
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStepwise reduction of a base-stabilised ferrocenyl aluminium(III) dihalide for the synthesis of structurally-diverse dialane species†
Debabrata
Dhara
 ab,
Felipe
Fantuzzi
c,
Marcel
Härterich
ab,
Rian D.
Dewhurst
ab,
Ivo
Krummenacher
ab,
Merle
Arrowsmith
ab,
Conor
Pranckevicius
ab and
Holger
Braunschweig
*ab
ab,
Felipe
Fantuzzi
c,
Marcel
Härterich
ab,
Rian D.
Dewhurst
ab,
Ivo
Krummenacher
ab,
Merle
Arrowsmith
ab,
Conor
Pranckevicius
ab and
Holger
Braunschweig
*ab
aInstitute for Inorganic Chemistry, Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany. E-mail: h.braunschweig@uni-wuerzburg.de
bInstitute for Sustainable Chemistry & Catalysis with Boron, Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany
cSchool of Physical Sciences, Ingram Building, University of Kent, Park Wood Rd, Canterbury CT2 7NH, UK
First published on 1st August 2022
Abstract
We report the reduction of bulky ferrocenyl-based NHC-stabilised aluminium(III) diiodide [Fc*(NHC)AlI2] (Fc* = 2,5-bis(3,5-di-tert-butylphenyl)-1-ferrocenyl) in different hydrocarbon solvents (hexane, benzene, toluene, and p-xylene), which results in different outcomes. Reduction in hexane with an equivalent amount of KC8 generates the diiododialane [(Fc*(NHC)AlI)2], whereas complete reduction in hexane leads to an unusual C–H activation at an N–Me group of one NHC unit. In contrast, reaction in aromatic solvents result in hitherto unknown Birch-type reductions of the corresponding solvent molecules by transient aluminium radicals of the type [LAlR2]˙, which is ultimately bound to two aluminium centers.
Introduction
In 1988, Uhl and coworkers first reported the dialane R2Al–AlR2 (R = CH(SiMe3)2; I, Scheme 1) by employing steric encumbrance around the aluminium centre for kinetic stabilisation.1 This discovery was a landmark in the development of compounds containing Al–Al bonds. In 1994, Schnöckel et al. reported the donor-stabilised dialane Al2Br4(MeOPh)2 (II, Scheme 1).2 These two strategies, i.e. kinetic stabilisation by bulky substituents and electronic stabilisation by Lewis donors, paved the way for the synthesis of low-valent aluminium compounds.3 Following a similar strategy, a number of dialanes (aluminium in oxidation state +2, AlII) have been reported since.4 The reactivities of these dialanes towards Lewis bases,5 alkenes and alkynes,6 have now been explored. However, the parent dialane Al2H4 remains elusive, and its fleetingness has been predicted theoretically.7 In 2010, Jones, Stasch and coworkers reported the parent dialane (IDip2AlH2)2 (III; IDip = 1,3-bis(2,6-di-iso-propylphenyl)imidazole-2-ylidene; Scheme 1) stabilised by N-heterocyclic carbenes.4e By employing a similar strategy, low-valent aluminium compounds of other types such as masked dialumenes,8 dialumenes,9 aluminyl anions,10 and alumylenes11 have been reported. Some of these compounds have shown the potential for small-molecule activations and catalysis.3 However, compounds of these types remain extremely rare due to their general synthetic strategy and inherent tendency to disproportionate into Al0 (metallic aluminium) and AlIII (aluminium in +3 oxidation state) compounds.12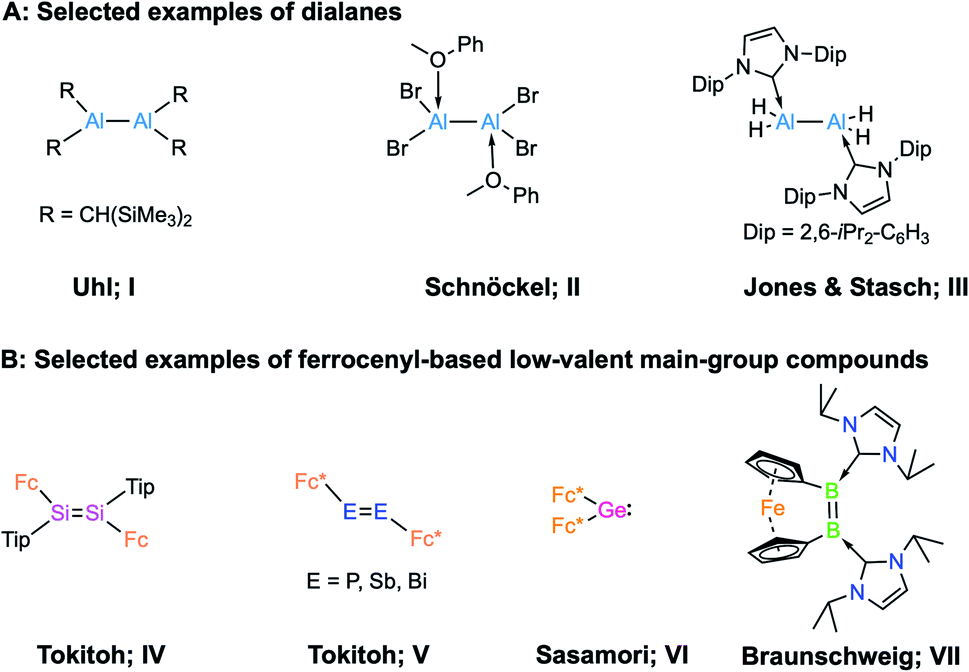 | ||
| Scheme 1 (A) Examples of selected dialanes (I–III); (B) examples of ferrocenyl-based low-valent main-group compounds (IV–VII). | ||
An interesting possibility for controlling the reactivity of Al–Al species is the introduction of redox-active ligands at the Al centres, and a handful of low-valent aluminium compounds have been prepared with redox-active organic ligands.13 However, the incorporation of classical redox-active organometallic substituents such as ferrocenyl groups has not been demonstrated. Redox-active ligands bound to a metal centre play an important role in controlling the reactivity of the complex,14 and their nature depends on redox-active moieties, either bound as ligands or in the second coordination sphere of a metal cofactor, to catalyse challenging reactions.15 We were therefore eager to investigate the effects of redox-active ligands on low-valent aluminium centres. It is worth noting that group 14 and group 15 multiply-bonded compounds stabilised by redox-active ferrocenyl ligands (type IV and V, Scheme 1) and their carbene analogue (VI) are known.16 In the case of group 13, in 1996 Wagner and co-workers investigated the geometry and electronic structure of borylferrocenes, FcBR2, revealing that there is an interaction between filled d-type orbitals at iron and the empty p orbital of boron, which causes bending of the BR2 substituent toward the central iron atom.17 In 2017, our group reported an NHC-stabilised dibora[2]ferrocenophane (VII, in Scheme 1)18 that showed a strained cis configuration. Electrochemical measurements revealed that dibora[2]ferrocenophane has a remarkably low oxidation potential of Epa = −1.56 V vs. the ferrocene/ferrocenium couple, corresponding to the one-electron oxidation of the diborene unit, suggesting that the iron centre has an effect on the electrochemical properties of the compound.18 However, there remains no report on low-valent aluminium compounds bearing redox-active ferrocenyl units.
Herein, we report the reduction of a carbene-stabilised, sterically encumbered ferrocenyl-based diiodoalane [Fc*AlI2(NHC)], which we expected to lead to either the redox-active dialumene [Fc*(NHC)Al![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Al(NHC)Fc*] or alternatively a dicoordinate alumylene ([Fc*(NHC)Al:]). Contrary to our expectations, we instead obtained a diiododialane when the reduction was performed in hexane, and highly unusual doubly aluminyl-substituted Birch-type aromatic reduction products when the reduction was performed in aromatic solvents (toluene and p-xylene). In our knowledge this is the first report where a transient aluminium radical of type [LAlR2]˙ can reduce the aromatic solvent in a Birch-reduction-like fashion.
Al(NHC)Fc*] or alternatively a dicoordinate alumylene ([Fc*(NHC)Al:]). Contrary to our expectations, we instead obtained a diiododialane when the reduction was performed in hexane, and highly unusual doubly aluminyl-substituted Birch-type aromatic reduction products when the reduction was performed in aromatic solvents (toluene and p-xylene). In our knowledge this is the first report where a transient aluminium radical of type [LAlR2]˙ can reduce the aromatic solvent in a Birch-reduction-like fashion.
Results and discussion
Lewis-base-coordinated ferrocenyl-alane precursor 1 was synthesised by combining Fc*Li (Fc* = 2,5-bis(3,5-di-tert-butylphenyl)-1-ferrocenyl)19 and NHCMe4 AlH3 (NHCMe4 = 1,3,4,5-tetramethylimidazol-2-ylidene)20 in equimolar amounts following our recently published strategy.21 The treatment of 1 with an excess (5 equiv) of methyl iodide led to formation of Lewis-base-coordinated alkyldihaloalane precursor 2 (Scheme 2).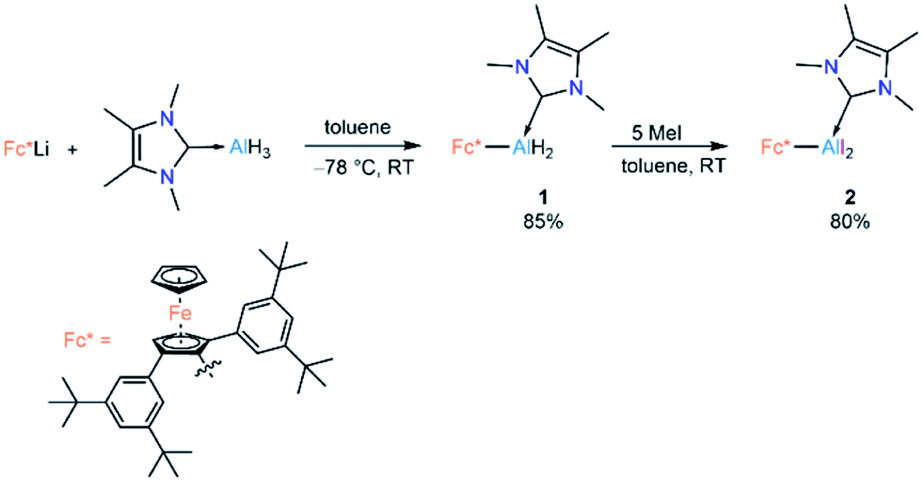 | ||
| Scheme 2 Synthesis of 1 and 2. | ||
Compounds 1 and 2 have been characterised by solution-state NMR and single-crystal X-ray diffraction (SCXRD). The most diagnostic differences in the 1H NMR spectra of 1 and 2 are in the signals for the unsubstituted (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4.35; 2:4.46 ppm) and substituted Cp ring protons of the Fc* unit (1:4.98; 2:4.86 ppm), which move in opposite directions upon iodination. SCXRD confirmed the attachment of the NHC to the aluminium centre in both cases (Fig. 1).
:4.35; 2:4.46 ppm) and substituted Cp ring protons of the Fc* unit (1:4.98; 2:4.86 ppm), which move in opposite directions upon iodination. SCXRD confirmed the attachment of the NHC to the aluminium centre in both cases (Fig. 1).
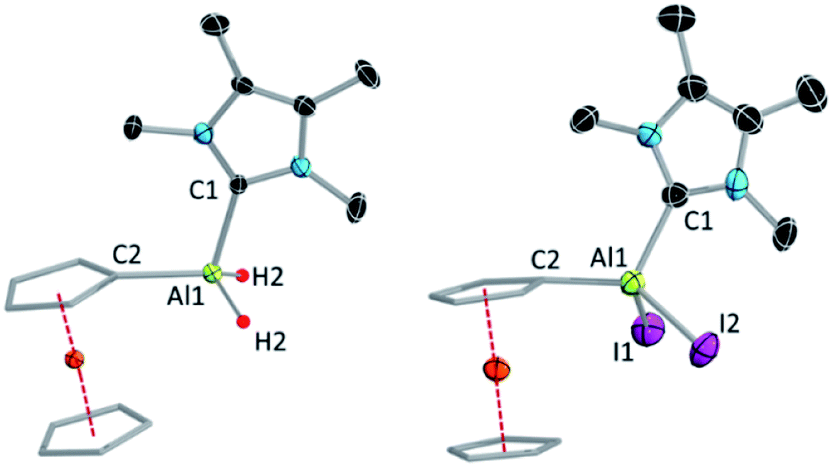 | ||
| Fig. 1 Molecular structures of 1 (left) and 2 (right) with ellipsoids at the 50% probability level. All hydrogen atoms except H1 and H2 in 1 and the aromatic rings (3,5-di-tert-butylphenyl moiety) attached to the Cp rings are omitted for clarity. A molecule of hexane is omitted from the structure of 1. Selected bond lengths [Å] and bond angles [°] for 1: Al1–C1 2.059(3), Al1–C2 1.985(2); C1–Al1–C2 108.07(10); for 2: Al1–C1 2.040(6), Al1–C2 1.967(6); C1–Al1–C2 115.0(2). | ||
Cyclic voltammetric measurement of 2 in difluorobenzene solution revealed a low reduction potential Epc = −1.33 V (Fig. 5) which indicates it could be easily reducible. We thus performed the stepwise reduction of 2 in different solvents (hexane, benzene, toluene, and p-xylene). Treatment of 2 in hexane with 1.5 equiv of potassium graphite (Scheme 3) led to the formation of dialane 3, which was isolated as orange crystals from a saturated hexane solution. The 1H NMR spectrum of 3 showed two doublets at 4.43 and 4.49 ppm (2J = 2.2 Hz, substituted Cp ring) along with a singlet at 4.38 ppm (unsubstituted Cp ring), both upfield of the corresponding proton signals of 2 (unsubstituted Cp ring: 4.46 ppm; substituted Cp ring: 4.86 ppm).
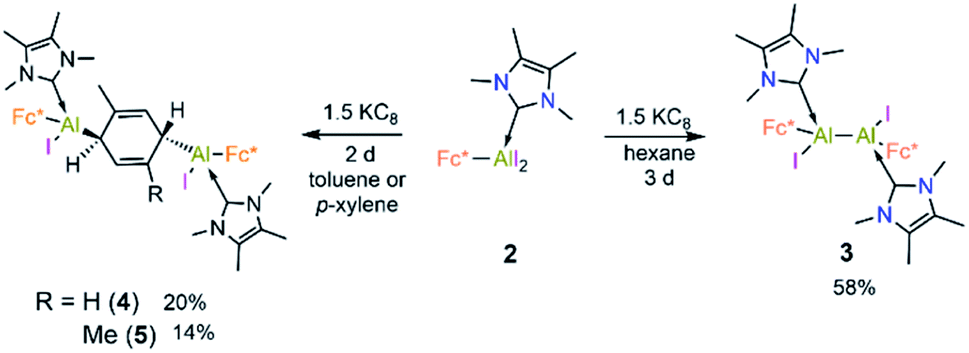 | ||
| Scheme 3 Synthesis of 3–5 from 2. | ||
The solid-state structure of 3 is shown in Fig. 2a. The Al–Al distance of 3 (2.648(2) Å) is longer than that of [Al2I2(η5-Cp*)2] (2.532(1) Å),20 R2Al2Br2 (2.592(3) Å; R = 2,6-bis[bis(trimethylsilyl)methyl]phenyl),4f II (2.527 (6) Å),2 within statistical uncertainty of that of the THF-stabilised dialumene [(L2−)(thf)AlII–AlII(thf)(L2−)] (2.658(2) Å; L = [(2,6-C6H3iPr2)NCMe]2),4h and in the typical range of other Al–Al single bond distances (2.5–2.95 Å).4
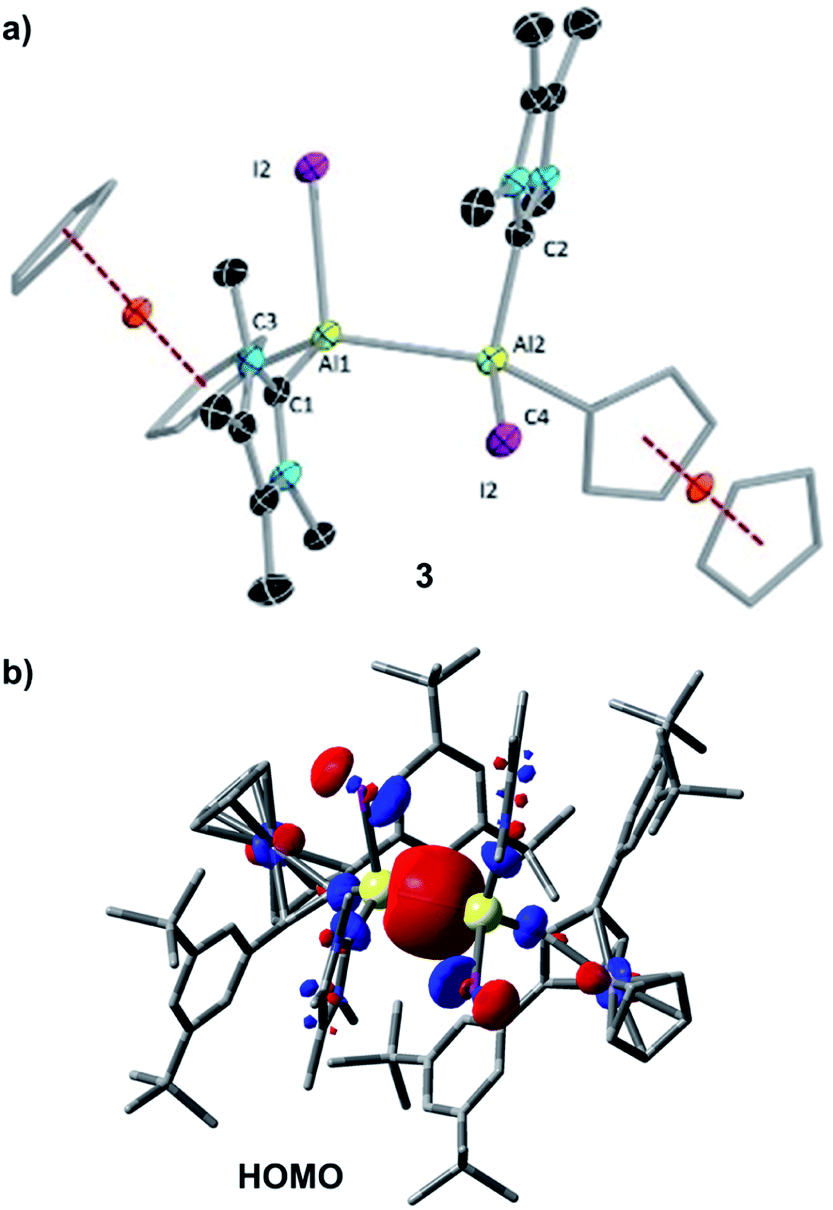 | ||
| Fig. 2 (a) Molecular structure of 3 with thermal ellipsoids at the 50% probability level. All hydrogen atoms, hexane (solvent of crystallisation) and aromatic rings (3,5-di-tert-butylphenyl moiety) attached to the Cp rings are omitted for clarity. Selected bond lengths [Å] and bond angles [°]: Al1–Al2 2.648(2), Al1–C1 2.057(5), Al1–C2 2.059(5), Al1–C3 1.996(5), Al2–C4 2.007(5), Al1–Al2–C2 133.08(15), Al1–Al2–C2 97.45(14), Al1–Al2–C1 95.94(15), C2–Al2–C4 108.1(2), C3–Al1–C1 107.6(2); I1–Al1–Al2–I2 110.77 (6), C1–Al1–Al2–C2 109.4(2), C3–Al1–Al2–C4 6.0(3); (b) HOMO of 3 at the PBE0-D3(BJ)/def2-SVP level of theory. Isovalue: 0.03 au. Hydrogen atoms are omitted for clarity. | ||
The single bond character of the Al–Al bond of 3 is also corroborated by its computed Mayer bond order (MBO) of 0.886. Accordingly, the HOMO of 3 (Fig. 2b) is dominated by the Al–Al σ bonding interaction. The aluminium-carbene carbon distances (Al1–C1 2.057(5) Å, Al1–C2 2.059(5) Å) are similar to that of the carbene-stabilised parent dialane [{(IDip)H2Al}2] (IDip = 1,3-bis(2,6-di-iso-propylphenyl)imidazole-2-ylidene).4e The I–Al–Al–I dihedral angle of 3 (110.77(6)°) was found to be similar to the corresponding Br–Al–Al–Br dihedral angle of [Al2Br2(η5-Cp*)2] (102.04(5)°)].6a The two Fc* substituents are almost in the same plane (C3–Al1–Al2–C4 6.0(3)°), while the two carbenes are staggered with a dihedral angle (C1–Al1–Al2–C2) of 109.4(2)°.
When the same reaction was performed in benzene, an unidentified mixture of products was obtained. However, by changing the solvent from benzene to toluene, we were able to isolate the toluene-activation product 4 (Scheme 3) in 20% yield, along with a few crystals of 3. The susceptibility of aromatic rings towards low-valent aluminium centres is well known.8,21,22,10e In 2003 and 2013 the groups of Power and Tokitoh respectively showed that transient dialumenes can readily be trapped by [2 + 4] cycloaddition with toluene and benzene.8 Very recently, the groups of Crimmin and Liu showed that base-stabilised alumylenes are capable of cleaving activated C–C bonds.22 From our group, we also revealed that a transient base-stabilised alumylene can break the C–C bonds of benzene and toluene.21 The synthesis of 4 is essentially a dialuminium analogue of the Birch reduction,23 a novel outcome in aluminium chemistry.
Like 3, compound 4 is somewhat unstable in benzene and toluene, liberating the ferrocenyl substituent as the protonated Fc*H,19 in addition to an unidentified white solid. The 1H NMR spectrum of 4 showed characteristic signals for the vinylic proton (4.83 ppm) and the methyl protons of the toluene unit (1.40 ppm), the latter being upfield of the methyl signal of free toluene (2.1 ppm). Unfortunately, no 27Al NMR signal could be observed. X-ray quality single crystals of 4 were obtained from a saturated hexane solution. The solid-state structure of 4 showed the presence of a 1,4-cyclohexadiene unit flanked by two aluminium centres (Fig. 3). The two aluminium centres are situated mutually trans to the ring and the cyclohexadiene ring is almost planar (dihedral angle C5–C6–C7–C8 = 4.41(7)°), sandwiched between the two carbene moieties. The Al1–C5 (2.01(2) Å) and Al2–C8 (2.074(7) Å) distances are similar to typical Al–C single bonds.8,24 However, the Al–Ccarbene distances are slightly longer (Al2–C3 2.064(3) Å; Al1–C1 2.059(3) Å) than the Al–CFc* distances (Al–C1 1.990(3) Å, Al2–C4 1.996(3) Å), suggesting donor–acceptor character of the former bonds (Fig. 3). To test the scope of this dialuminyl Birch reduction, we repeated the reaction while replacing the solvent toluene by the three isomers of xylene. In the case of p-xylene we obtained the solvent-activated product 5 (Scheme 3), analogous to the toluene product 4, while with m-xylene and o-xylene we only obtained a sample of crystals of 3 in addition to 1H NMR data suggesting the formation of Fc*H along with an unidentified mixture of products.
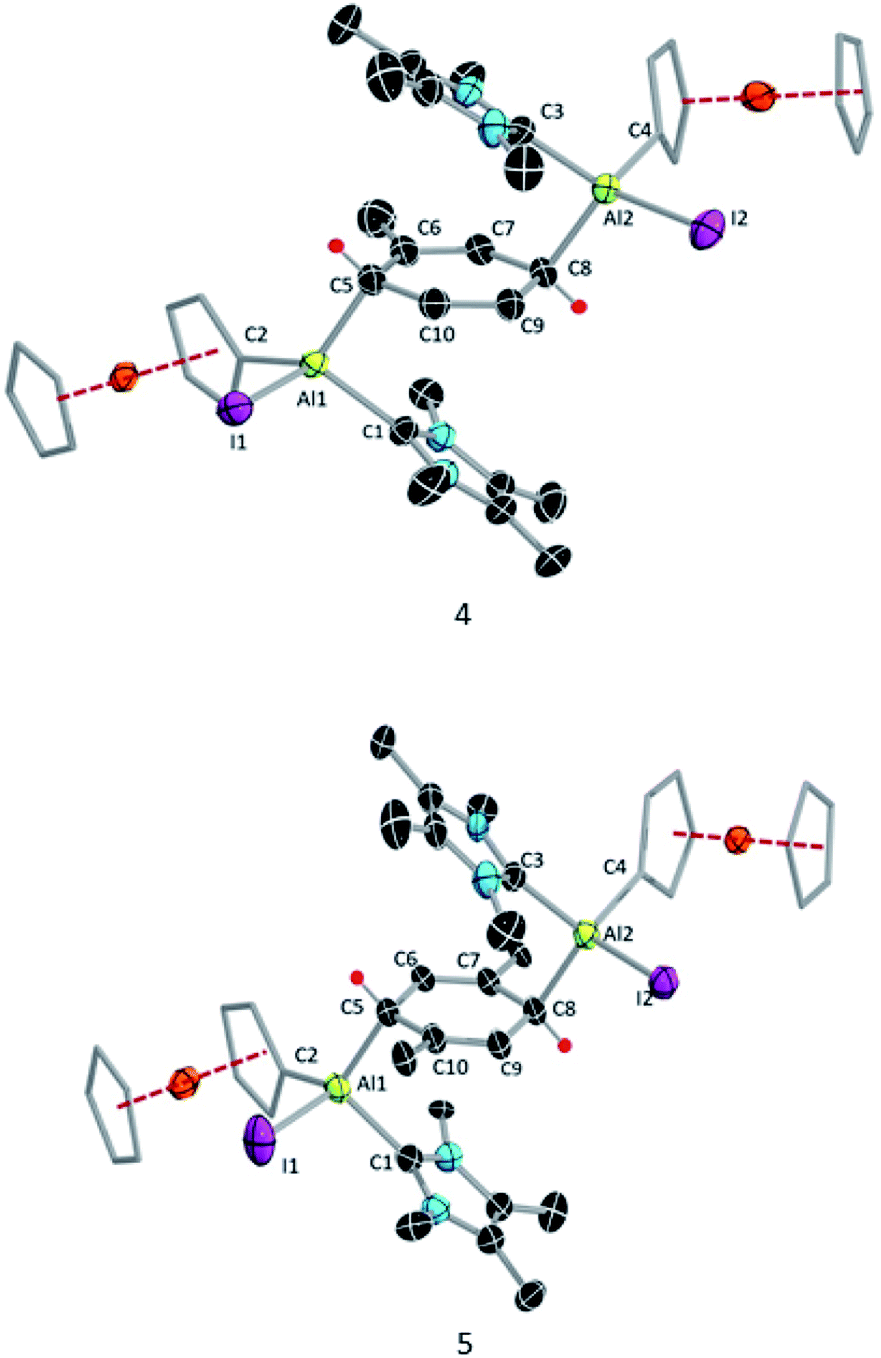 | ||
| Fig. 3 Molecular structures of 4 and 5 with thermal ellipsoids at the 50% probability level. All hydrogen atoms along except H1 and H2, aromatic rings (3,5-di-tert-butylphenyl) attached to the Cp rings and a hexane molecule as solvent of crystallisation in 5 are omitted for clarity. Selected bond lengths [Å] and bond angles [°]: For 4: Al2–C8 2.074(16), Al1–C5 2.01(2), Al1–C2 1.990(3), Al2–C4 1.996(3), Al1–C1 2.059(3), Al2–C3 2.064(3), C10–C9 1.340(13), C6–C7 1.339(12), C5–C10 1.493(16), C7–C8 1.502(13), C5–C6 1.495(16), C8–C9 1.513(15); C5–Al1–C2 121.7(4) C5–Al1–C1 100.6(6), C8–Al2–C3 99.4(5), C5–C6–C7–C8 4.41(7). For 5: Al1–C2 = Al2–C4 1.853(11), Al1–C1 = Al2–C4 2.065(6), Al1–C5 = Al2–C8 = 2.033(6), C10–C9 = C6–C7 = 1.336(8), C9–C8 = C5–C6 = 1.489(7), C5–C10 = C8–C7 = 1.494(8); C1–Al1–C5 = C3–Al2–C8 99.0(4), C1–Al1–C5–C6 62.1(4), C9–C10–C5–C6 9.1(7). | ||
The 1H NMR spectrum of 5 showed two signals for the vinylic xylyl protons (4.00 ppm and 5.23 ppm; confirmed by HSQC), as well as two xylyl methyl signals (1.55 and 1.84 ppm), suggesting that the two sides of the 1,4-cyclohexadiene unit are magnetically inequivalent. The solid-state structure of 5 (Fig. 3) confirmed its similarity to 4, with a near-planar cyclohexadiene ring (C9–C10–C5–C9 9.1(7)°) between two aluminium centres and sandwiched between the two carbene units. The structure also rationalises the observed inequivalence of the two sides of the 1,4-cyclohexadiene ring, as the Fc* substituents both align with the same xylyl methyl group.
The formation of 3–5 can be assumed to occur via reductive formation of the aluminium-centred radical of the type [LAlR2]˙ (6; Scheme 4), analogous to the established base-stabilised boryl radicals [LBR2]˙,25 which can either dimerise (to form 3) or react with an aromatic ring (to form 4/5). Our computations successfully located a potential minimum energy structure for 6. As expected, in its optimised geometry, the spin density (see Fig. 4) is mostly located at the Al centre (0.84), with a minor contribution from the NHC carbon atom (0.02). Attempts to detect radical 6 by EPR spectroscopy, or to trap it with TEMPO, were unsuccessful, likely due to its strong propensity to undergo dimerisation. Computational evidence for the latter is obtained from the higher negative enthalpy and free energy of dimerisation of the radical 6 to 3 (−59.5 and −42.1 kcal mol−1, respectively; see ESI† for details). Regarding the formation of 4/5, our computational results suggest that one molecule of radical 6 reacts with the aromatic solvent, leading to radical 6′ (Scheme 4); this step is only slightly exergonic (ΔG = −3.4 kcal mol−1). The spin density of 6′ is distributed around the substituted toluene unit, with its largest contribution (0.54) located at the carbon atom para to the C–Al bond (see Fig. 4). The combination of a second molecule of 6 to 6′, leading to 4, is considerably downhill in energy, and the overall free energy of the reaction is ΔG = −45.5 kcal mol−1. This value is slightly more negative than the formation of 3 and is in line with the experimental finding that 4 is the major product in toluene.
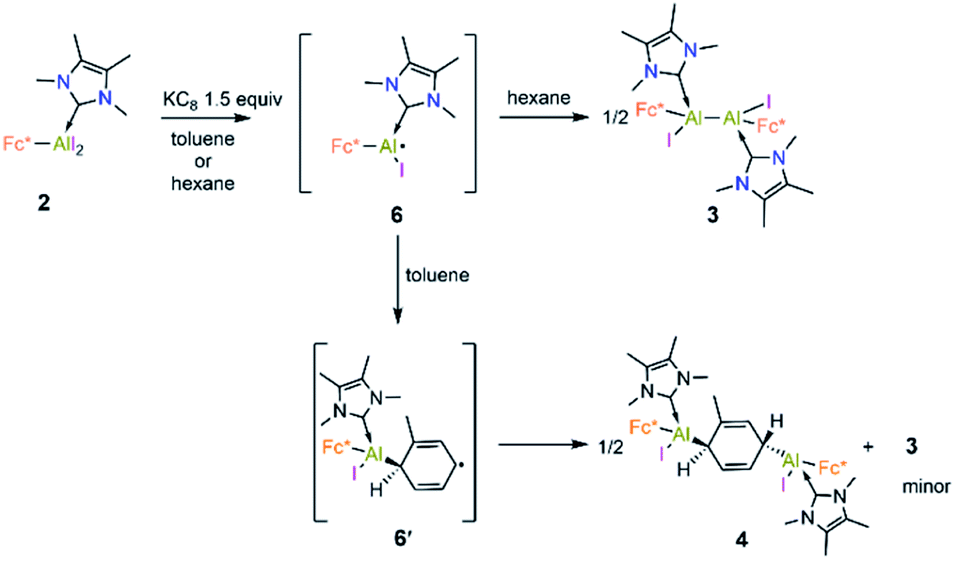 | ||
| Scheme 4 Proposed mechanisms for the formation of 3 and 4. | ||
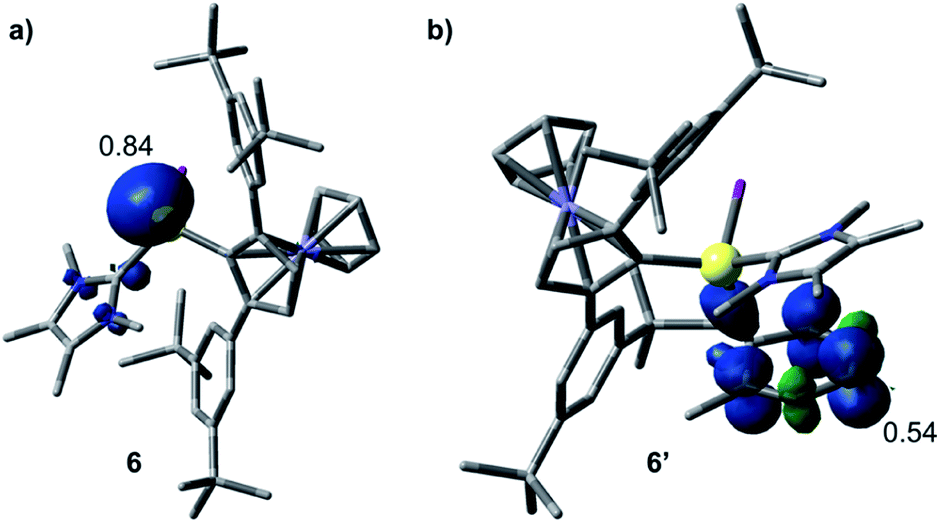 | ||
| Fig. 4 Optimised structures and Mulliken spin density plots of the radicals 6 (a) and 6′ (b). Level of theory: PBE0-D3(BJ)/def2-SVP. Isovalue: 0.005 au. Hydrogen atoms are omitted for clarity. | ||
It is well known that such dialanes are susceptible to reactions with unsaturated compounds such as alkenes, alkynes6a,b,d and carbodiimides,6c which insert into their Al–Al bonds. Consequently, diiododialane 3 was treated with bis(trimethylsilyl)acetylene, but this reaction led only to an intractable mixture of products. Additionally, 3 was dissolved in toluene to investigate whether toluene-insertion product 4 might be accessible from the dialane, however, only decomposition and formation of Fc*H was observed after 24 hours, suggesting that 3 does not undergo homolytic cleavage back to the radical 6. In Tokito and coworkers' dialumene benzene trapping reaction, it was shown that the aromatic ring attached to the dialumene can reversibly exchange with other aromatic molecules.8 However, we did not observe an analogous exchange by adding toluene to 5 or p-xylene to 4.
Compound 3 is not stable in THF, 1,2-difluorobenzene or CH3CN, hindering attempts to study its electrochemical properties. Nevertheless, we attempted the further reduction of 3 using 3 equiv of KC8 in both hexane and aromatic solvents (benzene, toluene, and p-xylene). The reactions in aromatic solvents led only to a mixture of intractable products, while that in hexane provided a product of C–H activation of an NHC ligand at its distal Al center (8; 50% isolated; Scheme 5). Interestingly, compound 8 was also obtained by reduction of 2 in the presence of 4 equiv KC8. In both reduction experiments, alongside a large amount of 8, a few crystals of 7 were observed in the reaction flask after different reaction periods (five days for 2, one day for 3; confirmed by SCXRD). Compound 7 is an isomer of 8; the formal product of C–H activation of an NHC ligand at its proximal Al centre. Unfortunately, subsequent attempts to generate 7 exclusively by varying the reaction time and temperature consistently failed.
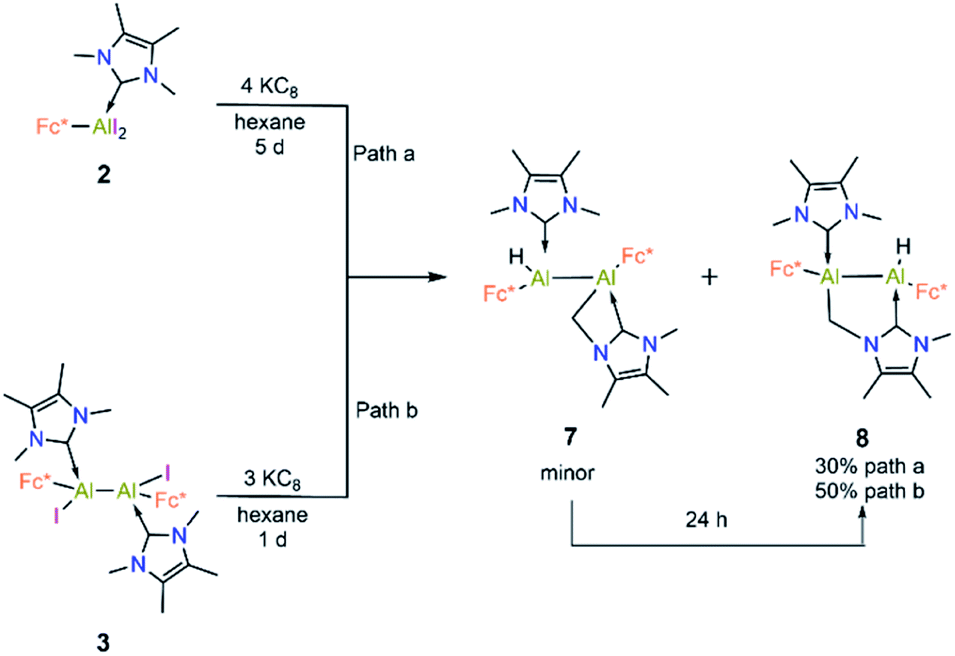 | ||
| Scheme 5 Complete reduction of 2 and 3 into 7 and 8. | ||
Compound 8 was characterised by solution-state NMR while the structures of 7 and 8 were confirmed by SCXRD (Fig. 6). The 1H NMR spectrum of 8 showed two distinct doublets arising from the diastereotopic methylene protons (2.20 ppm and 2.48 ppm; 2JHH = 12 Hz). The Al–Al distance (2.6035(15) Å) of 8 is significantly smaller than that of 3 (2.648(2) Å) and 7 (2.6259(8) Å), presumably due to its bridged nature. In 8 Al2–C2 bond distances is (2.060 (3) Å) a little shorter than Al1–C3 bond and Al2–C1 (2.089(3)) implying that pivotal bond. To test whether 7 spontaneously converts to 8, we stirred a mixture of 7 and 8 for 24 hours at room temperature, after which time the 1H NMR spectrum indicated the presence of 8 and an unidentified mixture of products. When a crystalline mixture of 7 and 8 was dissolved in deuterated benzene along with mesitylene as an internal standard, 1H NMR spectroscopic data indicated an increase of 8 over time along with a small amount of Fc*H formation after 12 hours. This suggests that 7 indeed converts into 8 over time. It should be noted that the insertion of low-valent main-group centres into C–H bonds of attached donor ligands is relatively common.26 In contrast, C–H activation at N-methyl groups of NHCs is not common with main-group elements, though it has been observed with transition metal NHC complexes.27
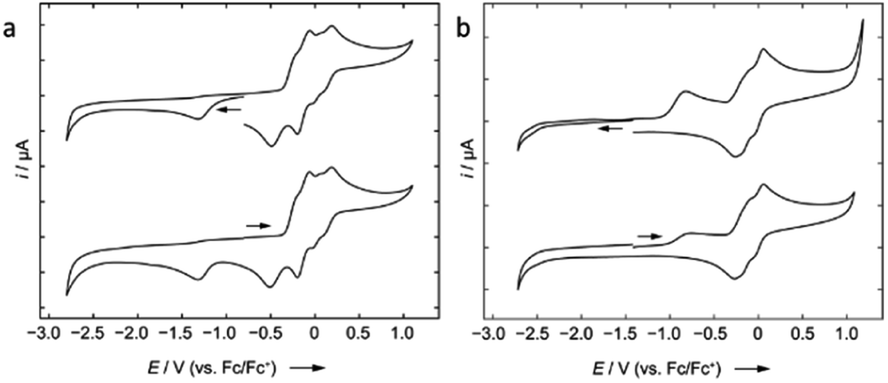 | ||
| Fig. 5 Cyclic voltammograms of 2 (a) and 8 (b) in 1,2-difluorobenzene/0.1 M [nBu4N][PF6] measured at 250 mV s−1. The voltammetric response for the positive and negative scan direction is shown. (a) Formal potentials for 2: Epc = −1.33 V, Epa (onset) = −0.34 V (oxidation of the ferrocene unit at ca. E1/2 = −0.15 V; relative to the Fc/Fc+ couple); (b) formal potentials for 8: Epa(onset) = −0.82 V (oxidation of the ferrocene units between ca. E1/2 = −0.2 and 0.0 V; relative to the Fc/Fc+ couple). | ||
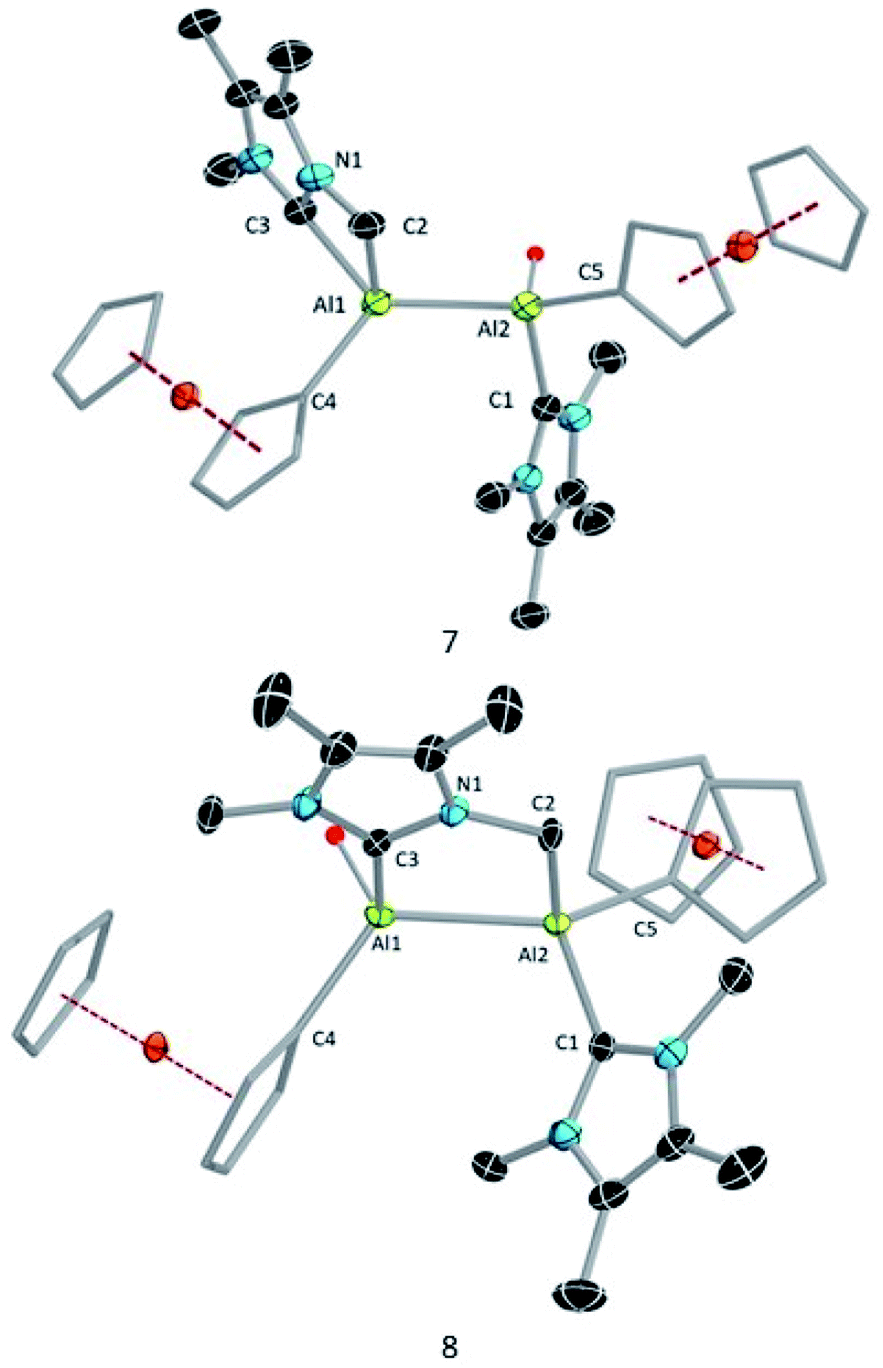 | ||
| Fig. 6 Molecular structures of 7 and 8 with thermal ellipsoids at the 50% probability level. All hydrogen atoms except H1 and the aromatic rings (3,5-di-tert-butylphenyl moiety) attached to the Cp rings are omitted for clarity in 7 and 8. Also, one molecule of hexane in the structure of 8 is omitted. Selected bond lengths [Å] and bond angles [°] for 7: Al2–Al2 2.6259(8), Al1–C3 2.0908(17), Al1–C2 2.0853(19), Al1–C4 2.057(8), Al2–C5 2.0162(17), Al2–C1 2.0708(17), C3–Al1–C2 68.47(7), C3–N1–C2 113.12(13), C5–Al2–Al1–C4 11.051(30). For 8: Al2–Al2 2.6035(15), Al1–C3 2.082(3), Al2–C2 2.060(3), Al1–C4 2.039(3), Al2–C5 1.998(3), Al2–C1 2.089(3). Al1–Al2–C2 92.84(9), Al2–Al1–C3 84.29(9), C3–N1–C2 125.5(2), Al2–Al1–C3–N1 10.8(5). | ||
Compound 8 is unstable in benzene and toluene over long periods. Electrochemical measurements of 8 in difluorobenzene solution revealed a low oxidation potential (Epa(onset) = −0.82 V, Fig. 5) which indicates 8 is more prone to oxidation. Subsequently, we probed the oxidation of 3 and 8 with iodine. Oxidation of 3 (Scheme 6) gave an unidentified mixture of products along with Fc*H, whereas oxidation of 8 (Scheme 6) led to 2, Fc*H, and Fc*I (33:43:23 relative product ratio as indicated by 1H NMR spectroscopic data).
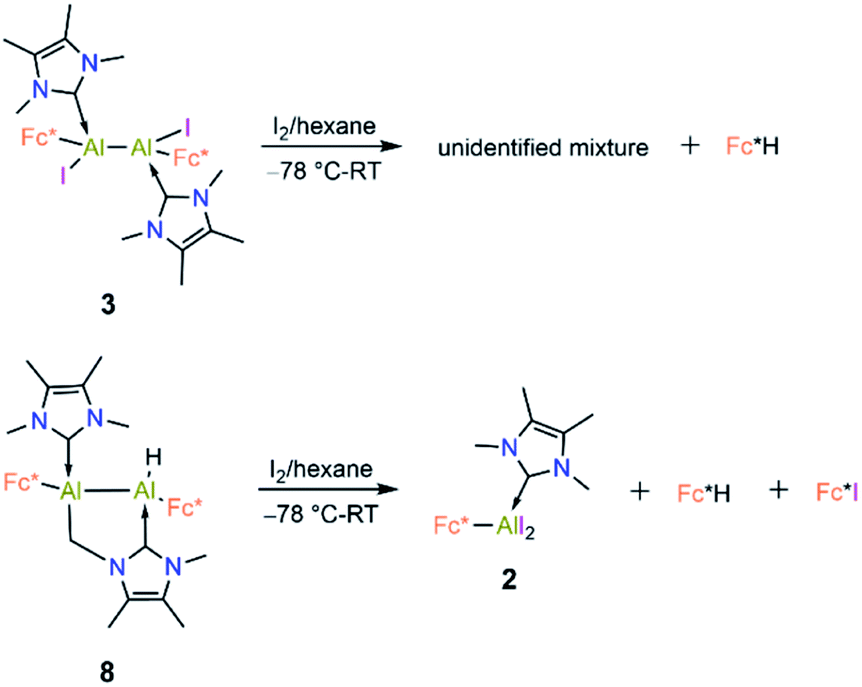 | ||
| Scheme 6 Reactions of 3 and 8 with iodine. | ||
The formation of products 7 and 8via C–H activation of the NHC moiety is quite unusual, therefore it was investigated in detail using DFT computations. Our main findings are summarised in Fig. 7.
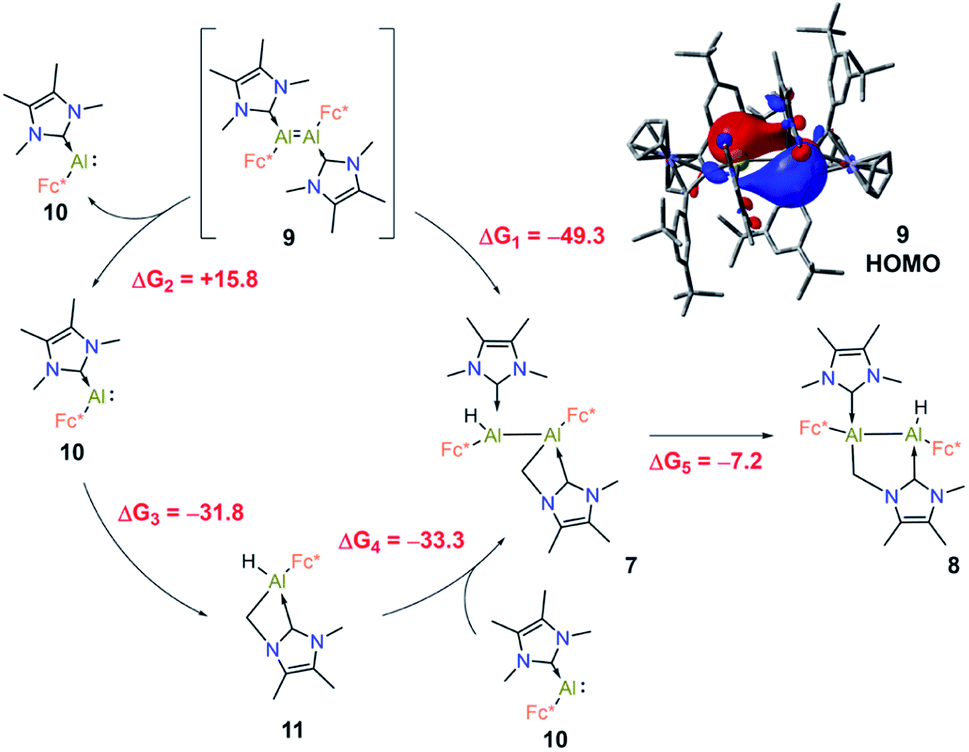 | ||
| Fig. 7 Proposed pathways for the formation of 7 and 8 from 9, along with their corresponding free energies (kcal mol−1) at the PBE0-D3(BJ)/def2-TZVP(SMD, n-hexane) level of theory. The HOMO of 9 is shown in the upper right corner (isovalue: 0.03 au). | ||
First, we suggest that the starting point of the reaction is the in situ generated dialumene Fc(NHC)AlAl(NHC)Fc (9). Inspection of the canonical Kohn–Sham molecular orbitals of 9 revealed that the Al–Al interaction has partial double bond character (see Fig. 7), a finding that is also corroborated by calculation of the corresponding MBO (1.11). Compound 9 is characterised by narrow HOMO-LUMO (2.07 eV) and vertical singlet-triplet (8.4 kcal mol−1) gaps in comparison to those of 3 (4.56 eV and 35.3 kcal mol−1, respectively). This agrees with the elusive and likely highly reactive nature of 9. Compound 7 could be further obtained from 9 by direct C–H insertion, this transformation having a computed free energy value of ΔG1 = −49.3 kcal mol−1. Alternatively, 9 could dissociate into its monomeric form Fc(NHC)Al: (10, ΔG2 = +15.8 kcal mol−1). The C–H insertion could then proceed from 10 to form 11 (ΔG3 = −31.8 kcal mol−1), and reaction with the second monomeric 10 is expected to form 7 (ΔG4 = −33.3 kcal mol−1). Formation of compound 7 through this process is more exergonic than the formation of two monomeric C–H-activated 11 by −1.6 kcal mol−1. The computed free energies indicated that the C–H insertion could occur either through the monomeric or dimeric Al species, as both pathways are energetically viable. Accordingly, DFT computations on the isomeric C–H activation products indicated that 8 is 8.1 kcal mol−1 lower in energy than 7 and that the formation of 8 from 7 is exergonic by ΔG5 = −7.2 kcal mol−1. These results are in line with the experimental findings and intuition given the geometrical distortion and presumed strain inherent in the four-membered AlCNC ring of 7.
Conclusions
A base-stabilised, aluminium(III) diiodide bearing a bulky ferrocenyl substituent has been synthesised and subjected to reduction in different hydrocarbon solvents. In hexane, one-electron reduction gave a dialane, while two-electron reduction provided two isomeric C–H activation products. In aromatic solvents, one-electron reduction led to reduction of the arene akin to a dialuminyl analogue of the Birch reduction. DFT calculations suggest that the dialane and arene-reduction products all arise via initial formation of a base-stabilised aluminyl radical, which undergoes radical homocoupling in the former case and sequential addition to the arene in the latter.Data availability
Full experimental and computational details are provided as part of the ESI.†Author contributions
H. B. supervised the study. D. D. carried out the synthetic work. I. K. carried out and analysed the EPR and CV experiments. D. D., M. H., M. A. and C. P. carried out the X-ray crystallographic analyses. F. F. carried out the computational studies. D. D., F. F. and R. D. D. prepared the manuscript and the ESI. All authors read and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Deutsche Forschungsgemeinschaft is gratefully acknowledged. D. D. thanks the Alexander von Humboldt Foundation for a Humboldt postdoctoral fellowship.References
- W. Uhl, Z. Naturforsch., B: J. Chem. Sci., 1988, 43, 1113–1118 CrossRef CAS.
- M. Mocker, C. Robl and H. Schnöckel, Angew. Chem., Int. Ed. Engl., 1994, 33, 862–863 CrossRef.
- (a) P. Bag, C. Weetman and S. Inoue, Angew. Chem., Int. Ed., 2018, 57, 14394–14413 CrossRef CAS PubMed; (b) K. Hobson, C. J. Carmalt and C. Bakewell, Chem. Sci., 2020, 11, 6942–6956 RSC.
- (a) A. Purath, C. Dohmeier, A. Ecker, H. Schnöckel, K. Amelunxen, T. Passler and N. Wiberg, Organometallics, 1998, 17, 1894–1896 CrossRef CAS; (b) A. Purath and H. Schnöckel, J. Organomet. Chem., 1999, 579, 373–375 CrossRef CAS; (c) M. Schiefer, N. D. Reddy, H. W. Roesky and D. Vidovic, Organometallics, 2003, 22, 3637–3638 CrossRef CAS; (d) C. Schnitter, H. W. Roesky, C. Röpken, R. Herbst-Irmer, H.-G. Schmidt and M. Noltemeyer, Angew. Chem., Int. Ed., 1998, 37, 1952–1955 CrossRef CAS; (e) S. J. Bonyhady, D. Collis, G. Frenking, N. Holzmann, C. Jones and A. Stasch, Nat. Chem., 2010, 2, 865–869 CrossRef CAS PubMed; (f) T. Agou, K. Nagata, H. Sakai, Y. Furukawa and N. Tokitoh, Organometallics, 2012, 31, 386–3809 Search PubMed; (g) I. L. Fedushkin, M. V. Moskalev, A. N. Lukoyanov, A. N. Tishkina, E. V. Baranov and G. A. Abakumov, Chem. – Eur. J., 2012, 18, 11264–11276 CrossRef CAS PubMed; (h) Y. Zhao, Y. Liu, L. Yang, J.-G. Yu, S. Li, B. Wu and X.-J. Yang, Chem. – Eur. J., 2012, 18, 6022–6030 CrossRef CAS PubMed; (i) R. L. Falconer, G. S. Nichol, I. V. Smolyar and M. J. Cowley, Angew. Chem., Int. Ed., 2021, 60, 2047–2052 CrossRef CAS PubMed; (j) C. Bakewell, K. Hobson and C. J. Carmalt, Angew. Chem., Int. Ed., 2022, e202205901 CAS.
- V. G. Sokolov, T. S. Koptseva, R. V. Rumyantcev, X.-J. Yang, Y. Zhao and I. L. Fedushkin, Organometallics, 2020, 39, 66–73 CrossRef CAS.
- (a) A. Hofmann, A. Lamprecht, O. F. González-Belman, R. D. Dewhurst, J. O. C. Jiménez-Halla, S. Kachel and H. Braunschweig, Chem. Commun., 2018, 54, 1639–1642 RSC; (b) Y. Zhao, Y. Liu, Y. Lei, B. Wub and X.-J. Yang, Chem. Commun., 2013, 49, 4546–4548 RSC; (c) L. Xiao, W. Chen, L. Shen, L. Liu, Y. Xue, Y. Zhao and X.-J. Yang, Chem. Commun., 2020, 56, 6352–6355 RSC; (d) Y. Zhao, Y. Lei, Q. Dong, B. Wu and X.-J. Yang, Chem. – Eur. J., 2013, 19, 12059–12066 CrossRef CAS PubMed.
- (a) K. Lammertsma, O. F. Güner, R. M. Drewes, A. E. Reed and P. v. R. Schleyer, Inorg. Chem., 1989, 28, 313–317 CrossRef CAS; (b) K. Lammertsma and J. Leszczyński, J. Phys. Chem., 1990, 94, 5543–5548 CrossRef CAS.
- (a) R. J. Wright, A. D. Phillips and P. P. Power, J. Am. Chem. Soc., 2003, 125, 10784–10785 CrossRef CAS PubMed; (b) T. Agou, K. Nagata and N. Tokitoh, Angew. Chem., Int. Ed., 2013, 52, 10818–10821 CrossRef CAS PubMed.
- (a) P. Bag, A. Porzelt, P. J. Altmann and S. Inoue, J. Am. Chem. Soc., 2017, 139, 14384–14387 CrossRef CAS PubMed; (b) C. Weetman, A. Porzelt, P. Bag, F. Hanusch and S. Inoue, Chem. Sci., 2020, 11, 4817–4827 RSC; (c) R. L. Falconer, K. M. Byrne, G. S. Nichol, T. Krämer and M. J. Cowley, Angew. Chem., Int. Ed., 2021, 60, 24702–24708 CrossRef CAS PubMed.
- (a) J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, Angew. Chem., Int. Ed., 2021, 60, 1702–1713 CrossRef CAS PubMed; (b) J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, Nature, 2018, 557, 92–95 CrossRef CAS PubMed; (c) K. Koshino and R. Kinjo, J. Am. Chem. Soc., 2020, 142(19), 9057–9062 CrossRef CAS PubMed; (d) R. J. Schwamm, M. D. Anker, M. Lein and M. P. Coles, Angew. Chem., Int. Ed., 2019, 58, 1489–1493 CrossRef CAS PubMed; (e) J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, J. Am. Chem. Soc., 2019, 141, 11000–11003 CrossRef CAS PubMed; (f) S. Kurumada, S. Takamori and M. Yamashita, Nat. Chem., 2020, 12, 36–39 CrossRef CAS PubMed.
- (a) C. Dohmeier, C. Robl, M. Tacke and H. Schnöckel, Angew. Chem., Int. Ed. Engl., 1991, 30, 564–565 CrossRef; (b) C. Cui, H. W. Roesky, H.-G. Schmidt, M. Noltemeyer, H. Hao and F. Cimpoesu, Angew. Chem., Int. Ed., 2000, 39, 4274–4276 CrossRef CAS; (c) A. Hofmann, T. Trӧster, T. Kupfer and H. Braunschweig, Chem. Sci., 2019, 10, 3421–3428 RSC; (d) S. K. Mellerup, Y. Cui, F. Fantuzzi, P. Schmid, J. T. Goettel, G. Bélanger-Chabot, M. Arrowsmith, I. Krummenacher, Q. Ye, V. Engel, B. Engels and H. Braunschweig, J. Am. Chem. Soc., 2019, 141, 16954–16960 CrossRef CAS PubMed; (e) J. D. Queen, A. Lehmann, J. C. Fettinger, H. M. Tuononen and P. P. Power, J. Am. Chem. Soc., 2020, 142, 20554–20559 CrossRef CAS PubMed; (f) X. Zhang and L. L. Liu, Angew. Chem., Int. Ed., 2021, 60, 27062–27069 CrossRef CAS PubMed; (g) A. Hinz and M. P. Müller, Chem. Commun., 2021, 57, 12532–12535 RSC.
- S. Aldridge and A. J. Downs, The Group 13 Metals Aluminium, Gallium, Indium and Thallium: Chemical Patterns and Peculiarities, Wiley, Chichester, 2011 Search PubMed.
- (a) R. Mondol and E. Otten, Inorg. Chem., 2019, 58, 6344–6355 CrossRef CAS PubMed; (b) W. Chen, V. A. Dodonov, V. G. Sokolov, L. Liu, E. V. Baranov, Y. Zhao, I. L. Fedushkin and X.-J. Yang, Organometallics, 2021, 40, 490–499 CrossRef CAS.
- O. R. Luca and R. H. Crabtree, Chem. Soc. Rev., 2013, 42, 1440–1459 RSC.
- V. K. K. Praneeth, M. R. Ringenberg and T. R. Ward, Angew. Chem., Int. Ed., 2012, 51, 10228–10234 CrossRef CAS PubMed.
- (a) N. Nagahora, T. Sasamori, N. Takeda and N. Tokitoh, Chem. – Eur. J., 2004, 10, 6146–6151 CrossRef CAS PubMed; (b) C. Moser, M. Nieger and R. Pietschnig, Organometallics, 2006, 25, 2667–2672 CrossRef CAS; (c) C. Moser, A. Orthaber, M. Nieger, F. Belaj and R. Pietschnig, Dalton Trans., 2006, 3879–3885 RSC; (d) T. Sasamori, A. Yuasa, Y. Hosoi, Y. Furukawa and N. Tokitoh, Organometallics, 2008, 27, 3325–3327 CrossRef CAS; (e) A. Yuasa, T. Sasamori, Y. Hosoi, Y. Furukawa and N. Tokitoh, Bull. Chem. Soc. Jpn., 2009, 82, 793–805 CrossRef CAS; (f) T. Sasamori, H. Miyamoto, H. Sakai, Y. Furukawa and N. Tokitoh, Organometallics, 2012, 31, 3904–3910 CrossRef CAS; (g) T. Sasamori, M. Sakagami, M. Niwa, H. Sakai, Y. Furukawa and N. Tokitoh, Chem. Commun., 2012, 48, 8562–8564 RSC; (h) M. Sakagami, T. Sasamori, H. Sakai, Y. Furukawa and N. Tokitoh, Chem. – Asian J., 2013, 8, 690–693 CrossRef CAS PubMed; (i) M. Sakagami, T. Sasamori, H. Sakai, Y. Furukawa and N. Tokitoh, Bull. Chem. Soc. Jpn., 2013, 86, 1132–1143 CrossRef CAS; (j) Y. Suzuki, T. Sasamori, J.-D. Guo and N. Tokitoh, Chem. – Eur. J., 2018, 24, 364–368 CrossRef CAS PubMed.
- A. Appel, F. Jäkle, T. Priermeier, R. Schmid and M. Wagner, Organometallics, 1996, 15, 1188–1194 CrossRef CAS.
- H. Braunschweig, I. Krummenacher, C. Lichtenberg, J. D. Mattock, M. Schäfer, U. Schmidt, C. Schneider, T. Steffenhagen, S. Ullrich and A. Vargas, Angew. Chem., Int. Ed., 2017, 56, 889–892 CrossRef CAS PubMed.
- T. Sasamori, Y. Suzuki and N. Tokitoh, Organometallics, 2014, 33, 6696–6699 CrossRef CAS.
- N. Kuhn and T. Kratz, Synthesis, 1993, 561–562 CrossRef CAS.
- D. Dhara, A. Jayaraman, M. Härterich, R. D. Dewhurst and H. Braunschweig, Chem. Sci., 2022, 13, 5631–5638 RSC.
- (a) R. Y. Kong and M. R. Crimmin, Angew. Chem., Int. Ed., 2021, 60, 2619–2623 CrossRef CAS PubMed; (b) X. Zhang and L. L. Liu, Angew. Chem., Int. Ed., 2022, 61, e202116658 CAS.
- (a) A. J. Birch, J. Chem. Soc., 1944, 430–436 RSC; (b) A. J. Birch, Pure Appl. Chem., 1996, 68, 553–556 CrossRef CAS.
- (a) S. G. Minasian and J. Arnold, Chem. Commun., 2008, 4043–4045 RSC; (b) K. Nagata, T. Agou, T. Sasamori and N. Tokitoh, Chem. Lett., 2015, 44, 1610–1612 CrossRef CAS.
- (a) S.-H. Ueng, A. Solovyev, X. Yuan, S. J. Geib, L. Fensterbank, E. Lacâte, M. Malacria, M. Newcomb, J. C. Walton and D. P. Curran, J. Am. Chem. Soc., 2009, 131, 11256–11262 CrossRef CAS PubMed; (b) P. Bissinger, H. Braunschweig, A. Damme, I. Krummenacher, A. K. Phukan, K. Radacki and S. Sugawara, Angew. Chem., Int. Ed., 2014, 53, 7360–7363 CrossRef CAS PubMed; (c) M.-A. Légaré, G. Bélanger-Chabot, R. D. Dewhurst, E. Welz, I. Krummenacher, B. Engels and H. Braunschweig, Science, 2018, 359, 896–900 CrossRef PubMed; (d) M.-A. Légaré, G. Bélanger-Chabot, M. Rang, R. D. Dewhurst, I. Krummenacher, R. Bertermann and H. Braunschweig, Nat. Chem., 2020, 12, 1076–1080 CrossRef PubMed.
- T. Chu and G. I. Nikonov, Chem. Rev., 2018, 118, 3608–3680 CrossRef CAS PubMed.
- (a) M. A. Esteruelas, M. Pilar Gay and E. Oňate, Organometallics, 2018, 37, 3412–3424 CrossRef CAS; (b) Q. Liang, A. Salmon, P. J. Kim, L. Yan and D. Song, J. Am. Chem. Soc., 2018, 140, 1263–1266 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, NMR and UV-vis spectra, X-ray crystallographic and computational details. CCDC 2166939, 2166948, 2166949, 2166951, 2166952, 2166953 and 2166954. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc02783f |
| This journal is © The Royal Society of Chemistry 2022 |