
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh-yield production of liquid fuels in CO2 hydrogenation on a zeolite-free Fe-based catalyst†
Lisheng
Guo
 *a,
Xinhua
Gao
b,
Weizhe
Gao
c,
Hao
Wu
a,
Xianbiao
Wang
a,
Song
Sun
*a,
Yuxue
Wei
a,
Yasuharu
Kugue
c,
Xiaoyu
Guo
c,
Jian
Sun
*d and
Noritatsu
Tsubaki
*c
*a,
Xinhua
Gao
b,
Weizhe
Gao
c,
Hao
Wu
a,
Xianbiao
Wang
a,
Song
Sun
*a,
Yuxue
Wei
a,
Yasuharu
Kugue
c,
Xiaoyu
Guo
c,
Jian
Sun
*d and
Noritatsu
Tsubaki
*c
aSchool of Chemistry and Chemical Engineering, Anhui University, Hefei, Anhui 230601, China. E-mail: lsguo@ahu.edu.cn; suns@ustc.edu.cn
bState Key Laboratory of High-Efficiency Utilization of Coal and Green Chemical Engineering, College of Chemistry & Chemical Engineering, Ningxia University, Yinchuan 750021, PR China
cDepartment of Applied Chemistry, School of Engineering, University of Toyama, Gofuku 3190, Toyama 930-8555, Japan. E-mail: tsubaki@eng.u-toyama.ac.jp
dDalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China. E-mail: sunj@dicp.ac.cn
First published on 16th November 2022
Abstract
Catalytic conversion of CO2 to long-chain hydrocarbons with high activity and selectivity is appealing but hugely challenging. For conventional bifunctional catalysts with zeolite, poor coordination among catalytic activity, CO selectivity and target product selectivity often limit the long-chain hydrocarbon yield. Herein, we constructed a singly cobalt-modified iron-based catalyst achieving 57.8% C5+ selectivity at a CO2 conversion of 50.2%. The C5+ yield reaches 26.7%, which is a record-breaking value. Co promotes the reduction and strengthens the interaction between raw CO2 molecules and iron species. In addition to the carbide mechanism path, the existence of Co3Fe7 sites can also provide sufficient O-containing intermediate species (CO*, HCOO*, CO32*, and 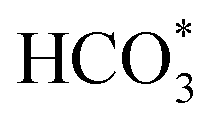 ) for subsequent chain propagation reaction via the oxygenate mechanism path. Reinforced cascade reactions between the reverse water gas shift (RWGS) reaction and chain propagation are achieved. The improved catalytic performance indicates that the KZFe–5.0Co catalyst could be an ideal candidate for industrial CO2 hydrogenation catalysts in the future.
) for subsequent chain propagation reaction via the oxygenate mechanism path. Reinforced cascade reactions between the reverse water gas shift (RWGS) reaction and chain propagation are achieved. The improved catalytic performance indicates that the KZFe–5.0Co catalyst could be an ideal candidate for industrial CO2 hydrogenation catalysts in the future.
Introduction
With the extensive use of carbon-based fossil fuels, the concentration of CO2 in the atmosphere has reached a record high, and this rising level creates a series of ecological problems such as ocean acidification and global warming.1,2 Carbon capture, utilization and storage (CCUS) is regarded as a powerful strategy for reducing the CO2 concentration.2–5 Chemical valorization of CO2 into valuable chemicals or fuels is a promising route to turn waste into treasure and reduce the detrimental effects of greenhouse gas. Long-chain hydrocarbons, as carbon-neutral fuels, have high energy density and facile mobile storage capacity, and have attracted wide attention.6–9 Owing to the inertia of CO2 molecules and imprecise control of carbon chain propagation during CO2 hydrogenation, efficient catalytic conversion of CO2 into long-chain hydrocarbons remains a huge challenge.10,11Iron-based catalysts can form two kinds of active sites in situ (Fe3O4 for RWGS, and FexCy for chain propagation) during reaction, and thus have been widely used in CO2 hydrogenation.12 To increase the C5+ selectivity or break the Anderson–Schulz–Flory (ASF) distribution, various strategies have been adopted to regulate the activity and product selectivity over iron catalysts. Doping with alkali promoters (K, Na, Mn, etc.) is a typical case.6,13 The existence of alkali promoters is beneficial for improving CO2 adsorption behaviors and Fe carbonization. Especially in recent years, metal oxides have been combined with zeolite to form bifunctional catalysts that show benign performance for CO2 hydrogenation. It is worth mentioning that although these novel composite catalysts (e.g., Na–Fe3O4/HZSM-5 and In2O3/HZSM-5) generally exhibit a high C5+ selectivity, their low catalytic activity (10–30%) and high CO by-product (20–60%) selectivity generally limit the effective yield of C5+ or lower the catalyst (metal oxide + zeolite) time yield.7,8
In addition, the introduction of a second active metal has been adopted.14,15 The second active metal is generally involved in the reverse water gas shift (RWGS) and/or chain propagation processes.14,16,17 Currently, the incorporation of a second active metal has been extensively used to improve the CO2 hydrogenation process.14–16,18,19 In terms of conventional Fischer–Tropsch synthesis (FTS), Co catalysts present higher chain growth ability than iron catalysts. In contrast, single Co catalysts produce large amounts of CH4 instead of long-chain products in CO2 hydrogenation. Interestingly, the introduction of an appropriate amount of Co into Fe-based catalysts can result in unique catalytic behavior. Xu et al. synthesized a ZnCoxFe2−xO4 catalyst for the selective conversion of CO2 into light olefins (36.1% for C=2–4), and ascribed the phenomenon to the optimized generation of iron–cobalt carbide, Co2C, and h-Fe3C phases.15 Similarly, Kim et al. suggested that the high olefin selectivity (39.0% for C=2–4) is due to the facile formation of a unique bimetallic alloy carbide (Fe1−xCox)5C2.19 Zhang et al. reported that a Na-modified CoFe alloy catalyst using a layered double-hydroxide (LDH) precursor is conducive to the formation of C8–C16 jet-fuel-range hydrocarbons.14 However, although the C5+ selectivity reaches 72.9%, the CO2 conversion in this case is only 10.2%. Guo et al. adopted Co doping to increase conversion activity (60.4%) and reduce CO selectivity (4.5%) with moderately decreased C5+ selectivity (20.7%).16 Based on the above inspiration, we wondered whether the introduction of Co metal would be promising to maintain high activity while maintaining high selectivity of C5+ products without the utilization of zeolite. Therefore, it is significant to understand tailor-made Fe catalysts with Co regulation for the efficient conversion of CO2 to long-chain hydrocarbons with high catalytic activity.
Herein, different from conventional composite catalysts for oriented synthesis of heavy hydrocarbons, we report how iron-based catalysts can be optimized via cobalt incorporation to realize efficient catalysts for CO2 hydrogenation to long-chain hydrocarbons with high-yield production. The Co-modified iron catalyst exhibits a low CO selectivity (8.1%) and a high selectivity toward C5+ hydrocarbons (57.8%) at a CO2 conversion of 50.2%, in which the yield of C5+ reaches 26.7%. With the introduction of Co, the cascade reactions between RWGS and chain propagation are notably reinforced, and the product distribution deviates from the ASF model via an oxygen-containing intermediate (CO*, HCOO*, CO32*, and 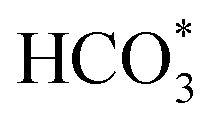 ) pathway.
) pathway.
Results and discussion
The molar ratio of Co/Fe is summarized in Table S1.† The results are close to the designed value. CO2 hydrogenation performance tests over different catalysts were conducted, and the results are depicted in Fig. 1a and S1.† The KZFe catalyst exhibits a high C5+ selectivity of 49.9% at a CO2 conversion of 48.3%, and the main products are olefin-rich hydrocarbons. Previous studies have also demonstrated that a ZnFe2O4 catalyst containing K as a promoter is conducive to the formation of heavy olefins.16 Alkali K promotes the carburization of iron species and enhances chain propagation ability. To further demonstrate this promotional effect derived from the spinel ZnFe2O4 structure and K, ZFe, KFe and KFe + Z (PM) catalysts were evaluated, as listed in Table S2.† The catalytic performance of the ZFe or KFe + Z (PM) catalyst alone is inferior to that of the KZFe catalyst. The presence of K promotes the formation of long-chain hydrocarbons. Apparently, the presence of K and Zn formed by spinel structure play a crucial role in improving the CO2 hydrogenation performance, e.g., improved adsorption and carburization ability, which is consistent with previous reports. The results also indicate that spinel KZFe is a promising catalyst for efficiently catalyzing the hydrogenation of CO2 to long-chain hydrocarbons. Co-based catalysts have strong chain growth capacity and are generally used to produce long-chain hydrocarbons during traditional FTS. In view of this, a small amount of cobalt metal species was introduced into the iron-based catalyst system (KZFe–2.5Co), and it is found that the catalytic activity is improved while the selectivity of the target C5+ products is further improved from 49.9% to 55.2%. With further increase of the Co content (KZFe–5.0Co), the long-chain hydrocarbon selectivity reaches 57.8%, while the CO2 conversion is as high as 50.2%, and the CO selectivity is as low as 8.1%. As the Co content is further increased, the selectivity of C5+ hydrocarbons decrease despite a slight increase in catalytic activity (KZFe–10.0Co and KZFe–20.0Co). Meanwhile, the CH4 selectivity gradually increases. When the catalyst contains only Co metal, the product is mainly CH4, since the Co species usually results in methanation in CO2 hydrogenation (Table S2†). For comparison, a physical mixture of Co and KZFe catalyst was prepared to obtain KZFe + 5.0Co (PM). The long-chain hydrocarbon selectivity from KZFe + 5.0Co was inferior to that of KZFe–5.0Co, indicating that the manner of the incorporation of Co plays a crucial role in the catalytic performance. However, for the reference KZFe–SiO2, the catalytic performance including activity and C5+ selectivity is rather lower than that of the KZFe catalyst, demonstrating that inert SiO2 is harmful to improving the CO2 hydrogenation performance.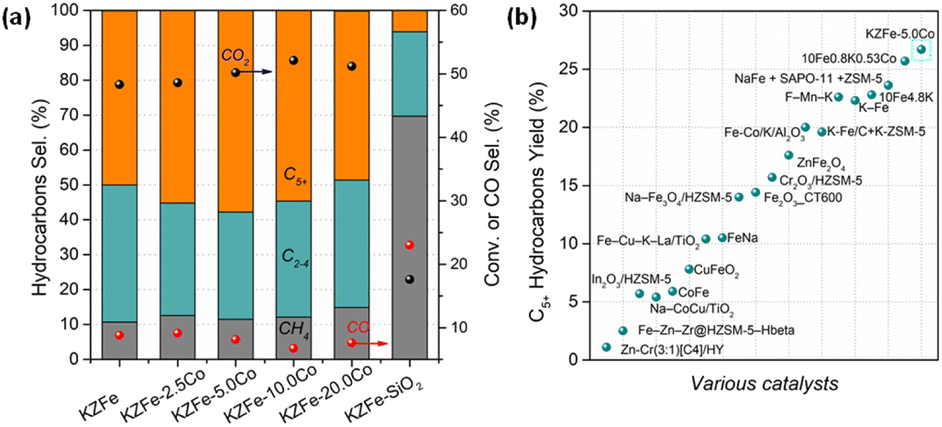 | ||
| Fig. 1 (a) Catalytic performances of as-prepared catalysts for CO2 hydrogenation at 320 °C, 2.0 MPa, and 6000 mL g−1 h−1. (b) Catalytic yields of C5+ hydrocarbons over various catalysts. Yield = CO2 conv. × (1 − CO sel.) × C5+ sel. Catalysts (in (b)) from left to right were cited from ref. 6–8, 14 and 20–32. | ||
In recent years, many research teams have focused on CO2 hydrogenation to produce long-chain hydrocarbons and have developed a series of catalysts, such as promoter-modified Fe-based catalysts (K–Fe, Fe–Mn–K, etc.), composite catalysts (Na–Fe3O4 + ZSM-5, In2O3 + ZSM-5, etc.), and alloy catalysts (CoFe, etc.).6–8,14,23,33,34 However, poor balance among catalytic activity, CO selectivity and target product selectivity often limit the yield of the target product (C5+). In this case, the directionally designed bimetallic or alloy catalyst (ZnFe2O4–5.0Co) can maintain high catalytic activity (50.2% CO2 conversion) while having high C5+ selectivity (57.8%) and low CO selectivity (8.1%), thus presenting a high yield of C5+, which is higher than that of previous catalysts (26.7% C5+ yield for KZFe–5.0Co, Fig. 1b). In addition, the CO2 hydrogenation activity and C5+ selectivity remain stable for 60 h (Fig. S2†). This indicates that KZFe–5.0Co is a promising catalyst for efficiently catalyzing CO2 into long-chain hydrocarbons with high yield.
The XRD patterns of the as-prepared and spent catalysts are shown in Fig. 2. For KZFe catalysts, the main phases are ZnFe2O4 (JCPDS, 77-0011) and ZnO (JCPDS, 89-0510) species. With the introduction of Co species, the characteristic diffraction peak of ZnFe2O4 moves towards the lower diffraction angle in XRD patterns, indicating that Co metal enters the system of ZnFe2O4 and forms Zn(FexCo2−x)O4. When the Co content is low, the presence of Fe2O3 (JCPDS, 89-8103) species can also be observed (KZFe–2.5Co). After reaction, for the KZFe catalyst, there exist three main phases, Fe3O4 (JCPDS, 89-3854), ZnO, and Fe5C2 (JCPDS, 20-0509) respectively.35,36 However, for KZFe–2.5Co and KZFe–10.0Co, the peaks ascribed to Fe3O4 disappear, while the peaks corresponding to Co3Fe7 (JCPDS, 48-1817) appear. In contrast, the diffraction intensity of KZFe–5.0Co and KZFe–20.0Co is lower that of KZFe–2.5Co and KZFe–10.0Co, probably due to benign dispersion or the formation of small particles. However, no peaks corresponding to the CoFe alloy phase are observed for the KZFe + 5.0Co (PM) catalyst (Fig. S3†). For the hydrogenation of CO2 to hydrocarbons, the first step is the RWGS reaction to form CO intermediates over Fe3O4, and then FTS occurs to form hydrocarbons over the surface of Fe5C2.2 Yang et al. constructed Fe5C2/Co heterostructured nanoparticles to improve catalytic yield and suggested that Co with a lower energy barrier for CO dissociation can provide enough C1 building blocks while Fe5C2 is responsible for the chain growth.37 Zhang et al. reported that a CoFe alloy is beneficial for the formation of jet fuels in CO2 hydrogenation owing to the existence of metallic CoFe alloy phases.14 It can be inferred that iron-based catalysts with different cobalt contents show great differences (Fig. 1 and Table S2†). Obviously, the presence of Co, which interacts with Fe, can efficiently tune the CO2 hydrogenation performance. Additionally, the N2 adsorption–desorption isotherms of different catalysts indicate that Co can affect the specific surface area (SSA) without a specific trend (Fig. S4 and Table S3†).
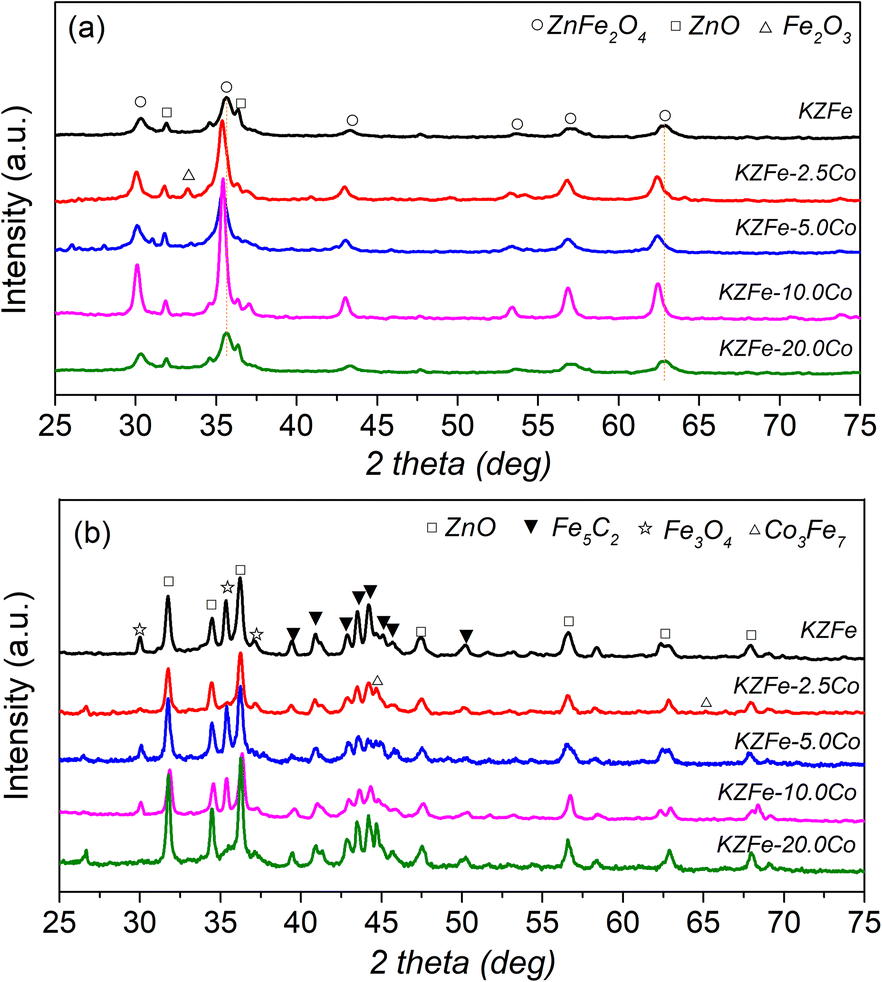 | ||
| Fig. 2 (a) Powder XRD patterns of as-prepared KZFe, KZFe–2.5Co, KZFe–5.0Co, KZFe–10.0Co, and KZFe–20.0Co. (b) XRD patterns of the catalysts after CO2 hydrogenation for 8 h. | ||
TEM was employed to characterize the spent catalysts (Fig. 3 and S5–S8†). For the KZFe catalyst, the lattice spacings of 0.296 nm and 0.265 nm can be attributed to the (220) plane of Fe3O4 and the (−311) plane of Fe5C2, respectively (Fig. S5†), which matches XRD pattern results well. With the introduction of Co (KZFe–2.5Co), a lattice spacing of 0.202 nm ascribed to the (110) plane of Co3Fe7 alloy can be observed (Fig. S6†). Similarly, specific lattice spacings corresponding to the (310) plane of Fe3O4, (−311) plane of Fe5C2, and (220) plane of Co3Fe7 phases are also observed for KZFe–5.0Co (Fig. 3a and b). Additionally, EDS line scanning profiles were used to investigate the distributions of different elements (Fig. 3c and d). It can be found that the Co species are highly adjacent to the Fe species (Fig. 3d). The line scanning at different positions also indicates the same phenomenon (Fig. S9†). To some extent, the overlapping of the Fe and Co signals further indicates the alloying of the elements Fe and Co. EDS elemental mapping depicts the uniform spatial surface distributions of the elements K, Fe, Zn, Co, and O (Fig. 3g–l). These results indicate that Fe and Co can effectively contact each other for catalyzing CO2 hydrogenation (Fig. 3b, S6 and S8†). It is worth noting that although the specific surface area of KZFe–5.0Co is lower than that of KZFe–2.5Co and KZFe–10.0Co, the active phases can still disperse evenly, indicating that the specific surface area is not the key factor; instead, the formation of the active phase is the key factor. The introduction of a moderate amount of Co can reduce the formation of other non-ideal species while forming a sufficient amount of Co3Fe7 species.
 | ||
| Fig. 3 (a and b) TEM and HR-TEM images, (c and d) line scanning, and (e–k) TEM image and corresponding EDS elemental maps of the spent KZFe–5.0Co catalyst after reaction. | ||
EXAFS experiments were also conducted to study the fine structure of the spent catalysts (Fig. 4). Data reduction, data analysis, and EXAFS fitting were performed according to the standard procedures using the ATHENA and ARTEMIS program integrated within the Demeter packages.38 The energy calibration of the sample was conducted using a standard Fe foil, which was simultaneously measured as a reference. For EXAFS modeling, the k3-weighted EXAFS spectra were obtained via subtracting the post-edge background from the overall absorption, normalization with respect to the edge-jump step, and Fourier transformation to real (R) space using Hanning windows (dk = 1.0 Å) ranging from 3.0–11.0 Å−1. The EXAFS of the Fe foil was fitted, and the value of the obtained amplitude reduction factor S02 (0.762) was set in the EXAFS analysis to determine the coordination numbers (CNs) in the Fe–O/Fe/Co scattering path in the sample. With the incorporation of Co, the Fe K-edge for KZFe–5.0Co shifts to slightly lower energy and the white line concomitantly increases, which clearly reflects strong interaction between Fe and Co. The Fe–Fe(Co) and Fe–O coordination shells are observed, which indicates that the Fe3O4 active phases and Co3Fe7 alloy structure are formed (Fig. S10†). Fitting of the Fe K-edge EXAFS data also reveals these results (Table S4†).
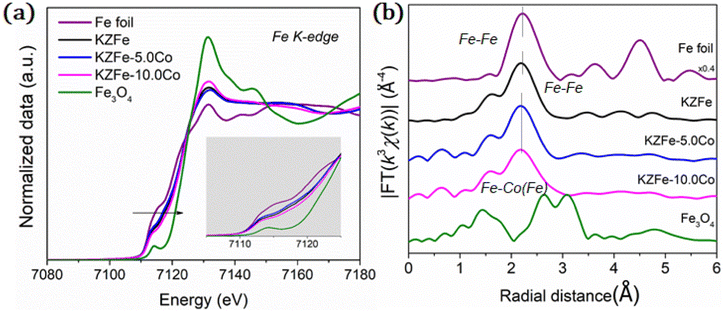 | ||
| Fig. 4 (a) XANES spectrum of the Fe K-edge for different spent catalysts. (b) Fourier transformed EXAFS data for spent catalysts. | ||
H2-TPR experiments were performed to compare the reducibilities of KZFe and Co-modified KZFe (Fig. S11†). For the KZFe catalyst, the overlapping peak can be divided into three stages, ZnFe2O4 to Fe3O4, Fe3O4 to FeO, and FeO to Fe. As shown, with the introduction of small Co species, the reduction temperature shifts towards low temperature (KZFe–2.5Co). The reduction temperature is further reduced with increasing Co content (KZFe–5.0Co). However, the reduction temperature changes slightly with further increasing the Co content (KZFe–10.0Co and KZFe–20.0Co). Apparently, the presence of Co is conducive to the reduction of iron oxide species to metallic Fe, which is consistent with previous reports.16 The formed Fe will be converted into active iron species under the reaction conditions. Thus, improved reduction ability is beneficial for subsequent active phase formation. The adsorption of raw CO2 molecules was investigated using CO2-TPD experiments (Fig. S12†). Compared to the KZFe catalyst, the adsorption intensity of CO2 increases over KZFe–2.5Co. With the further increase of Co, the adsorption intensity of CO2 increases evidently, and new peaks at 241 °C and 323 °C appear (KZFe–5.0Co). A similar phenomenon can be also observed for KZFe–10.0Co and KZFe–20.0Co. This illustrates that the existence of Co can not only enhance the adsorption of acidic CO2 molecules but also effectively strengthen the interaction between metal and CO2 molecules, which is beneficial for the C1 species formation and chain propagation under real reaction conditions. Notably, for the inert SiO2-supported KZFe catalyst, the reducing capacity of iron species is significantly reduced, which indicates that there is a strong interaction between metal species and the support (Fig. S13†). Additionally, with the utilization of SiO2, the adsorption capacity of COx species was also reduced at the reaction temperature (Fig. S13†). These results indicate that the strong interaction between metal species and the inert SiO2 support hinders the formation of active species and the adsorption of reaction molecules.
XPS was applied to investigate the phase composition and content of surface species (Fig. S14 and Table S5†). The binding energy peaks at 708.5, 710.9, and 712.0 eV in the Fe 2p spectrum are ascribed to Fe–C, Fe(II), and Fe(III).39 For the SiO2-supported KZFe catalysts, the main component is Fe(III), which also indicates that the strong metal–support interactions are not conducive to iron species reduction and active phase formation, which is accordance with the previous discussion (Table S5 and Fig. S13†). Inert SiO2, which can form strong interactions with metal species, is not conducive to the formation of heavy hydrocarbons (Fig. 1). However, with the introduction of Co, the surface phase composition changes significantly. The Fe 2p1/2 peak intensity of Fe(III) decreases significantly, while those of Fe–C and Fe(II) increase significantly. Additionally, the binding energy position of the Fe(II) bond shifts toward low binding energy with the incorporation of Co, indicating the formation of electron-rich iron species (Fig. S14†). It has also been reported that increased electron density of the iron species during reaction can strengthen the Fe–C bond and weaken the C–O bond.16,40 As summarized in Table S5,† it can be inferred that the introduction of Co can promote the transformation of Fe(III) into Fe(II) and Fe–C, which is essential to improve the CO2 hydrogenation performance. The Co 2p3/2 spectra of the different spent catalysts were also compared (Fig. S15†). The introduction of a small amount of Co results in a higher Co2+ content (Table S6†). In part, this suggests that small amounts of Co are more likely to form alloying species. Additionally, the characteristic C 1s spectra of the spent catalysts can be deconvoluted into C–C/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C at 298.5 eV, C–OH at 296.6 eV, CO at 298.9, and O–COH at 299.7 eV.41–43 As can be seen from the spectra, the surface of the KZFe catalyst is mainly composed of C–C/CC and C–OH species. For all catalysts, C–C/CC species are the dominant surface species, which is consistent with the CO2 hydrogenation results (Fig. 1a). However, with the introduction of Co, more surface CO and O–COH species appear (Fig. S16†). Interestingly, the peak intensity assigned to C–OH species from the Co-modified ones is rather lower than that of the KZFe catalyst. It is easy to infer that the presence of Co regulates the surface species of iron-based catalysts, producing more CO and O–COH species. In addition, the surface analysis from the O 1s spectra also demonstrates similar results (Fig. S17†). The peaks at 298.3 eV, 230.1 eV, 531.4 eV, and 532.8 eV can be assigned to Co–O, Fe–O, Fe–OH, and H2O (ad), respectively.41,44–46 In addition, as a structure-sensitive reaction, the particle size has a significant impact on the reaction, especially the small-size particles (6–20 nm).47,48 Based on TEM and refinement of XRD data, it can be found that the particle size is between 60 and 100 nm (Fig. 3 and S5–S8†), and the influence of these particle sizes on the CO2 hydrogenation can be ignored. Additionally, refinement of XRD also showed that with the introduction of Co, the carbide content increased from 48.3 wt% to 50.2 wt% and the Co3Fe7 content was about 1.7 wt% (Fig. S18†).
C at 298.5 eV, C–OH at 296.6 eV, CO at 298.9, and O–COH at 299.7 eV.41–43 As can be seen from the spectra, the surface of the KZFe catalyst is mainly composed of C–C/CC and C–OH species. For all catalysts, C–C/CC species are the dominant surface species, which is consistent with the CO2 hydrogenation results (Fig. 1a). However, with the introduction of Co, more surface CO and O–COH species appear (Fig. S16†). Interestingly, the peak intensity assigned to C–OH species from the Co-modified ones is rather lower than that of the KZFe catalyst. It is easy to infer that the presence of Co regulates the surface species of iron-based catalysts, producing more CO and O–COH species. In addition, the surface analysis from the O 1s spectra also demonstrates similar results (Fig. S17†). The peaks at 298.3 eV, 230.1 eV, 531.4 eV, and 532.8 eV can be assigned to Co–O, Fe–O, Fe–OH, and H2O (ad), respectively.41,44–46 In addition, as a structure-sensitive reaction, the particle size has a significant impact on the reaction, especially the small-size particles (6–20 nm).47,48 Based on TEM and refinement of XRD data, it can be found that the particle size is between 60 and 100 nm (Fig. 3 and S5–S8†), and the influence of these particle sizes on the CO2 hydrogenation can be ignored. Additionally, refinement of XRD also showed that with the introduction of Co, the carbide content increased from 48.3 wt% to 50.2 wt% and the Co3Fe7 content was about 1.7 wt% (Fig. S18†).
To further clarify the promotional effect of Co and adsorbed reaction intermediates in CO2 hydrogenation, in situ DRIFT spectra were recorded for the KZFe and KZFe–5.0Co catalysts during CO2 hydrogenation (Fig. 5). Prior to reaction, the as-prepared catalysts were reduced at 400 °C under 5 vol% H2 in Ar (50 mL min−1) for 2 h. The in situ DRIFT spectra were collected from 1 min to 60 min at 1.0 MPa, 320 °C, and 20 mL min−1 of a gas mixture (24 vol% CO2/70 vol% H2/6 vol% Ar). For the KZFe catalyst, the bands at 2386 and 2351 cm−1 are assigned to gas-phase CO2, and the bands appearing at 2174 and 2100 cm−1 are assigned to adsorption-state CO or gas-phase CO.49 Additionally, bands ascribed to H2O species can be observed at 3630 and 3595 cm−1.49 Apparently, the RWGS reaction occurs over KZFe and KZFe–5.0Co to form CO intermediates. In contrast, the adsorbed intermediates over the surface of KZFe–5.0Co are more complex than those over KZFe. For KZFe–5.0Co, the intensity corresponding to CO species increases as the reaction proceeds, indicating that Co promotes the RWGS reaction. Additionally, distinct peaks ascribed to intermediates are observed in the 1600–1200 cm−1 region. The bands at 1587 and 1391 cm−1, 1508 and 1340 cm−1, and 1438 cm−1 are assigned to the OCO vibration of formate (HCOO*), the asymmetric and symmetric vibrations of the monodentate carbonate (CO32*), and the OH vibration of bicarbonate 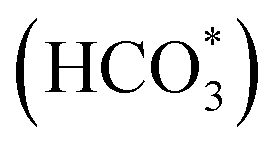 or ionic carbonate (CO32*), respectively.50–52 This indicates that the presence of Co will promote surface O-containing species formation. Additionally, evident characteristic peaks in the region of 2900–3100 cm−1 appear. The bands at 2925 and 2852 cm−1 both correspond to the CH vibrations of CH2 .50 Compared to KZFe, Co incorporation into KZFe (KZFe–5.0Co) efficiently promotes the formation of more surface active hydrocarbons, and these species will be further converted into long-chain hydrocarbons.
or ionic carbonate (CO32*), respectively.50–52 This indicates that the presence of Co will promote surface O-containing species formation. Additionally, evident characteristic peaks in the region of 2900–3100 cm−1 appear. The bands at 2925 and 2852 cm−1 both correspond to the CH vibrations of CH2 .50 Compared to KZFe, Co incorporation into KZFe (KZFe–5.0Co) efficiently promotes the formation of more surface active hydrocarbons, and these species will be further converted into long-chain hydrocarbons.
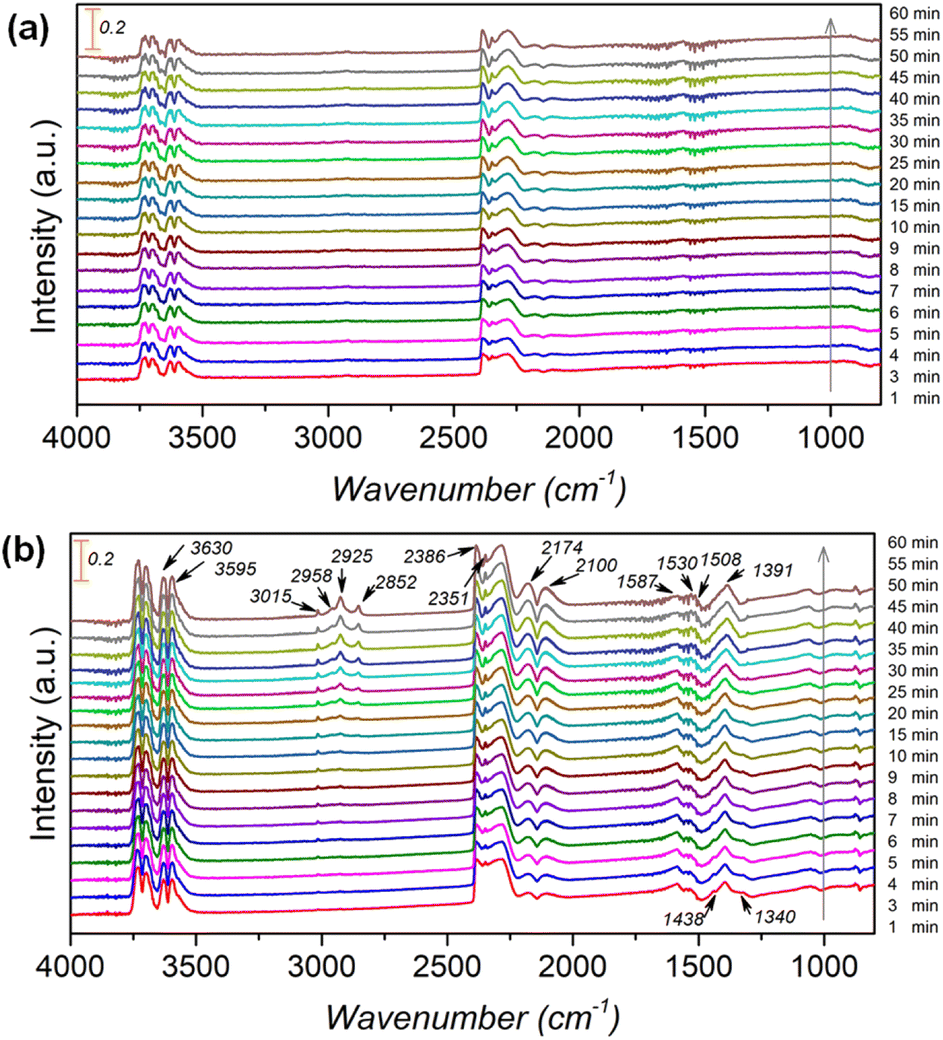 | ||
| Fig. 5 In situ DRIFT spectra obtained during CO2 hydrogenation over (a) KZFe and (b) KZFe–5.0Co (1.0 MPa, 320 °C, 20 mL min−1). | ||
For CO2 hydrogenation, it is widely accepted that the CO formed from Fe3O4 generates hydrocarbons via a typical FTS process. For KZFe, the formed CO product can produce active 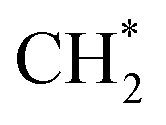 fragments, which will then be converted into hydrocarbons via the carbide mechanism (Fig. 6a and c). However, with the incorporation of Co, oxygen-bearing intermediate species appeared (Fig. 5). The XPS results also indicate that more O–CH and OC species were produced over KZFe–5.0Co than KZFe (Fig. S16†). Except for Co3Fe7, the KZFe and KZFe–5.0Co catalysts have similar active phase types (Fig. 2, 3 and S5–S8†). However, the surface species on the two catalysts differed markedly (Fig. 5 and S16†), suggesting that the difference is due to the Co3Fe7 structure. It is therefore reasonable to assume that the O-containing species come from the surface of Co3Fe7 structure. As a consequence, a reaction scheme over the KZFe–5.0Co catalysts is proposed based on the above characterization and reaction results (Fig. 6c and d). For KZFe–5.0Co, Co and Fe form alloy species (Co3Fe7, Fig. 2–4) during the reaction. Correspondingly, the improved CO2 adsorption behavior and the existence of bimetallic sites promote the formation of O-containing species (CO*, HCOO*, CO32*, and
fragments, which will then be converted into hydrocarbons via the carbide mechanism (Fig. 6a and c). However, with the incorporation of Co, oxygen-bearing intermediate species appeared (Fig. 5). The XPS results also indicate that more O–CH and OC species were produced over KZFe–5.0Co than KZFe (Fig. S16†). Except for Co3Fe7, the KZFe and KZFe–5.0Co catalysts have similar active phase types (Fig. 2, 3 and S5–S8†). However, the surface species on the two catalysts differed markedly (Fig. 5 and S16†), suggesting that the difference is due to the Co3Fe7 structure. It is therefore reasonable to assume that the O-containing species come from the surface of Co3Fe7 structure. As a consequence, a reaction scheme over the KZFe–5.0Co catalysts is proposed based on the above characterization and reaction results (Fig. 6c and d). For KZFe–5.0Co, Co and Fe form alloy species (Co3Fe7, Fig. 2–4) during the reaction. Correspondingly, the improved CO2 adsorption behavior and the existence of bimetallic sites promote the formation of O-containing species (CO*, HCOO*, CO32*, and 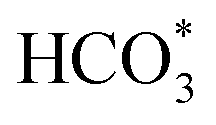 ). Unlike in the carbide mechanism over the KZFe catalyst, these abundant oxygen-bearing intermediate species can further support the chain propagation reaction continuously via an oxygenate mechanism, and thus present high heavy hydrocarbon selectivity (Fig. 6b–d). For example, the HCOO* species undergo hydrogenation and C–O scission leading to CH3O groups, and chain growth occurs by CO insertion into those R–O groups.53 The HCOO* species could come from either the direct hydrogenation of adsorbed CO2 or the interaction of CO intermediates and surface hydroxyl groups. In addition, CO* intermediates formed over Fe3O4 sites also undergo chain propagation via a carbide mechanism, and the active CO* is also involved in an oxygenation mechanism. The conversion of these surface species to long-chain hydrocarbons further drives the conversion of feedstock CO2 molecules into these C1 species. Thus, the KZFe–5.0Co catalyst shows a high catalytic activity and low CO selectivity (Fig. 1). Furthermore, the harmful competition between the methanation reaction and O-containing species formation reaction will hinder the production of long-chain products with the excess addition of Co.
). Unlike in the carbide mechanism over the KZFe catalyst, these abundant oxygen-bearing intermediate species can further support the chain propagation reaction continuously via an oxygenate mechanism, and thus present high heavy hydrocarbon selectivity (Fig. 6b–d). For example, the HCOO* species undergo hydrogenation and C–O scission leading to CH3O groups, and chain growth occurs by CO insertion into those R–O groups.53 The HCOO* species could come from either the direct hydrogenation of adsorbed CO2 or the interaction of CO intermediates and surface hydroxyl groups. In addition, CO* intermediates formed over Fe3O4 sites also undergo chain propagation via a carbide mechanism, and the active CO* is also involved in an oxygenation mechanism. The conversion of these surface species to long-chain hydrocarbons further drives the conversion of feedstock CO2 molecules into these C1 species. Thus, the KZFe–5.0Co catalyst shows a high catalytic activity and low CO selectivity (Fig. 1). Furthermore, the harmful competition between the methanation reaction and O-containing species formation reaction will hinder the production of long-chain products with the excess addition of Co.
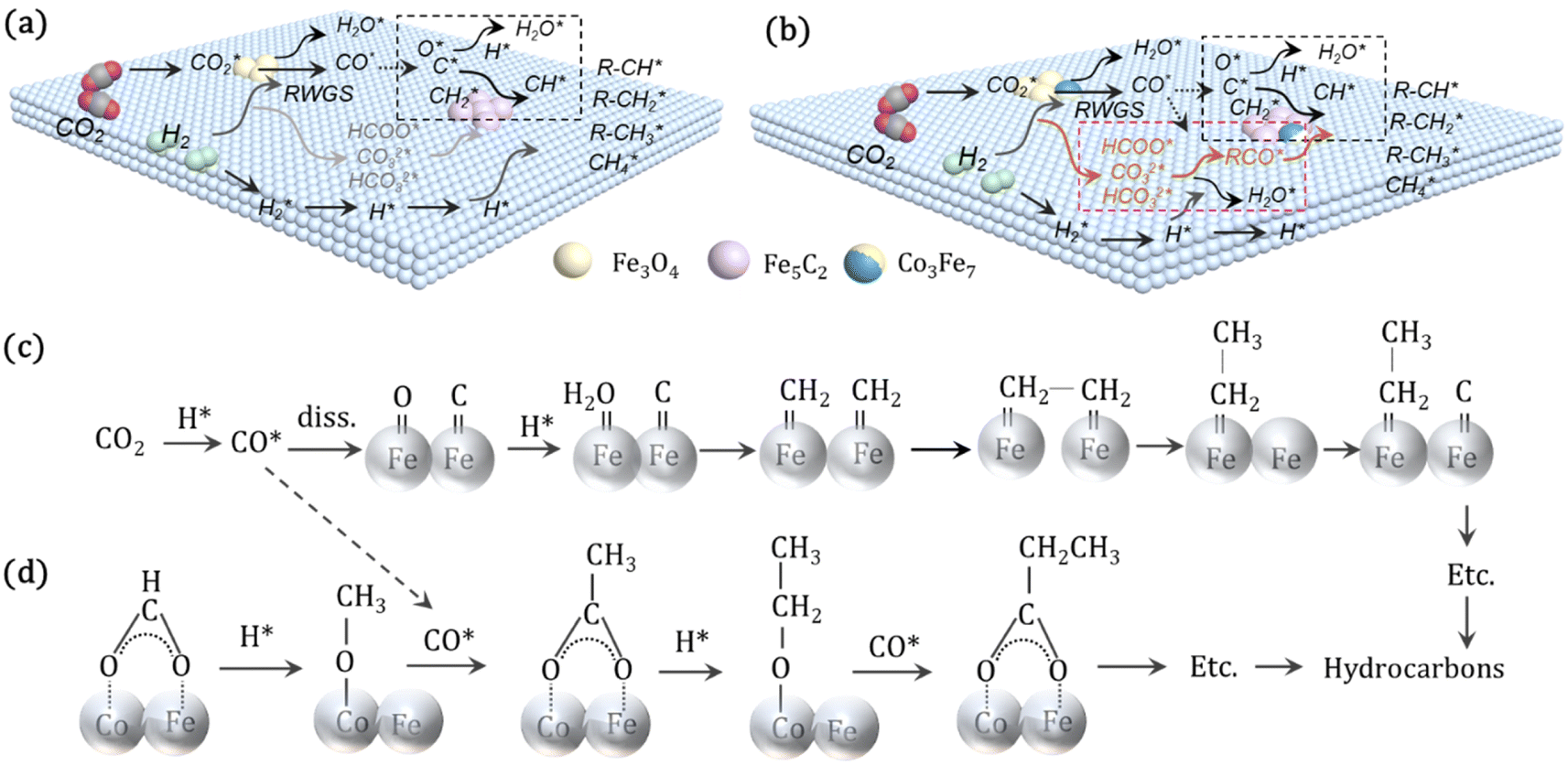 | ||
| Fig. 6 Reaction scheme over the (a) KZFe and (b) KZFe–5.0Co catalysts for the directional synthesis of long-chain hydrocarbons from CO2 hydrogenation. (c) Reaction path for long-chain hydrocarbon formation via the carbide mechanism, represented by black boxes in (a and b). (d) Oxygenate mechanism path for long-chain hydrocarbon formation, represented by the pink box in (b). | ||
Conclusions
In summary, a series of Fe-based catalysts with Co modification were prepared by the sacrificial template method for the efficient conversion of CO2 into long-chain hydrocarbons. The incorporation of Co improved the reduction and raw CO2 molecule adsorption properties, thus facilitating metallic Fe formation and the carburization of Fe to generate active Fe3O4 and Fe5C2 phases. Additionally, Co has much a lower energy barrier for CO dissociation and can provide enough active C1 species (CO*, HCOO*, CO32*, and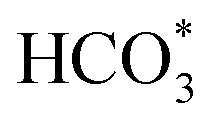 ). Subsequently, the adsorbed C1 species undergo chain propagation via the oxygenate mechanism and carbide mechanism over the bimetallic sites and carbide sites, achieving high C5+ selectivity. The driving force from the bimetallic FeCo alloy sites is beneficial for enhancing CO2 hydrogenation activity and decreasing CO selectivity in turn. Therefore, the tailor-made KZFe–5.0Co catalyst exhibits high activity (50.2%), low CO selectivity (8.1%), and high C5+ selectivity (57.8%), thus achieving a 26.7% yield of C5+. The designed bimetallic catalyst sheds light on the highly efficient conversion of CO2 to valuable hydrocarbons for practical industrial application, and provides new insights for the rational design of bimetallic or alloy catalysts.
). Subsequently, the adsorbed C1 species undergo chain propagation via the oxygenate mechanism and carbide mechanism over the bimetallic sites and carbide sites, achieving high C5+ selectivity. The driving force from the bimetallic FeCo alloy sites is beneficial for enhancing CO2 hydrogenation activity and decreasing CO selectivity in turn. Therefore, the tailor-made KZFe–5.0Co catalyst exhibits high activity (50.2%), low CO selectivity (8.1%), and high C5+ selectivity (57.8%), thus achieving a 26.7% yield of C5+. The designed bimetallic catalyst sheds light on the highly efficient conversion of CO2 to valuable hydrocarbons for practical industrial application, and provides new insights for the rational design of bimetallic or alloy catalysts.
Data availability
Data are available from the corresponding author upon reasonable request.Author contributions
L. G. conducted all experiments and characterized the catalysts. L. G., S. S. and J. S. designed the experiments. L. G., S. S., J. S., and N. T. wrote the manuscript. L. G., Y. W., and S. S. were responsible for funding application. G. G., H. W., X. W. Y. K., and X. G. contributed to the analysis and interpretation of the data.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The National Natural Science Foundation of China (U1832165, 21902001, 22102001), Key Research and Development Program of Anhui Province (202004a05020015, 006233172019), National Key Research and Development Program of China (2022YFA1604101), and DICP (Grant: DICP I202012) are greatly appreciated.Notes and references
- C. Hepburn, E. Adlen, J. Beddington, E. A. Carte, S. Fuss, N. M. Dowell, J. C. Minx, P. Smith and C. K. Williams, The technological and economic prospects for CO2 utilization and removal, Nature, 2019, 575, 87 CrossRef CAS PubMed.
- J. Wei, R. Yao, Y. Han, Q. Ge and J. Sun, Towards the development of the emerging process of CO2 heterogenous hydrogenation into high-value unsaturated heavy hydrocarbons, Chem. Soc. Rev., 2021, 50, 10764–10805 RSC.
- H. Li, C. Qiu, S. Ren, Q. Dong, S. Zhang, F. Zhou, X. Liang, J. Wang, S. Li and M. Yu, Na+-gated water-conducting nanochannels for boosting CO2 conversion to liquid fuels, Science, 2020, 367, 667–671 CrossRef CAS.
- H. Wang, Q. Lei, P. Li, C. Liu, Y. Xue, X. Zhang, C. Li and Z. Yang, Key CO2 capture technology of pure oxygen exhaust gas combustion for syngas-fueled high-temperature fuel cells, Int. J. Coal Sci. Technol., 2021, 8(3), 383–393 CrossRef CAS.
- Q. Li, The view of technological innovation in coal industry under the vision of carbon neutralization, Int. J. Coal Sci. Technol., 2021, 8(6), 1197–1207 CrossRef.
- B. Yao, T. Xiao, O. A. Makgae, X. Jie, S. Gonzalez-Cortes, S. Guan, A. I. Kirkland, J. R. Dilworth, H. A. Al-Megren, S. M. Alshihri, P. J. Dobson, G. P. Owen, J. M. Thomas and P. P. Edwards, Transforming carbon dioxide into jet fuel using an organic combustion-synthesized Fe-Mn-K catalyst, Nat. Commun., 2020, 11(1), 6395 CrossRef CAS.
- J. Wei, Q. Ge, R. Yao, Z. Wen, C. Fang, L. Guo, H. Xu and J. Sun, Directly converting CO2 into a gasoline fuel, Nat. Commun., 2017, 8, 15174–15181 CrossRef PubMed.
- P. Gao, S. Li, X. Bu, S. Dang, Z. Liu, H. Wang, L. Zhong, M. Qiu, C. Yang, J. Cai, W. Wei and Y. Sun, Direct conversion of CO2 into liquid fuels with high selectivity over a bifunctional catalyst, Nat. Chem., 2017, 9, 1019–1024 CrossRef CAS.
- X. Ye, C. Yang, X. Pan, J. Ma, Y. Zhang, Y. Ren, X. Liu, L. Li and Y. Huang, Highly Selective Hydrogenation of CO2 to Ethanol via Designed Bifunctional Ir1-In2O3 Single-Atom Catalyst, J. Am. Chem. Soc., 2020, 142(45), 19001–19005 CrossRef CAS PubMed.
- Y. Ni, Z. Chen, Y. Fu, Y. Liu, W. Zhu and Z. Liu, Selective Conversion of CO2 and H2 into Aromatics, Nat. Commun., 2018, 9, 3457 CrossRef.
- W. Zhou, K. Cheng, J. Kang, C. Zhou, V. Subramanian, Q. Zhang and Y. Wang, New horizon in C1 chemistry: breaking the selectivity limitation in transformation of syngas and hydrogenation of CO2 into hydrocarbon chemicals and fuels, Chem. Soc. Rev., 2019, 48, 3193–3228 RSC.
- R.-P. Ye, J. Ding, W. Gong, M. D. Argyle, Q. Zhong, Y. Wang, C. K. Russell, Z. Xu, A. G. Russell, Q. Li, M. Fan and Y.-G. Yao, CO2 hydrogenation to high-value products via heterogeneous catalysis, Nat. Commun., 2019, 10, 5698 CrossRef CAS PubMed.
- S. Wang, T. Wu, J. Lin, Y. Ji, S. Yan, Y. Pei, S. Xie, B. Zong and M. Qiao, Iron–Potassium on Single-Walled Carbon Nanotubes as Efficient Catalyst for CO2 Hydrogenation to Heavy Olefins, ACS Catal., 2020, 10(11), 6389–6401 CrossRef CAS.
- L. Zhang, Y. Dang, X. Zhou, P. Gao, A. Petrus van Bavel, H. Wang, S. Li, L. Shi, Y. Yang, E. I. Vovk, Y. Gao and Y. Sun, Direct conversion of CO2 to a jet fuel over CoFe alloy catalysts, The Innovation, 2021, 2(4), 100170 CrossRef CAS.
- Q. Xu, X. Xu, G. Fan, L. Yang and F. Li, Unveiling the roles of Fe-Co interactions over ternary spinel-type ZnCoxFe2-xO4 catalysts for highly efficient CO2 hydrogenation to produce light olefins, J. Catal., 2021, 400, 355–366 CrossRef CAS.
- L. Guo, J. Li, Y. Cui, R. Kosol, Y. Zeng, G. Liu, J. Wu, T. Zhao, G. Yang, L. Shao, P. Zhan, J. Chen and N. Tsubaki, Spinel-structure catalyst catalyzing CO2 hydrogenation to full spectrum alkenes with an ultra-high yield, Chem. Commun., 2020, 56, 9372–9375 RSC.
- X. Nie, H. Wang, M. J. Janik, Y. Chen, X. Guo and C. Song, Mechanistic Insight into C–C Coupling over Fe–Cu Bimetallic Catalysts in CO2 Hydrogenation, J. Phys. Chem. C, 2017, 121(24), 13164–13174 CrossRef CAS.
- L. Wang, L. Wang, J. Zhang, X. Liu, H. Wang, W. Zhang, Q. Yang, J. Ma, X. Dong, S. J. Yoo, J.-G. Kim, X. Meng and F.-S. Xiao, Selective Hydrogenation of CO2 to Ethanol over Cobalt Catalysts, Angew. Chem., Int. Ed., 2018, 130, 1–6 CrossRef.
- K. Y. Kim, H. Lee, W. Y. Noh, J. Shin, S. J. Han, S. K. Kim, K. An and J. S. Lee, Cobalt Ferrite Nanoparticles to Form a Catalytic Co–Fe Alloy Carbide Phase for Selective CO2 Hydrogenation to Light Olefins, ACS Catal., 2020, 10(15), 8660–8671 CrossRef CAS.
- M. Fujiwara, R. Kieffer, H. Ando and Y. Souma, Development of composite catalysts made of Cu-Zn-Cr oxide/zeolite for the hydrogenation of carbon dioxide, Appl. Catal., A, 1995, 121, 113–124 CrossRef CAS.
- X. Wang, G. Yang, J. Zhang, S. Chen, Y. Wu, Q. Zhang, J. Wang, Y. Han and Y. Tan, Synthesis of isoalkanes over a core (Fe-Zn-Zr)- shell (zeolite) catalyst by CO2 hydrogenation, Chem. Commun., 2016, 52, 7352–7355 RSC.
- Z. Shi, H. Yang, P. Gao, X. Chen, H. Liu, L. Zhong, H. Wang, W. Wei and Y. Sun, Effect of alkali metals on the performance of CoCu/TiO2 catalysts for CO2 hydrogenation to long-chain hydrocarbons, Chin. J. Catal., 2018, 39(8), 1294–1302 CrossRef CAS.
- Y. H. Choi, Y. J. Jang, H. Park, W. Y. Kim, Y. H. Lee, S. H. Choi and J. S. Lee, Carbon dioxide Fischer-Tropsch synthesis: A new path to carbon-neutral fuels, Appl. Catal., B, 2017, 202, 605–610 CrossRef CAS.
- N. Boreriboon, X. Jiang, C. Song and P. Prasassarakich, Fe-based bimetallic catalysts supported on TiO2 for selective CO2 hydrogenation to hydrocarbons, J. CO2 Util., 2018, 25, 330–337 CrossRef CAS.
- J. Wei, J. Sun, Z. Wen, C. Fang, Q. Ge and H. Xu, New insights into the effect of sodium on Fe3O4-based nanocatalysts for CO2 hydrogenation to light olefins, Catal. Sci. Technol., 2016, 6(13), 4786–4793 RSC.
- M. Albrecht, U. Rodemerck, M. Schneider, M. Bröring, D. Baabe and E. V. Kondratenko, Unexpectedly efficient CO2 hydrogenation to higher hydrocarbons over non-doped Fe2O3, Appl. Catal., B, 2017, 204, 119–126 CrossRef CAS.
- Y. Wang, L. Tan, M. H. Tan, P. P. Zhang, Y. Fang, Y. Yoneyama, G. H. Yang and N. Tsubaki, Rationally Designing Bifunctional Catalysts as an Efficient Strategy to Boost CO2 Hydrogenation Producing Value-Added Aromatics, ACS Catal., 2019, 9, 895–901 CrossRef CAS.
- Y. H. Choi, E. C. Ra, E. H. Kim, K. Y. Kim, Y. J. Jang, K.-N. Kang, S. H. Choi, J.-H. Jang and J. S. Lee, Sodium-Containing Spinel Zinc Ferrite as a Catalyst Precursor for the Selective Synthesis of Liquid Hydrocarbon Fuels, ChemSusChem, 2017, 10, 4764–4770 CrossRef CAS.
- T. Numpilai, T. Witoon, N. Chanlek, W. Limphirat, G. Bonura, M. Chareonpanich and J. Limtrakul, Structure–activity relationships of Fe-Co/K-Al2O3 catalysts calcined at different temperatures for CO2 hydrogenation to light olefins, Appl. Catal., A, 2017, 547, 219–229 CrossRef CAS.
- L. Guo, S. Sun, J. Li, W. Gao, H. Zhao, B. Zhang, Y. He, P. Zhang, G. Yang and N. Tsubaki, Boosting liquid hydrocarbons selectivity from CO2 hydrogenation by facilely tailoring surface acid properties of zeolite via a modified Fischer-Tropsch synthesis, Fuel, 2021, 306, 121684 CrossRef CAS.
- F. Jiang, B. Liu, S. Geng, Y. Xu and X. Liu, Hydrogenation of CO2 into hydrocarbons: enhanced catalytic activity over Fe-based Fischer–Tropsch catalysts, Catal. Sci. Technol., 2018, 8(16), 4097–4107 RSC.
- A. Noreen, M. Li, Y. Fu, C. C. Amoo, J. Wang, E. Maturura, C. Du, R. Yang, C. Xing and J. Sun, One-Pass Hydrogenation of CO2 to Multibranched Isoparaffins over Bifunctional Zeolite-Based Catalysts, ACS Catal., 2020, 10(23), 14186–14194 CrossRef CAS.
- W. D. Shafer, G. Jacobs, U. M. Graham, H. H. Hamdeh and B. H. Davis, Increased CO2 Hydrogenation to Liquid Products Using Promoted Iron Catalysts, J. Catal., 2019, 369, 239–248 CrossRef CAS.
- Y. Wang, G. Wang, L. I. v. d. Wal, K. Cheng, Q. Zhang, K. P. d. Jong and Y. Wang, Visualizing Element Migration over Bifunctional Metal-Zeolite Catalysts and its Impact on Catalysis, Angew. Chem., Int. Ed., 2021, 60(32), 17735–17743 CrossRef CAS PubMed.
- Y. Xu, J. Wang, G. Ma, J. Bai, Y. Du and M. Ding, Selective conversion of syngas to olefins-rich liquid fuels over core-shell FeMn@SiO2 catalysts, Fuel, 2020, 275, 117884 CrossRef CAS.
- J. Zhu, P. Wang, X. Zhang, G. Zhang, R. Li, W. Li, T. P. Senftle, W. Liu, J. Wang, Y. Wang, A. Zhang, Q. Fu, C. Song and X. Guo, Dynamic structural evolution of iron catalysts involving competitive oxidation and carburization during CO2 hydrogenation, Sci. Adv., 2022,(8), eabm3629 CrossRef CAS PubMed.
- C. Yang, B. Zhao, R. Gao, S. Yao, P. Zhai, S. Li, J. Yu, Y. Hou and D. Ma, Construction of Synergistic Fe5C2/Co Heterostructured Nanoparticles as an Enhanced Low Temperature Fischer-Tropsch Synthesis Catalyst, ACS Catal., 2017, 7, 5661–5667 CrossRef CAS.
- B. Ravel and M. Newville, ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT, J. Synchrotron Radiat., 2005, 12(Pt 4), 537–541 CrossRef CAS PubMed.
- L. Guo, J. Sun, X. Ji, J. Wei, Z. Wen, R. Yao, H. Xu and Q. Ge, Directly converting carbon dioxide to linear α-olefins on bio-promoted catalysts, Commun. Chem., 2018, 1, 11 CrossRef.
- J. Lu, L. Yang, B. Xu, Q. Wu, D. Zhang, S. Yuan, Y. Zhai, X. Wang, Y. Fan and Z. Hu, Promotion Effects of Nitrogen Doping into Carbon Nanotubes on Supported Iron Fischer–Tropsch Catalysts for Lower Olefins, ACS Catal., 2014, 4(2), 613–621 CrossRef CAS.
- J. M. Cho, B.-G. Kim, G. Y. Han, J. Sun, H.-K. Jeong and J. W. Bae, Effects of metal-organic framework-derived iron carbide phases for CO hydrogenation activity to hydrocarbons, Fuel, 2020, 281, 118779 CrossRef CAS.
- M. Fan, C. Zhu, Z.-Q. Feng, J. Yang, L. Liu and D. Sun, Preparation of N-doped graphene by reduction of graphene oxide with mixed microbial system and its haemocompatibility, Nanoscale, 2014, 6(9), 4882–4888 RSC.
- Y. Zhao, J. Zhang, X. Guo, H. Fan, W. Wu, H. Liu and G. Wang, Fe3C@nitrogen doped CNT arrays aligned on nitrogen functionalized carbon nanofibers as highly efficient catalysts for the oxygen evolution reaction, J. Mater. Chem. A, 2017, 5(37), 19672–19679 RSC.
- A. F. Lucrédio, G. T. Filho and E. M. Assaf, Co/Mg/Al hydrotalcite-type precursor, promoted with La and Ce, studied by XPS and applied to methane steam reforming reactions, Appl. Surf. Sci., 2009, 255(11), 5851–5856 CrossRef.
- P. Chen, T. Zhou, S. Wang, N. Zhang, Y. Tong, H. Ju, W. Chu, C. Wu and Y. Xie, Dynamic Migration of Surface Fluorine Anions on Cobalt-Based Materials to Achieve Enhanced Oxygen Evolution Catalysis, Angew. Chem., Int. Ed., 2018, 57(47), 15471–15475 CrossRef CAS.
- L. Cao, W. Liu, Q. Luo, R. Yin, B. Wang, J. Weissenrieder, M. Soldemo, H. Yan, Y. Lin, Z. Sun, C. Ma, W. Zhang, S. Chen, H. Wang, Q. Guan, T. Yao, S. Wei, J. Yang and J. Lu, Atomically dispersed iron hydroxide anchored on Pt for preferential oxidation of CO in H2, Nature, 2019, 565(7741), 631–635 CrossRef CAS PubMed.
- H. M. T. Galvis, J. H. Bitter, T. Davidian, M. Ruitenbeek, A. I. Dugulan and K. P. d. Jong, Iron particle size effects for direct production of lower olefins from synthesis gas, J. Am. Chem. Soc., 2012, 134(39), 16207–16215 CrossRef.
- L. Guo, P. Zhang, Y. Cui, G. Liu, J. Wu, G. Yang, Y. Yoneyama and N. Tsubaki, One-Pot Hydrothermal Synthesis of Nitrogen Functionalized Carbonaceous Material Catalysts with Embedded Iron Nanoparticles for CO2 Hydrogenation, ACS Sustainable Chem. Eng., 2019, 7, 8331–8339 CrossRef CAS.
- L. Guo, Y. Cui, H. Li, Y. Fang, R. Prasert, J. Wu, G. Yang, Y. Yoneyama and N. Tsubaki, Selective formation of linear-alpha olefins by catalyzing CO2 hydrogenation over a bimetallic catalyst, Catal. Commun., 2019, 130, 105759 CrossRef.
- J. Zhu, G. Zhang, W. Li, X. Zhang, F. Ding, C. Song and X. Guo, Deconvolution of the Particle Size Effect on CO2 Hydrogenation over Iron-Based Catalysts, ACS Catal., 2020, 10(13), 7424–7433 CrossRef CAS.
- H. Jo, M. K. Khan, M. Irshad, M. W. Arshad, S. K. Kim and J. Kim, Unraveling the role of cobalt in the direct conversion of CO2 to high-yield liquid fuels and lube base oil, Appl. Catal., B, 2022, 305, 121041 CrossRef CAS.
- Q. Sheng, R.-P. Ye, W. Gong, X. Shi, B. Xu, M. Argyle, H. Adidharma and M. Fan, Mechanism and catalytic performance for direct dimethyl ether synthesis by CO2 hydrogenation over CuZnZr/ferrierite hybrid catalyst, J. Environ. Sci., 2020, 92, 106–117 CrossRef CAS.
- A. Frennet, T. V. d. Bocarmé, J.-M. Bastin and N. Kruse, Mechanism and Kinetics of the Catalytic CO-H2 Reaction: An Approach by Chemical Transients and Surface Relaxation Spectroscopy, J. Phys. Chem. B, 2005, 109, 2350–2359 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc05047a |
| This journal is © The Royal Society of Chemistry 2023 |