Franke, D. Selent and A. Börner, Chem. Rev., 2012, 112, 5675–5732 CrossRef CAS PubMed.
- (a) Y. Ning, T. Ohwada and F.-E. Chen, Green Synth. Catal., 2021, 2, 247–266 CrossRef; (b) B. Breit and W. Seiche, Synthesis, 2001, 2001, 1–36 CrossRef.
- A. I. M. Keulemans, A. Kwantes and T. van Bavel, Recl. Trav. Chim. Pays-Bas, 1948, 67, 298–308 CrossRef CAS.
- For single entries of α-tetrasubstituted carbon synthesis via hydroformylation, see: (a) R. Tan, X. Zheng, B. Qu, C. A. Sader, K. R. Fandrick, C. H. Senanayake and X. Zhang, Org. Lett., 2016, 18, 3346–3349 CrossRef CAS PubMed; (b) X. Wang and S. L. Buchwald, J. Org. Chem., 2013, 78, 3429–3433 CrossRef CAS PubMed; (c) I. J. Krauss, C. C. Y. Wang and J. L. Leighton, J. Am. Chem. Soc., 2001, 123, 11514–11515 CrossRef CAS PubMed; (d) C. W. Lee and H. Alper, J. Org. Chem., 1995, 60, 499–503 CrossRef CAS.
- (a) D. Zhang, J. Wen and X. Zhang, Chem. Sci., 2022, 13, 7215–7223 RSC; (b) J. Eshon, F. Foarta, C. R. Landis and J. M. Schomaker, J. Org. Chem., 2018, 83, 10207–10220 CrossRef CAS PubMed; (c) Y. Ueki, H. Ito, I. Usui and B. Breit, Chem.–Eur. J., 2011, 17, 8555–8558 CrossRef CAS PubMed; (d) X. Sun, K. Frimpong and K. L. Tan, J. Am. Chem. Soc., 2010, 132, 11841–11843 CrossRef CAS PubMed; (e) M. L. Clarke and G. J. Roff, Chem.–Eur. J., 2006, 12, 7978–7986 CrossRef CAS PubMed; (f) C. Botteghi, S. Paganelli, L. Bigini and M. Marchetti, J. Mol. Catal., 1994, 93, 279–287 CrossRef CAS; (g) S. Gladiali and L. Pinna, Tetrahedron: Asymmetry, 1991, 2, 623–632 CrossRef CAS.
- (a) S. Brown, S. Clarkson, R. Grigg, W. A. Thomas, V. Sridharan and D. M. Wilson, Tetrahedron, 2001, 57, 1347–1359 CrossRef CAS; (b) S. Brown, S. Clarkson, R. Grigg and V. Sridharan, J. Chem. Soc., Chem. Commun., 1995, 1135–1136 RSC.
- Rh(I)-catalyzed intramolecular carboformylation has been invoked as a mechanistic step en route to substituted dihydronaphthalenes and naphthalenes: E. Okazaki, R. Okamoto, Y. Shibata, K. Noguchi and K. Tanaka, Angew. Chem., Int. Ed., 2012, 51, 6722–6727 CrossRef CAS PubMed.
- Y. H. Lee, E. H. Denton and B. Morandi, Nat. Chem., 2021, 13, 123–130 CrossRef CAS PubMed.
- W. Zhang and S. Lin, J. Am. Chem. Soc., 2020, 142, 20661–20670 CrossRef CAS PubMed.
- (a) S. Dongbang and J. A. Ellman, Angew. Chem., Int. Ed., 2021, 60, 2135–2139 CrossRef CAS PubMed; (b) Z. Shen, C. Li, B. Q. Mercado and J. A. Ellman, Synthesis, 2020, 52, 1239–1246 CrossRef CAS PubMed; (c) S. Dongbang, Z. Shen and J. A. Ellman, Angew. Chem., Int. Ed., 2019, 58, 12590–12594 CrossRef PubMed; (d) J. A. Boerth, S. Maity, S. K. Williams, B. Q. Mercado and J. A. Ellman, Nat. Catal., 2018, 1, 673–679 CrossRef CAS PubMed.
- J. Yang, D.-W. Ji, Y.-C. Hu, X.-T. Min, X. Zhou and Q.-A. Chen, Chem. Sci., 2019, 10, 9560–9564 RSC.
- For an example of Rh(III)-catalyzed sequential C–H bond addition to dienes and aldehydes, see: R. Li, C.-W. Ju and D. Zhao, Chem. Commun., 2019, 55, 695–698 RSC.
- For examples of other group 9 metal-catalyzed sequential C–H bond additions involving dienes, see: (a) D. S. Brandes and J. A. Ellman, Chem. Soc. Rev., 2022, 51, 6738–6756 RSC; (b) R. Mi, X. Zhang, J. Wang, H. Chen, Y. Lan, F. Wang and X. Li, ACS Catal., 2021, 11, 6692–6697 CrossRef CAS; (c) T. Pinkert, T. Wegner, S. Mondal and F. Glorius, Angew. Chem., Int. Ed., 2019, 58, 15041–15045 CrossRef CAS PubMed.
- J. R. Hummel and J. A. Ellman, J. Am. Chem. Soc., 2015, 137, 490–498 CrossRef CAS PubMed.
- P. Strazzolini, A. G. Giumanini and S. Cauci, Tetrahedron, 1990, 46, 1081–1118 CrossRef CAS.
- For examples of carbonylation reactions employing acetic formic anhydride as the carbon monoxide source, see: (a) A. V. Gadakh, D. Chikanna, S. S. Rindhe and B. K. Karale, Synth. Commun., 2012, 42, 658–666 CrossRef CAS; (b) S. Cacchi, G. Fabrizi and A. Goggiamani, J. Comb. Chem., 2004, 6, 692–694 CrossRef CAS PubMed.
- (a) R. M. Bellabarba, M. Neiuwenhuyzen and G. C. Saunnders, Organometallics, 2002, 21, 5726–5737 CrossRef CAS; (b) R. M. Bellabarba, M. Neiuwenhuyzen and G. C. Saunders, J. Chem. Soc., Dalton Trans., 2001, 512–514 RSC.
- (a) J. Bauer, H. Braunschweig, C. Hörl, K. Radacki and J. Wahler, Chem.–Eur. J., 2013, 19, 13396–13401 CrossRef CAS PubMed; (b) Y. Ohki, A. Murata, M. Imada and K. Tatsumi, Inorg. Chem., 2009, 48, 4271–4273 CrossRef CAS PubMed; (c) D. Buchholz, B. Gloaguen, J.-L. Fillaut, M. Cotrait and D. Astruc, Chem.–Eur. J., 1995, 1, 374–381 CrossRef CAS.
- For evidence of nitrogen bases facilitating concerted metalation-deprotonation in group 9 metal-catalyzed C–H activation, see: (a) J. Sanjosé-Orduna, J. M. S. Toro and M. H. Pérez-Temprano, Angew. Chem., Int. Ed., 2018, 57, 11369–11373 CrossRef PubMed; (b) M. E. Tauchert, C. D. Incarvito, A. L. Rheingold, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2012, 134, 1482–1485 CrossRef CAS PubMed.
- M. T. Robak, M. A. Herbage and J. A. Ellman, Chem. Rev., 2010, 110, 3600–3740 CrossRef CAS PubMed.
- G. K. S. Prakash, M. Mandal and G. A. Olah, Angew. Chem., Int. Ed., 2001, 40, 589–590 CrossRef CAS.
- D. A. Cogan, G. Liu and J. A. Ellman, Tetrahedron, 1999, 55, 8883–8904 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc05599f |
| This journal is © The Royal Society of Chemistry 2022 |