
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nickel-catalyzed regio- and enantio-selective Markovnikov hydromonofluoroalkylation of 1,3-dienes†
Ling
Liao‡
a,
Ying
Zhang‡
a,
Zhong-Wei
Wu
a,
Zhong-Tian
Ye
a,
Xue-Xin
Zhang
a,
Guangying
Chen
b and
Jin-Sheng
Yu
 *ab
*ab
aShanghai Engineering Research Center of Molecular Therapeutics and New Drug Development, Shanghai Key Laboratory of Green Chemistry and Chemical Processes, East China Normal University, Shanghai 200062, China. E-mail: jsyu@chem.ecnu.edu.cn
bKey Laboratory of Tropical Medicinal Resource Chemistry of Ministry of Education, Hainan Normal University, Haikou 571158, China
First published on 11th October 2022
Abstract
A highly enantio- and regio-selective Markovnikov hydromonofluoro(methyl)alkylation of 1,3-dienes was developed using redox-neutral nickel catalysis. It provided a facile strategy to construct diverse monofluoromethyl- or monofluoroalkyl-containing chiral allylic molecules. Notably, this represents the first catalytic asymmetric Markovnikov hydrofluoroalkylation of olefins. The practicability of this methodology is further highlighted by its broad substrate scope, mild base-free conditions, excellent enantio- and regio-selectivity, and diversified product elaborations to access useful fluorinated building blocks.
Introduction
The selective introduction of a fluorine or fluoroalkyl moiety into molecules often results in improved physical, chemical, and biological properties.1 In particular, the installation of a monofluoromethyl (CH2F) group as a bioisostere of various functional groups, such as methyl and hydroxymethyl, has been established as a robust and routine tactic in pharmaceutical chemistry and agrochemistry to tune the properties of bioactive compounds, including bioavailability and metabolic stability.2 Typical drugs or inhibitors featuring a CH2F unit are shown in Scheme 1a.3 However, despite the significant progress made in selective fluoroalkylation,4 the efficient incorporation of a CH2F group in a highly enantioselective manner remains challenging and very much in demand.5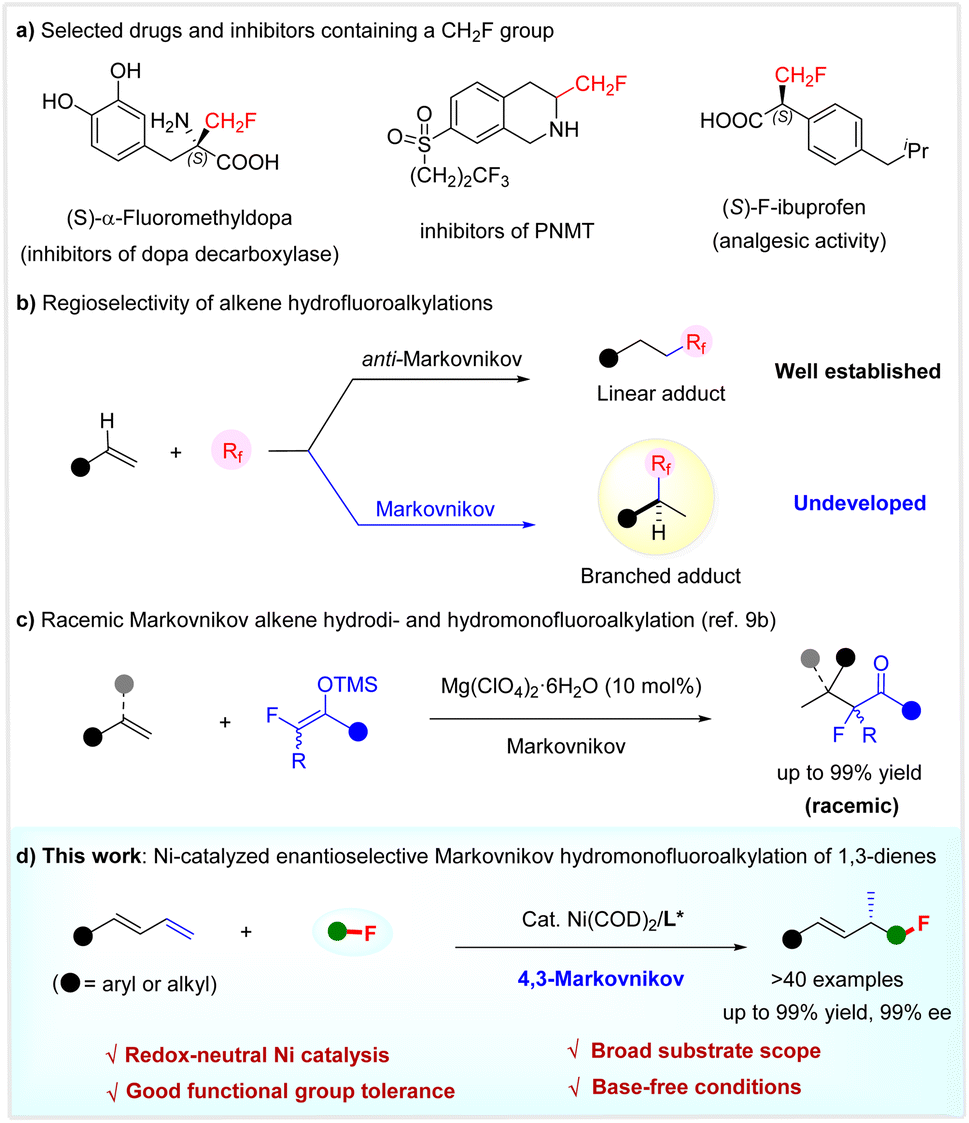 | ||
| Scheme 1 Regioselective hydrofluoroalkylation of alkenes. | ||
While the hydrofluoroalkylation of alkenes is a powerful strategy to introduce a fluoroalkyl group selectively,6 the catalytic enantioselective incorporation of a monofluoroalkyl group is unexplored. Notably, most known alkene hydrofluoroalkylations are based on radical processes, affording linear adducts with anti-Markovnikov regioselectivity.6,7 It is considered both interesting and urgent to develop the Markovnikov hydrofluoroalkylation of olefins. This not only offers the potential to develop catalytic enantioselective versions, but would afford branched adducts with a chemical space shape distinct from linear products, which are interesting targets for drug discovery because of the intimate relationship between the shape and their properties of organic molecules (Scheme 1b).8 Following our interest in selective fluoroalkylation,9 we recently developed the first Markovnikov hydrodi- and hydromonofluoroalkylation of simple alkenes using fluorinated enol silyl ethers, via an acid-catalyzed carbocationic process (Scheme 1c).9b Herein, we disclose a highly regio- and enantio-selective Markovnikov hydromonofluoro(methyl)alkylation reaction of 1,3-dienes by redox-neutral Ni catalysis (Scheme 1d).
Transition-metal-catalyzed regio- and enantio-selective hydrofunctionalization of 1,3-dienes 1 offers an efficient and atom-economical method to access chiral functionalized allylic compounds from readily available starting materials.10 Over the past few years, various highly enantioselective protocols have been used: hydroamination,11 hydroalkylation,12 hydroarylation,13 and hydrosulfonylation,14 among others.15 Despite the advances made, these reactions mainly rely on using chiral precious metal Pd and Rh catalysts. Since the landmark work of the Zhou group in 2018,12c the use of earth-abundant and low-cost chiral Ni catalysts for developing the asymmetric hydrofunctionalization of acyclic 1,3-dienes has gained increasing attention.12c,e,f,13b,c Despite ongoing achievements, the catalytic enantio- and regio-selective Markovnikov hydromonofluoromethylation of 1,3-dienes to construct functionalized chiral allylic compounds with a CH2F at the stereocenter is unexplored.
Inspired by these elegant advances, we speculated that the implementation of catalytic asymmetric 1,3-dienes hydromonofluoromethylation would provide a new direction for enantioselective monofluoromethylation and constitutes a new branch for the hydrofunctionalizations of 1,3-dienes. To reach this goal, the quest for a suitable monofluoromethyl reagent would be the key to success. Among various monofluoromethyl agents, fluorobis(phenyl-sulfonyl)methane (FBSM)16a,b2a proves to be a robust one in developing catalytic enantioselective monofluoromethylation reactions,5,16 since the landmark work of Shibata.16a On this basis, we determined to use FBSM 2a as a latent monofluoromethyl agent to explore the asymmetric Markovnikov hydromonofluoromethylation of 1,3-dienes 1 under the action of chiral nickel catalysis.
Results and discussion
Optimization of the reaction conditions
We commenced this study with 1-phenylbuta-1,3-diene 1a and FBSM 2a as the model substrates, in the presence of Ni(COD)2 (10 mol%) and N,N-diisopropylethylamine (DIPEA) (20 mol%). As shown in Table 1, a series of axially chiral bisphosphine ligands were first investigated. The use of (S)-Tol-BINAP afforded the 4,3-Markovnikov adduct 3a in 98% NMR yield with 37% ee within 14 h (entry 1), whilst the bulky (R)-DTBM-Segphos L2 afforded product 3a in only 9% NMR yield and 27% ee after 48 h (entry 2).|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Ligand | DIPEA (mol%) | Solvent | Time (h) | Yieldb (%) | eec (%) |
| a Reaction conditions: 1a (0.15 mmol), 2a (0.1 mmol), Ni(COD)2 (10 mol%), ligand (11 mol%), and DIPEA (20 or 0 mol%), run at 25 °C in the indicated solvent (1.0 mL), unless otherwise noted. b Determined by 1H NMR analysis of the crude product using 1,3,5-trimethoxybenzene as the internal standard. c Determined by chiral HPLC. d No reaction. e Run on a 0.25 mmol scale using Ni(COD)2 (5 mol%) and L8 (5.5 mol%). | ||||||
| 1 | L1 | 20 | EtOH | 14 | 98 | 37 |
| 2 | L2 | 20 | EtOH | 48 | 9 | 27 |
| 3 | L3 | 20 | EtOH | 24 | 4 | 22 |
| 4 | L4 | 20 | EtOH | 24 | nrd | — |
| 5 | L5 | 20 | EtOH | 14 | 79 | 22 |
| 6 | L6a | 20 | EtOH | 10 | 89 | 68 |
| 7 | L6a | 0 | EtOH | 10 | 90 | 67 |
| 8 | L6b | 0 | EtOH | 22 | 50 | 64 |
| 9 | L7 | 0 | EtOH | 22 | 50 | 78 |
| 10 | L8 | 0 | EtOH | 16 | 95 | 96 |
| 11 | L8 | 0 | Toluene | 24 | 84 | 70 |
| 12 | L8 | 0 | THF | 24 | 99 | 86 |
| 13 | L8 | 0 | CH2Cl2 | 24 | Trace | — |
| 14 | L8 | 0 | MeOH | 24 | Trace | — |
| 15 | L8 | 0 | iPrOH | 24 | Trace | — |
| 16e | L8 | 0 | EtOH | 72 | 86 | 96 |
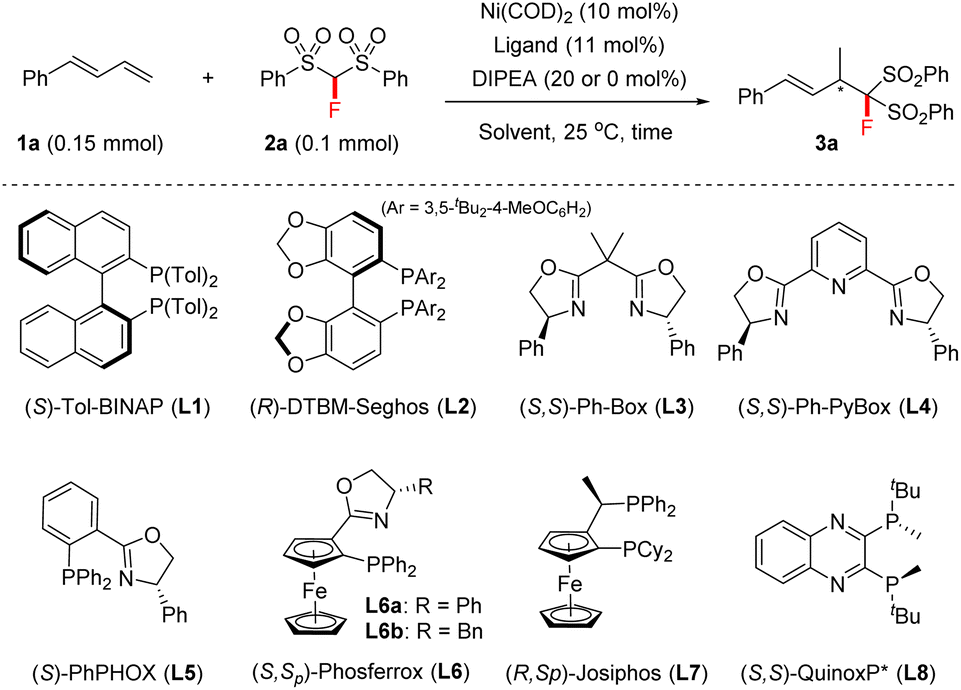
Encouraged by these results, we then tested the performance of chiral bisoxazoline ligands and P,N-based PHOX (entries 3–6), and found that the use of (S,Sp)-Ph-Phosferrox L6a could improve the ee of product 3a to 68% (entry 6). Interestingly, base DIPEA proved to be unnecessary in the current reaction. A comparable result was obtained in the absence of DIPEA (entries 6 vs. 7). The focus of further optimization was on chiral ferrocene-based chiral ligands, but there was no improvement in the ee values (entry 8, see the ESI† for details). Subsequently, we turned our attention to exploring P-chiral phosphine ligands because they usually exhibit distinct chirality-inducing ability.17 To our delight, P-chiral (S,S)-QuinoxP*18L8, never before used in hydrofunctionalizations of 1,3-dienes, proved to be efficient; it afforded 3a in 95% NMR yield with 96% ee within 16 h (entry 10). An examination of the solvent effect revealed that EtOH was still the best solvent (entries 10 vs. 11–15), although the use of THF also afforded the desired product 3a in 99% NMR yield, but with a slightly lower ee (entry 12). Moreover, the use of a 5 mol% Ni catalyst afforded the product 3a in 86% isolated yield with 96% ee, albeit within 72 h (entry 16).
Evaluation of substrate scope
With the optimized conditions in hand, we explored the generality of this Markovnikov hydromonofluoromethylation in EtOH under the catalysis of a 5 mol% or 10 mol% P-chiral (S,S)-QuinoxP* decorated Ni(COD)2 complex (Table 2).![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a
a
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | 1: substituent (●) | 2 | Time (d) | 3 | Yield (%) | ee (%) |
| a Conditions: 1 (0.375 mmol), 2 (0.25 mmol), and EtOH (2.5 mL), at rt, unless otherwise noted; yields of the isolated products are reported; the ee values were determined by chiral HPLC analysis. For 3a, 3c, 3o, and 3s: Ni(COD)2 (5 mol%) and L8 (5.5 mol%) were used; for the others: Ni(COD)2 (10 mol%) and L8 (11 mol%) were used. b At 50 °C. c At 60 °C. d At 70 °C. | ||||||
| 1 | 1a: C6H5 | 2a | 3 | 3a | 86 | 96 |
| 2 | 1b: 4-MeC6H4 | 2a | 4 | 3b | 99 | 94 |
| 3 | 1c: 4-MeOC6H4 | 2a | 3 | 3c | 97 | 97 |
| 4b | 1d: 3-MeOC6H4 | 2a | 3 | 3d | 86 | 98 |
| 5b | 1e: 2-MeOC6H4 | 2a | 3 | 3e | 93 | 98 |
| 6 | 1f: 3,5-MeO2C6H3 | 2a | 3 | 3f | 98 | 99 |
| 7b | 1g: 4-Me2NC6H4 | 2a | 4 | 3g | 48 | 93 |
| 8 |
1h: 4-CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH(CH2)2C6H4 CH(CH2)2C6H4 |
2a | 3 | 3h | 68 | 95 |
| 9 | 1i: 4-CF3C6H4 | 2a | 4 | 3i | 94 | 97 |
| 10 | 1j: 4-EtO2CC6H4 | 2a | 3 | 3j | 82 | 96 |
| 11 | 1k: 4-CNC6H4 | 2a | 3 | 3k | 90 | 98 |
| 12 | 1l: 4-COMeC6H4 | 2a | 3 | 3l | 90 | 98 |
| 13 | 1m: 4-CHOC6H4 | 2a | 3 | 3m | 71 | 90 |
| 14 | 1n: 4-ClC6H4 | 2a | 4 | 3n | 87 | 97 |
| 15 | 1o: 4-FC6H4 | 2a | 4 | 3o | 74 | 99 |
| 16c | 1p: 3-FC6H4 | 2a | 4 | 3p | 92 | 96 |
| 17c | 1q: 2-FC6H4 | 2a | 4 | 3q | 99 | 94 |
| 18 | 1r: 3,5-(CF3)2C6H3 | 2a | 3 | 3r | 49 | 95 |
| 19 | 1s: 2-naphthyl | 2a | 3 | 3s | 97 | 99 |
| 20c | 1t: 1-naphthyl | 2a | 3 | 3t | 98 | 91 |
| 21b | 1u: 2-furyl | 2a | 3 | 3u | 85 | 93 |
| 22b | 1v: 2-thienyl | 2a | 3 | 3v | 98 | 98 |
| 23 |
1w: (E)-Ph-CHCH |
2a | 5 | 3w | 99 | 97 |
| 24d | 1x: n-C5H11 | 2a | 3 | 3x | 67 | 93 |
| 25d | 1y: PhCH2CH2 | 2a | 5 | 3y | 81 | 90 |
| 26d |
|
2a | 4 | 3z | 69 | 94 |
| 27b | 1a: C6H5 | 2b | 5 | 3aa | 94 | 97 |
| 28b | 1a: C6H5 | 2c | 5 | 3ab | 99 | 95 |

|
||||||
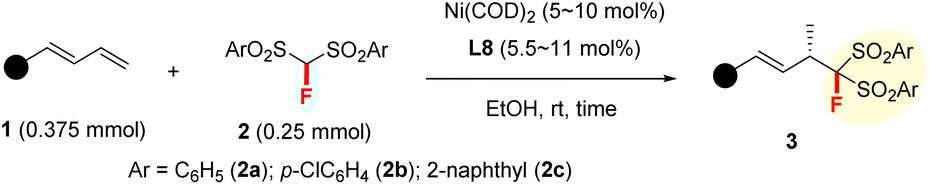
Various aromatic 1,3-dienes with different electron-donating and -withdrawing groups on the aryl ring were viable substrates, affording the corresponding 4,3-Markovnikov adducts 3b–3r in 48–99% yields with 90–99% ee (entries 2–18). The increase in reaction temperature was necessary to ensure full conversion in the cases of ortho- or meta-substituted aromatic 1,3-dienes. Of note is that the current reaction tolerated various functional groups on the aryl ring of 1,3-dienes: amine (3g), non-conjugated alkene (3h), ester (3j), nitrile (3k), ketone (3l), and aldehyde (3m). Naphthyl-, 2-furyl-, and 2-thienyl-substituted 1,3-dienes all worked smoothly with 2a to afford 3s–3v in excellent yields and ee values (entries 19–22). A conjugated triene was also tolerated; it afforded product 3w in 99% yield and with 97% ee (entry 23). Remarkably, the aliphatic 1,3-dienes, which are generally very challenging in terms of controlling both regio- and enantio-selectivity due to the small steric hindrance of the alkyl group,12c proved to be compatible in our reaction system. They afforded the adducts 3x–3z with up to 81% yields and 94% ee at slightly elevated temperatures (entries 24–26). Notably, the ketone functionalities attached in aliphatic 1,3-diene were also compatible well (3z). The differently substituted FBSM 2b and 2c also reacted efficiently with 1-phenylbuta-1,3-diene 1a at 50 °C to afford the targets 3aa (94% yield and 97% ee) and 3ab (99% yield and 95% ee). Furthermore, (S)-citronellal-derived alkyl 1,3-diene also reacted smoothly to afford adduct 3ac in moderate yield and with 95% de. X-ray diffraction (XRD) analysis revealed that the absolute configuration of 3a was (S). Subsequently, (S) was assigned to all other products 3 by analogy.
Unsurprisingly, the FBSM adduct 3a could efficiently undergo a reductive desulfonylation to access chiral α-monofluoromethyl (CH2F) allylic compound 4a with 96% ee under the action of Mg/MeOH19 (Scheme 2A). This result stimulated us to explore the assembly of deuterated monofluoromethyl (CD2F)-containing chiral allylic molecules, given that the incorporation of a deuterium atom in the bioactive molecules is emerging as a promising tactic to modulate the bioactivity or pharmacological properties in drug discovery programs since the first deuterated drug, Austedo, was approved by FDA in 2017.20a However, while the development of efficient approaches for preparing deuterated compounds is of current interest, the selective introduction of a CD2F group into the stereogenic center is still a challenging task and remains unexplored.21 To our delight, chiral deuterated allylic product D-4a featuring a CD2F group at the stereocenter, difficult to access by other methods, could be obtained smoothly by using CD3OD as the solvent in the desulfonylation step.
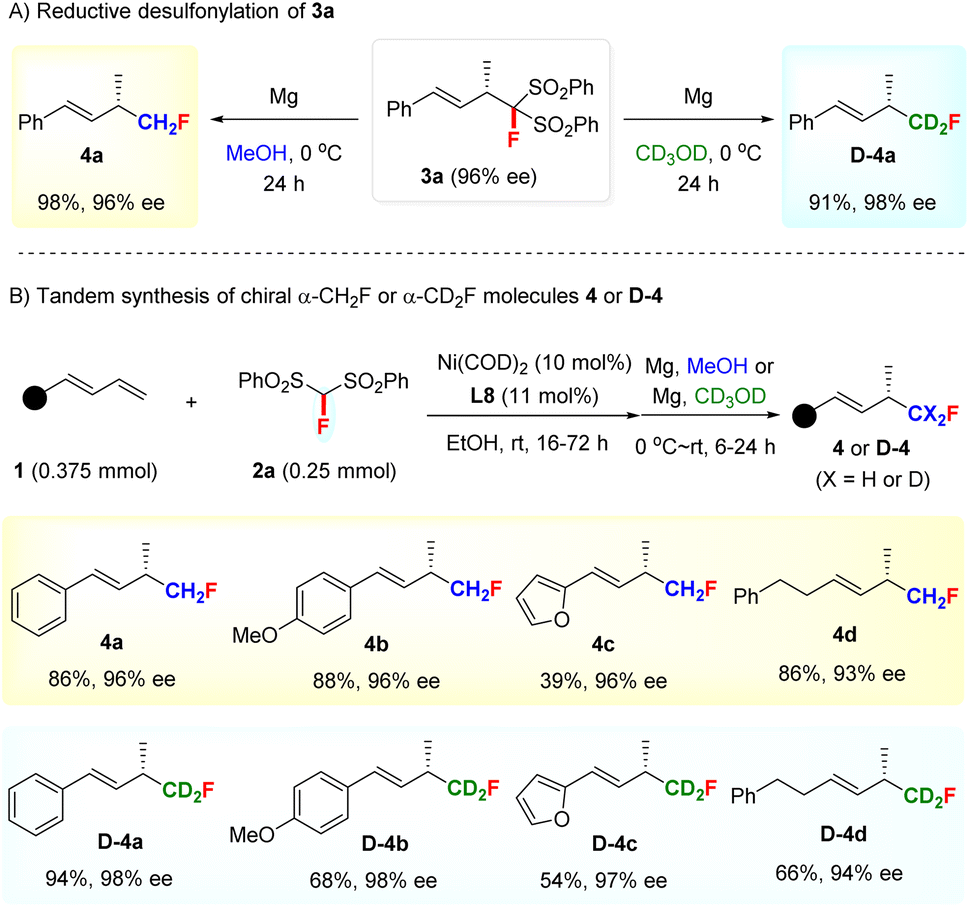 | ||
| Scheme 2 Synthetic applications of hydromonofluoromethylation. | ||
Furthermore, a tandem Ni-catalyzed asymmetric hydromonofluoro bis(phenylsulfonyl)methylation/reductive desulfonylation sequence was developed for the direct access of α-CH2F and α-CD2F substituted chiral allylic compounds 4 and D-4 (Scheme 2B). Both aryl- and alkyl-substituted 1,3-dienes were suitable partners for this tandem sequence, as exemplified by the preparation of 4a–4d and D-4a–4d with excellent ee values. It is worth mentioning that the facile synthesis of chiral allylic compounds bearing a CD2F-substituted stereocenter justified the use of FBSM as the monofluoromethylation reagent and further highlighted the value of our method.
The excellent regio- and enantio-selectivity of the above hydromonofluoromethylation inspired us to explore the realization of enantioselective hydromonofluoroalkylation with diethyl fluoromalonate4p,16d5 because of its ability to simultaneously incorporate a fluorine atom and two convertible ester groups,22 which allows the construction of functionalized chiral monofluorinated molecules with high structural complexity. After optimization of the reaction conditions (see the ESI† for details), the combination of Ni(COD)2 and (S,S)-QuinoxP* L8 still proved to be an optimal catalytic system.23 As illustrated in Scheme 3, the substrate scope was examined by running the reaction in EtOH at 50 °C using 5 mol% of Ni(COD)2-ligated (S,S)-QuinoxP* as the catalyst. Both (hetero)aromatic and aliphatic 1,3-dienes were suitable substrates, affording the 4,3-Markovnikov adducts 6 with excellent regio- and enantio-selectivity. Regardless of the nature and position of the substituent on the phenyl ring of aryl 1,3-dienes, all reacted well with 5 to afford the products 6a–6u in 68–99% yields with 93–99% ee. The XRD analysis of 6t confirmed its absolute configuration to be (S), and that of other products was assigned by analogy. Various functional groups, such as ester (6e), nitrile (6f), ketone (6g), aldehyde (6h), amine (6m), and non-conjugated alkene (6n), on the aryl ring of aromatic 1,3-dienes were well-tolerated under this hydromonofluoroalkylation as well. Heteroaromatic 2-furyl- and 2-thienyl-substituted 1,3-dienes, as well as conjugated triene, also afforded the adducts 6v–6x in 93–98% yields and 91–98% ee. Moreover, linear and branched alkyl-substituted 1,3-dienes were tolerated, affording 4,3-Markovnikov adducts 6y–6ab in 79–97% yields and 90–97% ee, with high to excellent regioselectivity, albeit with the generation of a small amount of 4,1-addition isomer in these cases.13c The use of a 10 mol% Ni catalyst was required to ensure excellent yields in the case of heteroaryl and alkyl 1,3-dienes. The protocol was also applied to the late-stage hydromonofluoroalkylation of (S)-citronellal and estrone derivatives, affording 6ac in 96% yield with 11:1 dr and 6ad in 82% yield with 9:1 dr.
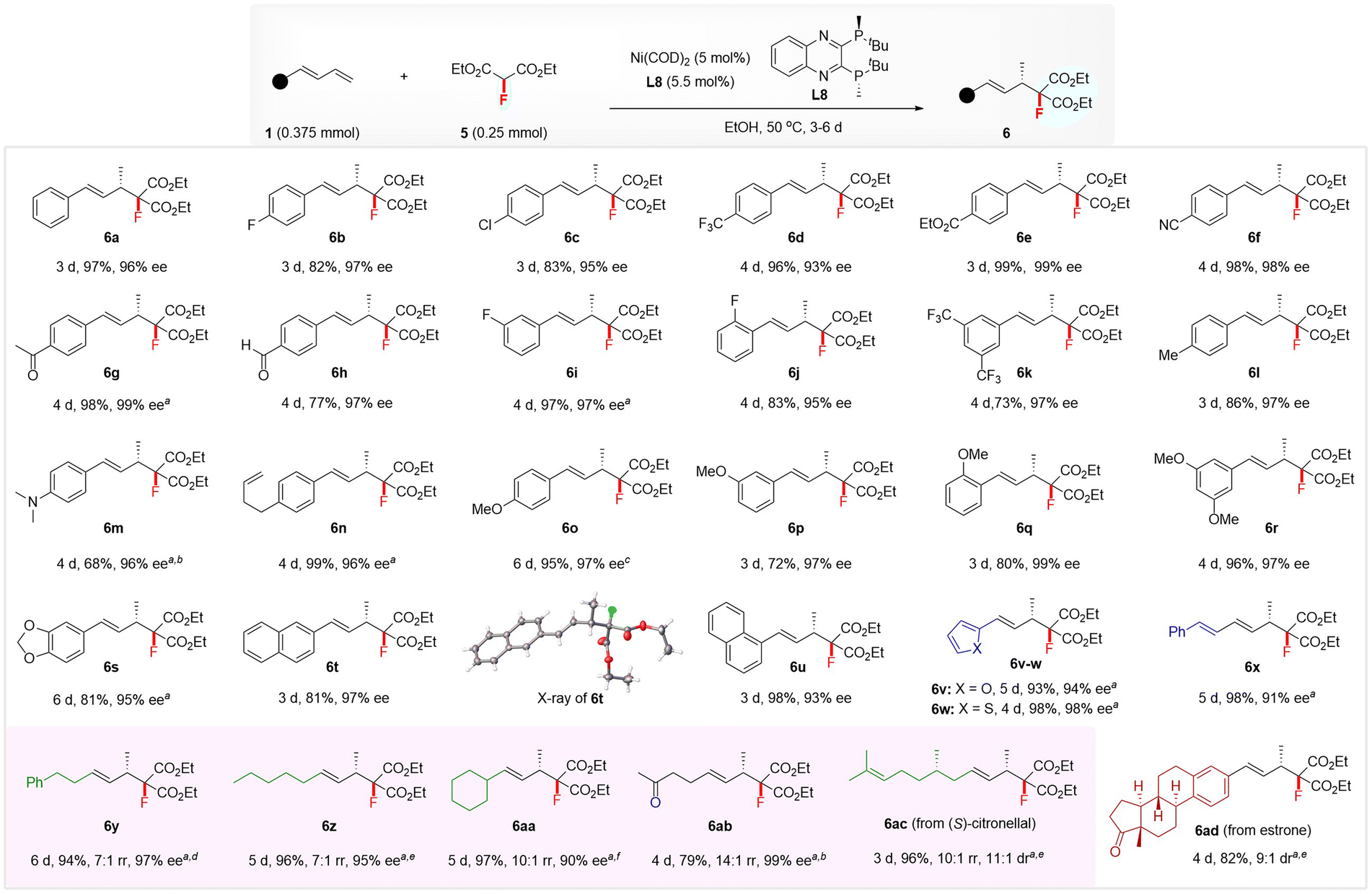 | ||
| Scheme 3 Scope of enantioselective hydromonofluoroalkylation of 1,3-dienes 1 with diethyl fluoromalonate 5. Conditions: 1 (0.375 mmol), 2 (0.25 mmol), Ni(COD)2 (5 mol%), and L8 (5.5 mol%) at 50 °C in EtOH (1.5 mL), unless otherwise noted. Yields of isolated products were reported and ee was determined by chiral HPLC analysis. a Using Ni(COD)2 (10 mol%) and L8 (11 mol%). b At 70 °C. c At 50–70 °C. d At 60 °C. e At 80 °C. f At 75 °C. The rr indicates the regioselectivity ratio of 4,3-Markovnikov isomer with another isomer, which was determined by 1H NMR analysis. The ee value of 6z and 6aa was determined by their derivatives; see the ESI† for details. The dr value of 6ac and 6ad was determined by 19F NMR analysis. | ||
Synthetic utility
To further highlight the practicality of the reaction, a gram-scale synthesis of product 6a and its synthetic elaborations toward structurally diversified fluorine-containing molecules was conducted. As shown in Scheme 4, starting from 1a (7.5 mmol) and 5 (5 mmol), 1.47 g of 6a could be readily generated in 95% yield and with 97% ee under the standard conditions. The two ester groups of 6a could be selectively hydrolyzed to fluorinated carboxylic acid or dicarboxylic acid, as demonstrated by the synthesis of 7 (94% yield and 12:1 dr) via a porcine liver esterase (PLE) enabled hydrolytic desymmetrization, and 8 (99% yield) via NaOH-mediated hydrolysis. The treatment of 6a with m-CPBA led to the epoxidation of the alkenyl and afforded chiral fluorinated epoxide 9 in 73% yield, albeit with modest dr. Compound 6a could also be selectively reduced with LiAl(OtBu)3 or NaBH4, affording a fluorinated hydroxy ester 10 in 67% yield with 1.4:1 dr and 96% ee, or a fluorinated diol 11 in 83% yield with 97% ee, respectively.
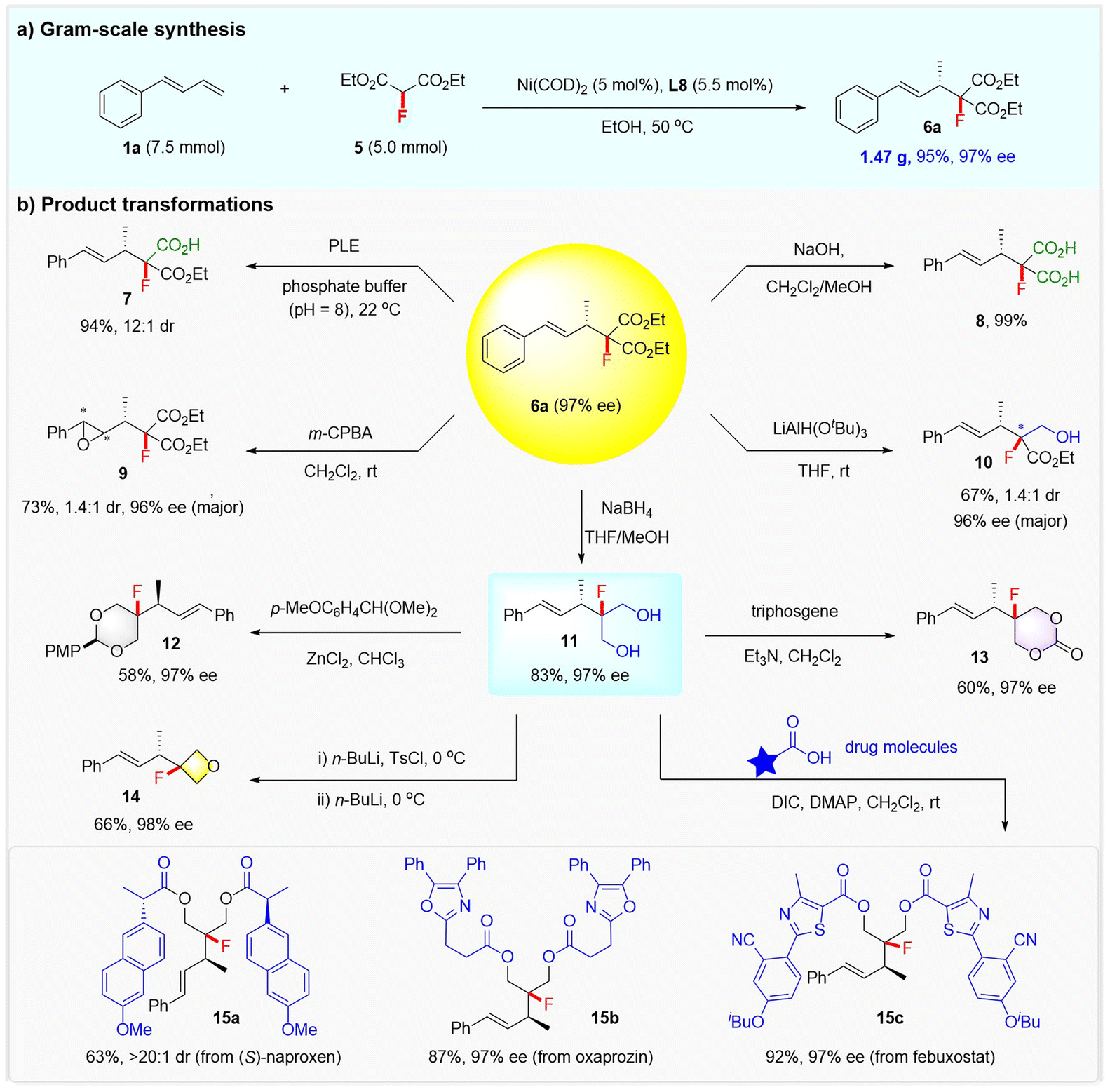 | ||
| Scheme 4 Synthetic utility. | ||
Notably, diol 11 was readily converted into a synthetically useful fluorinated 1,3-dioxane 12 or 1,3-dioxan-2-one 13 (ref.24) under the action of 1-(dimethoxymethyl)-4-methoxybenzene or triphosgene. A fluorinated oxetane 14 was also obtained from diol 11via a selective monotosylation and sequential cyclization process. The versatile diol 11 proved to be a very useful linker that can merge two drugs to form complex fluorine-containing molecules, as exemplified by the efficient installation of fluorinated compounds 15a–15c from drugs (S)-naproxen, oxaprozin, and febuxostat.
Conclusions
In summary, we have developed a highly enantioselective Markovnikov regioselective hydromonofluoroalkyl(methyl)ation of 1,3-dienes by using chiral Ni catalysis, allowing access to various functionalized chiral allylic compounds bearing a CH2F, CD2F or monofluoroalkyl group at the stereocenter. Remarkably, such a methodology provides a new direction for enantioselective monofluoroalkyl(methyl)ation, and it constitutes a new branch of asymmetric 1,3-diene hydrofunctionalizations. Moreover, this represents the first enantio- and regio-selective Markovnikov hydrofluoroalkylation of olefins. The salient features include broad substrate scope for both aromatic and aliphatic 1,3-dienes, excellent enantio- and regio-selectivity, good functional group tolerance, mild base-free conditions, and diverse product transformations. Further studies in our laboratory will focus on elucidating the reaction mechanism25 and developing other asymmetric Markovnikov regioselective hydrofluoroalkylation reactions.Data availability
All of the experimental data have been included in the ESI.† Crystallographic data can be obtained from the CCDC (2130032 and 2130034).Author contributions
L. Liao and Y. Zhang performed the experiments, and collected and analyzed the data; Z.-W. Wu, Z.-T. Ye, and X.-X. Zhang synthesized some of the 1,3-dienes. J.-S. Yu conceived the idea and directed the project; J.-S. Yu and G. Chen co-wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge Prof. Dr Jian Zhou at ECNU for his kind and valuable discussion. The financial support from the National Key Research and Development Program of China (2021YFC2102400), the National Natural Science Foundation of China (22171087 and 21901074), the Shanghai Science and Technology Innovation Action Plan (21N41900500 and and 20JC1416900), the Fundamental Research Funds for the Central Universities, and the Open Foundation of Key Laboratory of Tropical Medicinal Resource Chemistry of Ministry of Education (rdzh2020003) is highly appreciated. J.-S. Y. also acknowledges financial support from the “Zijiang Scholar Program” of East China Normal University.Notes and references
- (a) I. Ojima, Fluorine in Medicinal Chemistry and Chemical Biology, Wiley-Blackwell, Chichester, 2009 CrossRef; (b) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC; (c) D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308–319 RSC; (d) K. Müller, C. Faeh and F. Diederich, Science, 2017, 317, 1881–1886 CrossRef PubMed.
- (a) R. E. Banks, B. E. Smart and J. C. Tatlow, Organofluorine Chemistry: Principles and Commercial Applications, Plenum, New York, 1994, ch. 3 CrossRef; (b) V. P. Reddy, Organofluorine Compounds in Biology and Medicine, Elsevier, Amsterdam, 2020 Search PubMed; (c) B. E. Smart, J. Fluorine Chem., 2001, 109, 3–11 CrossRef CAS; (d) P. Jeschke, ChemBioChem, 2004, 5, 570–589 CrossRef CAS PubMed; (e) W. K. Hagmjann, J. Med. Chem., 2008, 51, 4359–4369 CrossRef PubMed; (f) J. Wang, M. Sánchez-Roselló, J. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS PubMed; (g) M. Reichel, B. Krumm, Y. V. Vishnevskiy, S. Blomeyer, J. Schwabedissen, H.-G. Stammler, K. Karaghiosoff and N. W. Mitzel, Angew. Chem., Int. Ed., 2019, 58, 18557–18561 CrossRef CAS PubMed; (h) M. Inoue, Y. Sumii and N. Shibata, ACS Omega, 2020, 5, 10633–10640 CrossRef CAS PubMed; (i) Y. Ogawa, E. Tokunaga, O. Kobayashi, K. Hirai and N. Shibata, iScience, 2020, 23, 101467 CrossRef CAS PubMed.
- (a) J. Kollonitsch, A. A. Patchett, S. Marburg, A. L. Maycock, L. M. Perkins, G. A. Doldouras, D. E. Duggan and S. D. Aster, Nature, 1978, 274, 906–908 CrossRef CAS PubMed; (b) G. L. Grunewald, M. R. Seim, R. C. Regier, J. L. Martin, C. L. Gee, N. Drinkwater and K. R. Criscione, J. Med. Chem., 2006, 49, 5424–5433 CrossRef CAS PubMed; (c) H. Su, Y. Xie, W.-B. Liu and S.-L. You, Bioorg. Med. Chem. Lett., 2011, 21, 3578–3582 CrossRef CAS PubMed.
- For selected reviews: (a) G. K. S. Prakash and A. K. Yudin, Chem. Rev., 1997, 97, 757–786 CrossRef CAS PubMed; (b) K. Uneyama, T. Katagiri and H. Amii, Acc. Chem. Res., 2008, 41, 817–829 CrossRef CAS PubMed; (c) J. Nie, H.-C. Guo, D. Cahard and J.-A. Ma, Chem. Rev., 2011, 111, 455–529 CrossRef CAS PubMed; (d) T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470–477 CrossRef CAS PubMed; (e) G. Valero, X. Companyó and R. Rios, Chem.–Eur. J., 2011, 17, 2018–2037 CrossRef CAS PubMed; (f) E. Merino and C. Nevado, Chem. Soc. Rev., 2014, 43, 6598–6608 RSC; (g) L. Chu and F.-L. Qing, Acc. Chem. Res., 2014, 47, 1513–1522 CrossRef CAS PubMed; (h) J. Charpentier, N. Früh and A. Togni, Chem. Rev., 2015, 115, 650–682 CrossRef CAS PubMed; (i) X. Yang, T. Wu, R. J. Phipps and F. D. Toste, Chem. Rev., 2015, 115, 826–870 CrossRef CAS PubMed; (j) C. Ni, M. Hu and J. Hu, Chem. Rev., 2015, 115, 765–825 CrossRef CAS PubMed; (k) C. Alonso, E. M. Martínez de Marigorta, G. Rubiales and F. Palacios, Chem. Rev., 2015, 115, 1847–1935 CrossRef CAS PubMed; (l) X. Liu, C. Xu, M. Wang and Q. Liu, Chem. Rev., 2015, 115, 683–730 CrossRef CAS PubMed; (m) V. Gouverneur and K. Seppelt, Chem. Rev., 2015, 115, 563–565 CrossRef CAS PubMed; (n) Z. Feng, Y.-L. Xiao and X. Zhang, Acc. Chem. Res., 2018, 51, 2264–2278 CrossRef CAS PubMed; (o) A. D. Dilman and V. V. Levin, Acc. Chem. Res., 2018, 51, 1272–1280 CrossRef CAS PubMed; (p) Y. Zhu, J. Han, J. Wang, N. Shibata, M. Sodeoka, V. A. Soloshonok, J. A. S. Coelho and F. D. Toste, Chem. Rev., 2018, 118, 3887–3964 CrossRef CAS PubMed; (q) D. Cahard and J.-A. Ma, Emerging Fluorinated Motifs: Synthesis, Properties, and Applications. Wiley-VC, 1st edn, 2020 Search PubMed; (r) T. W. Butcher, W. M. Amberg and J. F. Hartwig, Angew. Chem., Int. Ed., 2021, 61, e202112251 Search PubMed.
- For the leading review, see: M. Reichel and K. Karaghiosoff, Angew. Chem., Int. Ed., 2020, 59, 12268–12281 CrossRef CAS PubMed.
- (a) T. Chatterjee, N. Iqbal, Y. You and E. J. Cho, Acc. Chem. Res., 2016, 49, 2284–2294 CrossRef CAS PubMed; (b) S. Barata-Vallejo, M. V. Cooke and A. Postigo, ACS Catal., 2018, 8, 7287–7307 CrossRef CAS; (c) R. D. Laishram, J. Chen and B. Fan, Chem. Rec., 2021, 21, 69–86 CrossRef CAS PubMed; (d) T. Koike and M. Akita, Chem, 2018, 4, 409–437 CrossRef CAS; (e) D. E. Yerien, S. Barata-Vallejo and A. Postigo, Chem.–Eur. J., 2017, 23, 14676–14701 CrossRef CAS PubMed.
- For selected examples of perfluoroalkylations: (a) C.-M. Hu and Y.-L. Qiu, J. Org. Chem., 1992, 57, 3339 CrossRef CAS; (b) Z.-Y. Long and Q.-Y. Chen, J. Org. Chem., 1999, 64, 4775–4782 CrossRef CAS PubMed; (c) T. Yajima and S. Shigenaga, Org. Lett., 2019, 21, 138–141 CrossRef CAS PubMed; (d) C.-L. Tong, X.-H. Xu and F.-L. Qing, Org. Lett., 2019, 21, 9532–9535 CrossRef CAS PubMed; (e) W. Zhang, Z. Zou, Y. Wang, Y. Wang, Y. Liang, Z. Wu, Y. Zheng and Y. Pan, Angew. Chem., Int. Ed., 2019, 58, 624–627 CrossRef CAS PubMed , for selected examples of trifluoromethylations:; (f) S. Mizuta, S. Verhoog, K. M. Engle, T. Khotavivattana, M. O'Duill, K. Wheel-house, G. Rassias, M. Médebielle and V. Gouverneur, J. Am. Chem. Soc., 2013, 135, 2505–2508 CrossRef CAS PubMed; (g) X. Wu, L. Chu and F.-L. Qing, Angew. Chem., Int. Ed., 2013, 52, 2198–2202 CrossRef CAS PubMed; (h) D. J. Wilger, N. J. Gesmundo and D. A. Nicewicz, Chem. Sci., 2013, 4, 3160–3165 RSC; (i) P. Yu, J.-S. Lin, L. Li, S.-C. Zheng, Y.-P. Xiong, L.-J. Zhao, B. Tan and X.-Y. Liu, Angew. Chem., Int. Ed., 2014, 53, 11890–11894 CrossRef CAS PubMed; (j) N. J. W. Straathof, S. E. Cramer, V. Hessel and T. Noël, Angew. Chem., Int. Ed., 2016, 55, 15549–15553 CrossRef CAS PubMed; (k) J.-X. Xiang, Y. Ouyang, X.-H. Xu and F.-L. Qing, Angew. Chem., Int. Ed., 2019, 58, 10320–10324 CrossRef CAS PubMed , for selected difluoroalkylations:; (l) C. Yu, N. Iqbal, S. Park and E. Cho, Chem. Commun., 2014, 50, 12884–12887 RSC; (m) G. Ma, W. Wan, J. Li, Q. Hu, H. Jiang, S. Zhu, J. Wang and J. Hao, Chem. Commun., 2014, 50, 9749–9752 RSC; (n) Q.-Y. Lin, X.-H. Xu and F.-L. Qing, Org. Biomol. Chem., 2015, 13, 8740–8749 RSC; (o) X.-J. Tang, Z. Zhang and W. R. Dolbier Jr, Chem.–Eur. J., 2015, 21, 18961–18965 CrossRef CAS PubMed; (p) J. Yu, J.-H. Lin, Y.-C. Cao and J.-C. Xiao, Org. Chem. Front., 2019, 6, 3580–3583 RSC; (q) W. Huang, W. Chen, G. Wang, J. Li, X. Cheng and G. Li, ACS Catal., 2016, 6, 7471–7474 CrossRef CAS; (r) W. Shu, E. Merino and C. Nevado, ACS Catal., 2018, 8, 6401–6406 CrossRef CAS; (s) H. Wang and N. T. Jui, J. Am. Chem. Soc., 2018, 140, 163–166 CrossRef CAS PubMed; (t) D. B. Vogt, C. P. Seath, H. Wang and N. T. Jui, J. Am. Chem. Soc., 2019, 141, 13203–13211 CrossRef CAS PubMed.
- (a) D. G. Brown and J. Boström, J. Med. Chem., 2016, 59, 4443–4458 CrossRef CAS PubMed; (b) K. W. Quasdorf and L. E. Overman, Nature, 2014, 516, 181–191 CrossRef CAS PubMed.
- For a review, see: (a) X.-S. Hu, J.-S. Yu and J. Zhou, Chem. Commun., 2019, 55, 13638–13648 RSC; (b) X.-S. Hu, J.-X. He, S.-Z. Dong, Q.-H. Zhao, J.-S. Yu and J. Zhou, Nat. Commun., 2020, 11, 5500 CrossRef CAS PubMed; (c) J.-S. Yu, F.-M. Liao, W.-M. Gao, K. Liao, R.-L. Zuo and J. Zhou, Angew. Chem., Int. Ed., 2015, 54, 7381–7385 CrossRef CAS PubMed.
- For leading reviews: (a) G.-L. Li, X.-H. Huo, X.-Y. Jiang and W.-B. Zhang, Chem. Soc. Rev., 2020, 49, 2060–2118 RSC; (b) N. J. Adamson and S. J. Malcolmson, ACS Catal., 2020, 10, 1060–1076 CrossRef CAS.
- Hydroamination: (a) O. Löber, M. Kawatsura and J. F. Hartwig, J. Am. Chem. Soc., 2001, 123, 4366–4367 CrossRef PubMed; (b) X.-H. Yang and V. M. Dong, J. Am. Chem. Soc., 2017, 139, 1774–1777 CrossRef CAS PubMed; (c) N. J. Adamson, E. Hull and S. J. Malcolmson, J. Am. Chem. Soc., 2017, 139, 7180–7183 CrossRef CAS PubMed; (d) G. Tran, W. Shao and C. Mazet, J. Am. Chem. Soc., 2019, 141, 14814–14822 CrossRef CAS PubMed.
- Hydroalkylation: (a) A. Leitner, J. Larsen, C. Steffens and J. F. Hartwig, J. Org. Chem., 2004, 69, 7552–7557 CrossRef CAS PubMed; (b) N. J. Adamson, K. C. E. Wilbur and S. J. Malcolmson, J. Am. Chem. Soc., 2018, 140, 2761–2764 CrossRef CAS PubMed; (c) L. Cheng, M.-M. Li, L.-J. Xiao, J.-H. Xie and Q.-L. Zhou, J. Am. Chem. Soc., 2018, 140, 11627–11630 CrossRef CAS PubMed; (d) Q. Zhang, H. Yu, L. Shen, T. Tang, D. Dong, W. Chai and W. Zi, J. Am. Chem. Soc., 2019, 141, 14554–14559 CrossRef CAS PubMed; (e) W. Shao, C. Besnard, L. Guénée and C. Mazet, J. Am. Chem. Soc., 2020, 142, 16486–16492 CrossRef CAS PubMed; (f) J. Xia, T. Hirai, S. Katayama, H. Nagae, W. Zhang and K. Mashima, ACS Catal., 2021, 11, 6643–6655 CrossRef CAS; (g) H. Wang, R. Zhang, Q. Zhang and W. Zi, J. Am. Chem. Soc., 2021, 143, 10948–10962 CrossRef CAS PubMed.
- Hydroarylation: (a) C. C. Roberts, J. S. Marcum, C. C. Roberts, R. S. Manan, T. N. Cervarich and S. J. Meek, J. Am. Chem. Soc., 2017, 139, 15580–15583 CrossRef PubMed; (b) X.-Y. Lv, C. Fan, L.-J. Xiao, J.-H. Xie and Q.-L. Zhou, CCS Chem., 2019, 1, 328–334 CrossRef CAS; (c) L. Cheng, M.-M. Li, M.-L. Li, L.-J. Xiao, J.-H. Xie and Q.-L. Zhou, CCS Chem., 2021, 3, 3260–3267 Search PubMed.
- (a) Q. Zhang, D. Dong and W. Zi, J. Am. Chem. Soc., 2020, 142, 15860–15869 CrossRef CAS PubMed; (b) M.-M. Li, L. Cheng, L.-J. Xiao, J.-H. Xie and Q.-L. Zhou, Angew. Chem., Int. Ed., 2021, 60, 2948–2951 CrossRef CAS PubMed.
- (a) J. W. Han and T. Hayashi, Tetrahedron: Asymmetry, 2010, 21, 2193–2197 CrossRef CAS; (b) M. Shirakura and M. Suginome, Angew. Chem., Int. Ed., 2010, 49, 3827–3829 CrossRef CAS PubMed; (c) S.-Z. Nie, R. T. Davison and V. M. Dong, J. Am. Chem. Soc., 2018, 140, 16450–16454 CrossRef CAS PubMed; (d) X.-H. Yang, R. T. Davison and V. M. Dong, J. Am. Chem. Soc., 2018, 140, 10443–10446 CrossRef CAS PubMed; (e) X.-H. Yang, R. T. Davison, S.-Z. Nie, F. A. Cruz, T. M. McGinnis and V. M. Dong, J. Am. Chem. Soc., 2019, 141, 3006–3013 CrossRef CAS PubMed; (f) J. Long, Y.-Q. Li, W.-N. Zhao and G.-Y. Yin, Chem. Sci., 2022, 13, 1390–1397 RSC.
- For pioneering work on the synthesis and application of FBSM: (a) T. Fukuzumi, N. Shibata, M. Sugiura, H. Yasui, S. Nakamura and T. Toru, Angew. Chem., Int. Ed., 2006, 45, 4973–4977 CrossRef CAS PubMed; (b) C. Ni, Y. Li and J. Hu, J. Org. Chem., 2006, 71, 6829–6833 CrossRef CAS PubMed , for reviews:; (c) A.-N. R. Alba, X. Companyo and R. Rios, Chem. Soc. Rev., 2010, 39, 2018–2033 RSC; (d) C. Ni, L. Zhu and J. Hu, Acta Chim. Sin., 2015, 73, 90–115 CrossRef CAS , see selected examples for the application of FBSM in asymmetric reactions:; (e) S. Mizuta, N. Shibata, Y. Goto, T. Furukawa, S. Nakamura and T. Toru, J. Am. Chem. Soc., 2007, 129, 6394–6395 CrossRef CAS PubMed; (f) T. Furukawa, N. Shibata, S. Mizuta, S. Nakamura, T. Toru and M. Shiro, Angew. Chem., Int. Ed., 2008, 47, 8051–8054 CrossRef CAS PubMed; (g) T. Furukawa, J. Kawazoe, W. Zhang, T. Nishimine, E. Tokunaga, T. Matsumoto, M. Shiro and N. Shibata, Angew. Chem., Int. Ed., 2011, 50, 9684–9688 CrossRef CAS PubMed; (h) A. N. Alba, X. Companyó, A. Moyano and R. Rios, Chem.–Eur. J., 2009, 15, 7035–7038 CrossRef CAS PubMed; (i) S. Zhang, Y. Zhang, Y. Ji, H. Li and W. Wang, Chem. Commun., 2009, 4886–4888 RSC; (j) F. Ullah, G. L. Zhao, L. Deiana, M. Zhu, P. Dziedzic, I. Ibrahem, P. Hammar, J. Sun and A. Córdova, Chem.–Eur. J., 2009, 15, 10013–10017 CrossRef CAS PubMed; (k) W. B. Liu, S. C. Zheng, H. He, X. M. Zhao, L. X. Dai and S. L. You, Chem. Commun., 2009, 6604–6606 RSC; (l) X. Companyó, G. Valero, V. Ceban, T. Calvet, M. Font-Bardia, A. Moyano and R. Rios, Org. Biomol. Chem., 2011, 9, 7986–7989 RSC; (m) W. Yang, X. Wei, Y. Pan, R. Lee, B. Zhu, H. Liu, L. Yan, K.-W. Huang, Z. Jiang and C.-H. Tan, Chem.–Eur. J., 2011, 17, 8066–8070 CrossRef CAS PubMed , also see ref. 5..
- (a) M. Dutartre, J. Bayardon and S. Jugé, Chem. Soc. Rev., 2016, 45, 5771–5794 RSC; (b) R.-Y. Zhu, K. Liao, J.-S. Yu and J. Zhou, Acta Chim. Sin., 2020, 78, 193–216 CrossRef CAS.
- T. Imamoto, K. Sugita and K. Yoshida, J. Am. Chem. Soc., 2005, 127, 11934–11935 CrossRef CAS PubMed.
- (a) G. K. S. Prakash, S. Chacko, S. Alconcel, T. Stewart, T. Mathew and G. A. Olah, Angew. Chem., Int. Ed., 2007, 46, 4933–4936 CrossRef CAS PubMed; (b) G. K. S. Prakash, S. Chacko, H. Vaghoo, N. Shao, L. Gurung, T. Mathew and G. A. Olah, Org. Lett., 2009, 11, 1127–1130 CrossRef CAS PubMed.
- (a) C. Schmidt, First Deuterated Drug Approved, Nat. Biotechnol., 2017, 35, 493–494 CrossRef CAS PubMed; (b) S. H. DeWitt and B. E. Maryanoff, Biochemistry, 2018, 57, 472–473 CrossRef CAS PubMed; (c) V. Jacques, A. W. Czarnik, T. M. Judge, L. H. T. Van der Ploeg and S. H. DeWitt, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E1471–E1479 CrossRef CAS PubMed.
- Shen and Lu installed a CD2F group in a few achiral molecules using their developed electrophilic CD2F-substituted sulfonium ylides: Y. Liu, L. Lu and Q. Shen, Angew. Chem., Int. Ed., 2017, 56, 9930–9934 CrossRef CAS PubMed.
- P. Xu and Z. Huang, Nat. Chem., 2021, 13, 634–642 CrossRef CAS PubMed.
- An obvious fluorine effect was observed in the current reaction, though the origin is still unclear. In the presence of 10 mol% Ni(COD)2/L8, the use of diethyl 2-fluoromalonate 5 afforded the hydromonofluoroalkylated product 6a in 94% with 96% ee; in sharp contrast, under the same conditions, no reaction occurred at all when replacing 5 with diethyl 2-chloromalonate and only 74% ee of the corresponding product with 98% yield was obtained in the case of diethyl 2-methylmalonate.
- The utility of fluorinated rings: (a) P. Kirsch, A. Hahn, R. Fröhlich and G. Haufe, Eur. J. Org. Chem., 2006, 2006, 4819–4824 CrossRef; (b) R. Mondal, M. Agbaria and Z. Nairoukh, Chem.–Eur. J., 2021, 27, 7193–7213 CrossRef CAS.
- To gain some insight into the reaction mechanism, we conducted preliminary experimental investigation and also proposed a reaction mechanism; see Section 7 of the ESI† for details.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2130032 and 2130034. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc03958c |
| ‡ L. Liao and Y. Zhang contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |