
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced cycling stability and rate capability of a graphene-supported commercialized Vat Blue 4 anode for advanced Li-ion batteries†
Hongwei
Kang‡
ab,
Quanwei
Ma‡
bc,
Rui
Wang
bc,
Longhai
Zhang
*abc,
Shuisheng
Chen
 *a,
Xinrui
Wang
a and
Chaofeng
Zhang
*abc
*a,
Xinrui
Wang
a and
Chaofeng
Zhang
*abc
aSchool of Chemistry and Materials Engineering, Anhui Provincial Key Laboratory for Degradation and Monitoring of Pollution of the Environment, Fuyang Normal University, Fuyang 236037, China. E-mail: zlhedu@163.com
bKey Laboratory of Advanced Energy Materials Chemistry (Ministry of Education), College of Chemistry, Nankai University, Tianjin 300071, China. E-mail: chenss@fynu.edu.cn
cInstitutes of Physical Science and Information Technology, Anhui University, Hefei 230601, China. E-mail: cfz@ahu.edu.cn
First published on 23rd September 2022
Abstract
Commercialized Vat Blue 4 (VB4) has attracted more attention as a promising anode for large-scale applications in Li-ion batteries (LIBs) due to its high electrochemical activity, low price, and large-scale production. However, its moderate solubility results in severe capacity decay and low utilization of active components. Herein, we present a graphene-supported VB4 composite (VB4/rGO) prepared by a facile sonication and hydrothermal process for long cycling stability and high-rate capability. This design can significantly enhance the Li-storage properties, including high capacity (1045 mA h g−1 at 0.1 A g−1), long cycling stability (537 mA h g−1 even over 1000 cycles at 1 A g−1), and rate capability (315 mA h g−1 at 5 A g−1). Strong π–π interaction derived from the aromatic rings within the π-conjugated system (graphene and VB4) and spatial confinement in-between graphene sheets both can suppress the high solubility of VB4 for superior capacity retention. Moreover, conductive graphene and channels in-between nanosheets can simultaneously facilitate the electron and Li+ transfer. This work demonstrates a simple and effective method to improve the electrochemical performance of commercialized Vat dyes and provides a low-cost and large-scale strategy to develop their practical application in the energy storage field.
Introduction
Lithium ion batteries (LIBs) have been widely used in portable electronics and electric vehicles,1,2 owing to their high energy density, low self-discharge, and long-term stability.3–5 However, explosive growth in the demand for LIBs has led to raw materials becoming more and more scarce and expensive. Especially, conventional electrode materials heavily rely on inorganic materials prepared from raw resources.6,7 Additionally, these conventional inorganic electrode materials usually present complex preparation processes and high energy consumption.8–10 Meanwhile, serious structural collapse and pulverization for these intercalations or alloying type inorganic materials always lead to fast capacity decay during charge/discharge processes.11–13 Therefore, it is critical to develop low-cost, sustainable, and high-performance electrode materials instead of conventional inorganic compounds for future LIBs.14–16Compared to conventional inorganic electrode materials, organic compounds not only present high capacity, abundant and eco-friendly resources, and facile synthesis, but also can easily control the electrochemical properties by molecular structure design.9,17–19 Moreover, based on the rearrangement of chemical bonds during the energy storage process, organic electrode materials can avoid severe structural change, which often exists in inorganic electrode materials due to the insertion/extraction of ions.20,21 Benefiting from these advantages, numerous organic electrode materials have been applied in LIBs, including conductive polymers,22 carbonyl compounds,19,23,24 imine compounds,25 oxynitride compounds, sulfur-containing organic compounds, and radical compounds.26 Among them, Zhang et al.27 reported conjugated carboxylate (Li4C8H2O6) and non-conjugated carboxylate (Li4C4H2O6), which shows high discharge capacities of 251.7 and 251.6 mA h g−1 at 0.5 A g−1, respectively. Xia et al.28 reported a heteroaromatic hexacarboxylate compound (Li6-HAT) with a high capacity of 1126.3 mA h g−1, as well as superior cycling stability. Additionally, numerous covalent organic frameworks have also been reported as electrode materials for advanced LIBs.29,30 However, it is usually complicated and time-consuming to prepare these products with high and uniform quality on a large scale for practical applications.31
By contrast, a commercial carbonyl product, Vat Blue 4 (VB4), can be easily obtained from plants or synthesized artificially. More importantly, rich carbonyl and conjugated structures inside simultaneously ensured high theoretical capacity and high electronic conductivity. These advantages demonstrate that VB4 is a desirable electrode and capable of mass production. Unfortunately, VB4 exhibits high solubility in organic electrolytes (carbonate and ether-based), which easily results in performance degradation.32,33 To address these problems, polymerization, salification, and hybridization with insoluble materials have been adopted and proved effective strategies.34,35 Among them, constructing a carbon-supported structure is an effective approach to suppress its severe solubility through the strong π–π interaction derived from the highly π-conjugated system and spatial confinement in-between the graphene sheets. Meanwhile, high conductivity graphene and channels between the nanosheets can further ensure fast electron/ion transport throughout the hybrid,36,37 thus promoting the stability and kinetics of LIBs, simultaneously.
Herein, we developed a graphene-supported VB4 hybrid (VB4/rGO) via a facile combination of sonication and hydrothermal processes. According to the strong π–π interaction between the aromatic rings within VB4 and graphene, VB4 is uniformly confined in-between the graphene sheets. Benefiting from the synergistic effects of strong π–π interaction and spatial confinement, the high solubility of VB4 can be effectively suppressed, thus enhancing the stability and utilization of active components. Additionally, conductive graphene and numerous channels in a hybrid can further ensure fast electron/ion transport. Accordingly, when VB4/rGO was evaluated as an anode for LIBs, the VB4/rGO hybrid exhibited enhanced capacity, long-term cycling stability, and high-rate capability.
Results and discussion
Characterization
In this paper, VB4/rGO was obtained by a facile combination of sonication and hydrothermal processes. VB4 is uniformly distributed between the graphene sheets during the procedure according to the strong π–π interaction derived from their aromatic and conjugated systems (Fig. 1a). The molecule structure of VB4 inside the hybrid is not changed after hydrothermal processes, just as shown in the Fourier transform infrared spectra (FT-IR, Fig. 1b). VB4/rGO and VB4 display the same characteristic peaks for C–N, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, and CC bond stretching in Fig. 1b, revealing the successful compounding between VB4 and rGO. Subsequently, scanning electron microscopy (SEM) and transmission electron microscopy (TEM) are performed to observe the morphology of VB4 with and without graphene. Before the hydration, the pristine VB4 monomer presents a densely stacked block consisting of numerous nanorods (Fig. 1c). After sonication and hydrothermal processes, the uniformly dispersed VB4 nanorods are tightly attached to the graphene nanosheets, just as shown in Fig. 1d and e. Importantly, no evident bulk VB4 is viewed, explaining that VB4 has been fully dispersed and absorbed on the surface of graphene sheets. Further energy dispersive spectroscopy (EDS) mapping of VB4/rGO also confirms that C (green), O (red), and N (blue) elements are evenly distributed on the graphene sheets, just as shown in Fig. 1f. This graphene-supported structure can ensure high utilization of active sites inside the VB4 skeleton and enhanced electron/ion transfer kinetics.38
O, and CC bond stretching in Fig. 1b, revealing the successful compounding between VB4 and rGO. Subsequently, scanning electron microscopy (SEM) and transmission electron microscopy (TEM) are performed to observe the morphology of VB4 with and without graphene. Before the hydration, the pristine VB4 monomer presents a densely stacked block consisting of numerous nanorods (Fig. 1c). After sonication and hydrothermal processes, the uniformly dispersed VB4 nanorods are tightly attached to the graphene nanosheets, just as shown in Fig. 1d and e. Importantly, no evident bulk VB4 is viewed, explaining that VB4 has been fully dispersed and absorbed on the surface of graphene sheets. Further energy dispersive spectroscopy (EDS) mapping of VB4/rGO also confirms that C (green), O (red), and N (blue) elements are evenly distributed on the graphene sheets, just as shown in Fig. 1f. This graphene-supported structure can ensure high utilization of active sites inside the VB4 skeleton and enhanced electron/ion transfer kinetics.38
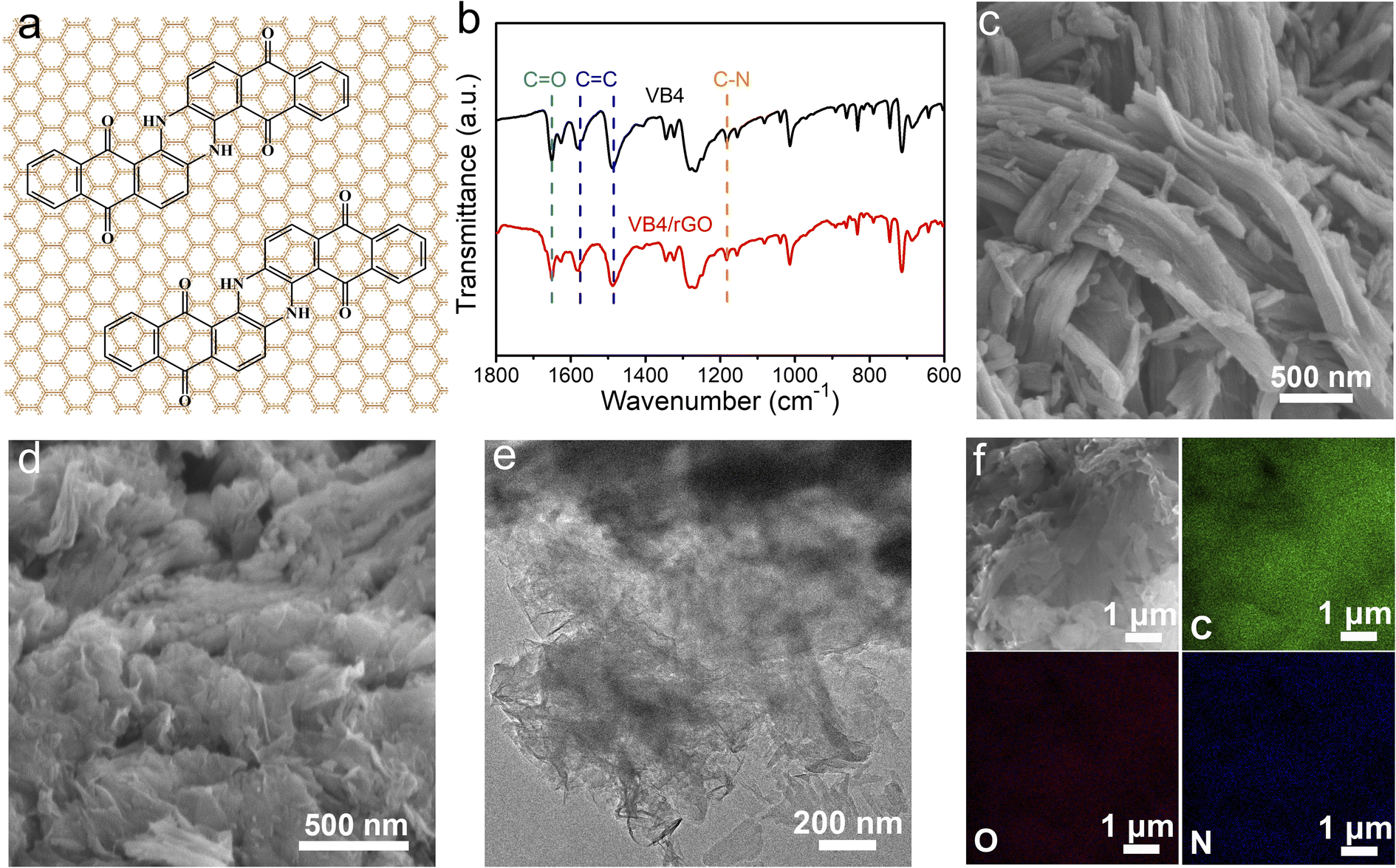 | ||
| Fig. 1 (a) Schematic illustration of the VB4/rGO composite preparation. (b) FT-IR spectra of the VB4 and VB4/rGO composites. (c) SEM images of VB4. (d) SEM, (e) TEM, and (f) elemental mapping images of the VB4/rGO composite. | ||
Afterward, the detailed composition and groups of these products are tested by X-ray photoelectron spectroscopy (XPS). Significantly, the existence of the N 1s signal at 400 eV in the XPS survey mainly contributes to the VB4 molecules, indicating the introduction of VB4 in the composite (Fig. S1†). Subsequently, the high-resolution C 1s XPS spectra analyze the evolution of the C–N group and C–O group originating from VB4 and rGO, respectively, as shown in Fig. 2a. The co-existence of C–N and C–O groups in the hybrid undoubtedly confirms the combination of VB4 and graphene. More importantly, a π–π* satellite peak at about 291 eV can be observed in the hybrid, which implies conjugated systems and π–π stacking,38,39 ensuring that VB4 strongly interacts with graphene. Moreover, the high-resolution N 1s and O 1s XPS spectra also prove the evolution of the C–N and C–O groups among them (Fig. 2b and c), which are well consistent with the C 1s XPS spectra. Additionally, the high-resolution C 1s and O 1s XPS spectra both show a decreased C–O group content in the composite after the hydrazine hydrate reduction process (Fig. 2a and c), indicating the reduction of GO.
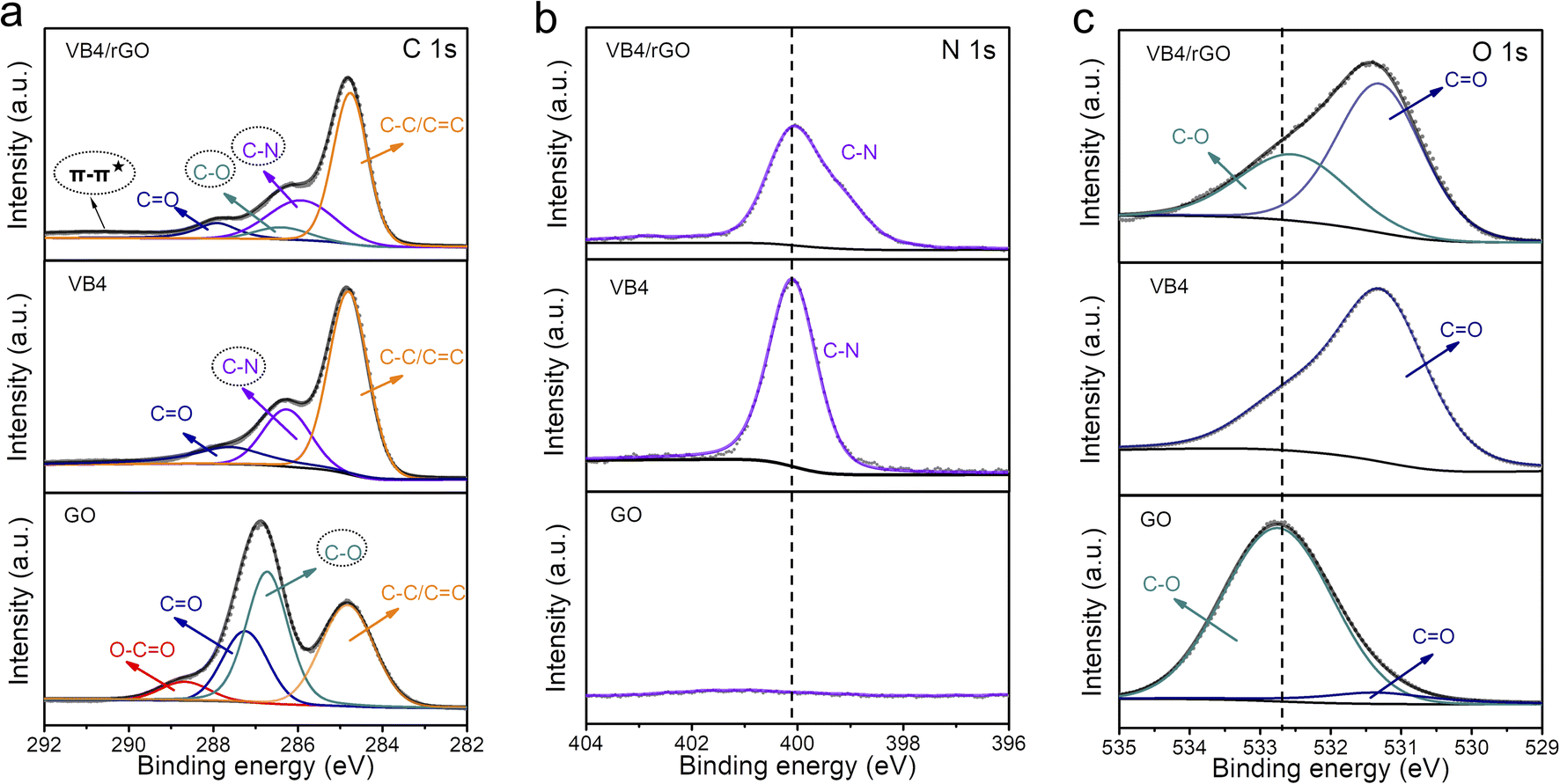 | ||
| Fig. 2 High-resolution XPS of (a) C 1s, (b) N 1s, and (c) O 1s spectra for VB4/rGO, VB4, and GO. | ||
Subsequently, the composition and interaction between VB4 and rGO were further explored. Fig. 3a exhibits the X-ray diffraction (XRD) patterns of VB4, rGO, and the VB4/rGO hybrid. For the VB4/rGO hybrid, the diffraction peaks located at 6.4°, 11.3°, 13.1°, 22.6°, and 27.2° can be attributed to the characteristic peaks of VB4 and rGO, further revealing the successful compounding between VB4 and rGO without any other peaks for impurities. Note that the above diffraction peaks of the hybrid significantly shift towards a low degree compared to those of VB4, demonstrating that rGO interacted with the VB4 molecule. Meanwhile, the Raman spectra of VB4, rGO, and the VB4/rGO hybrid present broad D and G bands (Fig. 3b). Compared with VB4 and rGO, the hybrid also presents an obvious shift of the D and G bands towards lower wavenumbers, which highly supports the charge transfer from VB4 to graphene, thus revealing the π–π interaction between VB4 and graphene.28 Owing to the strong π–π interaction and spatial confinement in-between graphene, this graphene-supported structure can effectively suppress the solubility of VB4 in the electrolyte, just as shown in Fig. 3c. Significantly, VB4 is dissolved in the electrolyte while VB4/rGO is insoluble even over four months, confirming the stability of VB4/rGO in the electrolyte. Subsequently, to analyze the structure–property relationships of VB4/rGO in-depth, density functional theory (DFT) simulation is carried out (Fig. S2†). Fig. 3d presents a high VB4 adsorption energy of −15.34 eV for the graphene-supported structure, proving the strong interaction against the high solubility of VB4 as well. Moreover, the charge density difference is introduced to explore the charge distribution of VB4 adsorption models. The charge accumulation region around the interfaces is more pronounced (Fig. 3d and e), indicating strong VB4 binding with graphene. Meanwhile, Fig. 3f represents different models’ density of states (DOS). The DOS of VB4/rGO around the Fermi level is greatly enhanced compared to that of VB4, revealing a high electronic conductivity.29 Thus, benefiting from the high VB4 adsorption ability and electronic conductivity, the graphene-supported VB4 hybrid could achieve a higher stability and faster reaction kinetics compared to pristine VB4.
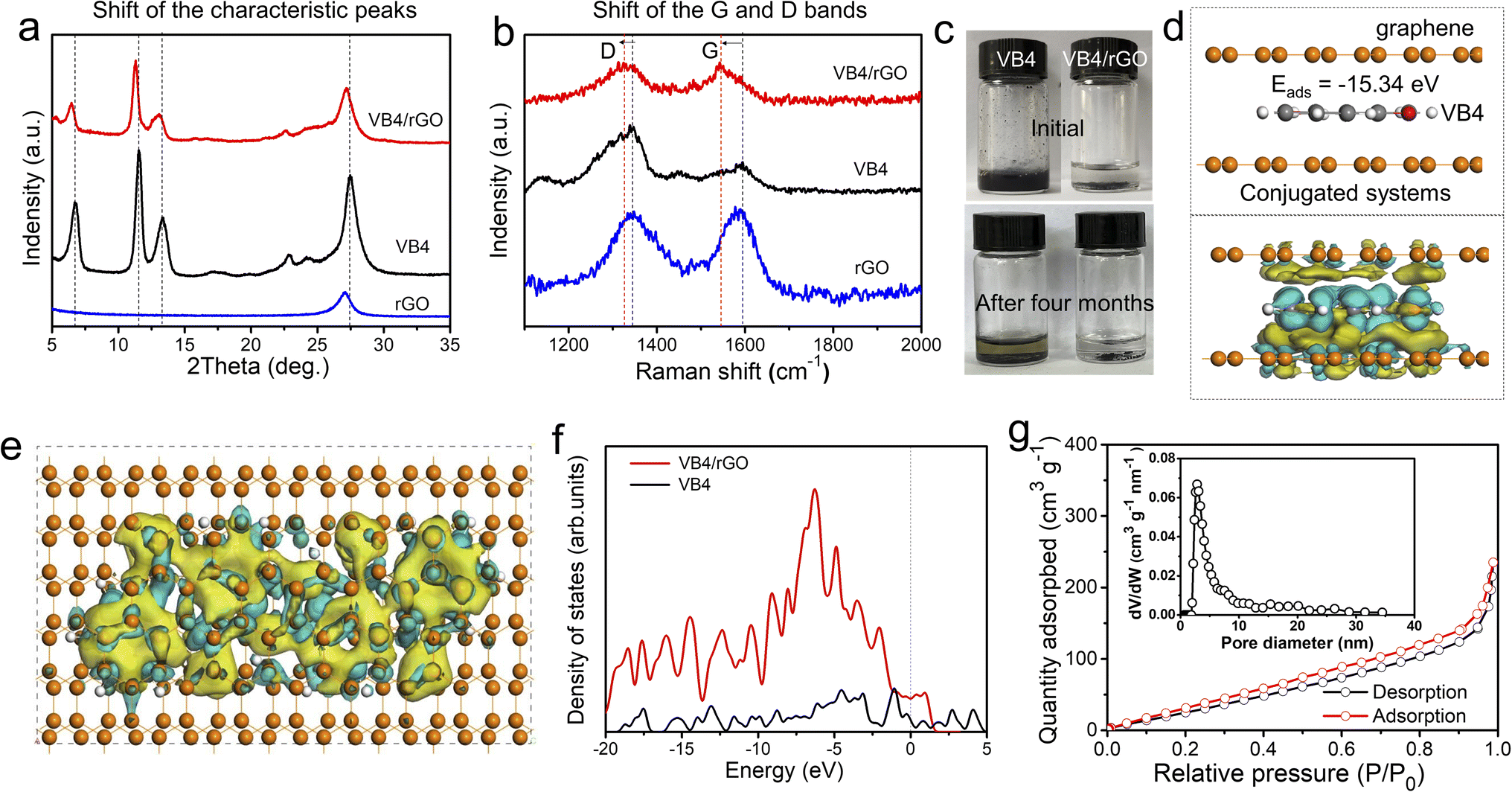 | ||
| Fig. 3 (a) XRD patterns and (b) Raman spectra of rGO, VB4, and VB4/rGO. (c) Photograph of VB4 and VB4/rGO soaked in the electrolyte. (d and e) Adsorption energy and charge density differences of VB4 molecules in-between graphene sheets. (f) Density of states of VB4 and VB4/rGO models. (g) N2 sorption isotherms and pore size distribution curve of VB4/rGO. | ||
Fig. 3g shows the nitrogen adsorption–desorption isotherms of VB4/rGO, which reveal a Brunauer–Emmett–Teller (BET) surface area of 184.85 m2 g−1. Moreover, the features of type IV and H3 hysteresis loops illustrate the existence of mesoporous structures. Afterward, an average pore size of 3–5 nm is confirmed according to the pore-size distribution of VB4/rGO (the inset of Fig. 3g). By contrast, owing to the stacked block morphology (Fig. 1c), the surface area of VB4 is calculated to be only 20.05 m2 g−1 (Fig. S3†). Therefore, the construction of a graphene-supported structure can form fast ion-diffusion channels, and provides large contact areas between the electrolyte and active components, finally enabling higher reaction kinetics and more active sites compared to pristine VB4.
Electrochemical performance
Benefiting from these advantages, the resultant hybrid could possess improved electrochemical performances. Subsequently, the CV curves of the VB4/rGO anode for LIBs are first collected. As shown in Fig. 4a, two redox peaks are observed in the first cycle. Among them, an irreversible peak appears at 1.4–1.6 V due to solid electrolyte interface (SEI) formation and disappears in the subsequent cycles.30,40,41 Note that the CV curves during the cycles all present obvious couples of redox peaks at 0.75/1.2 V, indicating good reversibility and great stability during the lithiation/delithiation processes.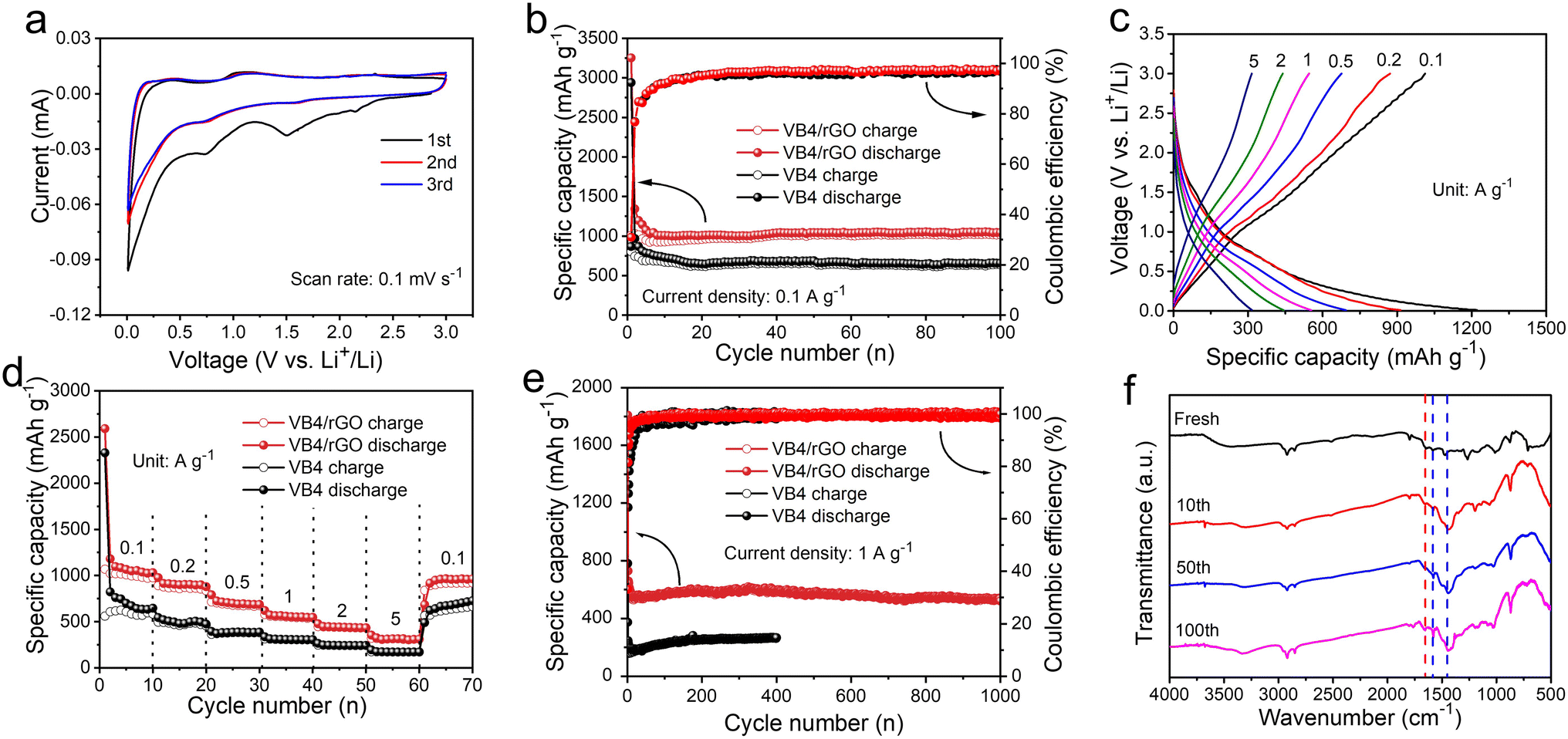 | ||
| Fig. 4 (a) The CV curves of VB4/rGO anodes at a scan rate of 0.1 mV s−1. (b) The cycling performance of VB4 and VB4/rGO anodes at 0.1 A g−1. (c) Charge–discharge voltage profiles of VB4/rGO at various current densities. (d) Rate performances of VB4 and VB4/rGO anodes. (e) Cycling performance of VB4 and VB4/rGO anodes at 1 A g−1. (f) Ex situ FT-IR spectra of the VB4/rGO anode in different cycles. | ||
Fig. 4b shows the cycling performance of the VB4 and VB4/rGO anodes at 0.1 A g−1. The VB4/rGO anode presents a high reversible discharge capacity of 1045 mA h g−1 even after 100 cycles, which is far superior to that of VB4 (626 mA h g−1) and other reported electrode materials.28,42–44 Subsequently, the rate performances of the VB4 and VB4/rGO anodes at different current densities ranging from 0.1 A g−1 to 5 A g−1 are explored (Fig. 4c and d). With the increase of current densities from 0.1 A g−1 to 5.0 A g−1, VB4/rGO exhibits a high capacity of 1097, 906, 714, 565, 444, and 315 mA h g−1, respectively. Particularly, the capacity of VB4/rGO can still recover to 1000 mA h g−1, when the current density is back to 0.1 A g−1. By contrast, the capacity of pristine VB4 drops quickly from 617 to 172 mA h g−1 owing to its high solubility. These enhanced properties of VB4/rGO can be attributed to the introduction of a graphene-supported structure and multi-electroactive centers inside the VB4 skeleton.
Moreover, the long-term cycling stability of these products at a high current density of 1.0 A g−1 is displayed in Fig. 4e. VB4/rGO exhibits an excellent reversible capacity of 537 mA h g−1 even over 1000 cycles. Meanwhile, an average coulombic efficiency of 100% can be obtained throughout the cycles. In contrast, the discharge capacity of VB4 is only 265 mA h g−1 after 400 cycles, much lower than that of VB4/rGO, indicating that the introduction of graphene is beneficial for Li-storage properties. To explore the stability of VB4/rGO, electrochemical impedance spectroscopy (EIS) plots upon cycling are collected, as shown in Fig. S4.† With increasing cycle numbers, the Rct corresponding to charge transfer impedance significantly decreases, indicating a fast charge transfer process throughout the cycling. Additionally, the slope of the EIS plots within the low frequency region is always related to the Li ion diffusion. In the initial cycles, the decreased slope usually corresponds to sluggish ion diffusion, which is related to the initial activation process. Afterward, the slopes remain basically unchanged, indicating stable Li ion diffusion, which is well consistent with the evolution of cycling performance. Impressively, compared to the reported electrodes (Table S1†), VB4/rGO can still exhibit competitive cycling stability and rate performance. Afterward, to further investigate this cycling stability, the ex situ FT-IR spectra of VB4/rGO in different cycles are collected in Fig. 4f. Throughout the long-term cycling, these characteristic peaks attributed to VB4 remain consistent, ensuring excellent reversibility and stability. These remarkably enhanced performances of VB4/rGO are mainly attributed to its unique graphene-supported structure, which can significantly suppress the high solubility of VB4 according to the strong interaction and enhance its electronic conductivity. Additionally, the large specific surface area and the porous structure can increase the contact area with the electrolyte and expose numerous electroactive sites of the VB4 molecule.
Afterward, to explore the reaction kinetics of VB4/rGO, EISis first performed to analyze the electronic/ionic conductivity. As shown in Fig. 5a, EIS curves are composed of a semicircle and sloping line. The smaller diameter of the semicircle assigned to VB4/rGO indicates a lower charge transfer resistance (Rct) than that of VB4, demonstrating that the introduction of graphene is beneficial for the improved conductivity of VB4. Additionally, the plot of VB4/rGO in the low-frequency region is closer to the real impedance axis, confirming the better Li+ diffusion compared to VB4. Afterward, the detailed Li+ diffusion coefficients are estimated according to the galvanostatic intermittent titration technique (GITT, Fig. 5b) and the following equation,45
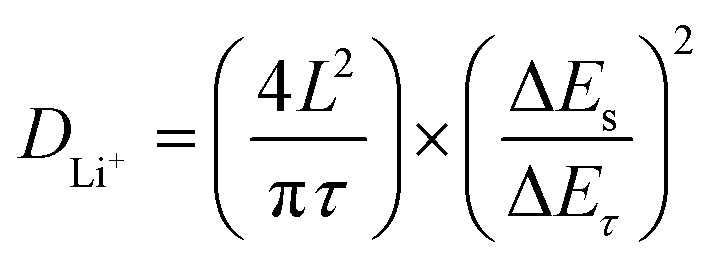 | (1) |
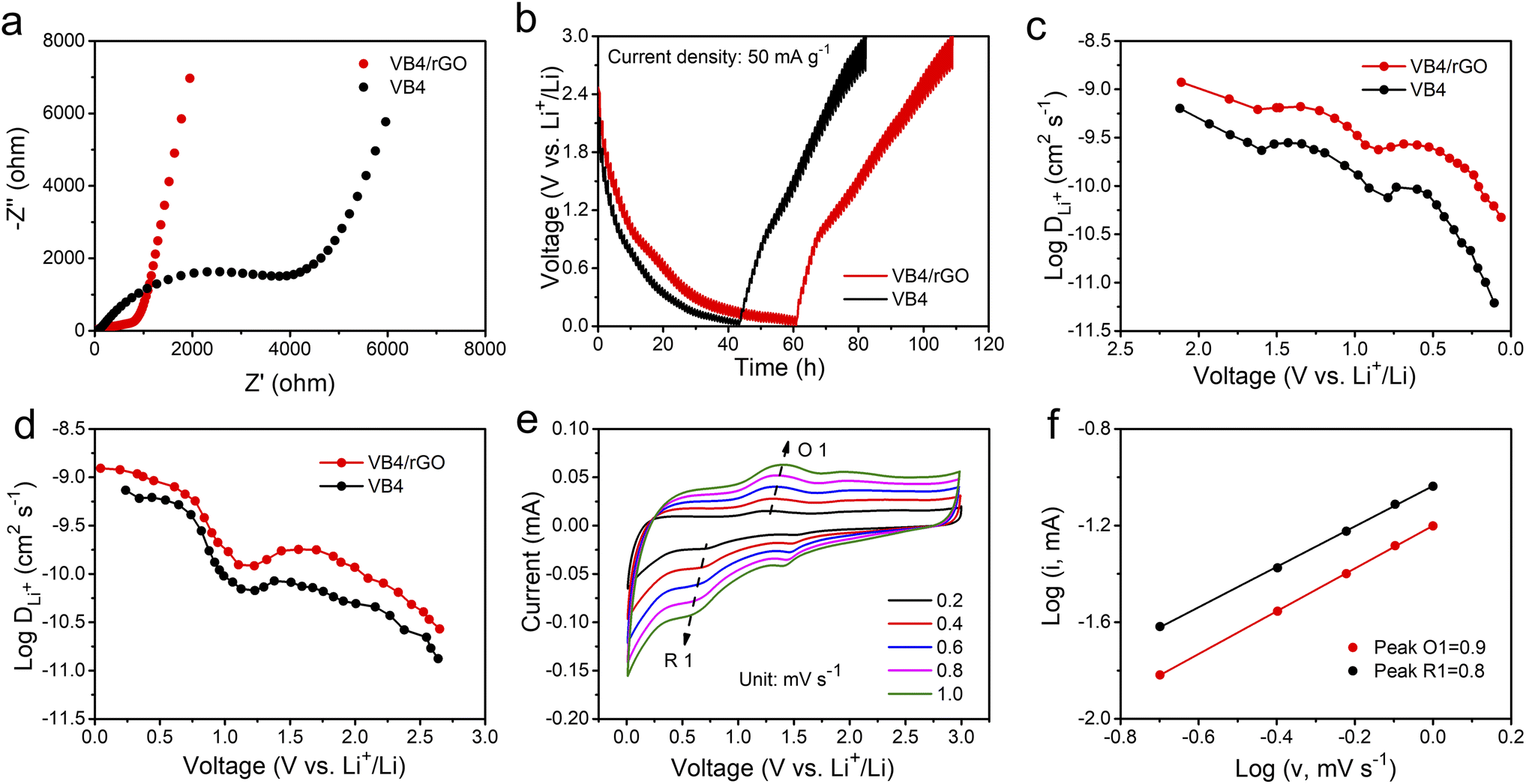 | ||
Fig. 5 (a) EIS plots and (b) GITT curves of VB4 and VB4/rGO. (c and d) Li+ diffusion coefficients during charge/discharge processes for VB4 and VB4/rGO. (e) CV curves of VB4/rGO at different scan rates. (f) log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) i vs. logv profiles of VB4/rGO. i vs. logv profiles of VB4/rGO. | ||
