
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBetween imide, imidyl and nitrene – an imido iron complex in two oxidation states†
Sascha
Reith
 a,
Serhiy
Demeshko
b,
Beatrice
Battistella
c,
Alexander
Reckziegel
a,
Christian
Schneider
a,
Andreas
Stoy
a,
Crispin
Lichtenberg
a,
Franc
Meyer
b,
Dominik
Munz
*de and
C. Gunnar
Werncke
*a
a,
Serhiy
Demeshko
b,
Beatrice
Battistella
c,
Alexander
Reckziegel
a,
Christian
Schneider
a,
Andreas
Stoy
a,
Crispin
Lichtenberg
a,
Franc
Meyer
b,
Dominik
Munz
*de and
C. Gunnar
Werncke
*a
aPhilipps-University Marburg, Department of Chemistry, Hans-Meerwein-Str. 4, D-35037 Marburg, Germany. E-mail: gunnar.werncke@chemie.uni-marburg.de
bUniversity of Göttingen, Institute of Inorganic Chemistry, Tammannstr. 4, D-37077 Göttingen, Germany
cHumboldt-University, Berlin Institute for Chemistry, Brook-Taylor-Str. 2, D-12489 Berlin, Germany
dSaarland University, Inorganic Chemistry: Coordination Chemistry, Campus C4.1, D-66123 Saarbrücken, Germany. E-mail: dominik.munz@uni-saarland.de
eFriedrich-Alexander University Erlangen-Nürnberg, Inorganic Chemistry, Egerlandstr. 1, D-91058 Erlangen, Germany
First published on 9th June 2022
Abstract
Imidyl and nitrene metal species play an important role in the N-functionalisation of unreactive C–H bonds as well as the aziridination of olefines. We report on the synthesis of the trigonal imido iron complexes [Fe(NMes)L2]0,− (L = –N{Dipp}SiMe3); Dipp = 2,6-diisopropyl-phenyl; Mes = (2,4,6-trimethylphenyl) via reaction of mesityl azide (MesN3) with the linear iron precursors [FeL2]0,−. UV-vis-, EPR-, 57Fe Mössbauer spectroscopy, magnetometry, and computational methods suggest for the reduced form an electronic structure as a ferromagnetically coupled iron(II) imidyl radical, whereas oxidation leads to mixed iron(III) imidyl and electrophilic iron(II) nitrene character. Reactivity studies show that both complexes are capable of H atom abstraction from C–H bonds. Further, the reduced form [Fe(NMes)L2]− reacts nucleophilically with CS2 by inserting into the imido iron bond, as well as electrophilically with CO under nitrene transfer. The neutral [Fe(NMes)L2] complex shows enhanced electrophilic behavior as evidenced by nitrene transfer to a phosphine, yet in combination with an overall reduced reactivity.
Introduction
Imido complexes of the late 3d-transition metals play a crucial role in nitrene transfer catalysis.1,2 As such there is an innate interest in understanding the structural and electronic features of the central imido metal unit to control and predict its reactivity.3–5 The imido ligand is classically regarded as a dianionic imide [NR]2− that forms either a double or triple bond, as it is the case for early transition metals.2,6 For the 3d-metals, the metal–imido bond becomes more covalent and p → d donation is reduced due to (partially) filled d-orbitals. This may result in intriguing bonding situations, namely metal stabilized imidyl radical [NR]˙− or even electrophilic nitrene [NR]0 ligands, that are highly reactive.4,5 The former was observed in a few instances for iron,7,8 cobalt9,10 and nickel.11,12 3d-Metal nitrenes are mostly known for copper,13–15 with only two isolated examples.13,15 Given the overall reactive nature of late 3d-metal imido species, the impact of oxidation state changes onto the imido unit is only partially understood. For low-spin complexes, mostly found for isolable and rather unreactive late 3d-metal imidos, few reports of one-electron oxidation showed contraction of the imido metal bond.12,16–20 This can be explained by a metal centred oxidation, accompanied with ion radius contraction as well as increased p → d donation and depopulation of anti-bonding molecular orbitals.5,21 Contrastingly, in the only two reports of oxidation of high-valent high-spin 3d-metal imidos M–N bond elongation took place (Fig. 1).9,22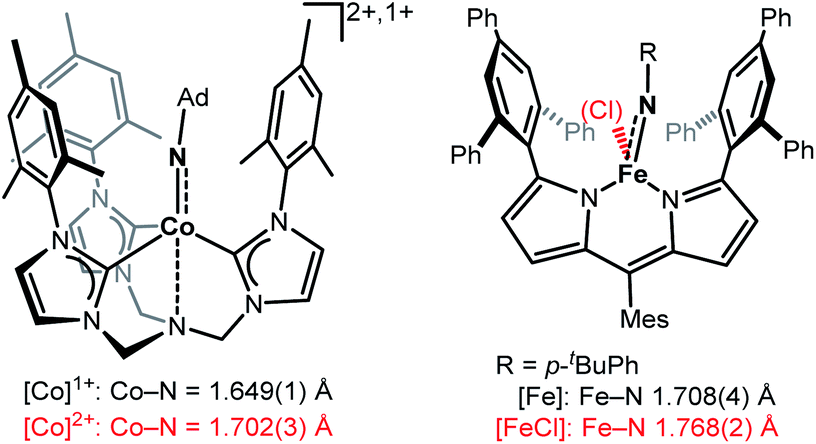 | ||
| Fig. 1 Examples of imido metal complex oxidation leading to M–N bond lengthening and imidyl character (Ad = adamantyl, Mes = 2,4,6-trimethylphenyl).7–9 | ||
This was attributed to a ligand-centred oxidation and spin density increase at the imido ligand, that resulted in an imidyl radical character. For a pseudo-trigonal bipyramidal imido cobalt complex (Fig. 1, left) oxidation led to a switch from the high-spin to the low-spin configuration (d5, S = 1/2).9 For a trigonal planar dipyrromethane (dpm) based imido iron complex (Fig. 1, right) the high-spin state persisted, with an increase of the coordination number.22
Building on recent examples of low-coordinate imido cobalt silylamides in higher spin-states23,24 we report herein the trigonal iron imido complexes [Fe(NMes)L2]0,− (L = –N(Dipp)SiMe3; Dipp = 2,6-diisopropyl-phenyl; Mes = 2,4,6-trimethylphenyl) in the formal oxidation states +III and +IV, which can be interconverted by oxidation/reduction. Detailed spectroscopic and computational analyses reveal the ambiguous electronic structure of both complexes. The anionic [Fe(NMes)L2]− is described best as an iron(II) imidyl complex whereas the oxidized neutral [Fe(NMes)L2]0 shares iron(III) imidyl and iron(II) nitrene character. The electronic ambiguity is reflected best by [Fe(NMes)L2]− which shows ambiphilic, i.e. combined nucleophilic and electrophilic behaviour, whereas the nucleophilicity is lost upon oxidation to [Fe(NMes)L2]0.
Results and discussion
The imido iron complex K{m}[Fe(NMes)L2], K{m}[1] (m = crypt.222), was obtained as a green crystalline solid (yield 86%) by treatment of a solution of the linear iron(I) precursor K{m}[FeL2] in a mixture of thf/Et2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) with MesN3 at −40 °C (Scheme 1). X-ray diffraction analysis revealed for the complex anion [1]− (Fig. 2) a Fe–Nimido bond length of 1.7744(15) Å and a Fe–N–CAryl bond angle of 173.6(13)°. Usually, imido–iron bond lengths of low-spin complexes range from 1.65 to 1.70 Å.16–18,25,26 Longer Fe–Nimido bonds are rarely observed, but are found for high-spin iron imidos with7,8,27 or without28 imidyl character on the nitrogen atom as well as an anionic iron(II) imide.20 [1]− shows a short N–CAryl bond of 1.339(2) Å, which is in line with aforementioned high-spin imido iron complexes with C–N double bond character. The observed structural metrics closely resemble the related high-spin imidyl cobalt(II) complex [Co(NDipp)L2]− (e.g. M–Nimide 1.751(2) Å, N–CAryl 1.347(2) Å),23 which suggested for [1]− also an iron(II) imidyl-like bonding situation on a structural level.
:3) with MesN3 at −40 °C (Scheme 1). X-ray diffraction analysis revealed for the complex anion [1]− (Fig. 2) a Fe–Nimido bond length of 1.7744(15) Å and a Fe–N–CAryl bond angle of 173.6(13)°. Usually, imido–iron bond lengths of low-spin complexes range from 1.65 to 1.70 Å.16–18,25,26 Longer Fe–Nimido bonds are rarely observed, but are found for high-spin iron imidos with7,8,27 or without28 imidyl character on the nitrogen atom as well as an anionic iron(II) imide.20 [1]− shows a short N–CAryl bond of 1.339(2) Å, which is in line with aforementioned high-spin imido iron complexes with C–N double bond character. The observed structural metrics closely resemble the related high-spin imidyl cobalt(II) complex [Co(NDipp)L2]− (e.g. M–Nimide 1.751(2) Å, N–CAryl 1.347(2) Å),23 which suggested for [1]− also an iron(II) imidyl-like bonding situation on a structural level.
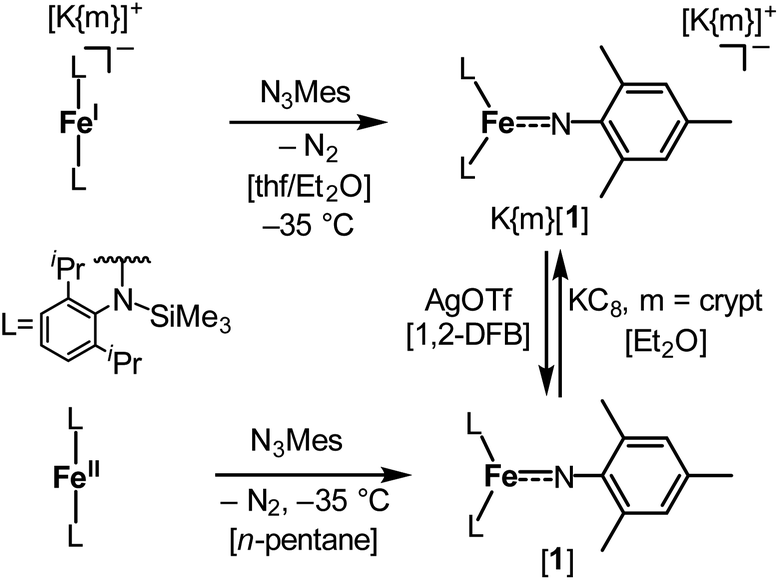 | ||
| Scheme 1 Synthesis of complexes K{m}[Fe(NMes)L2], K{m}[1] (m = crypt.222), and [Fe(NMes)L2], [1], as well as their interconversion by oxidation and reduction. | ||
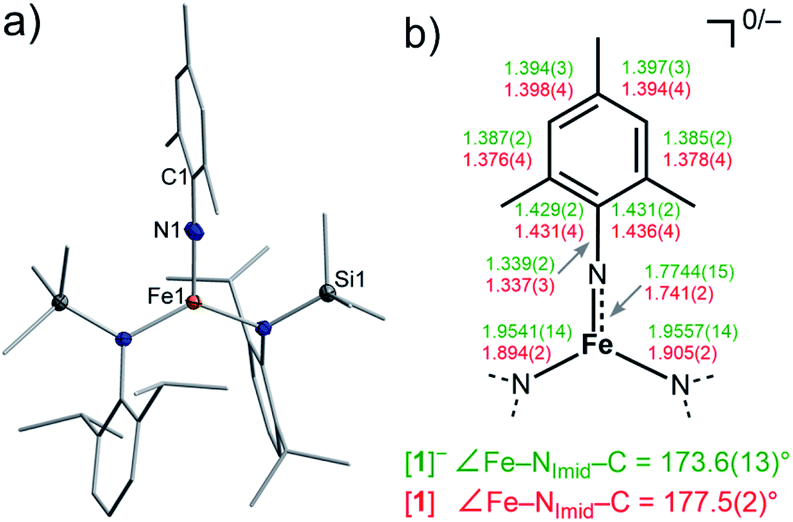 | ||
| Fig. 2 (a) Solid state structure of [1]. The H atoms are omitted for clarity. (b) Selected bond metrics for [1]− (green) and [1] (red) [Å]. | ||
Given the scarcity of unambiguously identified iron bound imidyl units, namely only tetrahedral iron(III) imidyls of the type [(dpm)Fe(Cl)(NR)] from Betley and co-workers (Fig. 1, right),7,8,22 we sought further insights into the electronic situation of [1]−. Magnetic measurements (SQUID) on solid material yielded an χMT value of 4.4 cm3 mol−1 K (corresponding to an effective magnetic moment of μeff = 5.93 μB) at 210 K (Fig. 3a).
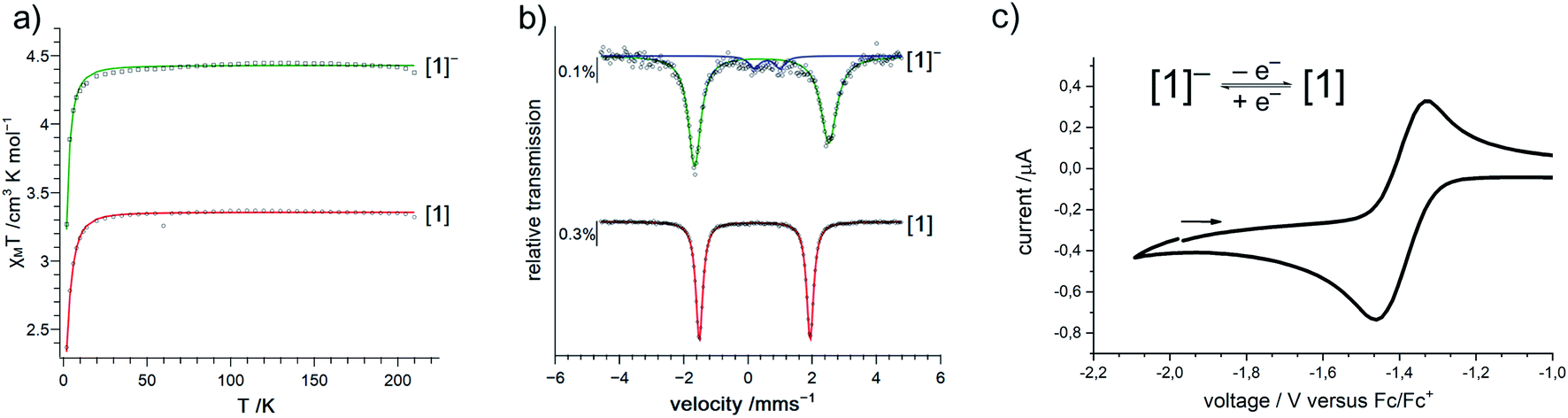 | ||
| Fig. 3 (a) Variable-temperature magnetic susceptibility of solid K{m}[1] (green) and [1] (red) in the range of 2–210 K. (b) Zero-field 57Fe Mössbauer spectra of solid K{m}[1] (green) and [1] (red) at 80 K. Parameters for K{m}[1]: δ = 0.43 mm s−1, |ΔEQ| = 4.18 mm s−1 (92%); [1]: δ = 0.21 mm s−1, |ΔEQ| = 3.45 mm s−1. (c) Cyclic voltammogram of K{m}[1] (500 mV s−1, thf, 0.1 m nBu4 [PF6] vs. Fc/Fc+). | ||
This is close to the spin-only value of a sextet state (μs.o. = 5.92 μB) and can be rationalized either by an iron(II) ion ferromagnetically coupled to an imidyl radical anion, or by a high-spin iron(III) imide. Upon cooling, a sharp drop is observed below 20 K, which is attributed to zero-field splitting (simulation of the χMT data gave g = 2.01, D = 2.5 cm−1; see ESI† for more details).
To assess the oxidation state of the metal in [1]−, zero-field 57Fe Mössbauer spectroscopy was employed. It revealed a doublet with an isomer shift (IS) of δ = 0.43 mm s−1 (at 80 K) and a large quadrupole splitting (QS) of |ΔEQ| = 4.18 mm s−1 (Fig. 3b). The IS is similar to that of the related three-coordinate high-spin iron(III) imide ([(dpm)Fe(N{p-tBu-Ph})]: δ = 0.44 mm s−1, |ΔEQ| = 0.00 mm s−1, at 200 K; Fig. 1, right),8,27,28 but also to that of the anionic iron(II) imide [(Ph2B{tBuIM}2)Fe(NDipp)]− (δ = 0.46 mm s−1, |ΔEQ| = 1.45 mm s−1, at 80 K).20 The IS of K{m}[1] ranges between those of the diferric and diferrous silylamide complexes [(L2Fe)2(μ-S)]2−,0 at 80 K with unambiguous trigonal iron(II) (δ = 0.59 mm s−1) and iron(III) ions (δ = 0.29 mm s−1).29 The large QS of K{m}[1] is remarkable and would generally point to divalent iron. However, also for a number of planar iron(III) complexes large QS have been reported.30 Moreover, related (pseudo-)trigonal iron silylamides exhibit substantially larger QS in the ferric compared to the ferrous state (e.g. [(L2Fe)2(μ-S)]2−,0: |ΔEQ| = 0.22 mm s−1 (Fe2+), 3.70 mm s−1 (Fe3+), at 80 K;29 [Fe(N(SiMe3)2)3]−,0: |ΔEQ| (Fe2+) = 0.60 mm s−1 at 3 K,31 |ΔEQ| (Fe3+) 5.12 mm s−1 at 77 K32).
X-band EPR spectroscopic examination of [1]− at 13 K yielded an axial signal with g1 = 6.5, g2 = 4.28, g3 = 4.18 (A1 (14N) = 107 G, A2 (14N) = 1.7 G, A3 (14N) = 20.5 G; see ESI†), the large 14N hyperfine coupling implicating substantial spin density on nitrogen. This is consistent with substantial imidyl character in [1]−, and also aligns with its structural features.
Next, we wanted to elaborate if we could electrochemically attenuate the electronic situation of [1]−. Cyclic voltammetry examination showed a reversible redox event at E1/2 = −1.4 V at 500 mV s−1 (THF, vs. Fc/Fc+, Fig. 3c), that becomes irreversible upon lowering the scan rate to 50 mV (Fig. S24†). The potential is in line with those for the oxidation of imido iron(III) and imido iron(II) species (≈ −1.3 to −0.7 V).16,17,33 Gratifyingly, chemical oxidation of [1]− in 1,2-difluorobenzene with AgOTf (OTf− = F3CSO3−) led to the formation of a new paramagnetic species which we attributed to neutral [1]. [1] could not be isolated via this pathway due to partial degradation, but was independently synthesized by reacting [FeL2] with MesN3 in n-pentane (yield 64%, Scheme 1). It can be reduced back to [1]− by KC8 in the presence of crypt.222, evidencing the possibility of their chemical interconversion. X-ray diffraction analysis of [1] showed in comparison with [1]− that all metal–nitrogen bonds contract upon oxidation with otherwise little changes in the Fe–N–C unit (Fe–Nimide 1.741(2) Å, Fe–Nimide–CAryl 177.5(2)°, Nimide–CAryl 1.337(3) Å; Fig. 2, right), where the contraction of the Fe–N amide bonds was slightly more pronounced. Thus, it was suggestive of at least partial metal based oxidation. This was corroborated by 57Fe Mössbauer spectroscopy showing an IS of δ = 0.21 mm s−1 for [1] (vs. [1]−: 0.43 mm s−1). It is in line with shorter Fe–ligand bonds and by that with the decrease of the radial extension of the 4 s wave function in case of the oxidized species [1]. Further, a negative correlation of the IS with the Fe oxidation state is usually valid for high spin complexes.34 The QS for [1] is with |ΔEQ| = 3.56 mm s−1 (vs. [1]−: 4.18 mm s−1) only slightly reduced, and indicates a similar electron density distribution (electric field gradient) in both complexes. The IS value of [1] is distinct from those of iron(IV) imides, which are found between −0.09 and −0.52 mm s−1.26,35 This precludes for [1] a formulation as an iron(IV) imide. Accordingly, its IS value is closer to those of high-spin iron(III) imides (δ = 0.37–0.47 mm s−1, |ΔEQ| ≈ 0 mm s−1 at 200 K),8,27,28 a trigonal intermediate-spin iron(III) imide (δ = 0.25 mm s−1, |ΔEQ| = 1.32 mm s−1, at 200 K)20 or four-coordinate high-spin iron(III) imidyls (e.g. [(dpm)Fe(N{p-tBu-C6H4})Cl] δ = 0.28 mm s−1, |ΔEQ| = 2.22 mm s−1, at 90 K).7,8,22 Solid state SQUID measurements on [1] (Fig. 3a) gave a χMT value of 3.3 cm3 mol−1 K (μeff = 5.14 μB) at 210 K with D = −3.5 cm−1 and g = 2.12. These values correspond to an overall S = 2 spin ground state that fits with the absence of pronounced X-band EPR features. [1] and [1]− are thus the first isostructural imido metal complexes in two oxidation states in which both exhibit a high-spin state.
To shed further light on the electronic structures of [1]− and [1], DFT-(PBE, TPSSh, PBE0) and CASSCF/NEVPT2 calculations were performed.36 All investigated computational methods afforded good to reasonable agreement with the XRD-, Mössbauer-, EPR- and SQUID data (bond lengths and angles, ΔEQ, δ, g, A, D; Tables S3–S6†). According to the CASSCF calculations, in [1]−, six electrons populate the two pairs of π/π*-interactions between the metal and imido ligand (Fig. 4, S43–S45†).
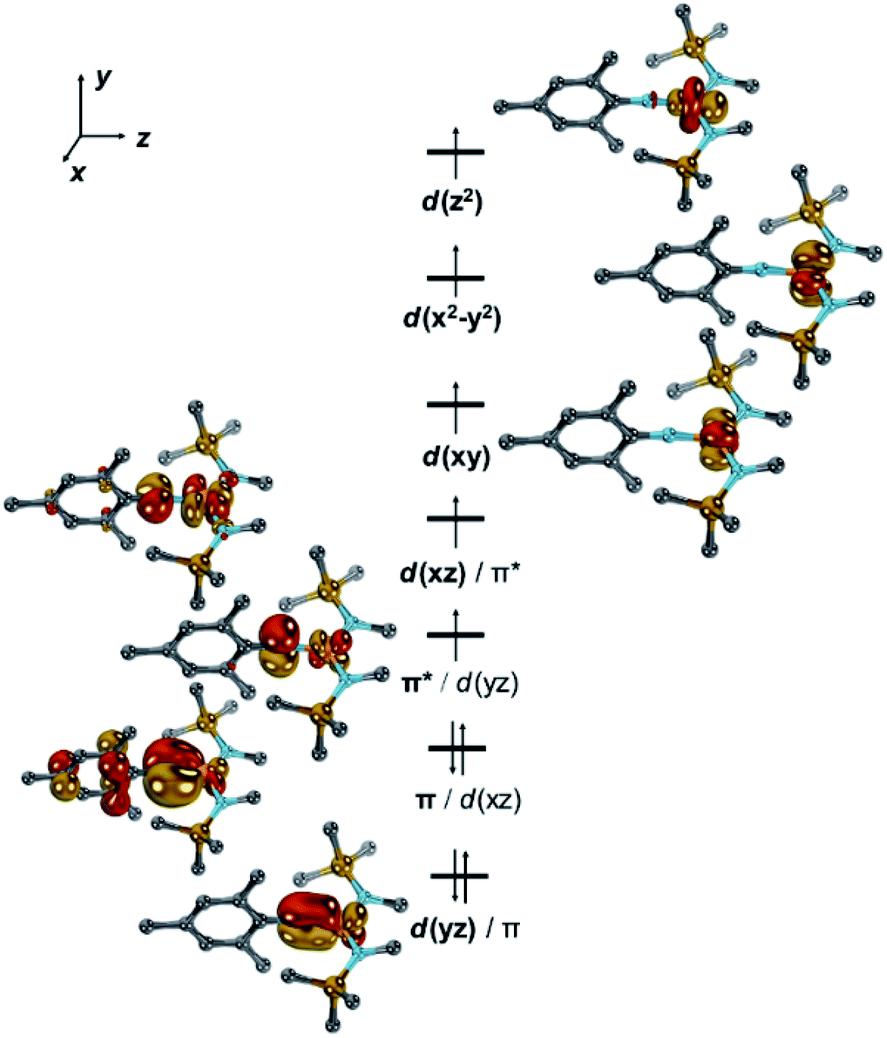 | ||
| Fig. 4 Electronic structure of [1]− by CASSCF(11,9). Dipp groups were truncated by methyl groups, hydrogen atoms are omitted for clarity, one set of bonding and antibonding Dipp-centred π-orbitals is omitted for clarity. | ||
The two unpaired electrons in the antibonding combinations are thereby once metal centred (dxz/π*; Fe:N = 0.8:0.2; Löwdin population analysis) and once rather located at the ligand (π*/dyz; N:Fe = 0.6:0.4). This tentatively gives rise to the electronic structure of an imidyl iron(II) complex for [1]−. An oxidation state of iron(+II) is also substantiated by the Löwdin spin density of +0.66 a.u. at the imido nitrogen atom.
Neutral [1] features a quintet ground state (ΔEq/t = +0.55 eV; ΔEq/sept = +1.16 eV). The main configuration (c = 0.63) predicts thereby four unpaired electrons in d-orbitals (Fig. 5 and S40–S42†). Further two electrons are paired in the dxy orbital, which combines with the imido ligand's px orbital (Fe:N = 7:3) and to a minor extent with the aromatic mesityl substituent. This gives overall rise to a formal d6 (FeII) electron configuration of the metal and an electrophilic nitrene-type ligand. However, the second most important configuration (c = 0.21) relates to the π–π* transition and thus suggests significant imidyl radical character.5 This is further corroborated by accumulation of spin density at the imido nitrogen atom (−0.21 a.u.), which is suggestive of considerable antiferromagnetic character. Accordingly, the doubly excited resonance structure is also of relevance (c = 0.13), which overall leads to a population of the LUMO by formally 0.49 e− and a shift of electron density to the ligand. Thus, when going from [1] to [1]− the additional electron populates the antibonding π* orbital, and weakens the metal–imido bond by formally −0.5. As such, the calculations highlight the electronic flexibility of the covalent iron–imido ligand interaction where the additional electron is not accepted by either the metal or the ligand, but by both. Since the electron is shared within the [FeNR] linkage, of course, rather similar Mössbauer spectroscopic features of [1] and [1]− are expected, which aligns well with the experimental (vide supra) and calculated (Table S4†) parameters. It is thus reasonable to consider the imido iron unit in both complexes in its entirety, as proposed for a linear imido cobalt(II) compound.37 Using the Enemark–Feltham notation for nitrosyl complexes38 this leads to notations {FeNR}8 for [1] and {FeNR}9 for [1]−, and by that avoids singling out or overemphasizing a particular resonance structure.
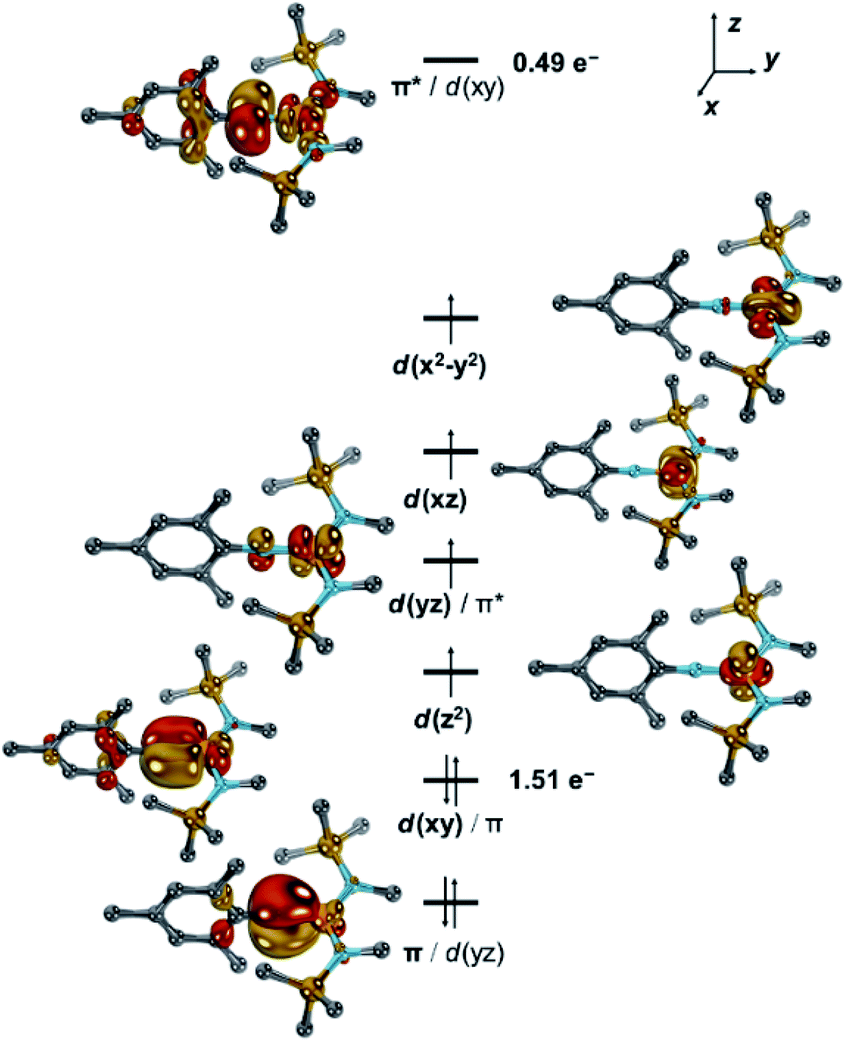 | ||
| Fig. 5 Electronic structure of [1] by CASSCF(10,9). Dipp groups were truncated by methyl groups, hydrogen atoms are omitted for clarity, one set of bonding and antibonding Dipp-centred π-orbitals is omitted for clarity. | ||
Given these computed electronic structures of the “iron(II) imidyl” [1]− as well as the “iron(II) nitrene” [1], we were curious if and how they would translate to differences in chemical reactivity. Both complexes showed only limited H-atom abstraction capabilities in case of 1,4-cyclohexadiene (after 24 h: [1]: 39%; [1]−: 73%). H atom abstraction from 1-hydroxy-2,2,6,6-tetramethyl-piperidine (TEMPO-H) was observed for [1] (24 h: 25% TEMPO by X-band EPR spectroscopy), whereas for [1]− an ill-defined transformation of the imido complex was detected, without significant formation of TEMPO. These observations can be explained by the steric congestion. Possibly also to the ambivalent electronic nature of both complexes may contribute to the observed differences in reactivity. Reaction products of the H atom abstraction, e.g. the corresponding amides, could neither be identified nor isolated. This is attributed to subsequent reaction pathways such as scrambling of the amide ligands, as observed in a related cobalt system.23 Subjecting [1] to an excess PEt3 resulted in initial disappearance of the 31P NMR signal of PEt3. This phenomenon pointed to reversible coordination of the phosphine to the paramagnetic metal centre leading to spin polarisation and disappearance of the 31P signal.39 Addition of a stronger donor (2,2′-bipyridine) to such a reaction mixture after 24 h led immediately to reoccurrence of the PEt3 signal, and more importantly small amounts of the nitrene transfer product MesN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) PEt3 (Scheme 2). This points to a slightly electrophilic behaviour of [1]. In contrast, [1]− is completely inert towards PEt3, as was also observed for the related cobalt complex.23 On the other hand [1]− reacts with CO to give MesNCO under nitrene transfer, whereas [1] does not.
PEt3 (Scheme 2). This points to a slightly electrophilic behaviour of [1]. In contrast, [1]− is completely inert towards PEt3, as was also observed for the related cobalt complex.23 On the other hand [1]− reacts with CO to give MesNCO under nitrene transfer, whereas [1] does not.
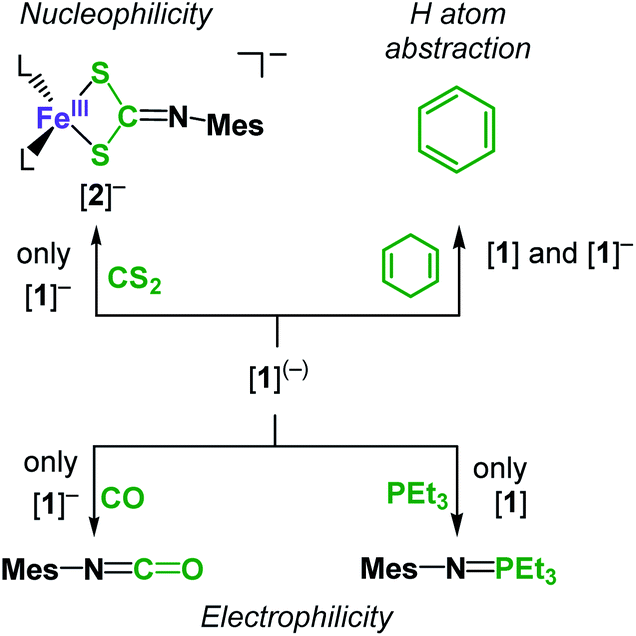 | ||
| Scheme 2 Diverging reactivity of [1] and K{m}[1] towards nucleophiles, electrophiles, and C–H bonds. | ||
Isonitriles (tBuNC, MesNC) interact with both complexes, however no reaction product could be identified (i.e., the presumed carbodiimide RNCNMes). Probing the nucleophilicity using CS2 as a substrate revealed that [1] is unaffected for a prolonged period of time. [1]− on the other hand reacted readily via insertion into the imido metal bond resulting in the dithiocarbonimidate complex [L2Fe (η2-S2CNMes)]−, [2]− (Fig. 6, Scheme 2). The assembly of a dithiocarbonimidate ligand via this reaction is remarkable, as in the coordination sphere of a metal it was only achieved by a [2 + 2] addition of organo isothiocyanates with transient terminal metal sulfides40 or oxidation of isonitriles with metal persulfides.41 The formation of [2]− resembles the insertion of a carbodiimide into the iron(II) imide bond of the nucleophilic [(Ph2B(tBuIM)2)Fe(NDipp)]−. In both cases it likely results from an initial [2 + 2] cycloaddition of one of the CX bonds with the M–N unit, in resemblance to the reaction of an imido nickel(II) complex with CO2 or an imido iron(IV) complex with a carbodiimide (PhNCNPh).26,42 [2]− exhibits in solution a magnetic moment of 5.39 μB that is close to the spin-value of an S = 5/2 ion (μs.o. = 5.92 μB). It shows that despite its mostly iron(II) imidyl character K{m}[1] reacts with an electrophile as an iron(III) imide, thus underscoring the electronic flexibility of the imido iron unit.
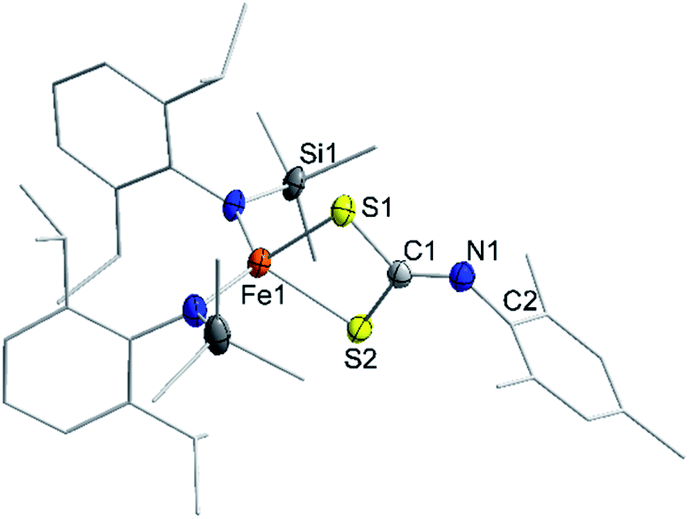 | ||
| Fig. 6 Solid state structure of K{m}[2]. The K{m} cation as well as H atoms are omitted for clarity. | ||
Conclusion
Concluding, we describe with [Fe(NMes)L2]0,− (L = –N(Dipp)SiMe3) a pair of isostructural imido iron complexes in two oxidation states. The electronic structures of the two compounds were comprehensively assessed, amongst others by EPR and 57Fe Mössbauer spectroscopy and magnetometry, and revealed for both complexes a high-spin state. In line with the computational analysis and structural features, the neutral compound shares iron(II) nitrene and iron(III) imidyl character, whereas the reduced form is described best as an iron(II) imidyl. The change of the complexes' oxidation state is thus ligand and metal based, revealing the electronic flexibility of the imido metal bond. The ambiguity of the [FeNR] unit is reflected best by [Fe(NMes)L2]− which shows a nucleophilic and electrophilic behaviour. Upon oxidation to [Fe(NMes)L2]0 the nucleophilicity is lost in accordance with a shift to a more nitrene-like electronic situation. We foresee these reactivity shifts to be relevant for directing NR group transfer reactivity of metal imidos towards different functionalities in organic substrates.Data availability
All experimental procedures, spectral data, and computational data are included in the ESI.† Xyz coordinates may be obtained via the ESI,† output files can be obtained by D. M upon request. NMR/EPR/IR/Mössbauer raw data is so far only available upon request to C. G. W.Author contributions
S. R. carried out the synthetic work and analytical characterization, including the crystallographic studies. S. D. recorded the SQUID and 57Fe Mössbauer data, and both S. D. and F. M. analyzed the data. B. B., A. R., A. S. and C. L. recorded and analyzed the EPR data. C. S. performed the cyclic voltammetric analysis. D. M. performed the quantum chemical calculations. S. R. and C. G. W. wrote the manuscript with contributions from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the DFG for funding (grant 5627/4-1 for C. G. W., INST 186/1329-1 FUGG (SQUID magnetometer) for F. M.). D. M. thanks the RRZ Erlangen for computational resources. This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 Research and Innovation Program (grant agreement no. 946184 for C. L. and no. 948185 for D. M.).Notes and references
- (a) P. Müller and C. Fruit, Chem. Rev., 2003, 103, 2905–2920 CrossRef PubMed; (b) L. Zhang and L. Deng, Chin. Sci. Bull., 2012, 57, 2352–2360 CrossRef CAS; (c) Y. Park, Y. Kim and S. Chang, Chem. Rev., 2017, 117, 9247–9301 CrossRef CAS PubMed; (d) Y. Liu, T. You, H.-X. Wang, Z. Tang, C.-Y. Zhou and C.-M. Che, Chem. Soc. Rev., 2020, 49, 5310–5358 RSC.
- K. Kawakita, Y. Kakiuchi, H. Tsurugi, K. Mashima, B. F. Parker, J. Arnold and I. A. Tonks, Coord. Chem. Rev., 2020, 407, 213118 CrossRef CAS PubMed.
- (a) J. Strähle, Angew. Chem., 1989, 101, 957 CrossRef; (b) P. F. Kuijpers, J. I. van der Vlugt, S. Schneider and B. de Bruin, Chem.–Eur. J., 2017, 23, 13819–13829 CrossRef CAS PubMed; (c) J. F. Berry, Comments Inorg. Chem., 2009, 30, 28–66 CrossRef CAS.
- K. Ray, F. Heims and F. F. Pfaff, Eur. J. Inorg. Chem., 2013, 2013, 3784–3807 CrossRef CAS.
- A. Grünwald, S. S. Anjana and D. Munz, Eur. J. Inorg. Chem., 2021, 2021, 4147–4166 CrossRef.
- (a) R. A. Eikey and M. M. Abu-Omar, Coord. Chem. Rev., 2003, 243, 83–124 CrossRef CAS; (b) N. Hazari and P. Mountford, Acc. Chem. Res., 2005, 38, 839–849 CrossRef CAS PubMed; (c) J. L. Martinez, S. A. Lutz, H. Yang, J. Xie, J. Telser, B. M. Hoffman, V. Carta, M. Pink, Y. Losovyj and J. M. Smith, Science, 2020, 370, 356–359 CrossRef CAS PubMed.
- E. R. King, E. T. Hennessy and T. A. Betley, J. Am. Chem. Soc., 2011, 133, 4917–4923 CrossRef CAS PubMed.
- M. J. T. Wilding, D. A. Iovan, A. T. Wrobel, J. T. Lukens, S. N. MacMillan, K. M. Lancaster and T. A. Betley, J. Am. Chem. Soc., 2017, 139, 14757–14766 CrossRef CAS PubMed.
- W. Mao, D. Fehn, F. W. Heinemann, A. Scheurer, D. Munz and K. Meyer, Angew. Chem., Int. Ed., 2021, 60, 16480–16486 CrossRef CAS PubMed.
- Y. Park, S. P. Semproni, H. Zhong and P. J. Chirik, Angew. Chem., Int. Ed., 2021, 60, 14376–14380 CrossRef CAS PubMed.
- Y. Dong, C. J. Lund, G. J. Porter, R. M. Clarke, S.-L. Zheng, T. R. Cundari and T. A. Betley, J. Am. Chem. Soc., 2021, 143, 817–829 CrossRef CAS PubMed.
- Y. Dong, J. T. Lukens, R. M. Clarke, S.-L. Zheng, K. M. Lancaster and T. A. Betley, Chem. Sci., 2020, 123, 4623 Search PubMed.
- K. M. Carsch, I. M. DiMucci, D. A. Iovan, A. Li, S.-L. Zheng, C. J. Titus, S. J. Lee, K. D. Irwin, D. Nordlund, K. M. Lancaster and T. A. Betley, Science, 2019, 365, 1138–1143 CrossRef CAS PubMed.
- (a) A. G. Bakhoda, Q. Jiang, Y. M. Badiei, J. A. Bertke, T. R. Cundari and T. H. Warren, Angew. Chem., Int. Ed., 2019, 58, 3421–3425 CrossRef CAS PubMed; (b) J. Moegling, A. Hoffmann, F. Thomas, N. Orth, P. Liebhäuser, U. Herber, R. Rampmaier, J. Stanek, G. Fink, I. Ivanović-Burmazović and S. Herres-Pawlis, Angew. Chem., Int. Ed., 2018, 57, 9154–9159 CrossRef CAS PubMed; (c) M. J. B. Aguila, Y. M. Badiei and T. H. Warren, J. Am. Chem. Soc., 2013, 135, 9399–9406 CrossRef CAS PubMed; (d) T. Corona, L. Ribas, M. Rovira, E. R. Farquhar, X. Ribas, K. Ray and A. Company, Angew. Chem., Int. Ed., 2016, 55, 14005–14008 CrossRef CAS PubMed; (e) S.-L. Abram, I. Monte-Pérez, F. F. Pfaff, E. R. Farquhar and K. Ray, Chem. Commun., 2014, 50, 9852–9854 RSC.
- F. Dielmann, D. M. Andrada, G. Frenking and G. Bertrand, J. Am. Chem. Soc., 2014, 136, 3800–3802 CrossRef CAS PubMed.
- C. M. Thomas, N. P. Mankad and J. C. Peters, J. Am. Chem. Soc., 2006, 128, 4956–4957 CrossRef CAS PubMed.
- S. D. Brown and J. C. Peters, J. Am. Chem. Soc., 2005, 127, 1913–1923 CrossRef CAS PubMed.
- I. Nieto, F. Ding, R. P. Bontchev, H. Wang and J. M. Smith, J. Am. Chem. Soc., 2008, 130, 2716–2717 CrossRef CAS PubMed.
- (a) L. Zhang, Y. Liu and L. Deng, J. Am. Chem. Soc., 2014, 136, 15525–15528 CrossRef CAS PubMed; (b) Y. Liu, J. Du and L. Deng, Inorg. Chem., 2017, 56, 8278–8286 CrossRef CAS PubMed.
- Y. Gao, V. Carta, M. Pink and J. M. Smith, J. Am. Chem. Soc., 2021, 143, 5324–5329 CrossRef CAS PubMed.
- J. R. Winkler and H. B. Gray, in Molecular Electronic Structures of Transition Metal Complexes I, ed. D. M. P. Mingos, P. Day and J. P. Dahl, Springer Berlin Heidelberg, Berlin, Heidelberg, 2012, vol. 142, pp. 17–28 Search PubMed.
- D. A. Iovan and T. A. Betley, J. Am. Chem. Soc., 2016, 138, 1983–1993 CrossRef CAS PubMed.
- A. Reckziegel, M. Kour, B. Battistella, S. Mebs, K. Beuthert, R. Berger and C. G. Werncke, Angew. Chem., Int. Ed., 2021, 60, 15376–15380 CrossRef CAS PubMed.
- (a) A. Reckziegel, C. Pietzonka, F. Kraus and C. G. Werncke, Angew. Chem., Int. Ed., 2020, 59, 8527–8531 CrossRef CAS PubMed; (b) R. Weller, L. Ruppach, A. Shlyaykher, F. Tambornino and C. G. Werncke, Dalton Trans., 2021, 50, 10947–10963 RSC.
- (a) A. K. Verma, T. N. Nazif, C. Achim and S. C. Lee, J. Am. Chem. Soc., 2000, 122, 11013–11014 CrossRef CAS; (b) S. D. Brown, T. A. Betley and J. C. Peters, J. Am. Chem. Soc., 2003, 125, 322–323 CrossRef CAS PubMed; (c) S. C. Bart, E. Lobkovsky, E. Bill and P. J. Chirik, J. Am. Chem. Soc., 2006, 128, 5302–5303 CrossRef CAS PubMed; (d) C. Ni, J. C. Fettinger, G. J. Long, M. Brynda and P. P. Power, Chem. Commun., 2008, 6045 RSC; (e) J. J. Scepaniak, J. A. Young, R. P. Bontchev and J. M. Smith, Angew. Chem., Int. Ed., 2009, 48, 3158–3160 CrossRef CAS PubMed; (f) R. E. Cowley, N. J. DeYonker, N. A. Eckert, T. R. Cundari, S. DeBeer, E. Bill, X. Ottenwaelder, C. Flaschenriem and P. L. Holland, Inorg. Chem., 2010, 49, 6172–6187 CrossRef CAS PubMed; (g) H. Zhang, Z. Ouyang, Y. Liu, Q. Zhang, L. Wang and L. Deng, Angew. Chem., Int. Ed., 2014, 53, 8432–8436 CrossRef CAS PubMed; (h) S. Kuppuswamy, T. M. Powers, B. M. Johnson, M. W. Bezpalko, C. K. Brozek, B. M. Foxman, L. A. Berben and C. M. Thomas, Inorg. Chem., 2013, 52, 4802–4811 CrossRef CAS PubMed; (i) B. P. Jacobs, P. T. Wolczanski, Q. Jiang, T. R. Cundari and S. N. MacMillan, J. Am. Chem. Soc., 2017, 139, 12145–12148 CrossRef CAS PubMed.
- L. Wang, L. Hu, H. Zhang, H. Chen and L. Deng, J. Am. Chem. Soc., 2015, 137, 14196–14207 CrossRef CAS PubMed.
- M. J. T. Wilding, D. A. Iovan and T. A. Betley, J. Am. Chem. Soc., 2017, 139, 12043–12049 CrossRef CAS PubMed.
- A. Sridharan, A. C. Brown and D. L. M. Suess, Angew. Chem., Int. Ed., 2021, 60, 12802–12806 CrossRef CAS PubMed.
- C. Schneider, S. Demeshko, F. Meyer and C. G. Werncke, Chem.–Eur. J., 2021, 27, 6348–6353 CrossRef CAS PubMed.
- Y. Sanakis, P. P. Power, A. Stubna and E. Münck, Inorg. Chem., 2002, 41, 2690–2696 CrossRef CAS PubMed.
- A. Eichhöfer, Y. Lan, V. Mereacre, T. Bodenstein and F. Weigend, Inorg. Chem., 2014, 53, 1962–1974 CrossRef PubMed.
- E. C. Alyea, D. C. Bradley, R. G. Copperthwaite, K. D. Sales, B. W. Fitzsimmons and C. E. Johnson, J. Chem. Soc. D, 1970, 1715–1716 RSC.
- K. E. Aldrich, B. S. Fales, A. K. Singh, R. J. Staples, B. G. Levine, J. McCracken, M. R. Smith and A. L. Odom, Inorg. Chem., 2019, 58, 11699–11715 CrossRef CAS PubMed.
- P. Gütlich, Mössbauer Spectroscopy and Transition Metal Chemistry. Fundamentals and Applications, Springer Berlin/Heidelberg, Berlin, Heidelberg, 2011 Search PubMed.
- (a) M. R. Anneser, G. R. Elpitiya, J. Townsend, E. J. Johnson, X. B. Powers, J. F. DeJesus, K. D. Vogiatzis and D. M. Jenkins, Angew. Chem., Int. Ed., 2019, 58, 8115–8118 CrossRef CAS PubMed; (b) K. Searles, S. Fortier, M. M. Khusniyarov, P. J. Carroll, J. Sutter, K. Meyer, D. J. Mindiola and K. G. Caulton, Angew. Chem., Int. Ed., 2014, 53, 14139–14143 CrossRef CAS PubMed.
- F. Neese, F. Wennmohs, U. Becker and C. Riplinger, J. Chem. Phys., 2020, 152, 224108 CrossRef CAS PubMed.
- X.-N. Yao, J.-Z. Du, Y.-Q. Zhang, X.-B. Leng, M.-W. Yang, S.-D. Jiang, Z.-X. Wang, Z.-W. Ouyang, L. Deng, B.-W. Wang and S. Gao, J. Am. Chem. Soc., 2017, 139, 373–380 CrossRef CAS PubMed.
- J. H. Enemark and R. D. Feltham, Coord. Chem. Rev., 1974, 13, 339–406 CrossRef CAS.
- R. Weller, A. Gonzalez, H. Gottschling, C. Hänisch and C. G. Werncke, Z. Anorg. Allg. Chem., 2022, e202100338 Search PubMed.
- (a) J. Ahmed, K. Itoh, I. Matsuda, F. Ueda, Y. Ishii and J. A. Ibers, Inorg. Chem., 1977, 16, 620–624 CrossRef CAS; (b) K.-E. Lee, X. Chang, Y.-J. Kim, H. S. Huh and S. W. Lee, Organometallics, 2008, 27, 5566–5570 CrossRef CAS; (c) Y.-J. Kim, H.-T. Jeon, K.-E. Lee and S. W. Lee, J. Organomet. Chem., 2010, 695, 2258–2263 CrossRef CAS; (d) R. O. Harris, J. Powell, A. Walker and P. V. Yaneff, J. Organomet. Chem., 1977, 141, 217–229 CrossRef CAS.
- R. D. Adams, B. Captain, O.-S. Kwon and S. Miao, Inorg. Chem., 2003, 42, 3356–3365 CrossRef CAS PubMed.
- D. J. Mindiola, R. Waterman, V. M. Iluc, T. R. Cundari and G. L. Hillhouse, Inorg. Chem., 2014, 53, 13227–13238 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: General, experimental, analytical (NMR, IR, UV/vis, 57Fe Mössbauer and EPR spectroscopy, SQUID magnetometry, cyclovoltammetry), computational and crystallographic details. CCDC 2130485–2130488. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc01088g |
| This journal is © The Royal Society of Chemistry 2022 |