
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiscovery strategy leads to the first melt-castable cocrystal based on an energetic oxidizing salt†
Michael K.
Bellas
ab and
Adam J.
Matzger
 *b
*b
aResearch Department, Chemistry Division, United States Navy – Naval Air Systems Command (NAVAIR), Naval Air Warfare Center, Weapons Division (NAWCWD), 1900 N. Knox Road, China Lake, California 93555, USA
bDepartment of Chemistry, University of Michigan, 930 North University Avenue, Ann Arbor, Michigan 48109-1055, USA. E-mail: matzger@umich.edu
First published on 10th October 2022
Abstract
Cocrystallization is a synthetic method employed across fields to improve functional materials while preserving properties inherent to the molecules/ions involved. However, there is no guarantee that cocrystals will demonstrate improved properties relative to the constituent materials. Oxygen balance, which is closely correlated to the performance of energetic materials, is an exception in that this attribute may be targetted with certainty. The combination of energetic oxidizing salts with small molecules presents a seemingly straightforward path to energetic materials with desirable performance properties. Unfortunately no general approach for the cocrystallization of salts and small molecules (salt cocrystallization) has yet emerged. Presented here is such an approach, focussing on ammonium salts, and applied to the energetic oxidizing salt ammonium dinitramide to achieve a melt-castable energetic material. Though focused on ammonium salts, this salt cocrystallization paradigm is a general approach that may be extended to other ions.
Introduction
In the past few decades cocrystallization has emerged as a powerful supramolecular synthesis tool that affords cocrystal products ranging from pharmaceuticals to explosives.1,2 Cocrystallization combines multiple unique components within a crystal lattice, often with defined stoichiometry, and, unlike salt formation, there is no requirement for charged species. Distinct advantages over covalent synthesis include achieving changes in materials properties without altering the chemical identity of the constituents, thus preserving properties inherent to the molecules or ions involved.3–6 This approach has been leveraged to address the poor materials properties (morphology, hygroscopicity, solubility, thermal stability, etc.) of otherwise promising candidate materials within an array of fields, e.g. preserving pharmacological properties of therapeutics while improving solubility/bioavailability thus enhancing efficacy.5,7,8 While approaches to the cocrystallization of neutral molecules have been developed and successfully applied (e.g., the supramolecular synthon approach),9–11 the cocrystallization of salts with neutral molecules (salt cocrystallization) has not progressed to the same extent. As a result, the data required to develop a widely applicable approach to salt cocrystallization are simply not available; simple database searching for supramolecular synthons is often an ineffective approach in salt cocrystallization. Additionally, the variety of ions in common use precludes the development of a single all-encompassing model for salt cocrystallization. Thus, each ion must be treated separately and salt cocrystallization models developed independently; however, the principles used to develop such models may themselves be broadly applicable. We present here an approach to salt cocrystallization, as it is applied to ammonium dinitramide (ADN), that employs a model ammonium salt in discovery efforts. ADN was developed as a chlorine/perchlorate free oxidant for use in composite propellant formulations and exhibits a positive oxygen balance (OB).12 OB relates the amount of oxygen present to that required for complete oxidation; positive values indicate excess oxygen, and the vast majority of positive OB materials are salts. OB is closely correlated with the performance of energetic materials.13 Despite its positive OB, ADN has failed to see broad implementation due to poor materials properties such as hygroscopicity, undesirable morphology, and thermal instability. As these impediments to ADN's deployment are properties known to be affected through cocrystallization, ADN presented an ideal target for this study and afforded the first melt-castable salt cocrystal incorporating an energetic oxidizing salt.Results and discussion
Our salt cocrystal discovery paradigm (Fig. 1) incorporates both existing solid-state data (crystal structures) and salt cocrystallization experiments. The application of this paradigm began with the identification of ADN as the target salt and hydrogen bonding with NH4+ as an exploitable interaction. Developing an ADN cocrystallization strategy based on interactions with NH4+ may provide a broader strategy for salt cocrystallization with energetic oxidizing salts, many of which contain ammonium functionality (Fig. 2). While NH4+ presents a distinct opportunity for charge-assisted hydrogen bonding interactions with neutral molecules (coformers) there is little available data concerning coformer⋯NH4+ interactions in the solid-state (i.e., few reported NH4+ salt cocrystals).5,14 These data, retrieved from the Cambridge Structural Database (CSD),15 were curated according to how frequently a given organic functional group (FG) is observed interacting with NH4+ and the average distance of interaction (Fig. 2b).16 The most frequently observed FGs in this data set are nitrogen heterocycles and amides; urea moieties are included under the amide heading because they contain a carbonyl-nitrogen subunit corresponding to the search term. Ranking the FGs according to how closely, on average, they are associated with NH4+ within their structures identified amide carbonyls as having the closest interactions with NH4+. This interaction ranks highly in both occurrence frequency and in possessing a short interatomic interaction distance, suggesting it is both strong and reliable. | ||
| Fig. 1 Flow chart diagram highlighting the ammonium salt cocrystal discovery paradigm presented here with ADN shown explicitly as the target salt. | ||
 | ||
| Fig. 2 (a) NH4+ balanced energetic oxidizing salts including ADN, ammonium perchlorate (AP), ammonium nitrate (AN), hydrazinium nitroformate (HNF), and hydroxylammonium nitrate (HAN). (b) Data from the CSD showing the occurrence and the average distance (as a function of van der Waals overlap) of NH4+⋯organic functional group interactions in the solid-state; van der Waals (VDW) overlap is calculated as the summation of VDW radii (ΣVDWradii) minus the observed interaction distance (d). (c) View along c-axis of ADN–PDO and AH–PDO crystal structures showing lamellar architecture in both cases, chemical structures of PDO and AH provided. | ||
To experimentally investigate amide⋯NH4+ interactions, a model salt that reduces the complexity of the system was employed. An immediate advantage of a model salt, in the context of energetic materials research, is the reduced exposure to energetic materials which is a key aspect safety protocols in such work. Our selection of a model salt was influenced by several criteria and ultimately balanced the need to generate data concerning this synthon with the need for conclusions drawn from these data to be applicable within our target system. Ammonium hexafluorophosphate (AH) represents a suitable selection as a model salt due to its similarities to ADN in solubility and size, as well as its ease of handling/stability, and economy/availability. Additionally, the simple, symmetric, weakly-coordinating nature of the anion (PF6−) affords a system where NH4+ interactions may be investigated while minimizing the impact of the counterion. Experimental validation of the selection of AH as an ADN surrogate was realized in an AH pyrazine-1,4-dioxide (PDO) salt cocrystal (AH–PDO); PDO is the only coformer outside of crown ether derivatives successfully cocrystallized with ADN.5,17 The ADN–PDO and AH–PDO salt cocrystals share several commonalities including 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 salt:PDO stoichiometry and a distinctive lamellar architecture (Fig. 2c).16
:1 salt:PDO stoichiometry and a distinctive lamellar architecture (Fig. 2c).16
In selecting amide-bearing coformers for cocrystallization experiments the transferability of the work to energetic systems was considered (i.e., how common is amide functionality in energetic molecules and how is it represented: free NH2, N-alkyl, cyclic, etc.). Cataloging the structures of known energetic molecules according to functional group revealed that amide functionality is rare, whereas cyclic urea moieties are reasonably well represented.18 Further analysis of our search results revealed that two-thirds of the cataloged amide⋯NH4+ interactions were actually urea moiety⋯NH4+ interactions. Thus, 2-imidizolidinone (2Im), being the simplest/smallest organic molecule containing a cyclic urea moiety, was selected as a coformer (Fig. 3a) and cocrystallized with AH.
 | ||
| Fig. 3 (a) Interactions between 2Im and AH in the crystal structure of AH–2Im, select interactions denoted with dashed lines. (b) Lamellar architecture, coordination environment of NH4+, packing with urea lamellae, and the urea-dimer synthon as in AH–urea. | ||
Single crystal X-ray diffraction reveals that within the AH–2Im cocrystal (AH–2Im) each NH4+ has close contacts with three 2Im carbonyl oxygen atoms and three PF6− ions. The fact that three coulombic NH4+⋯PF6− interactions have been displaced by NH4+⋯2Im interactions (pure AH has six NH4+⋯PF6− interactions per NH4+) suggests robustness of the NH4+⋯urea carbonyl synthon. Two NH4+ ions and two 2Im molecules form a hydrogen bonded cycle which interacts with two PF6− ions (Fig. 3a) via hydrogen bonding. These interactions result in a repeating tape motif in the structure which combines to form sheets via 2Im C–H hydrogen bond donation to PF6− ions. These sheets then layer via the third NH4+⋯2Im carbonyl interaction (Fig. 3a).
Seeking additional data concerning the NH4+⋯urea carbonyl synthon, urea was investigated as a coformer. Urea has demonstrated promise as an additive in energetic systems, acting as a combustion temperature modifier, burn rate modifier, and NOx inhibitor.19,20 The cocrystallization of AH with urea was successful and, like the PDO cocrystals, AH–urea presents lamellar architecture (Fig. 3b). In this structure each NH4+ is in an octahedral coordination environment with five PF6− close contacts and one NH4+⋯urea carbonyl interaction (Fig. 3b). Packing within the AH lamellae is reminiscent of that in pure AH, whereas the urea molecules pack in a manner not represented in known urea polymorphs. Urea molecules form dimers (Fig. 3b) that stack in a herringbone manner with both nitrogen and oxygen atoms acting as hydrogen bond acceptors for adjacent dimers. In this structure only one NH4+⋯PF6− interaction is replaced by an NH4+⋯urea carbonyl interaction (relative to pure AH). AH–urea highlights an additional, and necessary, consideration in cocrystallization experiments: the evaluation of all potential synthons in the system and the likelihood that they will form. In this system, urea dimerization is possible, and an arrangement is adopted that allows for this synthon while also maintaining the NH4+⋯urea carbonyl synthon.21 These data reveal that lamellar architecture is possible within these systems, which may afford additional avenues of property-driven salt cocrystal engineering, and that traditional molecular synthons are in competition with the desired FG⋯NH4+ interaction being exploited.
Transitioning from the model salt (AH) to the target salt (ADN) using the urea coformers affords insight into how the anion influences the system. The cocrystallization of ADN and 2Im (forming ADN–2Im) was accomplished without altering experimental conditions relative to AH–2Im. In addition, ADN–2Im forms in the same stoichiometry as AH–2Im and the solid-state interactions are analogous. The ease with which the transition from model salt to ADN was accomplished, both experimentally and regarding structural similarity, is due (at least in part) to the transition occurring without the introduction of competitive synthons. This was likely a factor leading to the similarities between AH–PDO and ADN–PDO as well. However, progressing from AH to ADN in the urea system will introduce a new potential synthon: the urea-nitro synthon (Fig. 4a). Though not considered a strong or reliable synthon in supramolecular synthesis, observed in only 13% of systems in which it can form (as determined through analysis of crystal structures deposited in the CSD, see ESI†),15,22 this synthon may be more of a concern here due to the potential for charge-assisted hydrogen bonding interactions with the dinitramide ion. This is an important consideration as formation of the urea-nitro synthon in this system may impede the urea dimer synthon observed in AH–urea resulting in a cocrystal with different stoichiometry and/or crystal packing.
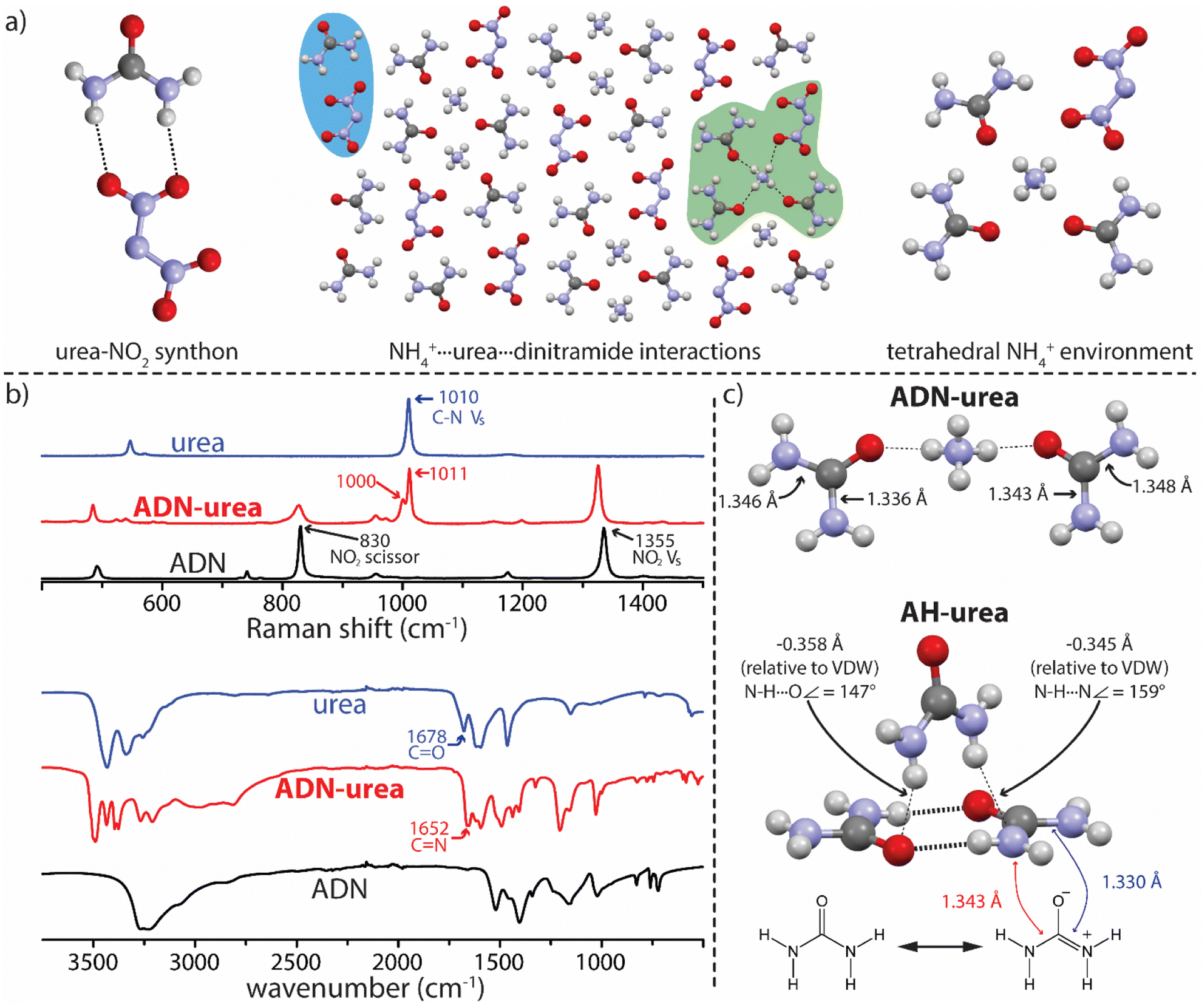 | ||
| Fig. 4 (a) Urea–nitro synthon, salt–molecule interactions, and coordination environment of NH4+ as in ADN–urea. (b) Raman and ATR-FTIR spectra for the ADN–urea system. (c) Diagram showing urea bond lengths in ADN–urea and AH–urea; structures collected at 100 K. | ||
The favorable NH4+⋯urea carbonyl synthon successfully exploited thus far again proved translatable from model to target system and an ADN–urea salt cocrystal was achieved. However, in ADN–urea the urea–nitro synthon is present rather than urea dimerization and the packing arrangement/stoichiometry therefore differs significantly from AH–urea. ADN–urea crystallizes in a 1ADN:2urea stoichiometry and each NH4+ has close contacts with three urea carbonyl oxygen atoms and one dinitramide ion in a tetrahedral orientation (Fig. 4a). Each dinitramide ion has close contacts with five urea molecules, one of which is via the urea-nitro synthon (Fig. 4a). While targeting NH4+ is a proven approach, ADN–urea highlights the importance of considering dinitramide interactions in future ADN cocrystallization endeavors.23
These experiments have provided insight into NH4+ salt cocrystallization in general, but have also provided a deeper understanding of the NH4+⋯urea carbonyl synthon. The Raman spectrum for ADN–urea shows signals at 1011 cm−1 and 1000 cm−1 where urea shows a single signal arising from a symmetrical C–N stretch (Fig. 4b).24 Splitting here indicates either that the symmetry of the urea molecule has been disturbed in the salt cocrystal and/or that urea exists in two distinct and non-equivalent environments. The infrared spectrum of ADN–urea displays an absorbance at 1652 cm−1 while the prominent carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) stretch for urea (1678 cm−1)25 is notably absent. Absorbance around 1650 cm−1 for binary systems incorporating urea is common and attributed to the increase in C–N double bond character (CN) associated with the greater contribution of zwitterionic resonance forms of urea which are favored in polar environments.26,27 Analysis of urea bond lengths in ADN–urea reveals non-equivalent C–N bond lengths within urea molecules which corroborates the spectroscopic analysis (Fig. 4c). A similar analysis of urea bond lengths and interactions in AH–urea also suggests a high degree of urea polarization in that structure. This explains the nitrogen atoms acting as hydrogen bond acceptors between urea dimers in AH–urea, which is rare for amide functionalities, but urea moieties appear more amenable given the ability to asymmetrically delocalize electron density into the carbonyl. These findings may influence other strategies for cocrystal design, such as the use of electrostatic potential mapping,28 where the assumed symmetry of urea moieties can influence the outcome.
O) stretch for urea (1678 cm−1)25 is notably absent. Absorbance around 1650 cm−1 for binary systems incorporating urea is common and attributed to the increase in C–N double bond character (CN) associated with the greater contribution of zwitterionic resonance forms of urea which are favored in polar environments.26,27 Analysis of urea bond lengths in ADN–urea reveals non-equivalent C–N bond lengths within urea molecules which corroborates the spectroscopic analysis (Fig. 4c). A similar analysis of urea bond lengths and interactions in AH–urea also suggests a high degree of urea polarization in that structure. This explains the nitrogen atoms acting as hydrogen bond acceptors between urea dimers in AH–urea, which is rare for amide functionalities, but urea moieties appear more amenable given the ability to asymmetrically delocalize electron density into the carbonyl. These findings may influence other strategies for cocrystal design, such as the use of electrostatic potential mapping,28 where the assumed symmetry of urea moieties can influence the outcome.
Given the promising attributes of both urea and ADN with respect to energetic materials, ADN–urea warranted further characterization. Raman spectroscopy shows an appreciable red-shift in prominent signals arising from dinitramide NO2 stretching and wagging frequencies (Fig. 4b).23 This suggests that dinitramide is better stabilized in this environment and/or has adopted a lower energy conformation than that in pure ADN. Previous studies have shown computationally that planar dinitramide is the lowest energy conformation yet the vast majority of compounds containing dinitramide fail to achieve planarity.23,29 The dinitramide torsion in ADN is 43.8° whereas that in ADN–urea is 5.1°, which may contribute to the red-shift in dinitramide absorbance. Stabilization of dinitramide is significant as interactions with the dinitramide ion are commonly invoked in theories concerning the thermal stability and hygroscopicity of ADN.30,31
The melt casting of an energetic material is the preferred processing method, though few energetic materials meet the requirements for melt casting which include melting temperatures between 70 °C and 120 °C and a large working window between melting and decomposition.13,32 Though far from widespread, melt-castable cocrystals are known33–35 and at least one energetic cocrystal was shown to impart melt-castability to a compound unsuitable for melt casing as a pure component; in this case, melt-phase stabilization was invoked based on vibrational spectroscopy.36 In other cases, phase separation has been observed upon melting, yielding a physical mixture of components with high impact sensitivity.37,38 Investigation of the thermal stability of both ADN salt cocrystals revealed that these materials recrystallize from the melt as the salt cocrystals rather than phase separating into ADN and urea/2Im (see ESI†). ADN–urea approaches the ideal lower limit in melting point (69.2 °C) for melt-castables and offers an excellent working window with decomposition onset at 134.6 °C. Cyclic DSC shows that melting and recrystallization is possible at least six times as evidenced by reproducible melting temperatures and negligible variance in ΔHfus (Fig. 5a). The salt cocrystal is also promising from an energetic performance perspective. ADN–urea is not impact-sensitive (in sharp contrast to ADN itself as determined using our in-house impact testing apparatus).6 The results of performance calculations, conducted using CHEETAH software (see ESI†), show ADN–urea to be competitive with contemporary state-of-the-art melt-castable energetic systems as both a stand-alone energetic material and as a component in currently fielded formulations18 The physical properties and calculated energetic performance parameters of ADN–urea demonstrate the promise of applying salt cocrystallization in energetic materials development.
 | ||
| Fig. 5 (a) Thermal characterization (DSC and TGA thermograms) of the ADN–urea system. (b) Cyclic-DSC thermogram for ADN–urea. (c) Calculated detonation pressures (PCJ) and velocities (DV) for several melt-castable energetic materials, including 2,4,6-trinitrotoluene (TNT) and 2,4-dinitroanisole (DNAN), and EM formulations. Entries bolded with an asterisk indicate an analogous formulation where the melting EM has been replaced by ADN–urea. See ESI† for the compositions of EM formulations included here. | ||
Conclusions
Here, an approach to NH4+ salt cocrystallization is presented and applied to the energetic oxidizing salt ADN. This approach afforded two novel ADN salt cocrystals, one of which is melt-castable. Though melt-casting was not a property targeted for improvement in this work, nor given the nature of the phenomenon could it have been, this observation highlights the ability of cocrystallization to imbue not only improved but novel properties in some instances. For ADN specifically, salt cocrystallization has proved capable of remediating major impediments to its application and now has facilitated its melt-casting.5 The approach put forth and applied here to achieve salt cocrystallization has considerable potential as a general methodology for the cocrystallization of energetic oxidizing salts, many of which are NH4+ salts. As this general counterion targeting approach is applied more broadly, it will expand the synthetic toolbox for properties driven synthesis of salt cocrystals.Data availability
Data is available upon request to corresponding author.Author contributions
MKB conducted and interpreted experiments/results. MKB and AJM contributed to project design, manuscript preparation/editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the ONR (Grant no. N00014-19-1-2086 and N00014-22-1-2101) for funding this research. M. K. B. acknowledges the Science, Mathematics, and Research for Transformation (SMART) program for their support.Notes and references
- O. N. Kavanagh, D. M. Croker, G. M. Walker and M. J. Zaworotko, Pharmaceutical cocrystals: from serendipity to design to application, Drug Discovery Today, 2019, 24(3), 796–804 CrossRef PubMed.
- D. Herrmannsdörfer, P. Gerber, T. Heintz, M. J. Herrmann and T. M. Klapötke, Investigation Of Crystallisation Conditions to Produce CL-20/HMX Cocrystal for Polymer-bonded Explosives, Propellants, Explos., Pyrotech., 2019, 44(6), 668–678 CrossRef.
- M. L. Cheney, D. R. Weyna, N. Shan, M. Hanna, L. Wojtas and M. J. Zaworotko, Coformer selection in pharmaceutical cocrystal development: A case study of a meloxicam aspirin cocrystal that exhibits enhanced solubility and pharmacokinetics, J. Pharm. Sci., 2011, 100(6), 2172–2181 CrossRef PubMed.
- Y. Huang, B. Zhang, Y. Gao, J. Zhang and L. Shi, Baicalein–Nicotinamide Cocrystal with Enhanced Solubility, Dissolution, and Oral Bioavailability, J. Pharm. Sci., 2014, 103(8), 2330–2337 CrossRef CAS PubMed.
- M. K. Bellas and A. J. Matzger, Achieving Balanced Energetics through Cocrystallization, Angew. Chem., Int. Ed., 2019, 58(48), 17185–17188 CrossRef CAS PubMed.
- J. C. Bennion, N. Chowdhury, J. W. Kampf and A. J. Matzger, Hydrogen Peroxide Solvates of 2,4,6,8,10,12-Hexanitro-2,4,6,8,10,12-hexaazaisowurtzitane, Angew. Chem., Int. Ed., 2016, 55(42), 13118–13121 CrossRef CAS.
- N. Schultheiss and A. Newman, Pharmaceutical Cocrystals and Their Physicochemical Properties, Cryst. Growth Des., 2009, 9(6), 2950–2967 CrossRef CAS.
- K. B. Pekar, J. B. Lefton, C. A. McConville, J. Burleson, D. Sethio and E. Kraka, et al., Mechanosynthesis of a Coamorphous Formulation of Creatine with Citric Acid and Humidity-Mediated Transformation into a Cocrystal, Cryst. Growth Des., 2021, 21(2), 1297–1306 CrossRef CAS.
- C. B. Aakeröy, A. M. Beatty and B. A. Helfrich, “Total Synthesis” Supramolecular Style: Design and Hydrogen-Bond-Directed Assembly of Ternary Supermolecules, Angew. Chem., Int. Ed., 2001, 40(17), 3240–3242 CrossRef.
- G. R. Desiraju, Supramolecular Synthons in Crystal Engineering—A New Organic Synthesis, Angew. Chem., Int. Ed. Engl., 1995, 34(21), 2311–2327 CrossRef CAS.
- G. R. Desiraju, Crystal engineering: solid state supramolecular synthesis, Curr. Opin. Solid State Mater. Sci., 1997, 2(4), 451–454 CrossRef CAS.
- M. B. Talawar, R. Sivabalan, T. Mukundan, H. Muthurajan, A. K. Sikder and B. R. Gandhe, et al., Environmentally compatible next generation green energetic materials (GEMs), J. Hazard. Mater., 2009, 161(2), 589–607 CrossRef CAS PubMed.
- T. M. Klapötke, Chemistry of High-Energy Materials, De Gruyter, 2015, Available from: DOI:10.1515/9783110439335.
- B. Q. Ma, H. L. Sun and S. Gao, The design and synthesis of a non-centric diamond-like network based NH4+ ion, Chem. Commun., 2003, 17, 2164–2165 RSC.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, The Cambridge Structural Database, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72(2), 171–179 CrossRef CAS.
- M. K. Bellas, L. V. MacKenzie and A. J. Matzger, Lamellar Architecture Affords Salt Cocrystals with Tunable Stoichiometry, Cryst. Growth Des., 2021, 21(6), 3540–3546 CrossRef CAS.
- Y. Wang, Y. Ma, H. Li, Y. Yu and Z. Yang, Preparation and Characterization of ADN/18C6 Eutectic, Chin. J. Energ. Mater., 2018, 26(6), 545–548 Search PubMed.
- R. Meyer, J. Köhler and A. Homburg. Explosives, Wiley-VCH, Weinheim, 6th edn, 2007, p. 421 Search PubMed.
- R. A. Pesce-Rodriguez, R. A. Fifer and J. M. Heimerl, “Clean burning” low flame temperature solid gun propellants, J. Energ. Mater., 1996, 14(3–4), 173–191 CrossRef.
- M. S. Miller and W. R. Anderson, Detailed Combustion Modeling as an Aid to Propellant Formulation: Two New Strategies, Army Research Laboratory, 2000, p. 49, Report No. ARL-TR-2167, Available from: https://apps.dtic.mil/sti/pdfs/ADA374698.pdf Search PubMed.
- V. Videnova-Adrabińska, The hydrogen bond as a design element in crystal engineering. Two- and three-dimensional building blocks of crystal architecture, J. Mol. Struct.: THEOCHEM, 1996, 374(1), 199–222 Search PubMed.
- I. J. Bruno, J. C. Cole, P. R. Edgington, M. Kessler, C. F. Macrae and P. McCabe, et al., New software for searching the Cambridge Structural Database and visualizing crystal structures, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58(3 Part 1), 389–397 CrossRef PubMed.
- K. O. Christe, W. W. Wilson, M. A. Petrie, H. H. Michels, J. C. Bottaro and R. Gilardi, The Dinitramide Anion, N(NO2)2−, Inorg. Chem., 1996, 35(17), 5068–5071 CrossRef PubMed.
- R. Keuleers, H. O. Desseyn, B. Rousseau and C. Van Alsenoy, Vibrational Analysis of Urea, J. Phys. Chem. A, 1999, 103(24), 4621–4630 CrossRef.
- Z. Piasek and T. Urbanski, The Infra-red Absorption Spectrum and Structure of Urea, Bull. Acad. Pol. Sci., 1960, X(3), 113–120 Search PubMed.
- A. R. Daniewski, U. Dąbrowska, Z. Piasek and T. Urbański, Infrared absorption spectra of some urea inclusion compounds, J. Chem. Soc., 1962, 2340–2343 RSC.
- Z. Piasek and T. Urbański, Tautomerism of urea, Tetrahedron Lett., 1962, 3(16), 723–727 CrossRef.
- R. V. Kent, R. A. Wiscons, P. Sharon, D. Grinstein, A. A. Frimer and A. J. Matzger, Cocrystal Engineering of a High Nitrogen Energetic Material, Cryst. Growth Des., 2018, 18(1), 219–224 CrossRef CAS.
- R. Gilardi, J. Flippen-Anderson, C. George and R. J. Butcher, A new class of flexible energetic salts: the crystal structures of the ammonium, lithium, potassium, and cesium salts of dinitramide, J. Am. Chem. Soc., 1997, 119(40), 9411–9416 CrossRef CAS.
- F. Wang, H. Liu and X. D. Gong, A theoretical study on the structure and hygroscopicity of ammonium dinitramide, Struct. Chem., 2013, 24(5), 1537–1543 CrossRef CAS.
- M. Bohn, Stabilization of the New Oxidizer Ammonium Dinitramide (ADN) in Solid Phase, in RTO AVT Specialists' Meeting on “Advances in Rocket Performance Life and Disposal”, NATO, Aalborg, Denmark, 2022, p. 20 Search PubMed.
- P. Ravi, D. M. Badgujar, G. M. Gore, S. P. Tewari and A. K. Sikder, Review on Melt Cast Explosives, Propellants, Explos., Pyrotech., 2011, 36(5), 393–403 CrossRef CAS.
- T. Yanwei, Y. Zongwei, S. Xiaole, L. Hailun, W. Lilun and L. Ping, TNB/TNCB cocrystal – an insensitive energetic cocrystal with low melting point, J. Energ. Mater., 2021, 1–14 CrossRef.
- D. Hong, Y. Li, S. Zhu, L. Zhang and C. Pang, Three Insensitive Energetic Co-crystals of 1-Nitronaphthalene, with 2,4,6-Trinitrotoluene (TNT), 2,4,6-Trinitrophenol (Picric Acid) and D-Mannitol Hexanitrate (MHN), Cent. Eur. J. Energ. Mater., 2015, 12(1), 47–62 Search PubMed.
- S. Qiao, J. Wang, Y. Yu, Y. Liu, Z. Yang and H. Li, Two novel TNB energetic cocrystals with low melting point: a potential strategy to construct melt cast explosive carriers, CrystEngComm, 2022, 24(16), 2948–2953 RSC.
- J. C. Bennion, Z. R. Siddiqi and A. J. Matzger, A melt castable energetic cocrystal, Chem. Commun., 2017, 53(45), 6065–6068 RSC.
- O. Bolton and A. J. Matzger, Improved Stability and Smart-Material Functionality Realized in an Energetic Cocrystal, Angew. Chem., Int. Ed., 2011, 50(38), 8960–8963 CrossRef.
- K. B. Landenberger and A. J. Matzger, Cocrystals of 1,3,5,7-Tetranitro-1,3,5,7-tetrazacyclooctane (HMX), Cryst. Growth Des., 2012, 12(7), 3603–3609 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2175805–2175811. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc03015b |
| This journal is © The Royal Society of Chemistry 2022 |