
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
ThC2@C82versus Th@C84: unexpected formation of triangular thorium carbide cluster inside fullerenes†
Yi
Shen‡
 a,
Xiaojuan
Yu‡
b,
Qingyu
Meng
a,
Yang-Rong
Yao
a,
Jochen
Autschbach
*b and
Ning
Chen
*a
a,
Xiaojuan
Yu‡
b,
Qingyu
Meng
a,
Yang-Rong
Yao
a,
Jochen
Autschbach
*b and
Ning
Chen
*a
aCollege of Chemistry, Chemical Engineering and Materials Science, State Key Laboratory of Radiation Medicine and Protection, Soochow University, Suzhou, Jiangsu 215123, P. R. China. E-mail: chenning@suda.edu.cn
bDepartment of Chemistry, University at Buffalo, State University of New York, Buffalo, NY 14260-3000, USA
First published on 20th October 2022
Abstract
Synthesis of the first thorium-containing clusterfullerenes, ThC2@Cs(6)–C82 and ThC2@C2(5)–C82, is reported. These two novel actinide fullerene compounds were characterized by mass spectrometry, single-crystal X-ray diffraction crystallography, UV–vis–NIR spectroscopy, and theoretical calculations. Crystallographic studies reveal that the encapsulated ThC2 clusters in both Cs(6)–C82 and C2(5)–C82 feature a novel bonding structure with one thorium metal center connected by a C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C unit, forming an isosceles triangular configuration, which has not been hitherto observed for endohedral fullerenes or for solid phase thorium carbides. Electronic structure calculations assign a formal electronic structure of [Th4+(C2)2−]2+@[C82]2−, with pronounced donation bonding from (C2)2− to Th4+, secondary backbonding from the fullerene to thorium and Th–C double bond character in both compounds. This work presents a new family of endohedral fullerenes, MC2@C2n−2, being unexpected isomers of MC2n, and provides broader understanding of thorium bonding.
C unit, forming an isosceles triangular configuration, which has not been hitherto observed for endohedral fullerenes or for solid phase thorium carbides. Electronic structure calculations assign a formal electronic structure of [Th4+(C2)2−]2+@[C82]2−, with pronounced donation bonding from (C2)2− to Th4+, secondary backbonding from the fullerene to thorium and Th–C double bond character in both compounds. This work presents a new family of endohedral fullerenes, MC2@C2n−2, being unexpected isomers of MC2n, and provides broader understanding of thorium bonding.
Introduction
Fullerenes are known for their ability to encapsulate clusters, which results in the formation of unique host–guest molecular compounds—endohedral clusterfullerenes.1–3 The unique interaction and mutual stabilization between the metal-containing clusters and fullerenes gave rise to fascinating electronic structures and potential applications of these compounds.4–7 To date, most lanthanides have been encapsulated in fullerene cages.8 Our recent research showed that novel actinide clusters can also be captured and stabilized by fullerene cages, such as U2C@Ih(7)–C80, U2C2@Ih(7)–C80, or UCN@C82.9–11 These systems exhibit substantially different electronic structures compared to known lanthanide-based analogs. In particular, the encapsulated uranium clusters reveal bonding properties that have never been observed in conventional uranium compounds. Thus, the exploration of novel actinide cluster fullerenes will not only expand the scope of endohedral fullerenes, but also have significance regarding the understanding of fundamental actinide chemistry. However, all of the actinide cluster fullerenes discovered thus far were based on uranium; other actinide cluster fullerenes have yet to be explored.12Thorium is arguably the new frontier of nuclear energy.13 Attempts have been made to synthesize and characterize thorium compounds for use as potential fuels in advanced reactors. Recently, thorium carbides have attracted increasing attention because these compounds are suitable for high-burnup and high-temperature operations with increased “margin to melting” in the framework of modern nuclear systems.14 Many advantages of thorium carbides, such as high melting points, corrosion resistivity, low thermal expansion coefficients and high thermal conductivity, have been reported in recent research.15,16 Therefore, understanding the behavior and properties of thorium carbides is essential to explore their potential application as nuclear reactor fuel materials.17,18
Thorium carbides (ThCn, n = 1–6) have been detected in vapors above solid carbides or metal alloys in graphite systems, and partial pressures of thorium carbides were measured by mass spectrometry.19–22 Thorium dicarbide (ThC2), as the main type of stoichiometric thorium carbides, exists in polymorphic modifications at ambient pressure.16,23–25 However, the structural and electronic properties of ThC2 have only been studied by theoretical calculations.18,23,26 Thus far, the molecular structure of ThC2 has not been observed in the condensed phase.
On the other hand, carbide cluster fullerenes (CCFs) are an important branch of endohedral cluster fullerenes and have been extensively investigated in the past two decades.12 The first reported CCF is Sc2C2@D2d(23)–C84, initially assigned as a di-metallofullerene (EMF), Sc2@C86.27 This discovery confirmed that the composition M2C2n could exist as M2C2n or as M2C2@C2n–2. Subsequent studies revealed a large family of CCFs with variable M2C2 clusters encapsulated inside different fullerene cages, such as Sc2C2@C2n,28–30 Gd2C2@C2n,31 Lu2C2@C2n32et al.2 In addition, composition Sc3C82 was also reassigned as Sc3C2@Ih–C80.33 Up to now, a large variety of CCFs entrapping multiple (2–4) metal atoms have been reported. However, monometallic carbide cluster fullerenes have not been yet available and whether M@C2n can exit as as MC2n or as MC2@C2n−2 has yet to be explored.
Herein, we report the first thorium-based cluster fullerenes, namely, ThC2@Cs(6)–C82 and ThC2@C2(5)–C82. Crystallographic studies reveal that, these two actinide endohedral fullerenes, initially assigned as isomers of Th@C84, are in fact thorium-based cluster fullerenes which contains a unique mononuclear thorium carbide cluster. Theoretical analyses confirm that ThC2@Cs(6)–C82 and ThC2@C2(5)–C82 can be described by a formal two-electron transfer from the ThC2 cluster to the C82 cage, which results in formal closed-shell electronic structures [Th4+(C2)2−]2+@[C82]2−.
Results and discussion
Synthesis and isolation of ThC2@Cs(6)–C82 and ThC2@C2(5)–C82
Thorium-based endohedral metallofullerenes were synthesized by a modified Krätschmer–Huffman DC arc discharge method. Graphite rods packed with U3O8/ThO2 and graphite powder (molar ratio of U![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Th:C = 1:1:30) were vaporized in the arcing chamber under a 200 Torr He atmosphere. The resulting soot was then collected and extracted with CS2 for 12 h. A series of thorium-containing endohedral metallofullerenes were generated from this process (Fig. S2†) and in this work, two novel isomers(I,II) of ThC84 (later assigned as ThC2@Cs(6)–C82 and ThC2@C2(5)–C82) were isolated by a multistage high-performance liquid chromatography (HPLC) separation process (Fig. S1†). The purity of the isolated compounds was confirmed by positive-ion-mode matrix-assisted laser/desorption ionization time-of-flight mass spectrometry (MALDI-TOF/MS), as shown in Fig. 1. The mass spectra of ThC2@Cs(6)–C82 and ThC2@C2(5)–C82 show peaks at m/z = 1240.196 and 1240.204. In addition, the experimental isotopic distributions of the two samples agree well with theoretical predictions.
:Th:C = 1:1:30) were vaporized in the arcing chamber under a 200 Torr He atmosphere. The resulting soot was then collected and extracted with CS2 for 12 h. A series of thorium-containing endohedral metallofullerenes were generated from this process (Fig. S2†) and in this work, two novel isomers(I,II) of ThC84 (later assigned as ThC2@Cs(6)–C82 and ThC2@C2(5)–C82) were isolated by a multistage high-performance liquid chromatography (HPLC) separation process (Fig. S1†). The purity of the isolated compounds was confirmed by positive-ion-mode matrix-assisted laser/desorption ionization time-of-flight mass spectrometry (MALDI-TOF/MS), as shown in Fig. 1. The mass spectra of ThC2@Cs(6)–C82 and ThC2@C2(5)–C82 show peaks at m/z = 1240.196 and 1240.204. In addition, the experimental isotopic distributions of the two samples agree well with theoretical predictions.
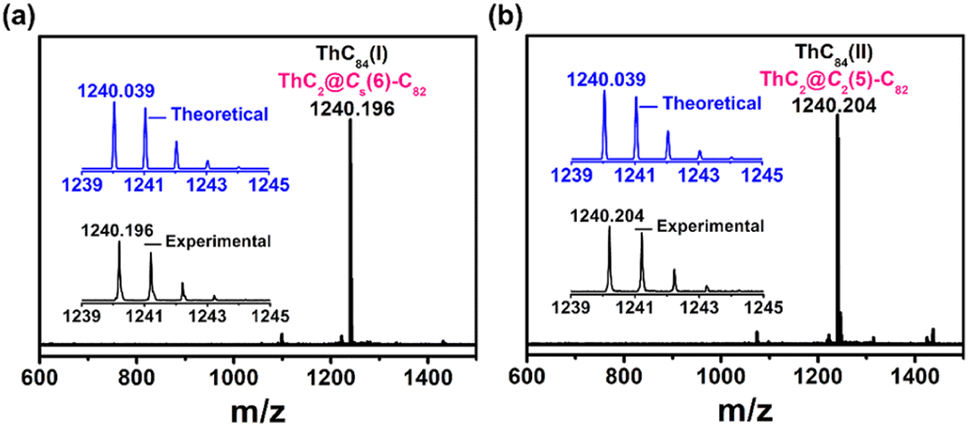 | ||
| Fig. 1 Positive-ion mode MALDI-TOF mass spectra of purified (a) ThC2@Cs(6)–C82 and (b) ThC2@C2(5)–C82. The insets show the experimental and theoretical isotopic distribution for compounds. | ||
Molecular and electronic structures of ThC2@Cs(6)–C82·[NiIIOEP] and ThC2@C2(5)–C82·[NiIIOEP]
Two black block cocrystals were obtained by slow diffusion of NiII(OEP) (OEP = 2,3,7,8,12,13,17,18-octaethylporphin dianion) in benzene into a CS2 solution of the corresponding compounds. NiII(OEP) was used to hinder rotation of fullerene molecules in the co-crystal. The molecular structures of ThC2@Cs(6)-C82·[NiIIOEP] and ThC2@C2(5)-C82·[NiIIOEP] were determined by single-crystal X-ray diffraction analysis, excluding other molecular structures with the same molecular weight, such as Th-based mono-metallofullerenes, Th@C84 isomers. The shortest Ni-cage distance was measured as 2.846(104) and 2.855(115) Å for ThC2@Cs(6)–C82·[NiIIOEP] and ThC2@C2(5)–C82·[NiIIOEP], indicating a strong π–π interaction between ThC2@C82 and NiII(OEP). Both ThC2@Cs(6)–C82·[NiIIOEP] and ThC2@C2(5)–C82·[NiIIOEP] were solved in the monoclinic space group C2/m.The whole molecule of ThC2@Cs(6)–C82, including the fullerene cage and the encapsulated cluster, shows two equivalent orientations with the same occupancy of 0.5, which is common in many analogous metallofullerene/NiII(OEP) cocrystal systems.1 The encapsulated Th ion shows only slight disorder with a total occupancy of 0.5 for the three disordered sites Th1–Th3. Th1 is assigned as the major Th site, as it has a much higher occupancy of 0.418 compared to those of the other two sites (0.0489 and 0.0326 for Th2 and Th3, respectively). Furthermore, Th1A, Th2A and Th3A are also generated via their mirror-related counterparts, Th1, Th2 and Th3, due to the same crystallographic mirror plane. Further structural analysis shows that Th1 is situated on the symmetry plane of the Cs(6)–C82 cage, while Th1A is located away from the symmetry plane (Fig. S4†). The density functional theory (DFT) calculation results also suggest that the Th1 site is approximately 13 kcal mol−1 lower in energy for all functionals than Th1A (Table S2†). In addition, previous studies suggest that the metal ion prefers to remain symmetrically aligned with interacting motifs that share one of the symmetry planes with the fullerene containing mirror planes.34 Therefore, we assign Th1 as the optimal position of ThC2@Cs(6)–C82 (Fig. 2a).
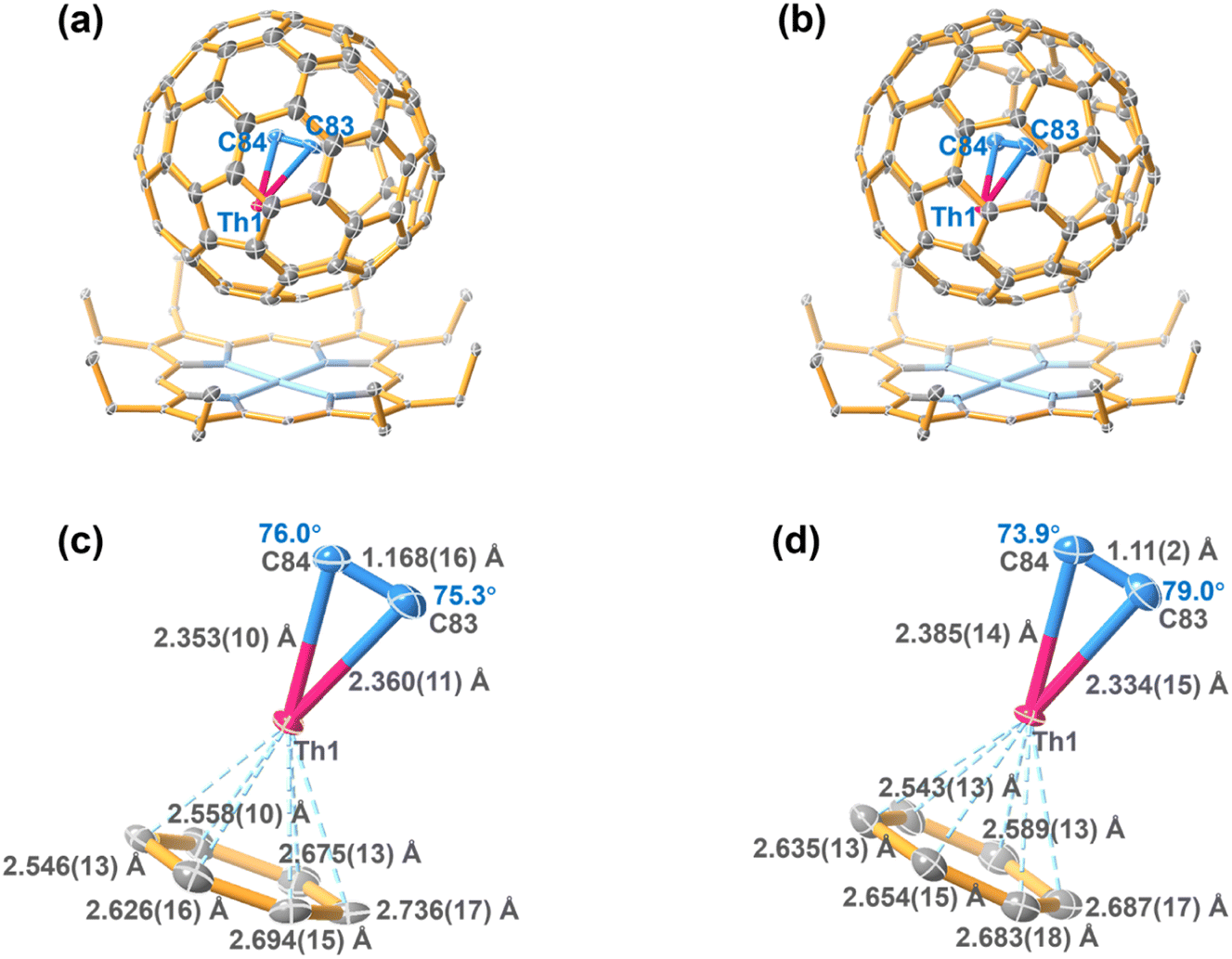 | ||
| Fig. 2 ORTEP drawing of ThC2@Cs(6)–C82·[NiIIOEP] (a) and ThC2@C2(5)–C82·[NiIIOEP] (b) with 20% thermal ellipsoids. Only the predominant Th (Th1) sites are shown. For clarity, the solvent molecules and minor metal sites (Fig. S3†) are omitted. Fragment view showing the interaction of the ThC2 clusters with the closest aromatic ring fragments of the Cs(6)–C82 cage (c) and C2(5)–C82 cage (d). | ||
The crystallographic results of ThC2@C2(5)–C82 also show two orientations of the fullerene molecule with equal occupancy of 0.5. These two orientations are related by the molecular crystallographic mirror. The Th1 site is the major Th position for ThC2@C2(5)–C82, with a fractional occupancy of 0.281. Th1A has the same occupancy of 0.281 and is symmetrical with Th1 through a crystallographic mirror. The rest of the minor sites are displayed in Fig. S3(b).† Th1 is located beneath the corresponding hexagon, with the shortest metal-cage distances of 2.542(12) Å (Th1–C3) and 2.589(13) Å (Th1–C2). Its mirror-related counterpart, Th1A, on the other hand, has the closest metal-cage of 2.475(13) (Th1A–C33) and 2.576(14) (Th1A–C55) Å, respectively (Fig. S5†). Thus, in this case, neither Th1 nor Th1A can be assigned as the optimal site only by crystallographic analysis, and their metal-cage distances are very similar. Therefore, theoretical calculations were employed to further determine the optimized position of the encapsulated ThC2 cluster relative to the selected cage orientation. The results show that the Th1 site has a lower energy than the Th1A site for ThC2@C2(5)–C82 (Table S2†). Thus, the optimal ThC2 cluster orientation can be accurately determined, as shown in Fig. 2b.
As shown in Fig. 2c and d, the Th–C distances are 2.360(11)/2.353(10) Å for ThC2@Cs(6)–C82 and 2.334(15)/2.385(14) Å for ThC2@C2(5)–C82, which are significantly shorter than the Th–C single bonds in thorium-based organometallic complexes (2.471–2.892 Å),35,36 as shown in Table 1. Moreover, for the encapsulated cluster ThC2, the C–C distances from the X-ray diffraction are 1.168(16) Å and 1.11(2) Å in the Cs(6)–C82 and C2(5)–C82 cages, respectively, as shown in Table S3.† These C–C bonds can be assigned as triple bonds, but they are approximately 0.1 Å shorter than the optimized distances obtained by theoretical calculations (1.252 and 1.251 Å, respectively) of the isolated cluster fullerenes. The unusual phenomenon of shrinking C–C bonds inside fullerene cages has also been observed for metal carbide cluster fullerenes such as Sc2C2@D3h(14246)–C74, Ga2C2@D3(85)–C92 and U2C2@D3h(5)–C78, in which the X-ray crystallographically determined C–C bond lengths (1.049(17) Å,37 1.04(2) Å (ref. 38) and 1.127(18) Å (ref. 10) respectively) are also notably shorter than the C–C triple bonds in alkyne compounds (1.21 Å). In addition, as shown in Fig. 2c andd, the metal–Ccage distances are 2.546(13)–2.736(17) Å for ThC2@Cs(6)–C82 and 2.543(13)–2.687(17) Å for ThC2@C2(5)–C82, which agree well with the theoretical calculations (Table S3†) (2.606–2.814 Å for ThC2@Cs(6)–C82 and 2.635–2.686 Å for ThC2@C2(5)–C82). These distances are similar to the Th–Cp(cent) distances in organometallic compounds; for example, the Th–Cp(cent) distances are 2.532(4)–2.649(8) Å in actinide phosphinidene metallocene (Cp = cyclopentadienyl ring).39,40 This result suggests that the coordination interaction between Th and the fullerene cage may be similar to that between Th and the cyclopentadienyl group in organometallic compounds.
The ThC2 cluster in Cs(6)–C82 features two almost identical Th–C distances, 2.360(11) and 2.353(10) Å, respectively, leading to an isosceles triangular configuration. ThC2 in C2(5)–C82 has a similar but slightly distorted isosceles triangular configuration, with a Th–C bond length difference of 0.05 Å. Note that the metal–Ccage distances in the two ThC2@C82 isomers, as mentioned already, are also somewhat different [2.546(13)–2.736(17) Å for ThC2@Cs(6)–C82 and 2.543(13)–2.687(17) Å for ThC2@C2(5)–C82]. This result suggests that the variable isomeric cage structure has a slight impact on the interaction between Th and cage carbon, which likely results in differences in the ThC2 cluster configurations inside the two C82 cage isomers.
The symmetric isosceles triangular structure configuration of the ThC2 cluster encapsulated in either C2(5)–C82 or Cs(6)–C82 is similar to a previously reported theoretically optimized structure of the ThC2 molecule:18 Kovacs and coworkers predicted that, for neutral ThC2, the symmetric triangular arrangement is much more stable than alternate linear or asymmetric triangular conformations.18,41 The Th–C distance in the symmetric triangular isolated ThC2 molecule obtained in the previous calculations is 2.281 Å,18,41 which is shorter than the experimentally obtained Th–C bond length for ThC2@C82 (2.360(11)/2.353(10) Å for ThC2@Cs(6)–C82 and 2.334(15)/2.385(14) Å for ThC2@C2(5)–C82). The variability of the Th–C distance may be rationalized by the fact that the coordination bonding between the Th and C2 moiety in ThC2@C82 is weakened by the coordination interaction between Th and the fullerene cage, as discussed later.
A closer look at the symmetric structure of ThC2@Cs(6)–C82 shows that, although the encapsulated ThC2 can have many possible orientations relative to the cage, both the metal atom, Th1, and the cluster, ThC2, are located right on the symmetry plane. Further analysis of the crystallographic data of other mononuclear cluster fullerene-containing symmetry planes, such as MCN@Cs(6)–C82 (M = U, Y and Dy), MCN@C2v(19138)–C76 (M = Tb, Lu and Y), DyCN@C2v(9)–C82 and DyCN@C2v(17)–C84, as show in Fig. 4 suggests that the encapsulated mononuclear clusters are all located on the mirror planes of fullerene cages (see Fig. 3).5,11,42–45 Previous studies of monometallic fullerenes (only one metal ion inside the cage) have found that in fullerene cages containing symmetry planes, the metal prefers to occupy a symmetric arrangement with respect to the interacting motifs, which share one of their symmetry planes with the fullerene.34 This observation further suggests that the endohedral mononuclear cluster also prefers to share a symmetry plane with the fullerene cages. That is, in general, as long as the fullerene encapsulating a mononuclear cluster possesses mirror planes, the entire molecule tends to be symmetric.
 | ||
| Fig. 3 Orbital isosurfaces (±0.03 au) and atomic orbital weight compositions (in %) obtained from NLMO analysis of the singlet ground state of ThC2@Cs(6)–C82. | ||
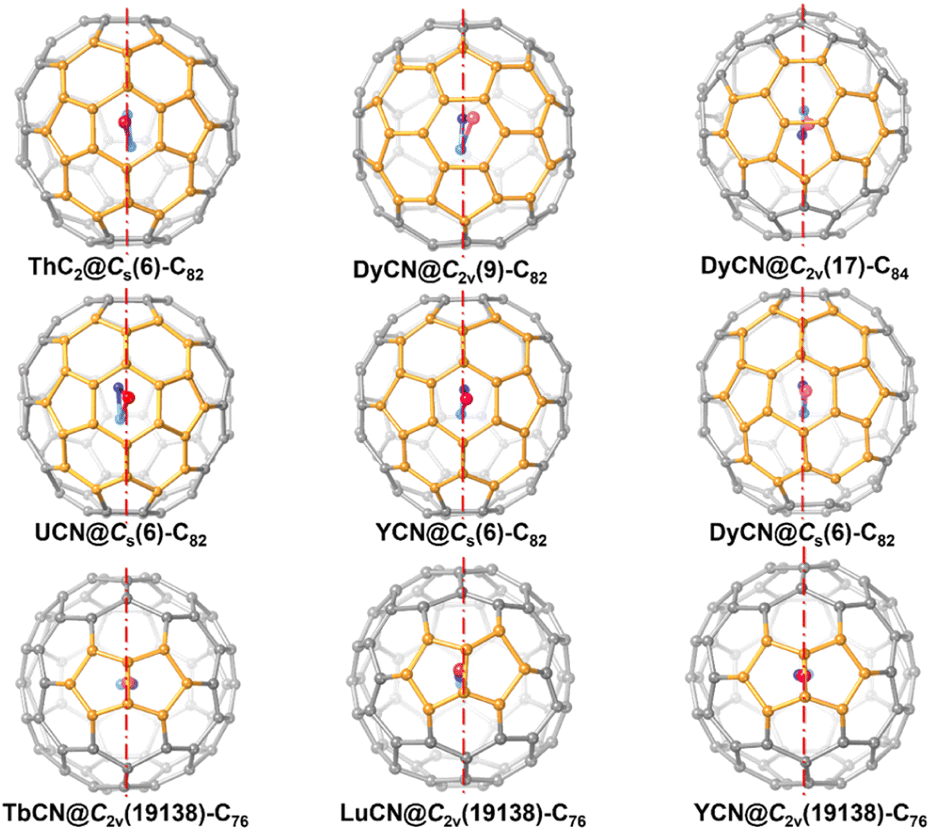 | ||
| Fig. 4 Structures of crystallographically characterized mononuclear cluster fullerenes with pristine cages that contain symmetry planes (highlighted with dotted red lines). The fullerene cage segments closest to the encapsulated metal ions are highlighted in light orange. | ||
The identification of the encapsulated ThC2 cluster expands our understanding of endohedral fullerenes. It represents a new type of endohedral cluster MC2, in which a single metal ion is coordinated to a CC unit. In previous fullerene studies, if a fullerene compound was identified by mass spectrometry as MC2n, it can be intuitively assigned as a mono-metallofullerene, i.e., M@C2n, in which only a single metal ion is encapsulated inside the cage. The discovery of MC2@C2n, however, breaks this paradigm and suggests that MC2n can also be the isostructural isomer of MC2@C2n−2. Moreover, it provides the first crystallographic observation of a discrete ThC2, which may be beneficial for the better understanding of those thorium carbide gas molecules generated in high temperature.
Theoretical investigation
DFT calculations for ThC2@Cs(6)–C82 and ThC2@C2(5)–C82 identified spin-singlet ground states. It is known that C82 can accept two electrons, for example, in the case of Sm@C82 and TbCN@C82 [Cs(6) and C2(5) isomers].46,47 Based on the frontier molecular orbitals in our calculations, we verified that there is a transfer of two electrons from the encapsulated ThC2 cluster to the C82 cage, i.e., the system adopts a formal closed-shell [Th4+(C2)2−]2+@[C82]2− electron configuration. Therefore, we interpret that ThC2@C82 isomers have similar two-electron transfer to those of Sm@C82 and TbCN@C82. In all three cases, isomeric structures of Cs(6)–C82 and C2(5)–C82 are stabilized by the metal/cluster-to-cage two electron transfer.46,47 For the spin-singlet states, the structural parameters optimized with the B3LYP hybrid functional match the experimental data better than other tested functionals. The following discussion is based on all-electron scalar relativistic B3LYP optimizations and the corresponding electronic structures.The metal–ligand bonding in ThC2@Cs(6)–C82 and ThC2@C2(5)–C82, was characterized in terms of natural localized molecular orbitals (NLMOs) and Wiberg Bond Orders (WBOs). In ThC2@Cs(6)–C82, as shown in Fig. 3, there are three pairs of NLMOs, one σ and two π, describing the formal triple bond of C22−. There is pronounced covalency with thorium. The two π NLMOs display three-center characteristics involving Th, with 10% and 9% weights of the orbital density associated with Th 6d–5f hybrids. The carbon lone pairs are even stronger donating, with 13% weight at thorium. The WBO for C22− in the cluster fullerene is 2.51, that is, a triple bond slightly weakened by the donation to the metal. In comparison with other known Th–C bonds, the Th–C interaction in the fullerene shows double bond character. The average WBO of 0.85 is close to the bond order of 0.91 for the formal Th![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond in complex [{(NR2)3}Th(CCCPh2)]− (R = SiMe3)),48 and nearly double the WBOs (0.47 and 0.49) of the single Th–Cipso bonds in [Li(DME)2(Et2O)]2[Li(DME)2][Th(C6Cl5)5]3 and [Li(DME)2(Et2O)][Li(Et2O)2][ThCl3(C6Cl5)3].49 These data help rationalizing the aforementioned short Th–C distances. Some of the NLMOs centered in the fullerene also have density at Th; the corresponding plots are shown in Fig. S9.† Among them, the strongest Th–C(cage) interaction has 6% Th weight. Therefore, the formal transfer of two electrons from ThC2 to C82 is accompanied by secondary cage-metal backbonding.
C double bond in complex [{(NR2)3}Th(CCCPh2)]− (R = SiMe3)),48 and nearly double the WBOs (0.47 and 0.49) of the single Th–Cipso bonds in [Li(DME)2(Et2O)]2[Li(DME)2][Th(C6Cl5)5]3 and [Li(DME)2(Et2O)][Li(Et2O)2][ThCl3(C6Cl5)3].49 These data help rationalizing the aforementioned short Th–C distances. Some of the NLMOs centered in the fullerene also have density at Th; the corresponding plots are shown in Fig. S9.† Among them, the strongest Th–C(cage) interaction has 6% Th weight. Therefore, the formal transfer of two electrons from ThC2 to C82 is accompanied by secondary cage-metal backbonding.
The main difference between ThC2@C2(5)–C82 and ThC2@Cs(6)–C82 (Fig. S8 and S10†) is the backbonding between Th and the fullerene, with only 4% for the largest Th weight in the former, which may rationalize the slightly higher energy of ThC2@C2(5)–C82 by 2 kcal mol−1.
Spectroscopic characterization
The UV–vis–NIR absorption spectra of ThC2@Cs(6)–C82 and ThC2@C2(5)–C82 dissolved in CS2 are shown in Fig. 5. ThC2@Cs(6)–C82 shows broad peaks at 673, 752, 905, 1040, and 1292 nm and a shoulder peak at 484 nm, similar to those of TbCN@Cs(6)–C82.46 For ThC2@C2(5)–C82, the characteristic absorption peaks were observed at 621, 650, 767, 907, and 1036 cm−1, almost identical to those of TbCN@C2(5)–C82 with the same fullerene cage.46 This indicates similar cage isomer and electronic transfer between ThC2@C82(Cs(6) and C2(5)) and TbCN@C82(Cs(6) and C2(5)), which is consistent with the results obtained from single-crystal X-ray diffraction and the computational results for [ThC2]2+@C822−.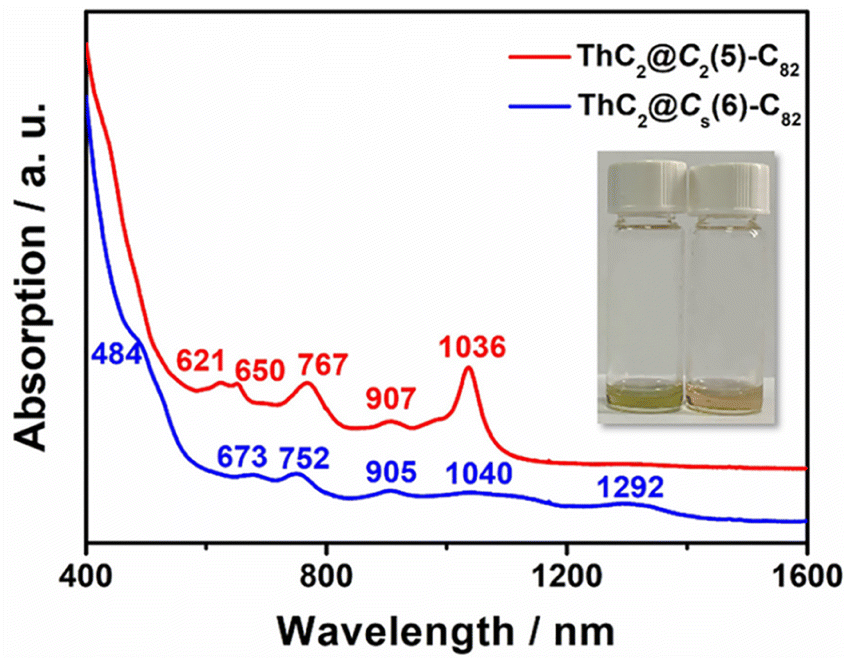 | ||
| Fig. 5 UV–vis–NIR spectra of ThC2@Cs(6)–C82 and ThC2@C2(5)–C82 dissolved in CS2. Insets: Photographs of ThC2@C2(5)–C82 (left) and ThC2@Cs(6)–C82 (right) dissolved in CS2. | ||
Conclusions
For the first time, thorium clusters were encapsulated inside fullerene cages. ThC2@Cs(6)–C82 and ThC2@C2(5)–C82 were synthesized and characterized by mass spectrometry, single-crystal X-ray diffraction crystallography, UV–vis–NIR spectroscopy and DFT calculations. Crystallographic studies reveal that a mononuclear carbide, which has never been found in endohedral fullerenes, is stabilized inside a C82 cage. The two Th–C bond lengths of the ThC2 cluster encapsulated in both Cs(6)–C82 and C2(5)–C82 are 2.360(11)/2.353(10) Å and 2.334(15)/2.385(14) Å, presenting isosceles triangular configurations, although the latter shows slight distortion, likely affected by the different cage isomeric structures.DFT calculations for two isomers of ThC2@C82 revealed that the electronic structure can be described as a spin singlet ground state, formally [Th4+(C2)2−]2+@[C82]2−, with pronounced donation bonding from (C2)2− to Th4+ and secondary backbonding from the fullerene to thorium. The triangular cluster [ThC2]2+ is more stable in the Cs(6)–C82 cage (1a) than in the C2(5)–C82 cage (2a), which is in part rationalized by a weaker backbonding in the latter. Theoretical analysis also shows a triple bond in the C22− fragment that is somewhat weakened by the donation to the metal. The calculations provide an intuitive description of the bonding of actinide and main group atoms as they are encapsulated in fullerenes.
This work expands the scope of both endohedral fullerenes and actinide compounds. ThC2@C82 represents a new family of endohedral fullerenes, which reveals for the first time that MC2n fullerenes, the most commonly observed endohedral fullerenes, may have two isomeric structures, namely, M@C2nversus MC2@C2n−2. Furthermore, identification of the unique bonding motif of ThC2 deepens our understanding of the chemical bonding of thorium.
Experimental section
Spectroscopic study
Positive-ion mode matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) (Bruker, Germany) was employed for mass characterization. The UV–vis–NIR spectra of the purified ThC2@C82 were measured in CS2 solution with a Cary 5000 UV–vis–NIR spectrophotometer (Agilent, USA).X-ray crystallographic study
The black block crystals of ThC2@Cs(6)–C82 and ThC2@C2(5)–C82 were obtained by slow diffusion of the CS2 solution of the corresponding metallofullerene compounds into the benzene solution of [NiII(OEP)]. Single-crystal X-ray data of ThC2@Cs(6)–C82 and ThC2@C2(5)–C82 were collected at 120 K on a diffractometer (Bruker D8 Venture) equipped with a CCD collector. The multiscan method was used for absorption correction. The structures were solved using direct methods50 and refined on F2 using full-matrix least-squares using the SHELXL2015 crystallographic software packages.51 Hydrogen atoms were inserted at calculated positions and constrained with isotropic thermal parameters. Crystal data for ThC2@Cs(6)–C82·[NiII(OEP)]·2C6H6 and ThC2@C2(5)–C82·[NiII(OEP)]·2C6H6 are provided in Table S4.†Computational details
Kohn–Sham density functional calculations were performed for ThC2@Cs(6)–C82 and ThC2@C2(5)–C82 structures with the 2017 release of the Amsterdam Density Functional (ADF) suite.52 Different functionals, including the Perdew–Burke–Ernzerhof (PBE) and Becke–Perdew (BP86) nonhybrid functional, a global hybrid based on PBE with 25% exact exchange (PBE0), and the popular B3LYP hybrid functionals, were used in conjunction with all-electron Slater-type atomic orbital (STO) basis sets of triple-ζ polarized (TZP) quality for the geometry optimizations and electronic structure analyses.53–58 Relativistic effects were considered by means of the scalar-relativistic Zeroth-Order Regular Approximation (ZORA) Hamiltonian.59 An atom-pairwise correction for dispersion forces was considered via Grimme's D3 model augmented with Becke–Johnson (BJ) damping.60 To quantify the compositions of the chemical bonds for selected optimized systems, natural localized molecular orbital (NLMO) analyses were carried out with the NBO program, version 6.0, interfaced with ADF.61Data availability
CCDC 2183932 and 2183933 contain the supplementary crystallographic data for this paper.† These data can be obtained free of charge viahttps://www.ccdc.cam.ac.uk/data_request/cif. All other data supporting the findings of this study are available from the corresponding authors on request.Author contributions
N. C. conceived and designed the experiments. Y. S. and Q. M. synthesized and isolated all the compounds. J. A. and X. Y. performed the computations and theoretical analyses. Y. S. and Q. M. performed the single-crystal measurements. Y. Y. and Y. S. performed the crystallographic analysis. Y. S. performed the spectroscopic measurements. N. C., J. A., Y. S., X. Y., Q. M. and Y. Y. co-wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
N. C. thanks the National Science Foundation China (NSFC No. 91961109, 52172051), the Natural Science Foundation of Jiangsu Province (BK20200041) and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD). J. A. acknowledges support for the theoretical component of this study from grant DE-SC0001136 from the US Department of Energy, Office of Basic Energy Sciences, Heavy Element Chemistry program. J. A. and X. Y. thank the Center for Computational Research (CCR) at the University at Buffalo for providing computing resources.Notes and references
- A. A. Popov, S. Yang and L. Dunsch, Chem. Rev., 2013, 113, 5989–6113 CrossRef CAS PubMed.
- S. Yang, T. Wei and F. Jin, Chem. Soc. Rev., 2017, 46, 5005–5058 RSC.
- W. Shen, S. Hu and X. Lu, Chem.–Eur. J., 2020, 26, 5748–5757 CrossRef CAS PubMed.
- G. Velkos, W. Yang, Y. R. Yao, S. M. Sudarkova, X. Liu, B. Buchner, S. M. Avdoshenko, N. Chen and A. A. Popov, Chem. Sci., 2020, 11, 4766–4772 RSC.
- F. Liu, S. Wang, C.-L. Gao, Q. Deng, X. Zhu, A. Kostanyan, R. Westerström, F. Jin, S.-Y. Xie, A. A. Popov, T. Greber and S. Yang, Angew. Chem., Int. Ed., 2017, 56, 1830–1834 CrossRef CAS PubMed.
- T. Li, S. Murphy, B. Kiselev, K. S. Bakshi, J. Zhang, A. Eltahir, Y. Zhang, Y. Chen, J. Zhu, R. M. Davis, L. A. Madsen, J. R. Morris, D. R. Karolyi, S. M. LaConte, Z. Sheng and H. C. Dorn, J. Am. Chem. Soc., 2015, 137, 7881–7888 CrossRef CAS PubMed.
- W. Li, F. Qu, L. Liu, Z. Zhang, J. Liang, Y. Lu, J. Zhang, L. Wang, C. Wang and T. Wang, Angew. Chem., Int. Ed., 2022, 61, e202116854 CAS.
- F. Liu, L. Spree, D. S. Krylov, G. Velkos, S. M. Avdoshenko and A. A. Popov, Acc. Chem. Res., 2019, 52, 2981–2993 CrossRef CAS PubMed.
- X. Zhang, W. Li, L. Feng, X. Chen, A. Hansen, S. Grimme, S. Fortier, D. C. Sergentu, T. J. Duignan, J. Autschbach, S. Wang, Y. Wang, G. Velkos, A. A. Popov, N. Aghdassi, S. Duhm, X. Li, J. Li, L. Echegoyen, W. H. E. Schwarz and N. Chen, Nat. Commun., 2018, 9, 2753 CrossRef PubMed.
- J. Zhuang, L. Abella, D. C. Sergentu, Y. R. Yao, M. Jin, W. Yang, X. Zhang, X. Li, D. Zhang, Y. Zhao, X. Li, S. Wang, L. Echegoyen, J. Autschbach and N. Chen, J. Am. Chem. Soc., 2019, 141, 20249–20260 CrossRef CAS PubMed.
- Q. Meng, L. Abella, W. Yang, Y. R. Yao, X. Liu, J. Zhuang, X. Li, L. Echegoyen, J. Autschbach and N. Chen, J. Am. Chem. Soc., 2021, 143, 16226–16234 CrossRef CAS PubMed.
- X. Lu, T. Akasaka and S. Nagase, Acc. Chem. Res., 2013, 46, 1627–1635 CrossRef CAS PubMed.
- P. Bagla, Science, 2015, 350, 726–727 CrossRef CAS PubMed.
- T. Abram and S. Ion, Energy Policy, 2008, 36, 4323–4330 CrossRef.
- D. R. J. Konings, T. R. Allen, R. E. Stoller and P. S. Yamanaka, Technical Report, Oak Ridge National Laboratory (ORNL), 2012 Search PubMed.
- H. Kleykamp, Thorium Carbides, in Gmelin Handbook of Inorganic and Organometallic Chemistry, 8th edn, Springer, Berlin, Germany, 1992, vol. C6 Search PubMed.
- R. J. Needs and C. J. Pickard, APL Mater., 2016, 4, 053210 CrossRef.
- P. Pogany, A. Kovacs, Z. Varga, F. M. Bickelhaupt and R. J. Konings, J. Phys. Chem. A, 2012, 116, 747–755 CrossRef CAS PubMed.
- K. A. Cingerich, Chem. Phys. Lett., 1978, 59, 136–139 CrossRef.
- D. D. Jackson, G. W. Barton, O. H. Krikorian and R. S. Newbury, J. Phys. Chem., 1964, 68, 1516–1523 CrossRef CAS.
- N. Sasaki, K. Kubo and M. Asano, J. Nucl. Sci. Technol., 1971, 8, 614–621 CrossRef.
- S. K. Gupta and K. A. Gingerich, J. Chem. Phys., 1980, 72, 2795–2800 CrossRef CAS.
- I. R. Shein and A. L. Ivanovskii, J. Nucl. Mater., 2009, 393, 192–196 CrossRef CAS.
- D. W. Jones, I. J. McColm, R. Steadman and J. Yerkess, J. Solid State Chem., 1987, 68, 219–226 CrossRef CAS.
- A. L. Bowman, N. H. Krikorian, G. P. Arnold, T. C. Wallace and N. G. Nereson, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1968, 24, 1121–1123 CrossRef CAS.
- I. R. Shein, K. I. Shein, N. I. Medvedeva and A. L. Ivanovskii, Phys. Status Solidi, 2007, 244, 3198–3205 CrossRef CAS.
- C.-R. Wang, T. Kai, T. Tomiyama, T. Yoshida, Y. Kobayashi, E. Nishibori, M. Takata, M. Sakata and H. Shinohara, Angew. Chem., Int. Ed., 2001, 40, 397–399 CrossRef CAS PubMed.
- Y. Iiduka, T. Wakahara, K. Nakajima, T. Tsuchiya, T. Nakahodo, Y. Maeda, T. Akasaka, N. Mizorogi and S. Nagase, Chem. Commun., 2006, 2057–2059 RSC.
- X. Lu, K. Nakajima, Y. Iiduka, H. Nikawa, N. Mizorogi, Z. Slanina, T. Tsuchiya, S. Nagase and T. Akasaka, J. Am. Chem. Soc., 2011, 133, 19553–19558 CrossRef CAS PubMed.
- X. Lu, K. Nakajima, Y. Iiduka, H. Nikawa, T. Tsuchiya, N. Mizorogi, Z. Slanina, S. Nagase and T. Akasaka, Angew. Chem., Int. Ed., 2012, 51, 5889–5892 CrossRef CAS PubMed.
- J. Zhang, F. L. Bowles, D. W. Bearden, W. K. Ray, T. Fuhrer, Y. Ye, C. Dixon, K. Harich, R. F. Helm, M. M. Olmstead, A. L. Balch and H. C. Dorn, Nat. Chem., 2013, 5, 880–885 CrossRef CAS PubMed.
- W. Shen, L. Bao, P. Yu, L. Yang, B. Li, P. Yu, P. Jin and X. Lu, Carbon, 2020, 164, 157–163 CrossRef CAS.
- Y. Iiduka, T. Wakahara, T. Nakahodo, T. Tsuchiya, A. Sakuraba, Y. Maeda, T. Akasaka, K. Yoza, E. Horn, T. Kato, M. T. H. Liu, N. Mizorogi, K. Kobayashi and S. Nagase, J. Am. Chem. Soc., 2005, 127, 12500–12501 CrossRef CAS PubMed.
- Y. R. Yao, Y. Rosello, L. Ma, A. R. Puente Santiago, A. Metta-Magana, N. Chen, A. Rodriguez-Fortea, J. M. Poblet and L. Echegoyen, J. Am. Chem. Soc., 2021, 143, 15309–15318 CrossRef CAS PubMed.
- C. N. Carlson, T. P. Hanusa and W. W. Brennessel, J. Am. Chem. Soc., 2004, 126, 10550–10551 CrossRef CAS PubMed.
- K. C. Jantunen, C. J. Burns, I. Castro-Rodriguez, R. E. Da Re, J. T. Golden, D. E. Morris, B. L. Scott, F. L. Taw and J. L. Kiplinger, Organometallics, 2004, 23, 4682–4692 CrossRef CAS.
- Y. Wang, Q. Tang, L. Feng and N. Chen, Inorg. Chem., 2017, 56, 1974–1980 CrossRef CAS PubMed.
- H. Yang, C. Lu, Z. Liu, H. Jin, Y. Che, M. M. Olmstead and A. L. Balch, J. Am. Chem. Soc., 2008, 130, 17296–17300 CrossRef CAS PubMed.
- C. Zhang, G. Hou, G. Zi, W. Ding and M. D. Walter, J. Am. Chem. Soc., 2018, 140, 14511–14525 CrossRef CAS PubMed.
- C. Zhang, G. Hou, G. Zi, W. Ding and M. D. Walter, Inorg. Chem., 2019, 58, 1571–1590 CrossRef CAS PubMed.
- P. Tecmer, K. Boguslawski and P. W. Ayers, Phys. Chem. Chem. Phys., 2015, 17, 14427–14436 RSC.
- S. Yang, C. Chen, F. Liu, Y. Xie, F. Li, M. Jiao, M. Suzuki, T. Wei, S. Wang, Z. Chen, X. Lu and T. Akasaka, Sci. Rep., 2013, 3, 1487 CrossRef PubMed.
- J. Xin, F. Jin, R. Guan, M. Chen, X.-M. Xie, Q. Zhang, S.-Y. Xie and S. Yang, Inorg. Chem. Front., 2021, 8, 1719–1726 RSC.
- W. Shen, Z. Hu, P. Yu, Z. Wei, P. Jin, Z. Shi and X. Lu, Inorg. Chem. Front., 2020, 7, 4563–4571 RSC.
- R. Guan, M. Chen, J. Xin, X.-M. Xie, F. Jin, Q. Zhang, S.-Y. Xie and S. Yang, J. Am. Chem. Soc., 2021, 143, 8078–8085 CrossRef CAS PubMed.
- F. Liu, C. L. Gao, Q. Deng, X. Zhu, A. Kostanyan, R. Westerstrom, S. Wang, Y. Z. Tan, J. Tao, S. Y. Xie, A. A. Popov, T. Greber and S. Yang, J. Am. Chem. Soc., 2016, 138, 14764–14771 CrossRef CAS PubMed.
- H. Yang, H. Jin, X. Wang, Z. Liu, M. Yu, F. Zhao, B. Q. Mercado, M. M. Olmstead and A. L. Balch, J. Am. Chem. Soc., 2012, 134, 14127–14136 CrossRef CAS PubMed.
- G. T. Kent, X. Yu, G. Wu, J. Autschbach and T. W. Hayton, Chem. Sci., 2021, 12, 14383–14388 RSC.
- O. Ordoñez, X. Yu, G. Wu, J. Autschbach and T. W. Hayton, Inorg. Chem., 2021, 60, 12436–12444 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- E. J. Baerends, T. Ziegler, A. Atkins, J. Autschbach, D. Bashford, O. Baseggio, A. Bérces, F. Bickelhaupt, C. Bo and P. Boerritger. ADF2017, SCM, Theoretical Chemistry, Vrije University, Amsterdam, The Netherlands, 2017 Search PubMed.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 34, 7406 CrossRef.
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Phys. Rev. B, 1992, 46, 6671–6687 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1986, 85, 7184–7187 CrossRef CAS.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- E. van Lenthe and E. J. Baerends, J. Comput. Chem., 2003, 24, 1142–1156 CrossRef CAS PubMed.
- E. van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1993, 99, 4597–4610 CrossRef CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- E. D. Glendening, C. R. Landis and F. Weinhold, J. Comput. Chem., 2013, 34, 1429–1437 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: HPLC profiles, experimental details, additional single crystal structural parameters and theoretical calculation results. CCDC 2183932 and 2183933. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc04846a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |