
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unified synthesis of multiply arylated alkanes by catalytic deoxygenative transformation of diarylketones†
Miki B.
Kurosawa
a,
Kenta
Kato
a,
Kei
Muto
 b and
Junichiro
Yamaguchi
*a
b and
Junichiro
Yamaguchi
*a
aDepartment of Applied Chemistry, Waseda University, 513 Wasedatsurumakicho, Shinjuku, Tokyo 162-0041, Japan. E-mail: junyamaguchi@waseda.jp
bWaseda Institute for Advanced Study, Waseda University, 513 Wasedatsurumakicho, Shinjuku, Tokyo 162-0041, Japan
First published on 22nd August 2022
Abstract
A deoxygenative transformation of diarylketones leading to multiply arylated alkanes was developed. Diarylketones were reacted with diphenylphosphine oxide resulting in a phospha-Brook rearrangement, followed by palladium-catalyzed cross-couplings or a Friedel–Crafts type alkylation to afford the corresponding multiply arylated alkanes. A variety of diarylketones can be converted to multiply arylated alkanes such as diarylmethanes, tetraarylethanes, and triarylmethanes by reduction, dimerization, and arylation in one pot. Furthermore, a one-pot conversion from arylcarboxylic acids to diarylmethanes and tetraarylethanes, and a synthesis of tetraarylmethane and triphenylethane using sequential coupling reactions are also presented.
Introduction
Multiply arylated alkanes are widely used as pharmaceuticals, natural products, and organic materials (Fig. 1A). A diarylmethane such as tofogliflozin is a representative motif among multiply arylated alkanes.1 A triarylmethane such as (+)-securidane A and a tetraarylethane such as Raptinal are also known bioactive products.2–4 CDP-840, a phosphodiesterase (PDE)-IV inhibitor,5 is classified as a triarylethane, and COF-102, known as a covalent organic framework, has a tetraarylmethane skeleton.6,7 Since these are important structures, the synthesis of multiply arylated alkanes has long been investigated.8–21 Particularly recently, excellent catalytic and efficient syntheses of these alkanes have been reported using readily available building blocks and state-of-the-art methodologies (Fig. 1B). For example, diarylmethanes are generally synthesized from benzyl electrophiles, but recently, a direct coupling from benzyl alcohol, which is a readily available starting material, has been reported. Shu and coworkers discovered a nickel-catalyzed direct cross-coupling of benzylic alcohols with aryl triflates in the presence of DMO (dimethyl oxalate) to afford the corresponding diarylmethanes.22 Masarwa and coworkers reported a unique synthetic method of triarylmethanes from aldehydes:23 an aldehyde was mixed with an arene and triphenylphosphine in the presence of TfOH to generate a diarylphosphonium salt, followed by another arene in one pot to furnish the corresponding triarylmethane. This simple operation allowed for a one-pot synthesis of triarylmethanes from aldehydes, but this method is limited to the use of electron-rich arenes. Li's group reported a deoxygenative dimerization from diaryl ketones, inspired by a classical Wolff–Kishner type of diarylmethane synthesis.24 This is an excellent method that can synthesize triarylethanes in two steps (2 pots) through the formation of hydrazone as an intermediate. Moran's group then developed a one-pot synthesis of triarylethanes from aryl epoxides.25 A ring-opening reaction of the epoxide by an electron-rich arene was followed by a Friedel–Crafts type alkylation of the resulting diarylethanol with another electron-rich arene via a phenonium intermediate. Although this method appears to be a classical synthetic method, a variety of electron-rich triarylethanes can be prepared. Walsh and coworkers demonstrated a straightforward synthesis of tetraarylmethanes from diarylmethanes.26 After deprotonation of the diarylmethane with a base (KOt-Bu), two further arenes can be introduced by palladium-catalyzed C–H arylations with aryl chlorides. A variety of tetraarylmethanes can be synthesized, although an electron-deficient heteroarene (a pyridine moiety) is required for one of the aromatic rings of the diarylmethane. While various syntheses of multiply arylated alkanes have been reported,27,28 thus far, there is no unified synthetic protocol from the same starting materials.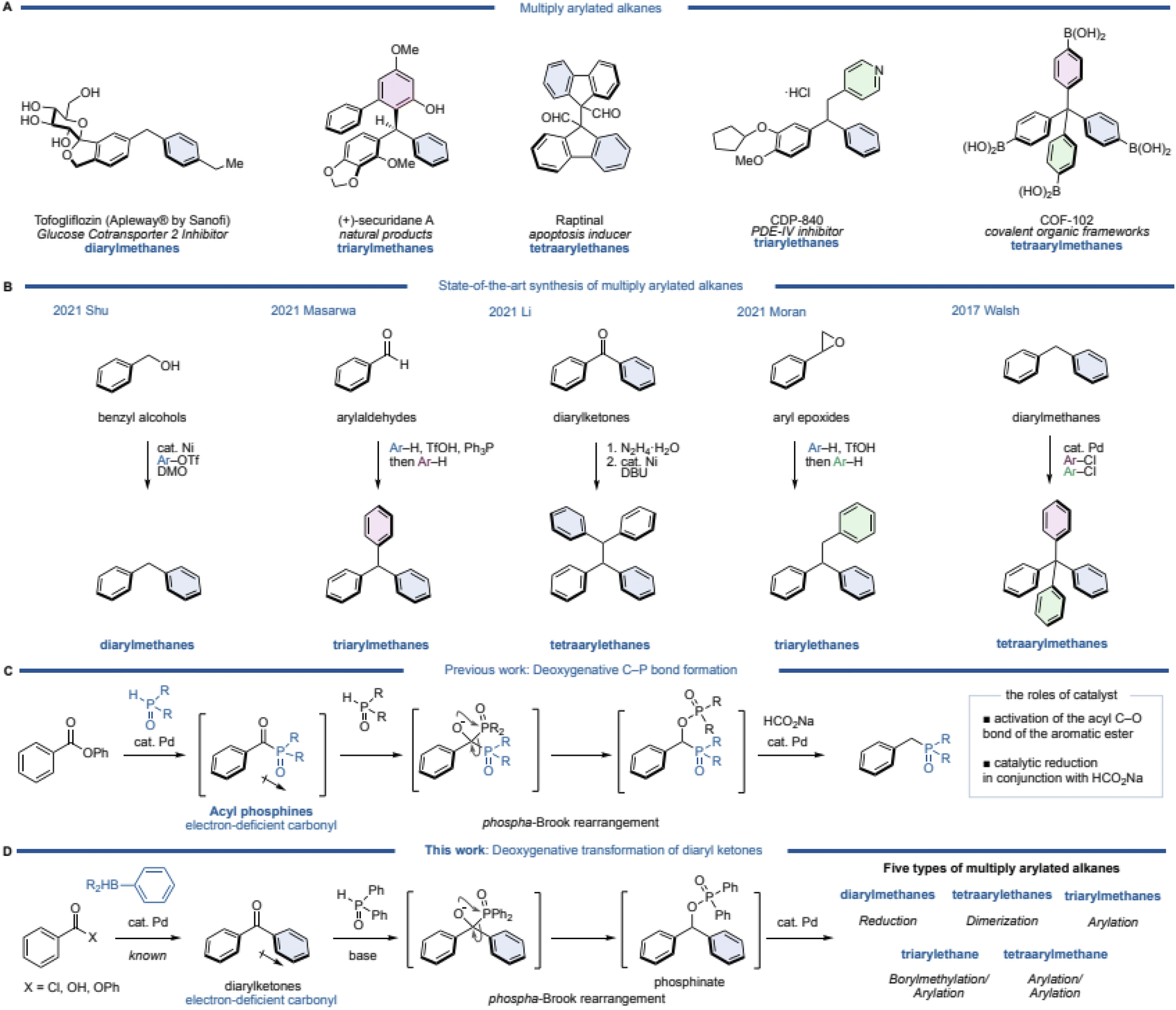 | ||
| Fig. 1 (A) Multiply arylated alkanes in pharmaceuticals, natural products, and organic materials. (B) State-of-the-art synthesis of multiply arylated alkanes. (C) Deoxygenative C–P bond formation of aromatic esters. (D) Deoxygenative transformation of diarylketones. | ||
Meanwhile, recent attention has been focused on the deoxygenative transformation of aromatic carbonyls, which has been extensively studied by the Li group and others.29–33 Regarding this field of research, we have also developed a deoxygenative C–P bond formation of aromatic esters (Fig. 1C).34 In this reaction, an aromatic ester reacts with phosphine oxide in the presence of a palladium catalyst to give an acylphosphine oxide species as the intermediate. The intermediate has a highly electron-deficient carbonyl, thus a nucleophilic attack of the phosphine oxide to the carbonyl occurs, followed by a [1,2]-phospha-Brook rearrangement and reduction of the carbon–oxygen bond using a palladium catalyst and sodium formate (HCO2Na) as a mild reductant to afford the corresponding deoxygenative products. Based on this mechanism, we envisioned that this reaction could be utilized as a unified synthesis of multiply arylated alkanes from diarylketones (Fig. 1D).
Diarylketones are inexpensive, readily available basic chemicals that can also be generated from aryl carboxylic acids, acid chlorides, and esters in a single step by palladium-catalyzed cross-coupling.35–38 In general, diarylketones have an electron-deficient carbonyl, and phosphine oxide is able to attack the carbonyl, which would then undergo a [1,2]-phospha-Brook rearrangement to produce diarylphosphinates.
Since this phosphinate would be a common synthetic intermediate similar to (pseudo)halogenated diarylmethane, we hypothesized that it could be transformed to diarylmethanes by reduction, tetraarylethanes by dimerization, and triarylmethanes by arylation. Furthermore, a homologation of the intermediate by diborylmethane, followed by arylation would afford triarylethanes, and tetraarylmethanes would be synthesized by arylation of triarylmethanes. Herein, we report a deoxygenative transformation of diarylketones with diphenyl phosphine oxide by using a palladium catalyst and HCO2Na, leading to five different types of multiply arylated arenes in one pot.
Results and discussion
Discovery and screening of optimal conditions for the synthesis of diarylmethanes and tetraarylethanes by deoxygenative transformations
Following our previous work on deoxygenative C–P bond formation,34 benzophenone (1) and diphenylphosphine oxide (1.5 equiv.) with 5.0 mol% PdCl2, 10 mol% dcype (1,2-bis(dicyclohexylphosphino)ethane), and sodium formate (HCO2Na, 1.5 equiv.) as a hydrogen source in 1,2-dimethoxyethane (DME) at 150 °C for 12 h were used as the initial conditions. As a result, these conditions successfully gave the desired diphenylmethane (2A) in 10% yield along with the unexpected tetraphenylethane (3A) in 4% yield. After extensive optimization of the reaction conditions (see the ESI for details†), we identified the optimal conditions for 2A. To our delight, when the conditions were changed to PdCl2/PPh3 as a catalyst and Cs2CO3 (2.0 equiv.), HCO2Na (2.0 equiv.) in DMSO at 150 °C for 1 h, the yield of 2A significantly improved to 93% and no 3A was observed (Table 1, entry 1).|
|
|||||
|---|---|---|---|---|---|
| entry | variationsb | 1/% | 2A/% | 3A/% | 2A′/% |
| a Conditions; 1 (0.20 mmol), PdCl2 (5.0 mol%), PPh3 (20 mol%), Cs2CO3 (2.0 equiv.), HCO2Na (1.5 equiv.), DMSO (1.0 mL), 150 °C, 1 h. Yields were determined by 1H NMR analysis. b Variations from standard conditions. c Cs2CO3 (0.40 equiv.) was used. | |||||
| 1 | None | 0 | 93 | 0 | 0 |
| 2 | w/o PdCl2 | 35 | 0 | 0 | 44 |
| 3 | w/o PPh3 | 14 | 56 | 0 | 15 |
| 4 | w/o HP(O)Ph2 | 86 | 0 | 0 | 12 |
| 5 | w/o Cs2CO3 | 27 | 31 | 12 | 28 |
| 6 | w/o HCO2Na | 64 | 13 | 4 | 14 |
| 7 | 100 °C | 4 | 83 | 0 | 0 |
| 8c | DME, 12 h | 0 | 13 | 76 | 0 |

Next, control experiments were undertaken. The palladium salt is essential for this reaction, because without it, the reaction furnished the “half-reduced” diphenylmethanol (2A′), and the starting material 1 was also recovered (Table 1, entry 2). Without the ligand, the yield was lower (Table 1, entry 3), and diphenylphosphine oxide was critical (Table 1, entry 4). The reaction can proceed without the base, but the yield of 2A and the selectivity for 2A and 3A was significantly decreased (Table 1, entry 5). The reaction resulted in further decreasing product yields without HCO2Na (Table 1, entry 6). Although this reaction is typically performed at 150 °C, it can proceed at 100 °C with only a slight decrease in yield of 2A (Table 1, entry 7). We then examined various conditions to improve the yield of 3A and found that simply changing the solvent to DME, and decreasing the amount of Cs2CO3 (0.40 equiv.) significantly improves the yield of 3A. Finally, by extending the reaction time to 12 h, we succeeded in preferentially producing 3A in 76% yield (Table 1, entry 8, and see the ESI for details†).
Substrate scope for diarylmethanes and tetraarylethanes
Under these optimized conditions, the substrate scope of diarylketones was investigated (Fig. 2). Changing one of phenyl groups of the benzophenone (1) to a naphthyl or a meta-tolyl group also afforded diarylmethanes 2B and 2C in moderate yields. Biphenyl group worked as well (2D), however, installing a methoxy group at the para-position of the biphenyl decreased the yield (2E) even after increasing the amounts of catalysts, since this resulted in a lot of accompanying diarylmethanol (54% yield). The low yield was caused by the failure of the Pd/HCO2Na reduction of the phosphinate formed by the phospha-Brook rearrangement due to the electron-donating group. As a result, the phosphinate was hydrolyzed after quenching by water, resulting in the alcohol. Fluorinated diarylketones gave the corresponding diarylmethanes 2F and 2G in moderate yields. Diarylketones with an alkyne, bis-trifluoromethyl groups, or dimethoxy groups were tolerated to furnish the corresponding diarylmethanes 2H–2J. When the phenyl group of 1 was replaced by heteroaromatics such as pyridine or quinoline, the corresponding diarylmethanes 2K–2N were obtained in good to high yields. Even for cyclic ketones, the corresponding diarylmethanes 2O–2S were afforded, regardless of the ring size or the presence of heteroatoms. We also attempted this reaction with ketoprofen as well as its precursor, giving the corresponding diarylmethanes 2T and 2U. Additionally, diarylketones, which can be readily derivatized in one step from carboxylic-acid-containing drugs such as probenecid and adapalene, successfully converted to the corresponding diarylmethanes 2V and 2W, albeit in low yields. Fenofibrate, which is a well-known pharmaceutical, succeeded in giving product 2X in a moderate yield.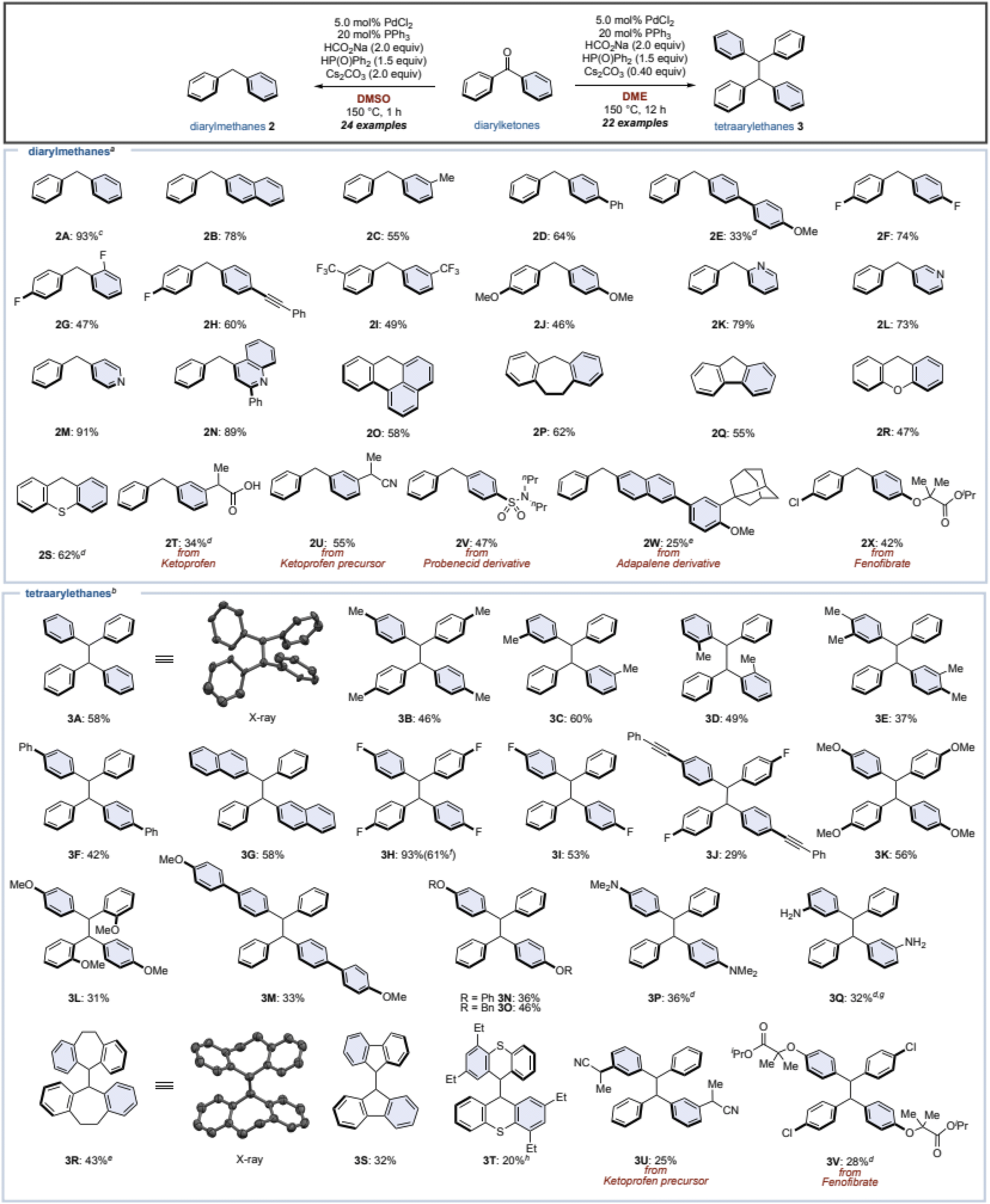 | ||
| Fig. 2 Substrate scope Conditions; a1 (0.40 mmol), diphenylphosphine oxide (1.5 equiv.), PdCl2 (5.0 mol%), PPh3 (20 mol%), HCO2Na (2.0 equiv.), Cs2CO3 (2.0 equiv.), DMSO (1.0 mL), 150 °C, 1 h. b1 (0.40 mmol), diphenylphosphine oxide (1.5 equiv.), PdCl2 (5.0 mol%), PPh3 (20 mol%), HCO2Na (2.0 equiv.), Cs2CO3 (0.40 equiv.), DME (1.0 mL), 150 °C, 12 h. c Yield was determined by 1H NMR analysis due to the volatile nature of 2A. d PdCl2 (10 mol%), PPh3 (40 mol%) were used. e Pn-Bu3 (20 mol%), K2CO3 (2.0 equiv.), MeCN, 12 h. f 6.0 mmol scale. g Isolated as an acetamide by treating with Ac2O. h 20 mol% MePPh2, K2CO3 (2.0 equiv.), MeCN, 12 h. | ||
Next, we examined the substrate scope for the synthesis of tetraarylethanes 3 using the standard conditions (Table 1, entry 8). Benzophenone (1) gave 1,1,2,2-tetraphenylethane (3A) in 58% isolated yield, and the structure of 3A was assigned by X-ray crystallographic analysis. Mono- or di-tolylmethanones with methyl groups in the para-, meta-, or ortho-positions, as well as xylylmethanone smoothly generated the corresponding tetraarylethanes 3B–3E. Diarylketones bearing biphenyl and naphthyl groups afforded the corresponding tetraarylethanes 3F and 3G in moderate yields. Note that when unsymmetrical diarylketones are used, the resulting tetraarylethanes 3 become a mixture of diastereomers with a ratio of almost 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. A ketone with bis-4-fluorophenyl groups reacted well to give the desired product 3H in a high yield, and this reaction also proceeds on gram scale (61% yield). In contrast, reactions with a mono-fluorophenyl group gave 3I and 3J in moderate to low yields. Diarylketones with methoxy, alkoxy, dimethylamino, and NH2 groups were tolerated to furnish the corresponding tetraarylethanes 3K–3Q. In the case of low yields, the corresponding diarylmethanes and the diarylmethanols were obtained as by-products. Cyclic ketones such as dibenzosuberone did not progress at all under our standard conditions. Therefore, we re-optimized the reaction and found that tributyl phosphine was effective as a ligand to give 3R in 43% yield (the structure of 3R was assigned by X-ray crystallographic analysis). However, the reaction conditions were ineffective with other cyclic ketones (3S and 3T). Ketoprofen precursor and fenofibrate were applicable to this reaction as well (3U and 3V).
:1. A ketone with bis-4-fluorophenyl groups reacted well to give the desired product 3H in a high yield, and this reaction also proceeds on gram scale (61% yield). In contrast, reactions with a mono-fluorophenyl group gave 3I and 3J in moderate to low yields. Diarylketones with methoxy, alkoxy, dimethylamino, and NH2 groups were tolerated to furnish the corresponding tetraarylethanes 3K–3Q. In the case of low yields, the corresponding diarylmethanes and the diarylmethanols were obtained as by-products. Cyclic ketones such as dibenzosuberone did not progress at all under our standard conditions. Therefore, we re-optimized the reaction and found that tributyl phosphine was effective as a ligand to give 3R in 43% yield (the structure of 3R was assigned by X-ray crystallographic analysis). However, the reaction conditions were ineffective with other cyclic ketones (3S and 3T). Ketoprofen precursor and fenofibrate were applicable to this reaction as well (3U and 3V).
Studies for elucidation of the reaction mechanism
We hypothesize the reaction mechanism as follows: (1) the diarylketone starting material could be converted to a diarylphosphinate via a phospha-Brook rearrangement; (2) then, reduction and dimerization by a Pd/HCO2Na catalytic system would lead to diarylmethanes and tetraarylethanes. However, their precise mechanisms and requirements for adequate conditions are not known. Therefore, we performed additional control experiments to elucidate the reaction mechanism for the synthesis of diarylmethane 2 and tetraarylethane 3 (Fig. 3). When benzophenone (1) was reacted with diphenylphosphine oxide in the presence of Cs2CO3 as a base without Pd/HCO2Na, phosphinate 4A was obtained quantitatively (Fig. 3A). Although stoichiometric amounts of metal carbonates (Cs2CO3 or K2CO3) are not required, the reaction did not proceed at all without the addition of metal carbonates. These results indicate that the base is critical for the formation of phosphinate 4A, but that high temperatures (150 °C), a palladium catalyst, and HCO2Na are not required (see the ESI for details†).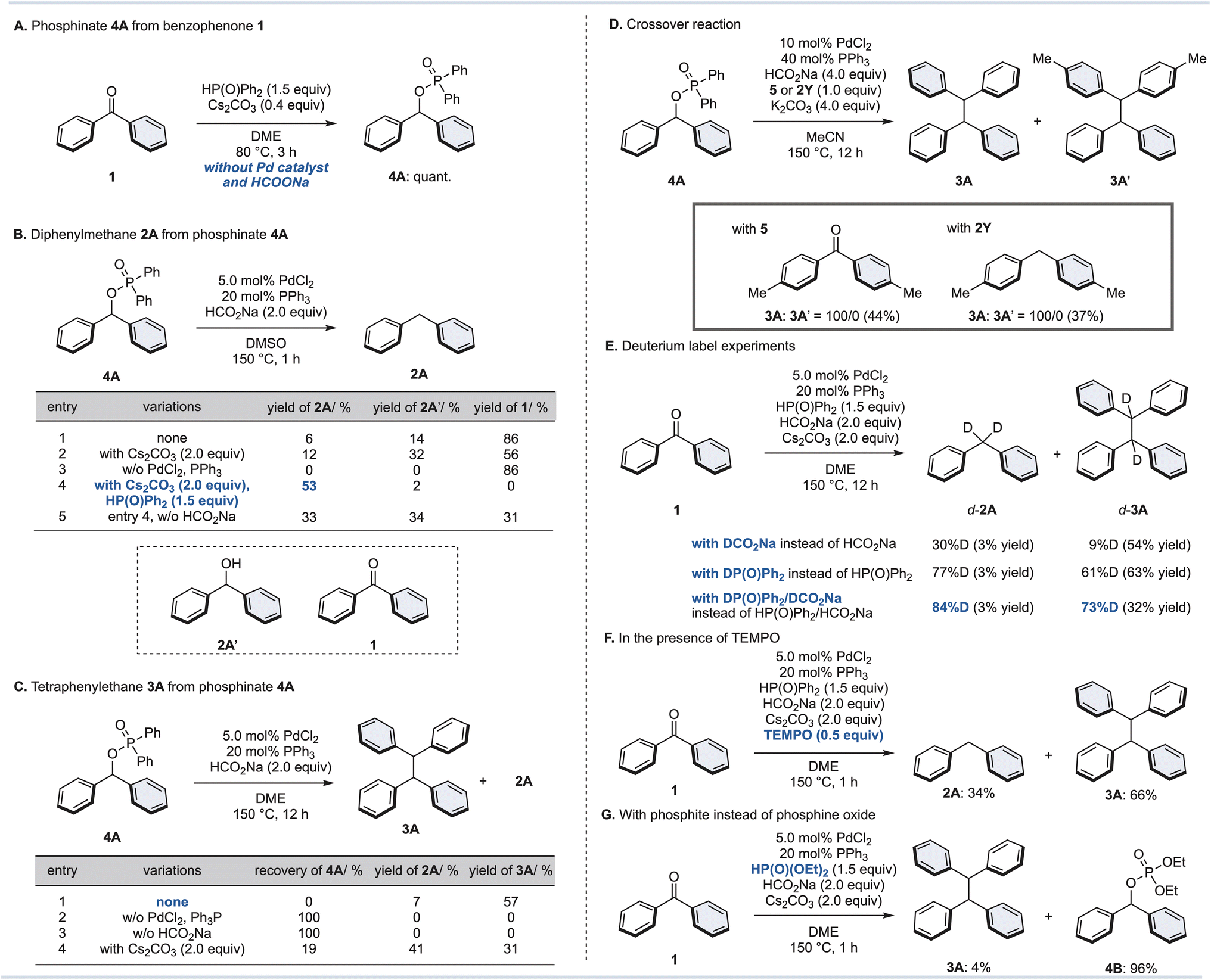 | ||
| Fig. 3 (A) Phosphinate 4A from benzophenone (1). (B) Diphenylmethane (2A) from phosphinate 4A. (C) Tetraphenylethane 3A from phosphinate 4A. (D) Crossover reaction. Numbers in the parentheses are yields of 3A. (E) Deuterium labeling experiments. (F) Standard conditions with TEMPO as a radical scavenger. (G) With phosphite instead of phosphine oxide. | ||
Next, we attempted the diphenylmethane (2A) synthesis from phosphinate 4A under our optimal conditions without addition of Cs2CO3 (Fig. 3B). Surprisingly, the desired diphenylmethane 2A was obtained in only 6% yield, and ketone 1 was obtained as the major product (entry 1). The addition of Cs2CO3 slightly improved the yield of 2A (12%), but increased the yield of alcohol 2A′ as well, which is an effect of phosphinate 4A being partially hydrolyzed (entry 2). Without the addition of the palladium catalyst, 2A was not produced at all and was completely converted to ketone 1 (entry 3). It was finally realized that the desired diphenylmethane 2A was obtained in 53% yield by adding diphenylphosphine oxide and Cs2CO3 (entry 4). In the absence of HCO2Na (entry 5), the yield of 2A decreased, but the yield increased for alcohol 2A′ and ketone 1. The existing equilibrium between ketone 1 and phosphinate 4A in DMSO suggests that reduction might be proceeding as soon as the phosphinate 4A is produced by the Pd/HCO2Na catalytic system.
Additional control experiments for the synthesis of tetraphenylethane 3A from phosphinate 4A were performed (Fig. 3C). Phosphinate 4A in DME in the presence of PdCl2/PPh3 and HCO2Na gave the desired tetraphenylethane 3A in 57% yield, along with 7% diphenylmethane 2A as a byproduct. This result is mostly consistent with the results from ketone 1 (Table 1, entry 8), but the requirements were further elaborated. Without the addition of a palladium catalyst or HCO2Na, the reaction hardly proceeded (entries 2 and 3). Furthermore, an excess amount of Cs2CO3 was found to inhibit the dimerization reaction (entry 4). Hence, the amount of Cs2CO3 in the dimerization of 1 was kept minimal (0.40 equiv., see Fig. 2).
Furthermore, the following crossover experiment was performed to confirm whether dimerization of the phosphinate 4A or cross-coupling with the diphenylketone (1)/diphenylmethane (2A) occurs (Fig. 3D). To this end, phosphinate 4A was reacted under our standard conditions for dimerization with dip-tolyl ketone 5 or diarylmethane 2Y (1.0 equiv. of each), respectively. If a crossover reaction had occurred, 3A′ would have been detected in addition to 3A, however 3A′ was not detected at all. These results suggest that a dimerization reaction of phosphinate 4A is proceeding.
Moreover, deuterium labelling experiments were conducted (Fig. 3E). Under optimal conditions for 2A synthesis, deuterated DCO2Na was added instead of HCO2Na to afford d-2A (30% D) and d-3A (9% D). When diphenylphosphine oxide (DP(O)Ph2) was used instead of diphenylphosphine oxide, this led to a dramatic increase in the deuterated ratio of d-2A (77% D) and d-3A (61% D). Finally, both deuterated agents were combined to give 2A (84% D) and 3A (73% D). These results suggest that diphenylphosphine oxide works as a hydrogen source in this reaction.
We also attempted this reductive transformation under a radical scavenger (Fig. 3F): in the presence of TEMPO, the reaction gave both 2A and 3A. This result confirmed that these reactions do not proceed through a radical pathway. Lastly, the same reaction was attempted using diethyl phosphite instead of diphenylphosphine oxide (Fig. 3G). This is because generally the phospha-Brook rearrangement is almost exclusively reported for phosphite esters.39–42 When this reaction was performed with diethyl phosphite, the phosphate ester 4B was obtained in 96% yield, but the desired tetraphenylethane 3A was formed in small amounts. Thus, the reaction proved to be effective only with diphenylphosphine oxide. Based on the results of these control experiments, it was concluded that the dimerization reaction also proceeds via diarylphosphinate. Although the exact reaction mechanism of dimerization remains unclear, similar dimerization reactions have proceeded when diaryl pseudohalides were used in the presence of Pd, and Ni catalysts,43–45 and we believe that dimerization occurs through disproportionation of the oxidative addition complexes of the Pd catalysts.
Synthesis of triarylmethanes
Next, we considered that the phosphinate is a (pseudo)halide, and that triarylmethane could be synthesized by screening for appropriate conditions. To the best of our knowledge, Friedel–Crafts-type arylations or cross-couplings of phosphite have been reported,46–48 but there is no report for phosphinates thus far. As a result of our investigation, we found the following two reaction conditions for the one-pot synthesis of triarylmethane from diarylketones (Fig. 4). | ||
| Fig. 4 Synthesis of triarylmethanes. Conditions: a diarylketone (0.40 mmol), diphenylphosphine oxide (1.5 equiv.), Cs2CO3 (0.40 equiv.), DME (2.0 mL), 80 °C, 3 h; then arene (1.0 mL), TfOH (2.0 equiv.), RT, 2 min. b Diarylketone (0.40 mmol), diphenylphosphine oxide (1.2 equiv.), Cs2CO3 (2.0 equiv.), 1,4-dioxane (2.0 mL), 150 °C, 1 h; then arylboronic acid (1.5 equiv.), Pd(OAc)2 (10 mol%), P (p-tolyl)3 (40 mol%), 150 °C, 1 h. c The ratio of isomers (para/ortho-position (6A) or C2/C3 position (6B)) is shown in parentheses. | ||
For electron-rich aromatic rings, Friedel–Crafts-type reaction conditions were effective. Benzophenone (1) was reacted with diphenyl phosphine oxide (1.5 equiv.) and Cs2CO3 (0.40 equiv.) in DME at 80 °C for 3 h to generate phosphinate 4Ain situ, followed by the addition of toluene, thiophene, or mesitylene with TfOH (2.0 equiv.) at room temperature for 2 min to give the corresponding triarylmethanes 6A–6C in moderate yields. Although small amounts of positional isomers were produced when generating 6A and 6B, triarylmethanes can be synthesized rapidly from benzophenone (1) without the use of metal catalysts. Not only benzophenone (1), but also an unsymmetrical diaryl ketone, a cyclic diaryl ketone, and a diarylketone with substituents such as a chlorine and an alkoxy could be converted to the corresponding triarylmethanes 6D–6F in moderate yields. When an aromatic ring with an electron-withdrawing substituent was used as the substrate, a Suzuki–Miyaura-type coupling reaction was effective. Similarly, after the preparation of phosphinate 4A, the corresponding triarylmethanes were formed simply by adding arylboronic acid (1.5 equiv.), Pd(OAc)2 (10 mol%), and P (p-tolyl)3 (40 mol%) to the reaction vessel and stirring at 150 °C for 1 h. In addition to triarylmethanes 6G–6I with trifluoromethyl, cyano, and ester groups, triarylmethanes 6J–6L with methoxy, phenyl, and phenoxy groups at the para position of the aromatic ring could be synthesized.
Synthetic utility and applications of this method
Finally, the synthetic utility and applications of this method were investigated (Fig. 5). As mentioned in the introduction, diaryl ketones can be readily prepared from aromatic carboxylic acids. Therefore, we demonstrated a one-pot diarylmethane/tetraarylethane synthesis from aryl carboxylic acids (Fig. 5A). The reaction of benzoic acid with phenylboronic acid and Boc2O in the presence of a palladium catalyst gave benzophenone (1) via a mixed acid anhydride.33 When 1 was subjected directly to the diverging conditions of Fig. 2 without isolation in one pot, diphenylmethane (2A) and tetraphenylmethane (3A) were successfully synthesized in 70% and 43% yields from benzoic acid, respectively. We also attempted a deoxygenative allylation of diarylketones (Fig. 5B). After phosphinate formation by a phospha-Brook rearrangement of naphthalen-2-yl(p-tolyl)methanone (7), allyltrifluoroborate with palladium catalyst/L1 led to benzyl substitution to form allyldiarylmethane 8 in 52% yield. In contrast, the use of naphthalen-2-yl (phenyl)methanone (9), which is a demethyl form of 7, afforded diarylmethane 10, which substituted at the para-position of the phenyl group. Diarylmethane 10 was likely formed from a π-benzyl palladium complex by a dearomatizing allylation reaction.49,50 | ||
| Fig. 5 Synthetic utility and applications of deoxygenative transformation of diarylketones. (A) One-pot synthesis from aryl carboxylic acid. (B) Allylation of diarylketone. (C) Intramolecular dimerization/oxidation. (D) Diarylmethane synthesis from arylaldehyde. (E) Triarylethane synthesis from diarylketone. (F) Tetraarylmethane synthesis from diarylketone. | ||
Next, an intramolecular dimerization was carried out (Fig. 5C). Applying the reaction conditions from the tetraarylethane synthesis to compound 11, which is essentially two diphenylketones linked together, the diphosphinate intermediate 12 becomes diphenylphenanthrene 13 as the coupling and subsequent auto-oxidation proceeds. Oxidation of 13 with DDQ in the presence of TfOH gave dibenzo [g,p]chrysene 14 in good yield. This method has the potential to be used in the synthesis of various polycyclic aromatic hydrocarbons (PAHs).
Furthermore, the phospha-Brook rearrangement can be used to make diarylmethane from aromatic aldehydes (Fig. 5D). For example, diphenylphosphine oxide and a base are applied to 2-naphthaldehyde (15) to form the corresponding phosphinate, followed by an sp3 Suzuki–Miyaura-type coupling with phenylboronic acid to afford diarylmethane 2B in moderate yield. Since aromatic aldehydes are also inexpensive and readily available, a variety of diarylmethanes may be synthesized in a practical fashion.
Finally, we attempted to synthesize triarylethanes and tetraarylmethanes from diarylketones (Fig. 5E and F). The coupling reaction of 9 with diborylmethane (1.5 equiv.) underwent phospha-Brook rearrangement, followed by palladium catalyst and base to afford diarylethylboronate 16. A one-pot alkyl-Suzuki–Miyaura type coupling with phenylboronic acid was carried out without isolation of 16, and then triarylethanes 17 were successfully produced in 37% yield from diarylketone 9. Furthermore, after subjecting benzophenone (1) to the reaction conditions of Fig. 5 for the synthesis of triarylmethane, the resulting triphenylmethane 18 was treated with benzofuran, DDQ and catalytic Sc(OTf)3 to give tetraarylmethane 19.51 The structure of tetraarylmethane 19 was determined by X-ray crystallography.
Conclusions
In summary, we developed a unified synthesis of multiply arylated alkanes from diarylketones. The key for this reaction is the readily generation of diarylphosphinates from diarylketones with diphenylphosphine oxide via a phospha-Brook rearrangement. Using this method, we have succeeded in synthesizing five different types of multiply arylated alkanes from diarylketones in a single step or one-pot. Expanding the range of nucleophiles for diarylphosphinates including enantioselective transformations is currently undergoing in our laboratory.52Data availability
All experimental data is available in the ESI.†Author contributions
J. Y. conceived and designed the study. M. B. K. performed the chemical experiments. M. B. K. and K. M. analyzed the data. K. K. performed the X-ray crystallography experiments and analyzed the obtained data. J. Y. wrote the manuscript and all authors discussed the results and commented on the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI Grant Number JP21K18966, JP21H05213 (to J.Y.), and JP21K05079 (to K.M.). This work was also partly supported by JST ERATO Grant Number JPMJER1901 (to J.Y.). We thank Mizuho Watanabe to help a part of experiment. We also thank Iwatani Corporation for providing Cs2CO3 as a gift. The Materials Characterization Central Laboratory in Waseda University is acknowledged for the support of HRMS measurement. We also would like to thank National Institute of Health Sciences in Japan for the support of NMR measurement.Notes and references
- Y. Ohtake, T. Sato, T. Kobayashi, M. Nishimoto, N. Taka, K. Takano, K. Yamamoto, M. Ohmori, M. Yamaguchi, K. Takami, S.-Y. Yeu, K.-H. Ahn, H. Matsuoka, K. Morikawa, M. Suzuki, H. Hagita, K. Ozawa, K. Yamaguchi, M. Kato and S. Ikeda, J. Med. Chem., 2012, 55, 7828–7840 CrossRef CAS PubMed.
- B. Zhou, D. X. Liu, X. J. Yuan, J. Y. Li, Y. C. Xu, J. Li, Y. Li and J. M. Yue, Research, 2018, 2018, 2674182 CrossRef CAS PubMed.
- R. Palchaudhuri, M. J. Lambrecht, R. C. Botham, K. C. Partlow, T. J. Van Ham, K. S. Putt, L. T. Nguyen, S.-H. Kim, R. T. Peterson and T. M. Fan, Cell Rep., 2015, 13, 2027–2036 CrossRef CAS PubMed.
- K. S. Schneider, C. J. Groß, R. F. Dreier, B. S. Saller, R. Mishra, O. Gorka, R. Heilig, E. Meunier, M. S. Dick, T. Ćiković, J. Sodenkamp, G. Médard, R. Naumann, J. Ruland, B. Kuster, P. Broz and O. Groß, Cell Rep., 2017, 21, 3846–3859 CrossRef CAS PubMed.
- R. P. Alexander, G. J. Warrellow, M. A. W. Eaton, E. C. Boyd, J. C. Head, J. R. Porter, J. A. Brown, J. T. Reuberson, B. Hutchinson, P. Turner, B. Boyce, D. Barnes, B. Mason, A. Cannell, R. J. Taylor, A. Zomaya, A. Millican, J. Leonard, R. Morphy, M. Wales, M. Perry, R. A. Allen, N. Gozzard, B. Hughes and G. Higgs, Bioorg. Med. Chem. Lett., 2002, 12, 1451–1456 CrossRef CAS PubMed.
- H. M. El-Kaderi, J. R. Hunt, J. L. Mendoza-Cortes, A. P. Côté, R. E. Taylor, M. O'Keeffe and O. M. Yaghi, Science, 2007, 316, 268–272 CrossRef CAS PubMed.
- H. Furukawa and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 8875–8883 CrossRef CAS PubMed.
- M. Rueping and B. J. Nachtsheim, Beilstein J. Org. Chem., 2010, 6, 6 Search PubMed.
- S. Mondal and G. Panda, RSC Adv., 2014, 4, 28317–28358 RSC.
- M. Nambo and C. M. Crudden, ACS Catal., 2015, 5, 4734–4742 CrossRef CAS.
- R. Kshatriya, V. P. Jejurkar and S. Saha, Eur. J. Org. Chem., 2019, 2019, 3818–3841 CrossRef CAS.
- X. Liu, X. Wu, L. Zhang, X. Lin and D. Huang, Synthesis, 2020, 52, 2311–2329 CrossRef CAS.
- U. Gulati, R. Gandhi and J. K. Laha, Chem.–Asian J., 2020, 15, 3135–3161 CrossRef CAS PubMed.
- J. Li, C.-Y. Huang, M. Ataya, R. Z. Khaliullin and C.-J. Li, Nat. Commun., 2021, 12, 4970 CrossRef CAS PubMed.
- X. Cheng, H. Lu and Z. Lu, Nat. Commun., 2019, 10, 3549 CrossRef PubMed.
- G. P. R. Freure, E. A. Skrotzki, J.-D. E. Lavertu and S. G. Newman, ACS Catal., 2021, 11, 12258–12263 CrossRef CAS.
- M. R. Harris, L. E. Hanna, M. A. Greene, C. E. Moore and E. R. Jarvo, J. Am. Chem. Soc., 2013, 135, 3303–3306 CrossRef CAS PubMed.
- M. Nambo and C. M. Crudden, Angew. Chem., Int. Ed., 2014, 53, 742–746 CrossRef CAS PubMed.
- I. N.-M. Leibler, M. A. Tekle-Smith and A. G. Doyle, Nat. Commun., 2021, 12, 6950 CrossRef CAS PubMed.
- C. M. Crudden, C. Ziebenhaus, J. P. G. Rygus, K. Ghozati, P. J. Unsworth, M. Nambo, S. Voth, M. Hutchinson, V. S. Laberge, Y. Maekawa and D. Imao, Nat. Commun., 2016, 7, 11065 CrossRef PubMed.
- B. Shrestha, P. Basnet, R. K. Dhungana, S. KC, S. Thapa, J. M. Sears and R. Giri, J. Am. Chem. Soc., 2017, 139, 10653–10656 CrossRef CAS PubMed.
- P. Guo, K. Wang, W.-J. Jin, H. Xie, L. Qi, X.-Y. Liu and X.-Z. Shu, J. Am. Chem. Soc., 2021, 143, 513–523 CrossRef CAS PubMed.
- K. N. Babu, F. Massarwe, I. Shioukhi and A. Masarwa, Angew. Chem., Int. Ed., 2021, 60, 26199–26209 CrossRef CAS PubMed.
- D. Cao, C.-C. Li, H. Zeng, Y. Peng and C.-J. Li, Nat. Commun., 2021, 12, 3729 CrossRef CAS PubMed.
- S. Zhang, M. Vayer, F. Noël, V. D. Vuković, A. Golushko, N. Rezajooei, C. N. Rowley, D. Lebœuf and J. Moran, Chem, 2021, 7, 3425–3441 CAS.
- S. Zhang, B.-S. Kim, C. Wu, J. Mao and P. J. Walsh, Nat. Commun., 2017, 8, 14641 CrossRef PubMed.
- J. P. G. Rygus and C. M. Crudden, J. Am. Chem. Soc., 2017, 139, 18124–18137 CrossRef CAS PubMed.
- C. Chen, H. Wang, T. Li, D. Lu, J. Li, X. Zhang, X. Hong and Z. Lu, Angew. Chem., Int. Ed., 2022, 61, e202205619 CAS.
- J. Li, C. Huang and C.-J. Li, Angew. Chem., Int. Ed., 2022, 61, e202112770 CAS.
- L. Wang, T. Zhang, W. Sun, Z. He, C. Xia, Y. Lan and C. Liu, J. Am. Chem. Soc., 2017, 139, 5257–5264 CrossRef CAS PubMed.
- D. Matheau-Raven, P. Gabriel, J. A. Leitch, Y. A. Almehmadi, K. Yamazaki and D. J. Dixon, ACS Catal., 2020, 10, 8880–8897 CrossRef CAS.
- W. Ou, X. Xiang, R. Zou, Q. Xu, K. P. Loh and C. Su, Angew. Chem., Int. Ed., 2021, 60, 6357–6361 CrossRef CAS PubMed.
- I. D. Alshakova and M. Albrecht, ACS Catal., 2021, 11, 8999–9007 CrossRef CAS.
- M. B. Kurosawa, R. I. K. Muto and J. Yamaguchi, J. Am. Chem. Soc., 2020, 142, 7386–7392 CrossRef CAS PubMed.
- L. J. Gooßen and K. Ghosh, Angew. Chem., Int. Ed., 2001, 113, 3566–3568 CrossRef.
- T. B. Halima, W. Zhang, I. Yalaoui, X. Hong, Y.-F. Yang, K. N. Houk and S. G. Newman, J. Am. Chem. Soc., 2017, 139, 1311–1318 CrossRef PubMed.
- A. H. Dardir, P. R. Melvin, R. M. Davis, N. Hazari and M. M. Beromi, J. Org. Chem., 2018, 83, 469–477 CrossRef CAS PubMed.
- R. G. Kinney and B. A. Arndtsen, Angew. Chem., Int. Ed., 2019, 58, 5085–5089 CrossRef CAS PubMed.
- A. Kondoh and M. Terada, Bull. Chem. Soc. Jpn., 2021, 94, 339–356 CrossRef CAS.
- M. Hayashi and S. Nakamura, Angew. Chem., Int. Ed., 2011, 50, 2249–2252 CrossRef CAS PubMed.
- M. A. Horwitz, N. Tanaka, T. Yokosaka, D. Uraguchi, J. S. Johnson and T. Ooi, Chem. Sci., 2015, 6, 6086–6090 RSC.
- Y. Qian, Q. Dai, Z. Li, Y. Liu and J. Zhang, Org. Lett., 2020, 22, 4742–4748 CrossRef CAS PubMed.
- L. H. Taylor, M. R. Harris and E. R. Jarvo, Angew. Chem., Int. Ed., 2012, 51, 7790–7793 CrossRef PubMed.
- M. Asai Miura and K. Hirano, J. Org. Chem., 2022, 87, 7436–7445 CrossRef PubMed.
- K. H. Asai and M. Miura, Eur. J. Org. Chem., 2022, e202101535 CAS.
- A. Kondoh, R. Ozawa, T. Aoki and M. Terada, Org. Biomol. Chem., 2017, 15, 7277–7281 RSC.
- Y. Yamamoto, T. Takase, E. Kuroyanagi and T. Yasui, Chem. Commun., 2021, 57, 3877–3880 RSC.
- A. Kondoh, N. Tasato, T. Aoki and M. Terada, Org. Lett., 2020, 22, 5170–5175 CrossRef CAS PubMed.
- S. Zhang, Y. Yamamoto and M. Bao, Adv. Synth. Catal., 2021, 363, 587–601 CrossRef CAS.
- M. Komatsuda, K. Muto and J. Yamaguchi, Org. Lett., 2018, 20, 4354–4357 CrossRef CAS PubMed.
- M. Nambo, J. Yim, K. Fowler and C. Crudden, Synlett, 2017, 28, 2936–2940 CrossRef CAS.
- A prior version of this work was deposited as a preprint: M. B. Kurosawa, K. Kato, K. Muto and J. Yamaguchi, ChemRxiv, 2022, DOI:10.26434/chemrxiv-2022-h1860.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2121107 (3A), 2121108 (3R) and 2159413 (18). For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc03720c |
| This journal is © The Royal Society of Chemistry 2022 |