
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNickel-catalysed diversification of phosphine ligands by formal substitution at phosphorus†
Sven
Roediger
 ,
Sebastian U.
Leutenegger
and
Bill
Morandi
*
,
Sebastian U.
Leutenegger
and
Bill
Morandi
*
Laboratorium für Organische Chemie, ETH Zürich, Vladimir-Prelog-Weg 3, HCI, 8093 Zürich, Switzerland. E-mail: bill.morandi@org.chem.ethz.ch
First published on 14th June 2022
Abstract
We report a diversification strategy that enables the direct substituent exchange of tertiary phosphines. Alkylated phosphonium salts, prepared by standard alkylation of phosphines, are selectively dearylated in a nickel-catalysed process to access alkylphosphine products via a formal substitution at the phosphorus center. The reaction can be used to introduce a wide range of alkyl substituents into both mono- and bisphosphines. We also show that the alkylation and dearylation steps can be conducted in a one-pot sequence, enabling accelerated access to derivatives of the parent ligand. The phosphine products of the reaction are converted in situ to air-stable borane adducts for isolation, and versatile derivatisation reactions of these adducts are demonstrated.
Introduction
Many of the recent advances in transition metal catalysis have been driven by the design of bespoke ancillary ligands that modulate the catalyst's reactivity in an unprecedented fashion.1 Despite the emergence of a large variety of ligand classes, phosphines remain the ligands of choice for many applications.2,3 A significant benefit of phosphines is that the ligand's electronic and steric properties can be tuned with precision by varying the substituents on the phosphorus centre. Taking advantage of a versatile toolbox of synthetic methods to access phosphines,4 many powerful phosphine ligand architectures have been developed.5–11 Phosphines have also been employed in numerous other applications such as organocatalysis,12,13 frustrated Lewis pair catalysis,14 or material sciences.15,16 It can be expected that the continued design of phosphines will lead to even more active ligands, opening further avenues for the application of this intriguing class of compounds.Traditionally, phosphine ligands are prepared by de novo synthetic approaches such as nucleophilic substitution reactions of halophosphine substrates with organometallic reagents.4 Alternative strategies include the reduction of phosphine oxides17–20 or reactions of hydrophosphines such as the hydrophosphination of unsaturated systems,21–23 cross-coupling with aryl halides,24,25 or substitution reactions with electrophiles in the presence of base.4,26 Combined, these methods allow access to a plethora of diverse phosphine architectures. However, they typically require multi-step procedures involving toxic or pyrophoric reagents and air-sensitive intermediates that are challenging to isolate. These drawbacks can make the preparation of phosphines arduous and restrict ligand optimisation campaigns to the evaluation of the limited collection of commercially available phosphines. However, if chemists only evaluate commercially available phosphines, they might fail to identify more active and selective catalysts. Therefore, new approaches towards the straightforward synthesis of phosphines and other phosphorus-containing compounds are in critical demand.27–29
A strategy to address this problem is the direct modification of tertiary phosphines (Scheme 1a). In this approach, substituents of phosphines are either altered or exchanged entirely, hence bypassing the need to handle toxic primary and secondary phosphines or even PH3. It is also an efficient way to quickly generate libraries of ligands, which is a central endeavour for the rapid discovery and optimisation of new reactions. The direct access to ligand derivatives from a hit result also becomes increasingly important in light of recent advances in statistical and machine learning-based approaches that facilitate the in silico optimisation of ligand structures.9,30,31 Most efforts directed at the modification of tertiary phosphines have focused on altering substituents, for instance by C–H functionalisation approaches.32–34 Reactions that entirely replace one of the substituents of a phosphine remain rare although they would arguably be the most versatile tools to modify a wide range of phosphines. Such a strategy would be particularly useful to access underexplored alkylated phosphines. Approaches towards this goal are however limited and mainly rely on the formation and subsequent reaction of metal phosphides by engaging phosphines with highly reactive alkali metals (Scheme 1b).35–37 As an alternative, Wang and co-workers reported that acyl phosphines can be used as surrogates for secondary phosphines in metal-catalysed alkylation and arylation reactions.38–41 While these methods expand the toolbox of phosphine modification reactions, they suffer from poor cleavage selectivity or limited scope, respectively. A general and selective strategy to introduce alkyl substituents into tertiary phosphines has thus remained elusive.
 | ||
| Scheme 1 Context of this work. | ||
Our group42–44 and others45–53 have used the ability of transition metals to oxidatively add into P–C bonds of phosphonium salts for catalytic reactions. In the context of phosphine modification, we have reported a palladium-catalysed process that scrambles aryl groups between two triarylphosphines (Scheme 1c top).42 The in situ formed phenylpalladium iodide catalyst undergoes C–P reductive elimination with a phosphine and subsequent C–P oxidative addition into another C–P bond of the formed phosphonium salt, leading to an exchange of the aryl group on the metal centre. Reaction with another phosphine results in further exchange of aryl groups between the different phosphine starting materials. While this process enables the formation of a large variety of triarylphosphines, it is not synthetically useful as the scrambled triarylphosphines are formed as a statistical mixture. Furthermore, the use of triarylphosphines as the source of the transferred aryl group is unpractical as separation of the desired product from the by-products becomes very demanding. To improve this process, we identified two key challenges that needed to be addressed. First, selective cleavage of one C–P bond over another in the intermediate phosphonium salt would be necessary to obtain a single product. Second, the use of simple R–X compounds instead of PAr3 as source of the introduced phosphine substituent would be more versatile and simplify purification.
Although the oxidative addition of transition metals into P–C(aryl) bonds is well established, only few examples are known in which a P–C(alkyl) bond of a phosphonium salt is cleaved.54–56 We hypothesized that this contrast in reactivity could provide a convenient entry to alkylated phosphines. A metal catalyst could undergo selective oxidative addition into a P–C(aryl) bond of an alkylarylphosphonium salt, retaining the alkyl group, to form the desired alkylphosphine (Scheme 1c bottom). The resulting metal aryl complex could then be engaged in a standard cross-coupling manifold to enable catalyst turnover and to avoid a challenging C–X reductive elimination.47,50,57 As the phosphonium salt starting material could be prepared by routine alkylation of a ubiquitous arylphosphine, the overall process would represent a formal substitution of aryl for alkyl groups at the phosphorus centre. Here, we report the realisation of this strategy as a versatile method that enables rapid diversification of commercial phosphines to access alkylphosphine ligand space (Scheme 1d).58
Results and discussion
After evaluation of a broad set of reaction conditions, we discovered that a combination of Ni(COD)2 as pre-catalyst, the ligand precursor IiPr·HBF4 (IiPr·HBF4 = 1,3-di(iso-propyl)imidazolium tetrafluoroborate), and potassium phosphate as base enabled the desired dearylation of a model phosphonium salt in high yield by trapping the cleaved aryl group in a Suzuki-type coupling with phenylboronic acid (see ESI† for details).47,50 The resulting biphenyl by-product from the Suzuki coupling can be easily separated from the desired product. For convenience, the reactions were typically set up in an argon-filled glovebox. Of note, a benchtop setup also provided the products in only slightly lower yield (see ESI† for details). Alternatively, the air-stable Ni(0) precatalysts developed by the groups of Cornella59,60 and Engle61 can be used instead of Ni(COD)2 to set the reaction up on the benchtop with no decrease in yield (see ESI†). With these results in hand, we investigated the scope of the reaction. Phosphonium salts containing different alkyl groups were prepared by alkylation of phosphines in good to high yields using standard methods4 (see ESI†) and then subjected to the dearylation reaction (Scheme 2). For ease of purification, the phosphine products were typically isolated after in situ conversion to the borane adducts. These adducts are air-stable, can be conveniently purified by column chromatography, and are easily deprotected (vide infra).62,63 | ||
Scheme 2 Scope of monophosphines. Yields refer to isolated compounds for the dearylation process after derivatisation to the air-stable BH3 adduct. adr = 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The starting material 1g had a dr of 9:1. bFrom the iodide salt. c14:1 mixture with Ph3P·BH3. dFrom the chloride salt. eSIMes·HCl used instead of IiPr·HBF4. :1. The starting material 1g had a dr of 9:1. bFrom the iodide salt. c14:1 mixture with Ph3P·BH3. dFrom the chloride salt. eSIMes·HCl used instead of IiPr·HBF4. | ||
Phosphines containing both activated and unactivated primary alkyl groups were prepared in high yield by our dearylation strategy (2a–d). A cyclohexyl group was incorporated in 73% yield (2e). This moiety as well as the tert-butyl and adamantyl groups are arguably the most widespread alkyl substituents in modern ligands. Our reaction not only tolerates the presence of these moieties (2e, 2i, 2q–r), but also allows to introduce other, less prevalent cyclic secondary alkyl groups (2f–g) or tertiary alkyl groups (2h). Alkyl groups containing coordinating moieties like pyridyl, primary alcohol, and ether groups were incorporated in good yield (2j–2l) and provide opportunities to use the reaction to prepare chelating ligands with two different coordinating atoms. A low, but synthetically useful yield was observed for a substrate containing a tertiary amine (2m, 24%). As an alternative, a phthalimide moiety, which can be used as a precursor for amines, was well tolerated (2n). The reaction can also be used to synthesize phosphines containing more than one alkyl group. Dialkylphosphonium salts with differing steric demand afforded the desired product in good yield (2o–r). Trialkylphosphine 2s was prepared in a lowered, but synthetically useful yield of 30%. In contrast, the caged trialkylphosphine 2t was obtained in high yield (84%).
Besides the introduced alkyl group, the nature of the removed aryl group can also be varied. Electron-rich and electron-poor aryl groups can be cleaved in the reaction in high yield (2u–2v). Notably, the reaction can also be conducted on a large scale, as demonstrated by the preparation of two grams of 2u. Besides the dearylation of phosphonium salts, the dealkylation is possible when the starting material does not contain an aryl group. Tetraalkylphosphonium salt 1w was selectively debenzylated in 19% yield without additional optimisation.
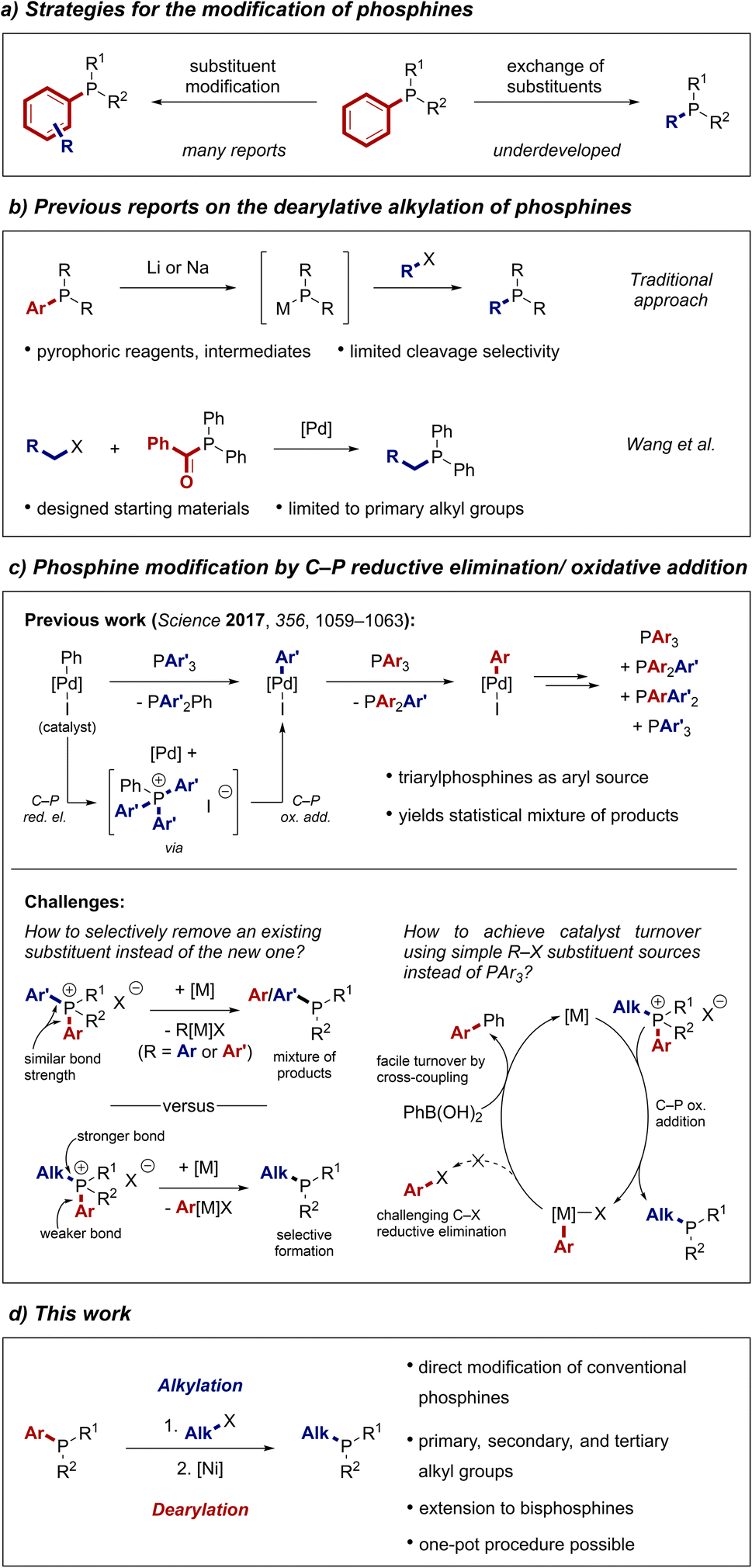
We next investigated reactions of phosphonium salts containing two different aryl groups (Scheme 3a). The transformation of phosphonium salt 1x, containing electron-donating and electron-withdrawing aryl groups, resulted in partial cleavage of both aryl groups to deliver the two alkylated phosphines 2v and 2x in useful yields in a single reaction. The products could be conveniently separated by column chromatography. In contrast, the 2-methoxyphenyl group was exclusively removed from phosphonium salt 1y, indicating a directing effect of the ortho-methoxy moiety. The reaction can also be used to modify Buchwald-type ligands, as demonstrated by the synthesis of the air-stable JohnPhos derivative 2z in good yield.
 | ||
| Scheme 3 Further applications. a8:1 mixture with 2-methoxybiphenyl. | ||
Results of further substrates that were tested in the reaction are summarized in Fig. S3.† Generally, substrates with high steric hindrance performed worse in the reaction than less sterically hindered substrates. Additionally, substrates with strongly coordinating moieties did not perform well or even led to cleavage of the alkyl group.
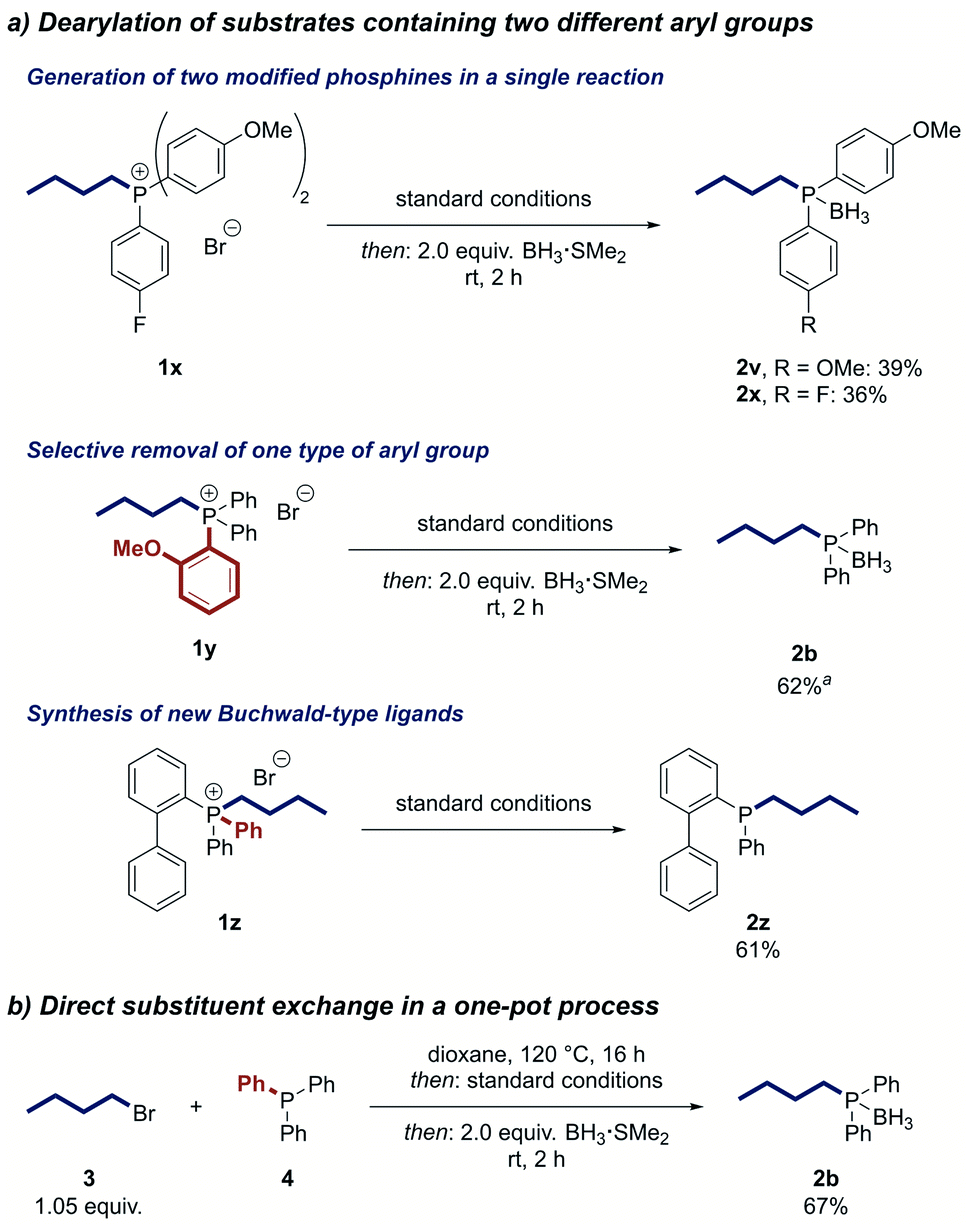
To further increase the utility of our strategy, we developed a one-pot protocol in which the phosphonium salt is first formed by the alkylation of a phosphine and then directly treated with the reagents for the nickel-catalysed dearylation. Applying this protocol, we were able to prepare n-butyldiphenylphosphine from triphenylphosphine (4) in 67% yield after converting the product to its air-stable borane adduct 2b (Scheme 3b). Notably, no intermediate workup or solvent change is required, making this process a direct substitution at the phosphorus centre and enabling rapid access to alkylated phosphine ligands.
Due to the high importance of bidentate ligands in catalysis,2 we attempted the synthesis of bisphosphines by the twofold dearylation of a bisphosphonium salt. However, no product was detected. Mechanistic experiments showed that the desired dearylation occurred, but the bidentate phosphine product L deactivated the catalyst by irreversibly coordinating to it in a NiL2 complex (see ESI†). We thus tested a range of metal scavengers to de-coordinate the product from the nickel centre of this complex and found that sodium cyanide is highly active for this process.64–67 This insight enabled us to develop a strategy for the dearylative alkylation of bisphosphines. After an alkylation step, the nickel complexes NiL2 of the desired dearylated ligand products were formed by a stoichiometric Suzuki reaction of the bisphosphonium salt and then directly exposed to sodium cyanide to afford the free bidentate phosphines. Notably, the protocol offers the possibility to modify the starting ligand selectively on both phosphorus centres or just one. 1,3-Bis(diphenylphosphino)propane (DPPP) (5) could be mono- or dialkylated in good yield, respectively, by simply changing the stoichiometry of the alkylation step (Scheme 4). The dearylation of the resulting phosphonium salts 6 and 8 proceeded smoothly to yield the symmetrically modified ligand 7 in 44% yield and the unsymmetrical ligand 9 in 70% yield, respectively. Other privileged ligand scaffolds such as 1,1′-bis(diphenylphosphino)ferrocene (DPPF) (10) can also be altered using this process. Alkylation of 10 selectively yielded the monophosphonium salt 12 potentially because of the hindered nature of the second phosphino moiety after the first alkylation. The dearylation of 12 occurred in good yield to furnish the unsymmetrical DPPF-type ligand 13 that would be difficult to prepare by traditional means. The reaction can not only be used to modify bidentate ligands but also to construct them. DPPP (5) could be prepared in 46% yield by the reaction of triphenylphosphine (4) with the alkyl dihalide 14 and subsequent two-fold dearylation.
 | ||
| Scheme 4 Modification of bidentate ligands. See ESI† for detailed reaction conditions. | ||
The phosphine borane adducts, as which the phosphine products were typically isolated, can be used in versatile derivatisation reactions (Scheme 5). The free phosphine 16 was accessed in nearly quantitative yield by treatment of the phosphine borane adduct 2u with DABCO and a subsequent simple filtration through Celite.68 Conversion to the HBF4 salt 17 was achieved in good yield.69,70 Such salts are air-stable and can be used directly as ligand precursors in catalysis by releasing the free phosphine in situ after treatment with a base.71
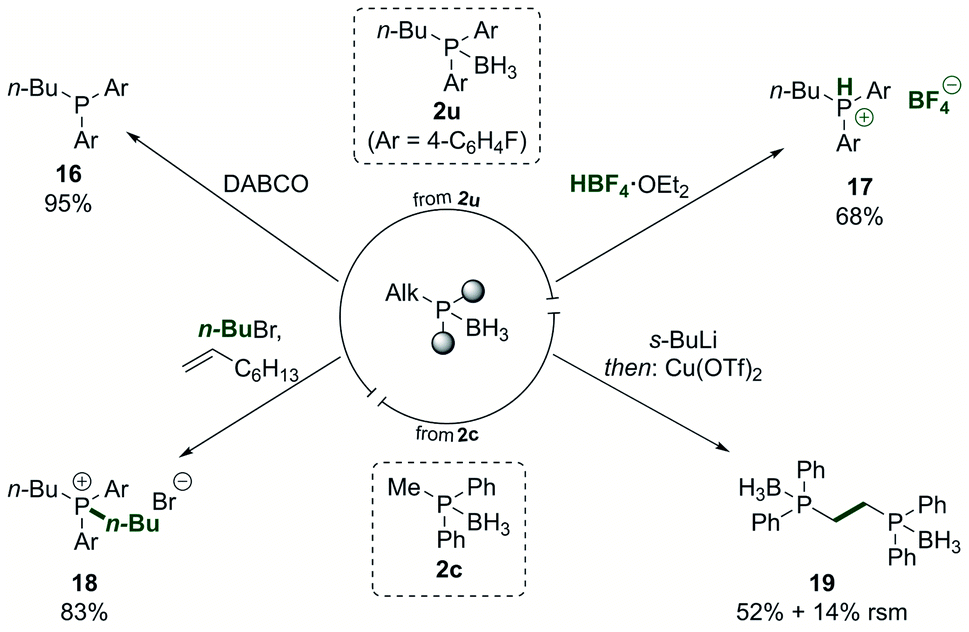 | ||
| Scheme 5 Derivatisation of the phosphine borane products. DABCO = 1,4-diazabicyclo[2.2.2]octane. RSM = recovered starting material. See ESI† for detailed reaction conditions. | ||
Phosphonium salt 18 was accessed in 83% yield from the phosphine borane adduct 2u by treating it with an alkyl halide in the presence of 1-octene.72 The product phosphonium salt could then be used in the nickel-catalysed dearylation reaction again, allowing to quickly introduce multiple alkyl groups in a phosphine in a programmed fashion. As an additional way to modify the products, the alkyl group in the phosphine borane adduct 2c was deprotonated in alpha-position to the phosphorus, and subsequent oxidative dimerisation yielded the bidentate product 19, providing a means to extend the ligand diversification beyond the scope of the dearylation reaction.73
Conclusions
In conclusion, we demonstrated a general strategy for the modification of phosphine ligands. The reaction enables the substitution of aryl groups in phosphines for alkyl groups in a protocol relying on the straightforward alkylation of a phosphine and a subsequent nickel-catalysed dearylation reaction. Besides the broad scope of the dearylation method, we demonstrate that the overall process can also be directly conducted as a one-pot protocol. Together with the development of a related strategy for the modification of bidentate ligands and versatile product derivatisation methods, this methodology provides a rapid entry into alkylphosphine ligand space. We expect that this strategy will streamline ligand optimisation campaigns and enable the identification of new powerful ligands for use in catalysis.Data availability
Full experimental and characterization data is available in the ESI.†Author contributions
S. R. conceived the project. S. R. and S. U. L. performed the experimental studies. B. M. supervised the research. S. R. wrote the original draft of the manuscript which was edited by all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
ETH Zürich and the FCI (scholarship to S. R.) are acknowledged for funding of this work. We thank the NMR service and the Molecular and Biomolecular Analysis Service (MoBiAS) of ETH Zürich for assistance. We thank Dr Ori Green for help with HPLC separations, our group for critical proofreading of the manuscript, and the groups of Prof. K. M. Engle and Prof. J. Cornella for providing their air-stable Ni(0) precursors.Notes and references
- J. F. Hartwig, Organotransition Metal Chemistry: From Bonding To Catalysis, University Science Books, Sausalito, CA, USA, 2010 Search PubMed.
- A. L. Clevenger, R. M. Stolley, J. Aderibigbe and J. Louie, Chem. Rev., 2020, 120, 6124–6196 CrossRef CAS PubMed.
- Phosphorus(III) Ligands in Homogeneous Catalysis: Design and Synthesis, ed. P. C. J. Kamer and P. W. N. M. van Leeuwen, John Wiley & Sons, Ltd, Chichester, UK, 2012 Search PubMed.
- E. I. Musina, A. S. Balueva and A. A. Karasik, in Organophosphorus Chemistry, ed. L. J. Higham, D. W. Allen and J. C. Tebby, Royal Society of Chemistry, 2021, vol. 50, pp. 37–114 Search PubMed.
- R. Martin and S. L. Buchwald, Acc. Chem. Res., 2008, 41, 1461–1473 CrossRef CAS PubMed.
- R. J. Lundgren and M. Stradiotto, Chem.–Eur. J., 2012, 18, 9758–9769 CrossRef CAS PubMed.
- P. C. J. Kamer, P. W. N. M. van Leeuwen and J. N. H. Reek, Acc. Chem. Res., 2001, 34, 895–904 CrossRef CAS PubMed.
- A. Sarbajna, V. S. V. S. N. Swamy and V. H. Gessner, Chem. Sci., 2021, 12, 2016–2024 RSC.
- K. Wu and A. G. Doyle, Nat. Chem., 2017, 9, 779–784 CrossRef CAS PubMed.
- L. Chen, P. Ren and B. P. Carrow, J. Am. Chem. Soc., 2016, 138, 6392–6395 CrossRef CAS PubMed.
- A. Zapf, A. Ehrentraut and M. Beller, Angew. Chem., Int. Ed., 2000, 39, 4153–4155 CrossRef CAS PubMed.
- H. Guo, Y. C. Fan, Z. Sun, Y. Wu and O. Kwon, Chem. Rev., 2018, 118, 10049–10293 CrossRef CAS PubMed.
- C. Xie, A. J. Smaligo, X.-R. Song and O. Kwon, ACS Cent. Sci., 2021, 7, 536–558 CrossRef CAS PubMed.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400–6441 CrossRef CAS PubMed.
- D. J. Young, S. W. Chien and T. S. A. Hor, Dalton Trans., 2012, 41, 12655 RSC.
- Y. Ren and T. Baumgartner, Dalton Trans., 2012, 41, 7792 RSC.
- Ł. Kapuśniak, P. N. Plessow, D. Trzybiński, K. Woźniak, P. Hofmann and P. I. Jolly, Organometallics, 2021, 40, 693–701 CrossRef PubMed.
- H.-C. Wu, J.-Q. Yu and J. B. Spencer, Org. Lett., 2004, 6, 4675–4678 CrossRef CAS PubMed.
- C. A. Busacca, R. Raju, N. Grinberg, N. Haddad, P. James-Jones, H. Lee, J. C. Lorenz, A. Saha and C. H. Senanayake, J. Org. Chem., 2008, 73, 1524–1531 CrossRef CAS PubMed.
- N. I. Rinehart, A. J. Kendall and D. R. Tyler, Organometallics, 2018, 37, 182–190 CrossRef CAS.
- Y.-B. Li, H. Tian and L. Yin, J. Am. Chem. Soc., 2020, 142, 20098–20106 CrossRef CAS PubMed.
- M. M. I. Basiouny, D. A. Dollard and J. A. R. Schmidt, ACS Catal., 2019, 9, 7143–7153 CrossRef CAS.
- S. Pullarkat, Synthesis, 2015, 48, 493–503 CrossRef.
- M. Murata and S. L. Buchwald, Tetrahedron, 2004, 60, 7397–7403 CrossRef CAS.
- J.-S. Zhang, T. Chen, J. Yang and L.-B. Han, Chem. Commun., 2015, 51, 7540–7542 RSC.
- P. B. Arockiam, U. Lennert, C. Graf, R. Rothfelder, D. J. Scott, T. G. Fischer, K. Zeitler and R. Wolf, Chem.–Eur. J., 2020, 26, 16374–16382 CrossRef CAS PubMed.
- A. Mondal, N. O. Thiel, R. Dorel and B. L. Feringa, Nat. Catal., 2022, 5, 10–19 CrossRef CAS.
- H. Deng, M. Wang, Y. Liang, X. Chen, T. Wang, J. J. Wong, Y. Zhao, K. N. Houk and Z. Shi, Chem, 2022, 8, 569–579 CAS.
- T. Barber, S. P. Argent and L. T. Ball, ACS Catal., 2020, 10, 5454–5461 CrossRef CAS.
- T. Gensch, G. dos Passos Gomes, P. Friederich, E. Peters, T. Gaudin, R. Pollice, K. Jorner, A. Nigam, M. Lindner-D'Addario, M. S. Sigman and A. Aspuru-Guzik, J. Am. Chem. Soc., 2022, 144, 1205–1217 CrossRef CAS PubMed.
- S. Zhao, T. Gensch, B. Murray, Z. L. Niemeyer, M. S. Sigman and M. R. Biscoe, Science, 2018, 362, 670–674 CrossRef CAS PubMed.
- X. Qiu, M. Wang, Y. Zhao and Z. Shi, Angew. Chem., Int. Ed., 2017, 56, 7233–7237 CrossRef CAS PubMed.
- J. Wen, B. Dong, J. Zhu, Y. Zhao and Z. Shi, Angew. Chem., Int. Ed., 2020, 59, 10909–10912 CrossRef CAS PubMed.
- M. R. Auth, K. A. McGarry and T. B. Clark, Adv. Synth. Catal., 2021, 363, 2354–2365 CrossRef CAS.
- A. M. Aguiar, J. Beisler and A. Mills, J. Org. Chem., 1962, 27, 1001–1005 CrossRef CAS.
- A. M. Aguiar, H. J. Greenberg and K. E. Rubenstein, J. Org. Chem., 1963, 28, 2091–2093 CrossRef CAS.
- J. Ye, J.-Q. Zhang, Y. Saga, S. Onozawa, S. Kobayashi, K. Sato, N. Fukaya and L.-B. Han, Organometallics, 2020, 39, 2682–2694 CrossRef CAS.
- R. Yu, X. Chen and Z. Wang, Tetrahedron Lett., 2016, 57, 3404–3406 CrossRef CAS.
- J. Yang, H. Wu and Z. Wang, J. Saudi Chem. Soc., 2018, 22, 1–5 CrossRef CAS.
- M. Zhang, Z. Ma, H. Du and Z. Wang, Tetrahedron Lett., 2020, 61, 152125 CrossRef CAS.
- X. Chen, H. Wu, R. Yu, H. Zhu and Z. Wang, J. Org. Chem., 2021, 86, 8987–8996 CrossRef CAS PubMed.
- Z. Lian, B. N. Bhawal, P. Yu and B. Morandi, Science, 2017, 356, 1059–1063 CrossRef CAS PubMed.
- Y. H. Lee and B. Morandi, Nat. Chem., 2018, 10, 1016–1022 CrossRef CAS PubMed.
- P. Boehm, T. Martini, Y. H. Lee, B. Cacherat and B. Morandi, Angew. Chem., Int. Ed., 2021, 60, 17211–17217 CrossRef CAS PubMed.
- F. Y. Kwong and K. S. Chan, Chem. Commun., 2000, 1069–1070 RSC.
- F. Y. Kwong and K. S. Chan, Organometallics, 2000, 19, 2058–2060 CrossRef CAS.
- L. K. Hwang, Y. Na, J. Lee, Y. Do and S. Chang, Angew. Chem., Int. Ed., 2005, 44, 6166–6169 CrossRef CAS PubMed.
- K. Baba, Y. Masuya, N. Chatani and M. Tobisu, Chem. Lett., 2017, 46, 1296–1299 CrossRef CAS.
- H. Fujimoto, M. Kusano, T. Kodama and M. Tobisu, Org. Lett., 2019, 21, 4177–4181 CrossRef CAS PubMed.
- X. Zhang and A. McNally, Angew. Chem., Int. Ed., 2017, 56, 9833–9836 CrossRef CAS PubMed.
- X. Zhang and A. McNally, ACS Catal., 2019, 9, 4862–4866 CrossRef CAS PubMed.
- Y. Che, Y. Yue, L. Lin, B. Pei, X. Deng and C. Feng, Angew. Chem., Int. Ed., 2020, 59, 16414–16419 CrossRef CAS PubMed.
- H. Wang, M. Yang, Y. Wang, X. Man, X. Lu, Z. Mou, Y. Luo and H. Liang, Org. Lett., 2021, 23, 8183–8188 CrossRef CAS PubMed.
- S. A. Macgregor, Chem. Soc. Rev., 2007, 36, 67–76 RSC.
- L. Wang, H. Chen and Z. Duan, Chem.–Asian J., 2018, 13, 2164–2173 CrossRef CAS PubMed.
- Y. H. Lee and B. Morandi, Coord. Chem. Rev., 2019, 386, 96–118 CrossRef CAS.
- M. Sakamoto, I. Shimizu and A. Yamamoto, Chem. Lett., 1995, 24, 1101–1102 CrossRef.
- After this manuscript was deposited on a preprint server, a related work appeared: M. Lei, X. Chen, Y. Wang, L. Zhang, H. Zhu and Z. Wang, Org. Lett., 2022, 24, 2868–2872 CrossRef CAS PubMed.
- L. Nattmann, R. Saeb, N. Nöthling and J. Cornella, Nat. Catal., 2020, 3, 6–13 CrossRef CAS.
- L. Nattmann and J. Cornella, Organometallics, 2020, 39, 3295–3300 CrossRef CAS.
- V. T. Tran, Z. Li, O. Apolinar, J. Derosa, M. V. Joannou, S. R. Wisniewski, M. D. Eastgate and K. M. Engle, Angew. Chem., Int. Ed., 2020, 59, 7409–7413 CrossRef CAS PubMed.
- J. M. Brunel, B. Faure and M. Maffei, Coord. Chem. Rev., 1998, 178–180, 665–698 CrossRef CAS.
- M. Ohff, J. Holz, M. Quirmbach and A. Börner, Synthesis, 1998, 1998, 1391–1415 CrossRef.
- T. A. DelDonno and W. Rosen, J. Am. Chem. Soc., 1977, 99, 8051–8052 CrossRef CAS.
- D. J. Brauer, F. Gol, S. Hietkamp, H. Peters, H. Sommer, O. Stelzer and W. S. Sheldrick, Chem. Ber., 1986, 119, 349–365 CrossRef CAS.
- C. D. Swor and D. R. Tyler, Coord. Chem. Rev., 2011, 255, 2860–2881 CrossRef CAS.
- B. P. Nell, C. D. Swor, E. A. Henle, L. N. Zakharov, N. I. Rinehart, A. Nathan and D. R. Tyler, Dalton Trans., 2016, 45, 8253–8264 RSC.
- H. Brisset, Y. Gourdel, P. Pellon and M. Le Corre, Tetrahedron Lett., 1993, 34, 4523–4526 CrossRef CAS.
- L. McKinstry and T. Livinghouse, Tetrahedron Lett., 1994, 35, 9319–9322 CrossRef CAS.
- S. E. Denmark and N. S. Werner, Org. Lett., 2011, 13, 4596–4599 CrossRef CAS PubMed.
- M. R. Netherton and G. C. Fu, Org. Lett., 2001, 3, 4295–4298 CrossRef CAS PubMed.
- J. Uziel, N. Riegel, B. Aka, P. Figuière and S. Jugé, Tetrahedron Lett., 1997, 38, 3405–3408 CrossRef CAS.
- T. Imamoto, T. Kusumoto, N. Suzuki and K. Sato, J. Am. Chem. Soc., 1985, 107, 5301–5303 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental and characterization data. See https://doi.org/10.1039/d2sc02496a |
| This journal is © The Royal Society of Chemistry 2022 |