Afterward, the electrochemical mechanism of VB4/rGO is tested by coupling the CV curves at different scan rates (Fig. 5e) with the following equations,38
| i = avb | (2) |
| logi(V) = blogv + loga | (3) |
Subsequently, ex situ FT-IR corresponding to the different potential states of discharge/charge profiles is studied to explore the Li+ storage mechanism of VB4/rGO (Fig. 6a and b). As shown in Fig. 6b, with the battery discharge from 3.0 to 0.01 V, the characteristic peaks of CC (1580 and 1450 cm−1) and CO (1640 cm−1) gradually weaken due to the Li+ reaction with CC and CO to form C–C–Li and C–O–Li, respectively.49 Then, during the charging process, the tendency of enhanced peaks of CC and CO can be observed, indicating the removal of Li+ from C–C–Li and C–O–Li. Such reversible evolutions can ensure the electroactive centers (CC and CO) and Li+ storage mechanism in the VB4 molecule. Besides, further ex situ XPS was also performed to analyze the Li-storage mechanism of VB4/rGO, just as shown in Fig. S7.† During the discharge process, the peaks of CO and CC gradually weaken in the high-resolution C 1s and O 1s spectra. By contrary, the peaks of C–C and C–O gradually increase inside, indicating the reaction of Li+ with CC and CO. During the subsequent charging process, the peaks of CC and CO bonds gradually recover, demonstrating the highly reversible process of Li-storage. Meanwhile, by DFT simulation, the results of electron density distribution around the CC and CO groups exhibit obvious negative values (Fig. 6c), further proving the highly reactive centers.50 In summary, the electrochemical merits of VB4/rGO are mainly due to its graphene-supported structure and molecular composition, as illustrated in Fig. 6d. (1) Strong π–π interaction within the conjugated systems and spatial confinement in-between graphene sheets both can suppress the high solubility of VB4 for high cycling stability. (2) Highly conductive graphene and the porous structure can facilitate electronic and Li+ transfer. (3) Multi-electroactive centers can coordinate Li+ for high theoretical capacity.
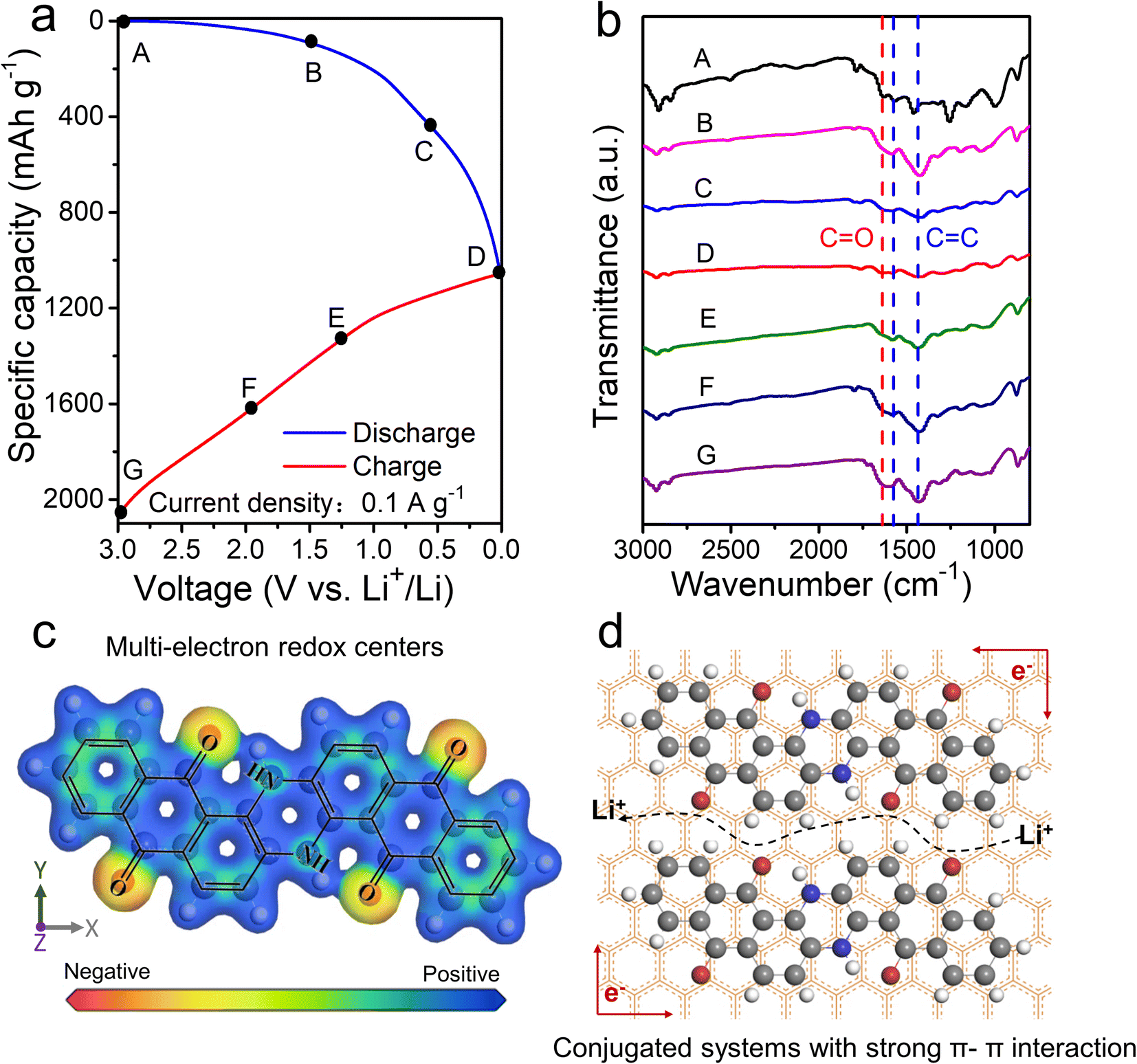 | ||
| Fig. 6 (a and b) The ex situ FT-IR spectra of the VB4/rGO anode under different charge/discharge states. (c) DFT calculated electronegativity of VB4. (d) Illustration of the structural advantages for the VB4/rGO hybrid. | ||
Conclusions
This paper reports a simple and universal strategy to prepare a graphene-supported VB4 anode for LIBs by assembling VB4 molecules and graphene. For this design, multi-electroactive centers within the VB4 skeleton can result in high Li-ion storage capacity. The introduction of conductive graphene can not only suppress the high solubility of VB4 according to the strong π–π interaction and spatial confinement in-between graphene sheets, but also exposes numerous electroactive sites and provide fast electron/ion transfer highways. Therefore, VB4/rGO presents outstanding electrochemical performance, such as a high reversible capacity of 1045 mA h g−1 at 0.1 A g−1, good cycling stability (537 mA h g−1 after 1000 cycles at 1 A g−1), and good rate performance (315 mA h g−1 even at 5 A g−1). More importantly, this work provides an effective method to improve the Li-ion storage performance of commercialized dyes and a low-cost, large-scale strategy for practical applications.Data availability
The data that support the findings of this study are openly available on line.Author contributions
H. W. K. and C. F. Z. designed and guided this experiment. Q. W. M., R.W., X.R.W. and H. W. K. carried out the experiment and materials characterization. S. S. C. and L.H.Z. contributed to the data analysis and co-wrote the paper. H. W. K., S. S. C., and C. F. Z. proposed and supervised the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the financial support from the National Natural Science Foundation of China (51872071 and 52172173), Anhui Provincial Natural Science Foundation for Distinguished Young Scholar (2108085J25), Anhui Province Key Laboratory of Environment-Friendly Polymer Materials, Open Project of Anhui Provincial Key Laboratory for Degradation and Monitoring of Pollution of the Environment (FSKFKT001D), Natural Science Research Projects of Universities in Anhui Province (KJ2020A0021 and KJ2020A0547), and Talent program of Fuyang Normal University (2020KYQD0015). This work was also supported by the Open Fund of Guangdong Provincial Key Laboratory of Advance Energy Storage Materials (AESM202106) and Science-Technology Development Program of Henan Province (202102210030). The authors also acknowledge the High-performance Computing Platform of Anhui University for providing computing resources.Notes and references
- Y.-Y. Wang, Z.-W. Zhao, Y. Liu, L.-R. Hou and C.-Z. Yuan, Rare Metals, 2020, 39, 1082–1091 CrossRef CAS.
- T. Song, C. Wang and C. S. Lee, Carbon Neutralization, 2022, 1, 68–92 CrossRef.
- Z. Cao, J. Fu, M. Wu, T. Hua and H. Hu, Energy Storage Mater., 2021, 40, 10–21 CrossRef.
- C. Zhang, Z. Wang, Z. Guo and X. W. Lou, ACS Appl. Mater. Interfaces, 2012, 4, 3765–3768 CrossRef CAS PubMed.
- C. Zhang, Z. Chen, Z. Guo and X. W. Lou, Energy Environ. Sci., 2013, 6, 974–978 RSC.
- Y. Bai, N. Muralidharan, Y.-K. Sun, S. Passerini, M. Stanley Whittingham and I. Belharouak, Mater. Today, 2020, 41, 304–315 CrossRef CAS.
- Q. Ma, J. Zheng, H. Kang, L. Zhang, Q. Zhang, H. Li, R. Wang, T. Zhou, Q. Chen, A. Liu, H. Li and C. Zhang, ACS Appl. Mater. Interfaces, 2021, 13, 43002–43010 CrossRef CAS PubMed.
- J. Cui, Z. Guo, J. Yi, X. Liu, K. Wu, P. Liang, Q. Li, Y. Liu, Y. Wang, Y. Xia and J. Zhang, ChemSusChem, 2020, 13, 2160–2185 CrossRef CAS PubMed.
- Y. Gao, G. Li, F. Wang, J. Chu, P. Yu, B. Wang, H. Zhan and Z. Song, Energy Storage Mater., 2021, 40, 31–40 CrossRef.
- J. Wu, Y. Cao, H. Zhao, J. Mao and Z. Guo, Carbon Energy, 2019, 1, 57–76 CrossRef CAS.
- G. Li, S. Guo, B. Xiang, S. Mei, Y. Zheng, X. Zhang, B. Gao, P. K. Chu and K. Huo, Energy Mater., 2022, 2, 200020 CrossRef.
- C.-C. Li, X.-S. Zhang, Y.-H. Zhu, Y. Zhang, S. Xin, L.-J. Wan and Y.-G. Guo, Energy Mater., 2021, 1, 100017 CrossRef.
- D. Zhang, L. Li, W. Zhang, M. Cao, H. Qiu and X. Ji, Chin. Chem. Lett., 2022 DOI:10.1016/j.cclet.2022.01.015.
- L. Zhang, R. Bao, S. Xu, L. Hou, C. Zhang and C. Yuan, Energy Technol., 2020, 9, 2000869 CrossRef.
- J. Mao, C. Ye, S. Zhang, F. Xie, R. Zeng, K. Davey, Z. Guo and S. Qiao, Energy Environ. Sci., 2022, 15, 2732–2752 RSC.
- H.-g. Wang, Y. Wang, Q. Wu and G. Zhu, Mater. Today, 2022, 52, 269–298 CrossRef CAS.
- Y. Zhao, Y. Huang, F. Wu, R. Chen and L. Li, Adv. Mater., 2021, 33, 2106469 CrossRef CAS PubMed.
- Y. Zhao, Y. Huang, R. Chen, F. Wu and L. Li, Mater. Horiz., 2021, 8, 3124–3132 RSC.
- Q. Yu, W. Tang, Y. Hu, J. Gao, M. Wang, S. Liu, H. Lai, L. Xu and C. Fan, Chem. Eng. J., 2021, 415, 128509 CrossRef CAS.
- X. Zhang, Z. Ju, Y. Zhu, K. J. Takeuchi, E. S. Takeuchi, A. C. Marschilok and G. Yu, Adv. Energy Mater., 2020, 11, 2000808 CrossRef.
- Z. Liu, K. Zhang, G. Huang, B. Xu, Y. L. Hong, X. Wu, Y. Nishiyama, S. Horike, G. Zhang and S. Kitagawa, Angew. Chem., Int. Ed., 2021, 60, 2–9 CrossRef.
- W. Wu, H.-Y. Shi, Z. Lin, X. Yang, C. Li, L. Lin, Y. Song, D. Guo, X.-X. Liu and X. Sun, Chem. Eng. J., 2021, 419, 129659 CrossRef CAS.
- Z. Luo, L. Liu, Q. Zhao, F. Li and J. Chen, Angew. Chem., Int. Ed., 2017, 56, 12561–12565 CrossRef CAS PubMed.
- M. Tang, H. Li, E. Wang and C. Wang, Chin. Chem. Lett., 2018, 29, 232–244 CrossRef CAS.
- Z. Ye, S. Xie, Z. Cao, L. Wang, D. Xu, H. Zhang, J. Matz, P. Dong, H. Fang, J. Shen and M. Ye, Energy Storage Mater., 2021, 37, 378–386 CrossRef.
- W. Wang, Z. Cao, G. A. Elia, Y. Wu, W. Wahyudi, E. Abou-Hamad, A.-H. Emwas, L. Cavallo, L.-J. Li and J. Ming, ACS Energy Lett., 2018, 3, 2899–2907 CrossRef CAS.
- H. Zhang, Y. Lin, L. Chen, D. Wang, H. Hu and C. Shen, ChemElectroChem, 2020, 7, 306–313 CrossRef CAS.
- S.-B. Xia, T. Liu, W.-J. Huang, H.-B. Suo, F.-X. Cheng, H. Guo and J.-J. Liu, J. Energy Chem., 2020, 51, 303–311 CrossRef.
- Z. Chen, C. Su, X. Zhu, R. Xu, L. Xu and C. Zhang, Polym. Chem., 2018, 56, 2574–2583 CrossRef CAS.
- Y. Zheng, H. Song, S. Chen, X. Yu, J. Zhu, J. Xu, K. A. I. Zhang, C. Zhang and T. Liu, Small, 2020, 16, e2004342 CrossRef PubMed.
- W. Yang, H. Liu, Z. Ren, N. Jian, M. Gao, Y. Wu, Y. Liu and H. Pan, Adv. Mater. Interfaces, 2019, 6, 1801613 Search PubMed.
- H. Wang, C.-J. Yao, H.-J. Nie, Ke-Z. Wang, Y.-W. Zhong, P. Chen, S. Mei and Q. Zhang, J. Mater. Chem. A, 2020, 8, 11906–11922 RSC.
- Q. Deng, Z. Luo, R. Yang and J. Li, ACS Sustainable Chem. Eng., 2020, 8, 15445–15465 CrossRef CAS.
- Y. Lu and J. Chen, Nat. Rev. Chem., 2020, 4, 127–142 CrossRef CAS.
- J. Xie, P. Gu and Q. Zhang, ACS Energy Lett., 2017, 2, 1985–1996 CrossRef CAS.
- H. Chang, Y.-R. Wu, X. Han and T.-F. Yi, Energy Mater., 2021, 1, 100003 CrossRef.
- J. Ruan, H. Sun, Y. Song, Y. Pang, J. Yang, D. Sun and S. Zheng, Energy Mater., 2021, 1, 100018 CrossRef.
- Y. Li, L. Liu, C. Liu, Y. Lu, R. Shi, F. Li and J. Chen, Chem, 2019, 5, 2159–2170 CAS.
- H. Zhuo, Y. Hu, Z. Chen and L. Zhong, Carbohydr. Polym., 2019, 215, 322–329 CrossRef CAS.
- Y. Zheng, S. Xia, F. Dong, H. Sun, Y. Pang, J. Yang, Y. Huang and S. Zheng, Adv. Funct. Mater., 2020, 31, 2006159 CrossRef.
- J. Chen, C. Wang, G. Wang, D. Zhou and L.-Z. Fan, Energy Mater., 2022, 2, 200023 CrossRef.
- L. Zhai, G. Li, X. Yang, S. Park, D. Han, L. Mi, Y. Wang, Z. Li and S. Y. Lee, Adv. Funct. Mater., 2021, 32, 2108798 CrossRef.
- E. A. Saverina, R. R. Kapaev, P. V. Stishenko, A. S. Galushko, V. A. Balycheva, V. P. Ananikov, M. P. Egorov, V. V. Jouikov, P. A. Troshin and M. A. Syroeshkin, ChemSusChem, 2020, 13, 3137–3146 CrossRef CAS PubMed.
- M. Jia, Y. Jin, P. Zhao, C. Zhao, M. Jia, L. Wang and X. He, Electrochim. Acta, 2019, 310, 230–239 CrossRef CAS.
- X. Chen, H. Zhang, C. Ci, W. Sun and Y. Wang, ACS Nano, 2019, 13, 3600–3607 CrossRef CAS PubMed.
- H. Zhao, D. Luo, H. Xu, W. He, B. Ding, H. Dou and X. Zhang, J. Mater. Sci., 2022, 57, 9980–9991 CrossRef CAS.
- O. Buyukcakir, J. Ryu, S. H. Joo, J. Kang, R. Yuksel, J. Lee, Y. Jiang, S. Choi, S. H. Lee, S. K. Kwak, S. Park and R. S. Ruoff, Adv. Funct. Mater., 2020, 30, 2003761 CrossRef CAS.
- H. Zhang, W. Sun, X. Chen and Y. Wang, ACS Nano, 2019, 13, 14252–14261 CrossRef CAS PubMed.
- X. Peng, Y. Xie, A. Baktash, J. Tang, T. Lin, X. Huang, Y. Hu, Z. Jia, D. J. Searles, Y. Yamauchi, L. Wang and B. Luo, Angew. Chem., Int. Ed., 2022, 61, 202203646 Search PubMed.
- W. Wang, V. S. Kale, Z. Cao, Y. Lei, S. Kandambeth, G. Zou, Y. Zhu, E. Abouhamad, O. Shekhah, L. Cavallo, M. Eddaoudi and H. N. Alshareef, Adv. Mater., 2021, 33, 2103617 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc03980j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |