
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An induced-fit model for asymmetric organocatalytic reactions: a case study of the activation of olefins via chiral Brønsted acid catalysts†
Ingolf
Harden
 a,
Frank
Neese
a and
Giovanni
Bistoni
*ab
a,
Frank
Neese
a and
Giovanni
Bistoni
*ab
aMax-Planck-Institut für Kohlenforschung, Kaiser-Wilhelm Platz 1, 45470 Mülheim an der Ruhr, Germany
bDepartment of Chemistry, Biology and Biotechnology, University of Perugia Via Elce di Sotto, 8, 06123 Perugia, Italy. E-mail: giovanni.bistoni@unipg.it
First published on 4th July 2022
Abstract
We elucidate the stereo-controlling factors of the asymmetric intramolecular hydroalkoxylation of terminal olefins catalyzed by bulky Brønsted acids [Science2018, 359 (6383), 1501–1505] using high-level electronic structure methods. The catalyst–substrate interaction is described using a dispersion-driven induced-fit model, in which the conformational changes of the catalyst and of the substrate in the transition states are governed to a large extent by London dispersion forces. The distortion energy of the catalyst is dominated by the change in the intramolecular dispersion interactions, while intermolecular catalyst–substrate dispersion interactions are the major stabilizing contribution in the transition state. This model provides a new general framework in which to discuss the stereoselectivity of transformations catalyzed by such confined organocatalysts.
Introduction
Catalysis is involved in the processing of most manufactured products and hence the design of new catalytic processes plays a central role in chemical research.1 The selectivity of a catalyst is one of the most important criteria in this context: selective reactions generate fewer undesired byproducts and are thus both environmentally sustainable and economical.In biocatalysis, reactions occur inside structurally elaborate pockets within the enzyme active site, leading to high degrees of selectivity and activity.2 This stimulated chemists of all disciplines to develop synthetic hosts that emulate these natural biological pockets. As a prominent example, Yoshizawa et al.3 carried out Diels–Alder reactions of anthracene and phthalimide guests in confined aqueous organopalladium cage complexes, which led to unprecedented regioselectivity. Hastings et al. used the tetrahedral assembly K12Ga4L6 (L = 1,5-biscatecholamidenaphthalene) to catalyze Nazarov cyclizations within a confined space. This led to an increase of rate constants of six orders of magnitude,4 showing a catalytic activity that is also reminiscent of that of enzymes. Similarly, under heterogeneous conditions, reactions in confined zeolite spaces are also well-known for their high activity and selectivity.5,6 Notably, in the field of asymmetric organocatalysis, the List group developed a series of confined chiral Brønsted acids with enzyme-like activity and selectivity. These encompass imidodiphosphates (IDP),7–10 imino-imidodiphosphates (iIDP),11 and imidodiphosphorimidates (IDPi) Brønsted acids,12–15 differing in their acidity as well as in the shape of their active site. As a recent example, IDPi catalysts were used to catalyze asymmetric Mukaiyama aldolizations of silylenolethers with Benzaldehyde and it was suggested that the size of the catalyst pocket determines the benzaldehyde enantiofacial preference.16
Based on these findings, List and coworkers suggested that “confinement”, i.e., the shaping of the active site, should be regarded as a unifying element in selective catalysis.17 But how does confinement induce selectivity, and how this information can be used in the design of new catalysts? In biocatalysis, a complex pattern of interactions within the active site forces the substrates into specific orientations, thus favoring and/or disfavoring specific reaction pathways. A similar situation is expected to occur in any kind of selective catalysis, and harnessing the power of such interactions is one of the key challenges in contemporary chemical research.18 These interactions can be either covalent or noncovalent, either attractive or repulsive, depending on the nature of the substrates and of the catalytic system.
A ubiquitous interaction energy component that is commonly associated with the concept of confinement is “steric repulsion”, i.e., the short-range repulsion of atoms, molecules or functional groups. This interaction has often been advocated as the key factor controlling the stereoselectivity of a large number of chemical transformations.19–23 The key idea behind this assumption is that the introduction of bulky groups in a catalytic system can be used to block all the undesirable reaction pathways. However, the increase of catalytic activity that is often associated with confined reaction spaces is difficult to explain using steric repulsion alone,24 and in fact several recent works have emphasized the importance of attractive noncovalent interactions (NCIs) in this context.25,26
In contrast to steric repulsion, attractive NCIs are expected to influence the selectivity of a chemical process by lowering the energy barrier for specific reaction pathways, rather than by blocking those that are undesirable. Among the various types of NCIs, London dispersion (LD) has recently emerged for its ubiquitous importance in selective catalysis.27–36 This attractive interaction is always present between atoms, molecules or groups, irrespective of their nature. Its magnitude increases with the polarizability of the interacting species and decreases with their distance. Its fundamental role in stereoselective catalysis has been emphasized by the group of Schreiner on the Dakin–West reaction32 and the Corey–Bakshi–Shibata reduction.33 The group of Houk has demonstrated its importance for intramolecular Aldol reactions34 as well as in the dual transition metal catalysis of α-allylation reactions.35 In addition, Fürstner and coworkers used London dispersion as a key design element for confined Bismuth–Rhodium catalysts with improved activity and selectivity.36
Thus, steric repulsion and LD are two ubiquitous interaction energy components of opposite sign that increase in absolute terms with the internuclear distance, and hence are expected to play an especially important role in reactions occurring in confined spaces. Their interplay was recently investigated by our group on challenging stereoselective organocatalytic Diels–Alder reactions catalyzed by List's enzyme-like Brønsted acids,37 and it was found that both components must be considered for a thorough understanding of the stereo-controlling factors.
In this paper, we seek to reveal how increasing confinement quantitatively influences dispersion and steric repulsion, and how these energy components can in principle be harnessed to increase the selectivity of asymmetric transformations. As a case study, we consider the intramolecular hydroalkoxylation of unactivated alkenes published by the List group in 2018.38 The reaction conditions and the catalyst used are reported in Scheme 1. Remarkably enough, an in-depth analysis of the key stereo-controlling factors for this reaction is still missing.
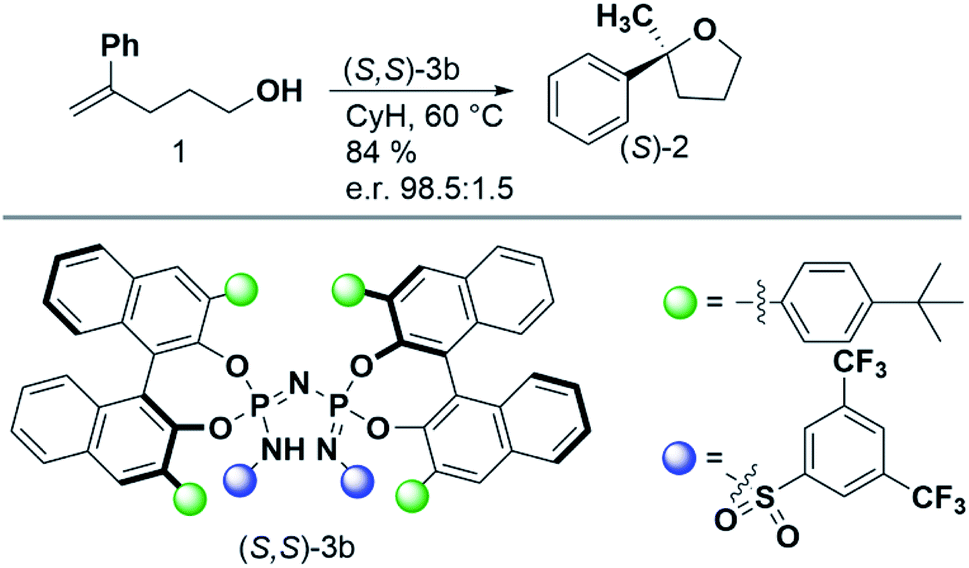 | ||
| Scheme 1 Reaction and experimental conditions considered in this work. Details can be found in ref. 38. | ||
Theoretical aspects
General considerations
Predicting the outcome of stereoselective reactions catalyzed by large and flexible catalysts would potentially require the calculation of a huge number of relative reaction rates with extremely high accuracy. Although several computational approaches have been suggested to this aim, none of them has so far established itself as the method of choice in this context.18 Among the most promising tools used in virtual screening for catalyst design, we mention CatVS39 (Catalyst Virtual Screening) AARON40,41 and ACE (Asymmetric Catalyst Evaluation).42 Alternatively, statistical and machine learning methods have also been introduced.43 In this work, we rely on a slightly modified version of the computational protocol developed in our group for the calculation of selectivities of asymmetric transformations,37 which combines advanced conformational sampling techniques, highly correlated wavefunction based methods and quantitative tools for the analysis of the key noncovalent interactions. Unless otherwise specified, all calculations were carried out with ORCA 4.2.1.44Computational protocol
Our computational protocol is illustrated in Scheme 2.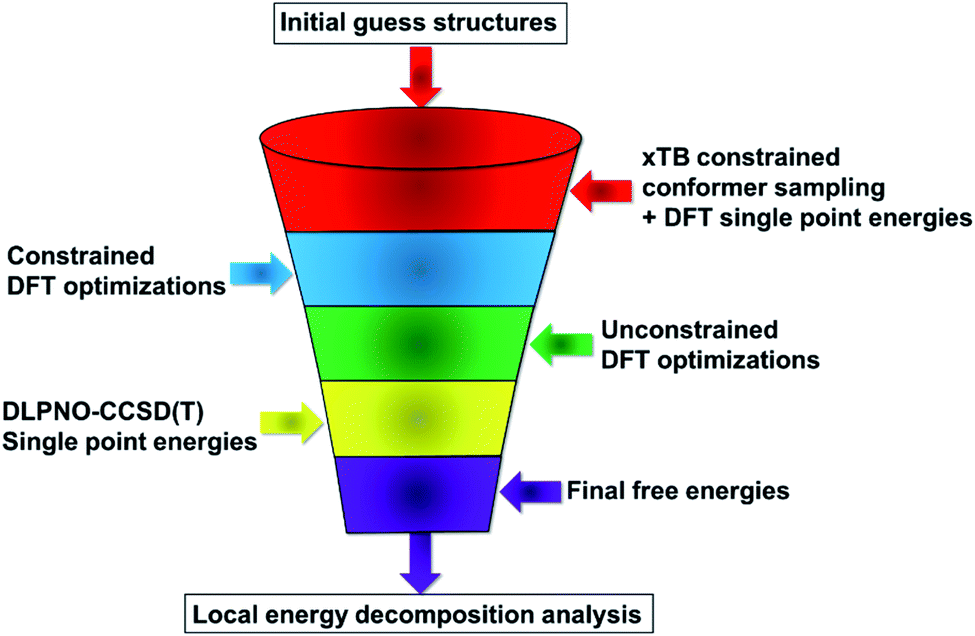 | ||
| Scheme 2 Computational protocol used in this work. | ||
As initial guess structures for the transition states leading to the major and minor enantiomers, we used those computed by List and coworkers in their original work.38 Importantly, the stereo-controlling factors of this important transformation were not discussed in this work, and no extensive conformational sampling procedure was carried out for the relevant transition states.
In the first step of our protocol, an initial conformational sampling at the transition states was carried out using the Conformer–Rotamer Ensemble Sampling Tool45–47 (CREST) at the xTB-GFN1 and xTB-GFN2 levels of theory, as implemented in the Extended Tight-Binding Program Package48–50 (xTB). Due to the large number of conformers obtained from various sampling runs, the protocol defined in ref. 37 was slightly changed to reduce the computational time. Instead of performing DFT geometry optimizations for the entire structural ensemble (which consists of 6485 structures), the conformers were initially sorted in energy by means of DFT single point calculations. The Perdew–Burke–Ernzerhof (PBE) functional51 was used in conjunction with the Ahlrichs double-zeta def2-SVP basis set.52 Grimme's dispersion correction D3 (ref. 53) was used together with Becke–Johnson damping.54 To speed up the calculation, the resolution of identity55 in the Split-RJ variant56 was used together with the appropriate Coulomb-fitting basis set.57 This computational protocol is denoted hereafter as PBE-D3/def2-SVP.
The low energy candidate structures were included in the subsequent constrained geometry optimization step. In particular, a cutoff value of 4 kcal mol−1 was used for the relative electronic energies. In addition, to make the protocol more robust, higher-energy structures with significantly different structural features were also included. This step reduced the number of candidate transition state structures from 6485 to 655. Constrained geometry optimizations at the same level of theory were performed for all the 655 structures that survived the initial screening. The N–H bond distance was selected as only constraint. Tight DFT grids were used for the geometry optimizations (GRID6). The relative energies of the structures that lead to the desired products (459 conformers) are shown in Fig. 1.
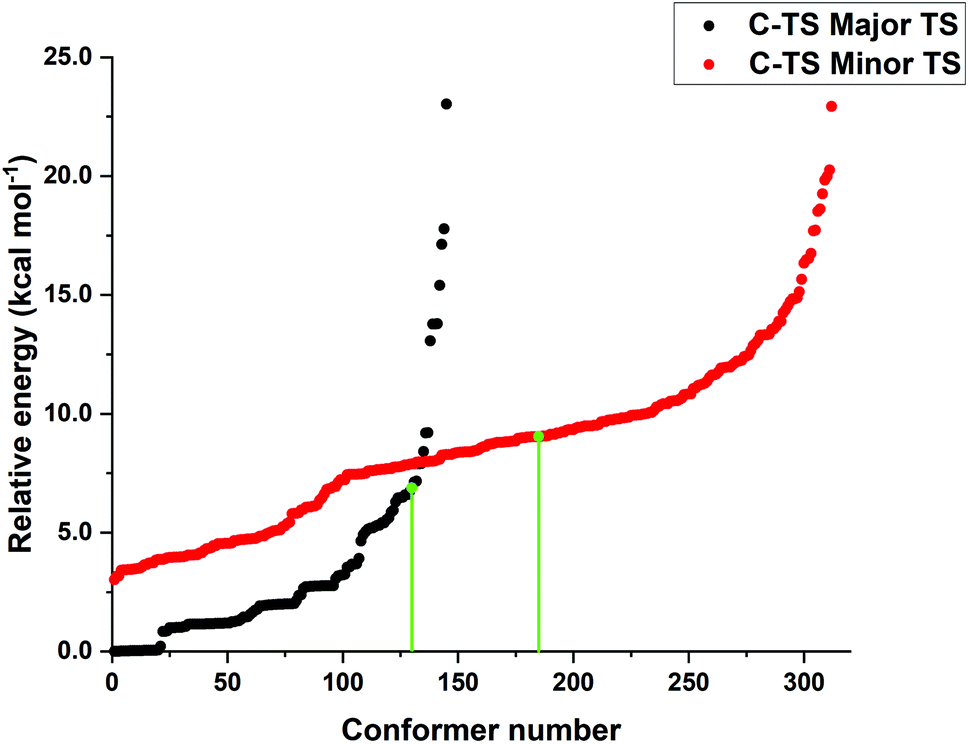 | ||
| Fig. 1 Relative energy obtained from constrained PBE-D3/def2-SVP geometry optimizations leading to the major (black) and minor (red) enantiomeric product for the energetically favored concerted mechanism “A”. The transition state energies of the conformers originally discussed in ref. 38 at the same level of theory are shown in green to emphasize the importance of the conformer sampling procedure. | ||
Remarkably enough, this protocol identifies more than 100 transition state structures lower in energy than those originally discussed in ref. 38 at the same level of theory. Note that, as discussed in detail in ref. 37, the energy difference between the “constrained” and fully relaxed transition states is typically very small when meaningful constraints are defined.
The low-energy conformers with significantly different structural features were optimized without constraints using the larger def2-TZVP(-f) basis set. Thermochemical corrections were obtained at the same level of theory. Final single point energy calculations were carried out with the domain-based-local-pair-natural-orbitals coupled cluster method with single, double and perturbative triple excitations (DLPNO-CCSD(T))58–63 together with the def2-TZVP basis set. The corresponding correlation-fitting basis set (def2-TZVP/C) was used. The NormalPNO settings with a tightened TCutPairs value of 1 × 10−5 Hartree were used. In addition, the RIJCOSX approximation64 to the exchange integrals was used together with the matching Coulomb-fitting basis set. Tight Grids (GRIDX7) and very tight SCF settings (VerytightSCF) were used. Solvation corrections were included at the B3LYP-D3/def2-TZVP51,65–67 level of theory with the solvation model based on density (SMD).68 Cyclohexane (CyH) was used as solvent. The final Gibbs free energy 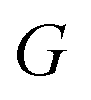 was calculated via:
was calculated via:
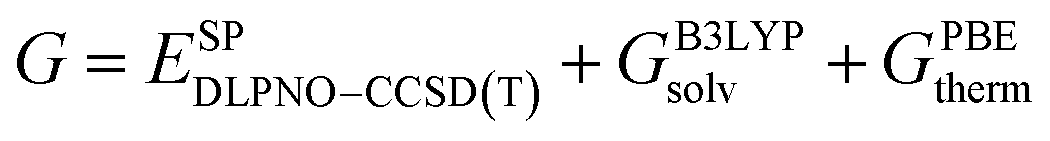 | (1) |
For the analysis of the key non-covalent interactions, the local energy decomposition (LED)69–74 was used to decompose free energy reaction barriers ΔG‡ into contributions associated with the interaction of the catalyst and the substrate:
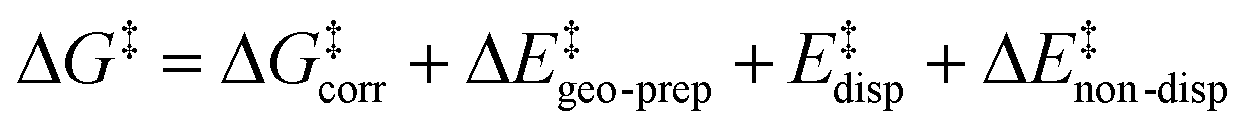 | (2) |
In which 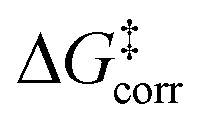 represents the thermostatistical correction to the barrier, including all thermal corrections, entropic and solvent contributions.
represents the thermostatistical correction to the barrier, including all thermal corrections, entropic and solvent contributions. 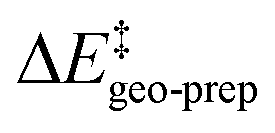 is the geometric preparation energy, that is, the needed energy in order to distort the catalyst and the substrate from their equilibrium geometries to the geometries they have in the transition state.
is the geometric preparation energy, that is, the needed energy in order to distort the catalyst and the substrate from their equilibrium geometries to the geometries they have in the transition state. 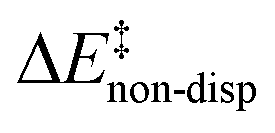 represents the non-dispersive part of the interaction between the catalyst and the substrate, including all steric and polarization effects, while
represents the non-dispersive part of the interaction between the catalyst and the substrate, including all steric and polarization effects, while 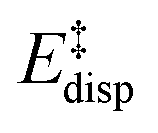 denotes the London dispersion component of the interaction.
denotes the London dispersion component of the interaction.
Steric maps were obtained with the SambVca 2.1 web tool.75 The protonated nitrogen atom of the catalyst was used as the center of the sphere with a radius of 7 Å. The 3D models are oriented in the same way as the steric maps, with the x-axis pointing to the right, the y-axis pointing upward and the z-axis pointing towards the reader. All distances in the steric maps are given in Å. The percentage of free volume within the sphere, % Vfree, is used in this work as a qualitative measure of “free” volume in the catalyst pocket that is accessible to the substrate.
For calculating the noncovalent interaction (NCI) plots, we used the NCIPLOT program.76–78 They were computed based on the electron density at the B3LYP-D3/def2-TZVP level of theory.
Results and discussion
Mechanism
For the reaction illustrated in Scheme 1, three different pathways were explored (Fig. 2).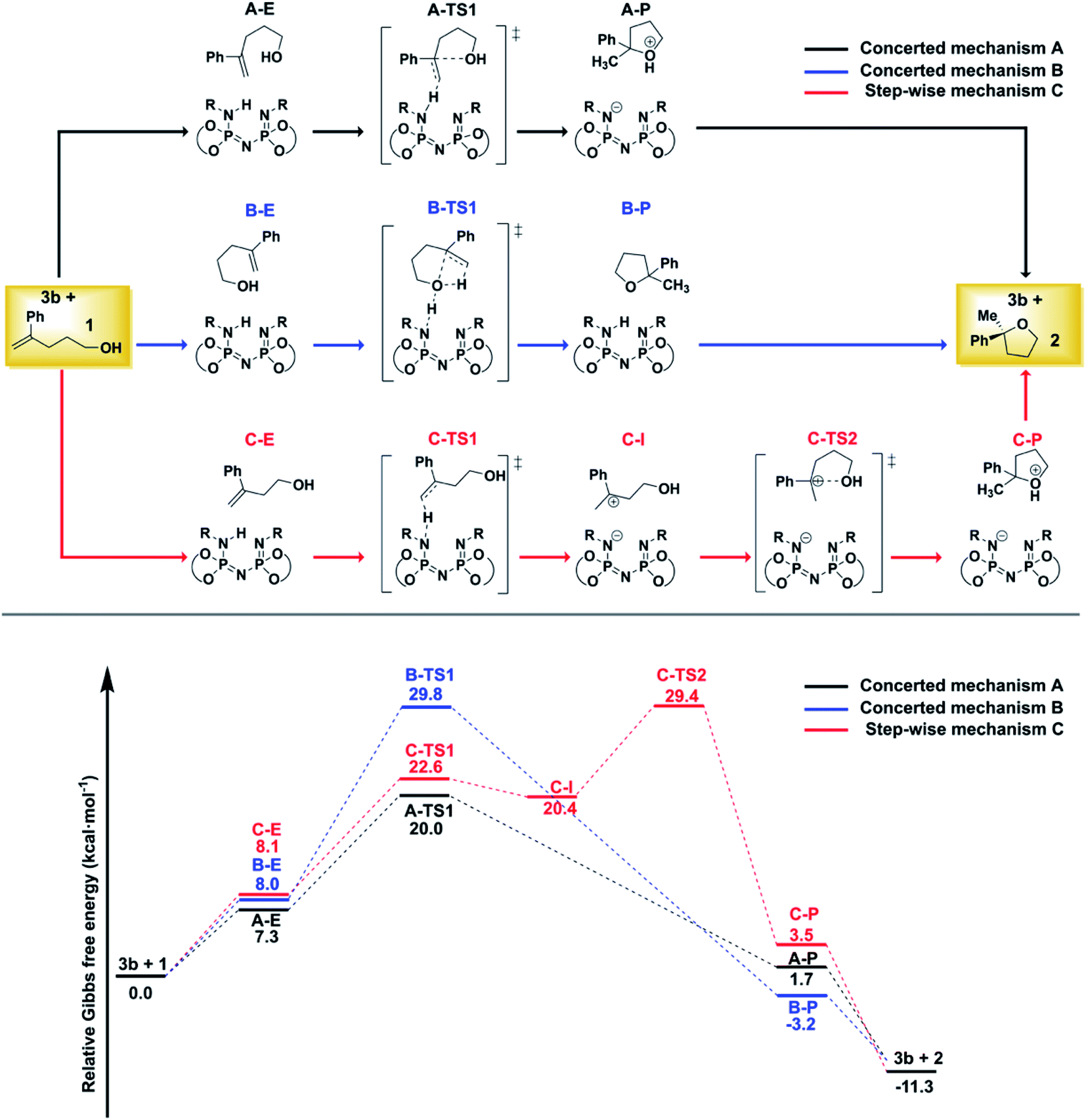 | ||
| Fig. 2 Computed mechanisms at the DLPNO-CCSD(T)/def2-TZVP level of theory. All energies are in kcal mol−1. See text for details. | ||
While the substrate–catalyst adducts A–E, B–E and C–E are all very close in energy, the different conformations of the substrate in these complexes lead to completely different reaction pathways. In the “concerted mechanism A”, the proton from the catalyst is transferred to the terminal methylene group of the folded substrate with simultaneous C–O bond formation, leading to the five-membered oxacycle in a single step. In contrast, the “step-wise mechanism C” involves the unfolded carbocationic species C–I as reaction intermediate. Finally, the “concerted mechanism B” pathway involves a hydrogen-bond assisted proton transfer from the O–H group of the folded substrate to the methylene group, with simultaneous C–O bond formation.
The comparison of the energy profiles associated with the three competing pathways indicates that the “concerted mechanism A” is preferred. As discussed in ref. 38, this pathway is consistent with the experimental observation that the non-terminal isomer of 1 does not react under the experimental conditions (both isomers would proceed through the same carbo-cationic intermediate within a step-wise mechanism).
For the “concerted mechanism A”, the lowest-in-energy transition states leading to the major and minor enantiomeric product are shown in Fig. 3.
 | ||
| Fig. 3 Low-energy transition state conformers leading to the major (panel a, TS1-1) and minor (panel b, TS1-1′, panel c, TS1-2′) enantiomeric products. The corresponding free energy barriers (ΔG‡) are given in kcal mol−1. On the left, a green-colored space filling model is used for the catalyst to emphasize the conformation of the substrate inside the catalyst pocket. On the right, dispersion interaction density (DID) surfaces are shown in light blue. The density value for the isosurface is 0.01 kcal (mol−1 bohr3). | ||
It is important to emphasize here that, for the pathway leading to the major enantiomeric product, all the low-energy TS conformers show the same qualitative structural features. Hence, only the lowest in energy structure is shown in Fig. 3 (TS1-1). In contrast, for the minor enantiomeric pathway, two transition states with significantly different structural features and similar energies were identified (TS1-1′, TS1-2′). This aspect is discussed in more detail in the next section. The higher in energy TS conformers are given in the ESI.†
Consistent with the experimental findings, the pathway leading to the major enantiomeric product is energetically favored. However, the difference between the computed free energy barriers ΔΔG‡ associated with the major and minor pathways amounts to ca. 8 kcal mol−1, which is significantly larger than the experimentally determined value of 2.8 kcal mol−1. This deviation might have different origins, including the details of the chosen computational settings, such as the use of an implicit solvation scheme. Importantly, even though the use of different electronic structure methods, solvation schemes or basis sets influences the absolute magnitude of ΔΔG‡, it remains the case that TS1-1 is always more stable than TS-1′ and TS1-2′, irrespective of the computational settings used (see Table S3, ESI†). Importantly, extrapolation of the coupled cluster electronic energies to the complete basis set limit reduces the ΔΔG‡ by around 1 kcal mol−1, thus shifting the computed stereoselectivity closer to the experimental one (see Tables S5 and S6, ESI†). In the following, we will provide an in-depth, quantitative analysis of the physical and chemical origin of this energy difference, which will allow us to achieve a thorough understanding of the stereo-controlling factors of this transformation.
Elucidating the stereo-controlling factors: induced fit vs. lock and key model
The aim of this section is to elucidate the mechanism through which the shape of the catalysts active site determines the selectivity of the transformation. The structure of the catalyst (3b) in its equilibrium geometry is shown in Fig. 4a. Steric maps are also shown in the same figure to emphasize its key structural features. In particular, the percentage of free volume within the catalyst pocket % Vfree is given: smaller (larger) values of % Vfree correlate with increasing (decreasing) confinement.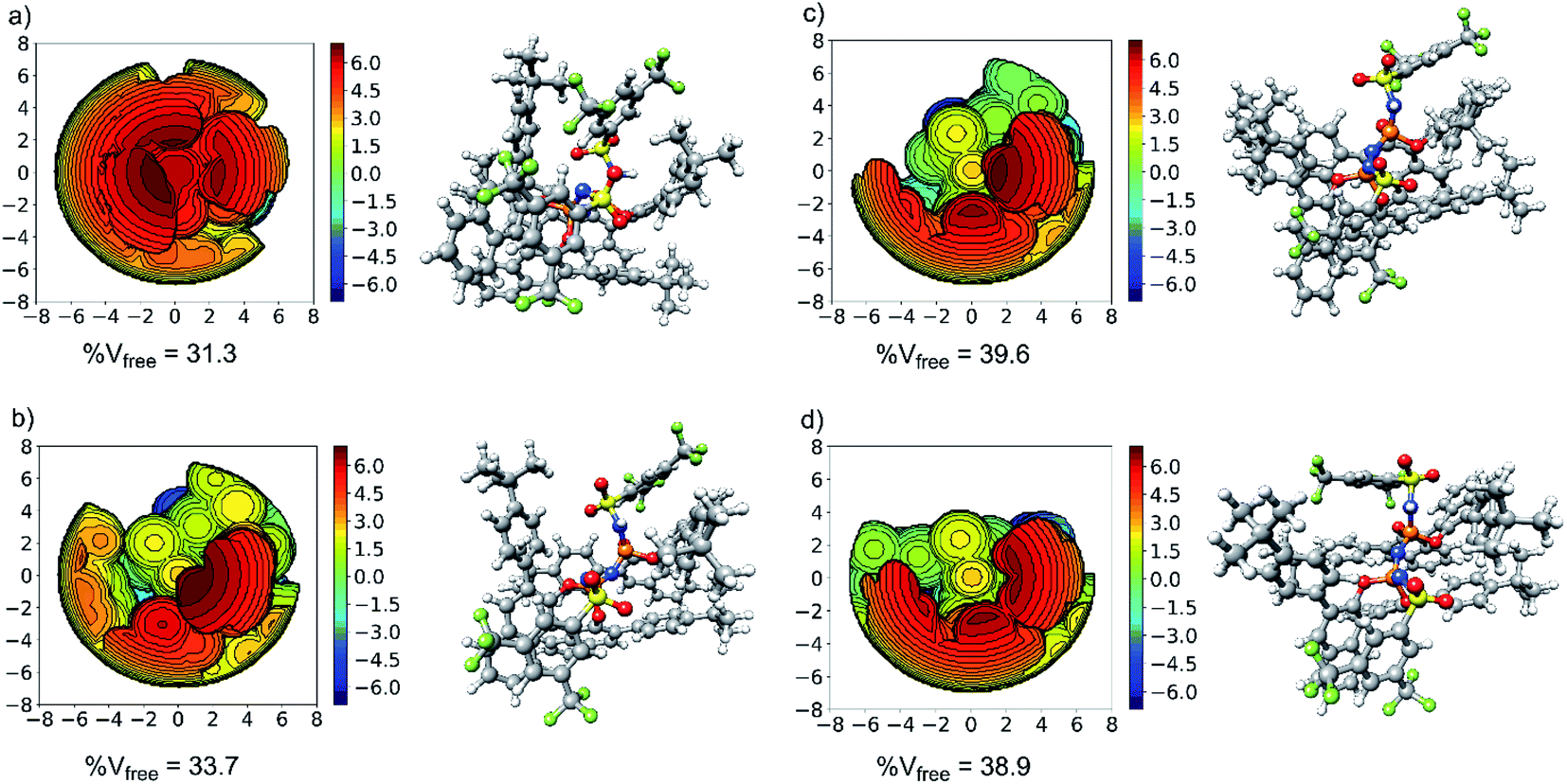 | ||
| Fig. 4 Steric map (left) and 3D model (right) for: (a) the catalyst in its equilibrium geometry; (b) the catalyst in its major transition state geometry; (c) the catalyst in its most stable minor transition state geometry (TS1-1′); (d) the catalyst in its second most stable minor transition state geometry (TS1-2′). | ||
Our results show that the catalyst in its ground state is described by a closed and compact structure, which shields the active site. To enable interaction with the substrate 1, the catalyst pocket has to open up, which raises the catalyst energy with respect to its ground state of an amount that is expected to be roughly proportional to % Vfree. The “open” structure assumed by the catalyst in the transition state is shown in Fig. 4b for TS1-1 and Fig. 4c and d for TS1-1′ and TS1-2′, respectively.
In the transition states, the energy needed to distort the catalyst might be, at least partially, compensated by weak noncovalent interactions between the catalyst and the substrate upon adduct formation. Hence, a thorough understanding of the stereo-controlling factors in this and similar transformations can only be achieved through an in-depth quantitative understanding of both effects. The LED analysis is clearly an extremely useful tool in this context. The LED decomposition of the activation barriers ΔG‡ (eqn (2)) for the three pathways just discussed is shown in Table 1.
| TS1-1 | TS1-1′ | TS1-2′ | TS1-1′-TS1-1 | TS1-2′-TS1-1 | |
|---|---|---|---|---|---|
| a Contribution associated with the catalyst distortion. b Contribution associated with the distortion of the substrate. | |||||
| ΔG‡ | 20.0 | 28.1 | 28.4 | 8.1 (exp. 2.8)38 | 8.4 (exp. 2.8)38 |
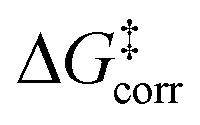
|
20.0 | 21.2 | 19.6 | 1.2 | −0.4 |
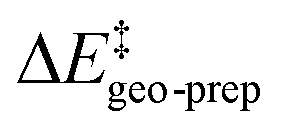
|
50.5 (41.3a + 9.2b) | 56.6 (43.2a + 13.4b) | 63.6 (50.0a + 13.6b) | 6.1 (1.9a + 4.2b) | 13.1 (8.7a + 4.4b) |
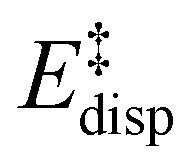
|
−38.2 | −43.2 | −45.4 | −5.0 | −7.2 |
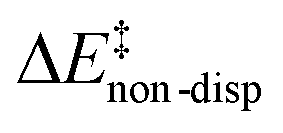
|
−12.2 | −6.5 | −9.5 | 5.7 | 2.8 |
The first important finding of this analysis is that the activation barrier for the pathway leading to the major enantiomeric product (ΔG‡ = 20.0 kcal mol−1) is identical in magnitude to the thermal and entropic contribution 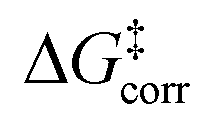 . This interesting effect originates from the fact that the increase in energy obtained when the catalyst structure is distorted from its ground state to the one that is optimal for interacting with the substrate (the dominant contribution in
. This interesting effect originates from the fact that the increase in energy obtained when the catalyst structure is distorted from its ground state to the one that is optimal for interacting with the substrate (the dominant contribution in 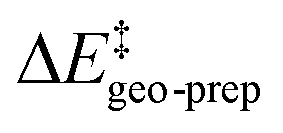 ) is entirely compensated by the energy gain due to attractive substrate–catalyst interactions. Importantly, London dispersion between the catalyst and the substrate is the dominant stabilizing factor in this case
) is entirely compensated by the energy gain due to attractive substrate–catalyst interactions. Importantly, London dispersion between the catalyst and the substrate is the dominant stabilizing factor in this case 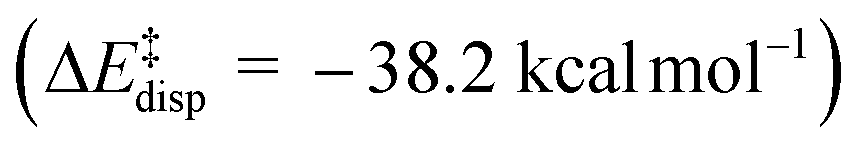 , while non-dispersive interactions provide a relatively minor but still noticeable contribution
, while non-dispersive interactions provide a relatively minor but still noticeable contribution 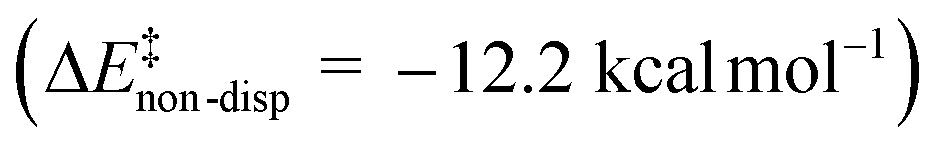 . In particular, there are strong dispersive interactions between the substrate and (i) both sulfonyl-groups of the catalyst, (ii) the aromatic rings of the aryl ligands and (iii) the tert-butyl groups of the pocket, as clearly shown via the DID plots69,80 in Fig. 3.
. In particular, there are strong dispersive interactions between the substrate and (i) both sulfonyl-groups of the catalyst, (ii) the aromatic rings of the aryl ligands and (iii) the tert-butyl groups of the pocket, as clearly shown via the DID plots69,80 in Fig. 3.
The pathways leading to the minor enantiomeric product show values of 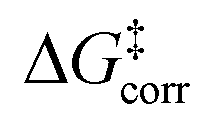 that are essentially identical to those obtained for the major pathway, while the overall free energy barriers are much larger. This effect originates from the fact that the
that are essentially identical to those obtained for the major pathway, while the overall free energy barriers are much larger. This effect originates from the fact that the 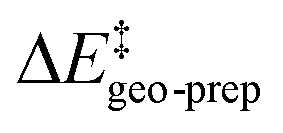 terms for the minor pathways are significantly larger compared to TS1-1. The interaction energy between the catalyst and the substrate is almost identical for TS1-1 and TS1-1′, while TS1-2′ features a stronger substrate–catalyst interaction, which partially compensates the corresponding increase in
terms for the minor pathways are significantly larger compared to TS1-1. The interaction energy between the catalyst and the substrate is almost identical for TS1-1 and TS1-1′, while TS1-2′ features a stronger substrate–catalyst interaction, which partially compensates the corresponding increase in 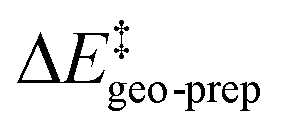 .
.
Thus, the catalyst–substrate interaction mechanism should be described as an “induced-fit” model, in which both the substrate and the catalyst adopt the conformation and orientation that is optimal for the interaction. The major pathway is the one that minimizes the energy penalty associated with the distortion of the catalyst and of the substrate 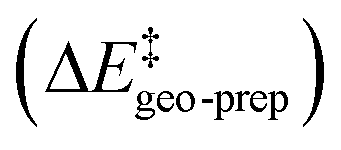 , while maximizing at the same time the catalyst–substrate interaction.
, while maximizing at the same time the catalyst–substrate interaction.
For the systems studied in this work, the distortion energy of the substrate correlates with the sp3-character of the methylene carbon atom. The factors contributing to the distortion energy of the catalyst, which is the dominant term in 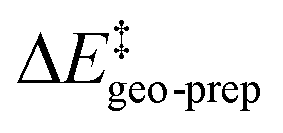 , are discussed in the next Section.
, are discussed in the next Section.
Understanding catalyst distortion within the induced fit model
We now seek to discuss the factors that determine the increase in energy associated with the catalyst distortion, as quantified by the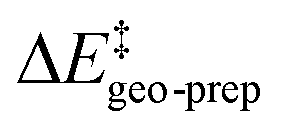 term in the LED, which can be regarded as the most important contribution influencing the stereoselectivity of the catalyst within the induced-fit model, alongside with the catalyst–substrate interaction.
term in the LED, which can be regarded as the most important contribution influencing the stereoselectivity of the catalyst within the induced-fit model, alongside with the catalyst–substrate interaction.
In order to understand the importance of individual groups or ligands, we performed a series of calculations in which we computed the catalyst contribution to 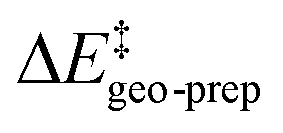 for a series of subsystems (see the ESI† for details). These subsystems were obtained by sequentially substituting the groups of the catalyst 3b with hydrogen atoms. During these calculations, the catalyst geometry was not optimized, and only the positions of the newly added hydrogen atoms were relaxed.
for a series of subsystems (see the ESI† for details). These subsystems were obtained by sequentially substituting the groups of the catalyst 3b with hydrogen atoms. During these calculations, the catalyst geometry was not optimized, and only the positions of the newly added hydrogen atoms were relaxed.
The results of this study are shown in Fig. 5. In this figure, the subsystems are numbered from 3b_1 to 3b_5; 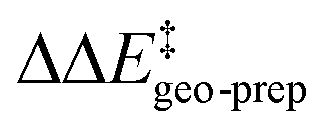 denotes the difference between the catalyst contribution to
denotes the difference between the catalyst contribution to 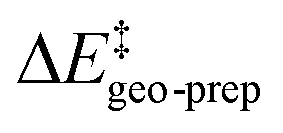 of the minor and major pathways.
of the minor and major pathways.
 | ||
| Fig. 5 The energy difference between the catalyst structure in (a) TS1-1′ and TS1-1 (b) TS1-2′ and TS1-1 for a series of “subsystems” (3b, 3b_1, 3b_2, 3b_3, 3b_4, 3b_5). (c) Definition of the subsystems. Calculations were performed at the B3LYP-D3/def2-TZVP level of theory. See text for details. | ||
Consistent with the results discussed in the previous section, 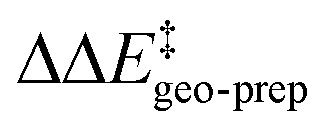 is positive for the original catalyst 3b, which indicates that the increase in energy associated with the distortion of the catalyst within the induced fit model is smaller for TS1-1 than for TS1-1′ and TS1-2′. Interestingly, by progressively replacing bulky groups with hydrogen atoms,
is positive for the original catalyst 3b, which indicates that the increase in energy associated with the distortion of the catalyst within the induced fit model is smaller for TS1-1 than for TS1-1′ and TS1-2′. Interestingly, by progressively replacing bulky groups with hydrogen atoms, 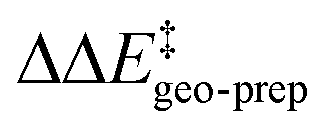 becomes smaller and smaller. For TS1-1′, the largest effect is associated with the substitution of the phenyl rings of the aryl-ligands (3b_1 → 3b_2), while for TS1-2′ it stems from the substitution of the Ph-(CF3)2 group at the catalyst's amine group (3b_2 → 3b_3). In both cases, the substitution leads to a change in the sign of
becomes smaller and smaller. For TS1-1′, the largest effect is associated with the substitution of the phenyl rings of the aryl-ligands (3b_1 → 3b_2), while for TS1-2′ it stems from the substitution of the Ph-(CF3)2 group at the catalyst's amine group (3b_2 → 3b_3). In both cases, the substitution leads to a change in the sign of 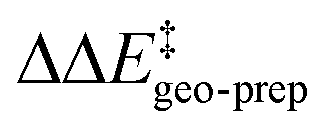 .
.
For TS1-1′, the 3b_1 → 3b_2 substitution reduces 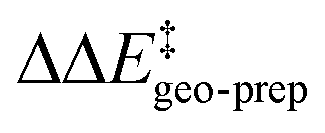 by −4.0 kcal mol−1. About 50% of this energy change (−1.9 kcal mol−1) is associated with the corresponding change in the intra-catalyst dispersion forces (see ESI† for details). For TS1-2′, the 3b_2 → 3b_3 substitution reduces
by −4.0 kcal mol−1. About 50% of this energy change (−1.9 kcal mol−1) is associated with the corresponding change in the intra-catalyst dispersion forces (see ESI† for details). For TS1-2′, the 3b_2 → 3b_3 substitution reduces 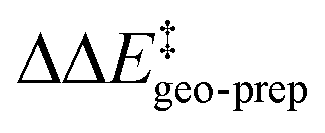 by −5.3 kcal mol−1. Again, intra-catalyst dispersion provides a fundamental contribution (−5.0 kcal mol−1). Thus, within the induced-fit model, intra-catalyst dispersion forces play an especially important role in determining the selectivity of this transformation, as they are largely responsible for the magnitude of
by −5.3 kcal mol−1. Again, intra-catalyst dispersion provides a fundamental contribution (−5.0 kcal mol−1). Thus, within the induced-fit model, intra-catalyst dispersion forces play an especially important role in determining the selectivity of this transformation, as they are largely responsible for the magnitude of 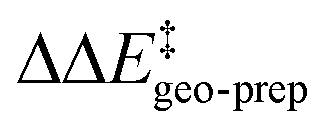 .
.
To illustrate the origin of this effect, we show in Fig. 6 the NCI plots obtained for the intramolecular interaction of the backbone of catalyst 3b with either the Ph ring at the aryl ligand (Fig. 6a and b) or the Ph-(CF3)2 group (Fig. 6c and d) in the relevant transition states. These plots show that interactions that are strong and attractive in TS1-1 are essentially absent in TS1-1′ (Fig. 6b) and TS1-2′ (Fig. 6d).
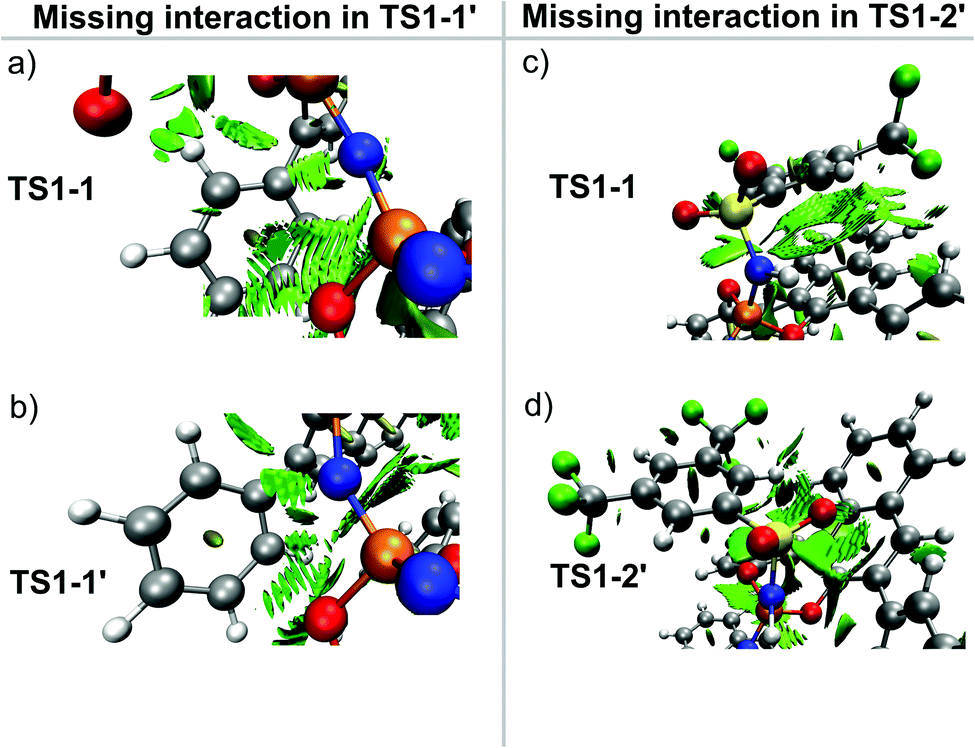 | ||
| Fig. 6 (a) NCI plot associated with the interaction between the Ph group and the catalyst backbone in TS1-1 (major pathway). (b) The analogous NCI plot for TS1-1′ (minor pathway). (c) NCI plot associated with the interaction between the Ph-(CF3)2 group and the catalyst backbone in TS1-1 (major pathway). (d) The analogous NCI plot for TS1-2′ (minor pathway). The density isovalue for the NCI plots is 0.5 a.u. | ||
Thus, for the major reaction pathway, the catalyst distortion associated with the interaction with the substrate does not disrupt the stabilizing Ph-⋯backbone or Ph-(CF3)2⋯backbone London dispersion interactions. In contrast, for each of the minor reaction pathways, some of the stabilizing intra-catalyst interactions are missing to facilitate the interaction with the substrate. In other words, only if the substrate and the catalyst are in the correct relative orientation (the one that leads to the formation of the major enantiomeric product) the energy penalty associated with the catalyst distortion can be minimized.
To summarize the results obtained so far, we have demonstrated that: (i) the energy required to distort the catalyst is a key factor controlling the selectivity of the transformation within an induced-fit model; (ii) this energy penalty is dominated by the change in the intra-catalyst dispersion interactions on going from the ground state of the catalyst to the structure that is optimal for the interaction with the substrate. Thus, a simple possible path towards rational catalyst design is the identification of confined catalysts that are largely stabilized by dispersion forces. This can be done by introducing bulky groups that act as dispersion energy donors,24,73,79 such as π systems, whose interaction with the catalyst backbone might or might not be disrupted upon the interaction with the substrate along specific reaction pathways.
In the next Section we will show how this simple argument can be used to rationalize catalyst selectivity in a series of previously published asymmetric transformations.
These results demonstrate that intra-catalyst dispersion forces are a fundamental factor contributing to the structural stability of these confined catalysts. In addition, as they are responsible for the magnitude of 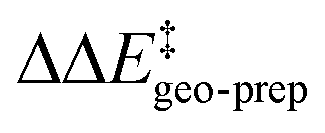 , they control to a large extent the selectivity of this transformation.
, they control to a large extent the selectivity of this transformation.
Testing the model
The results just discussed can be used to rationalize previously published experimental findings and will hopefully aid in the design of new catalysts with improved selectivity. As an example, we show in Fig. 7 three catalysts with different experimental selectivity for the same reaction investigated in this work but using a different substrate, i.e., 4-methylenedodecan-1-ol. In the figure, the catalyst equilibrium geometry is shown alongside with the associated steric maps and NCI plots.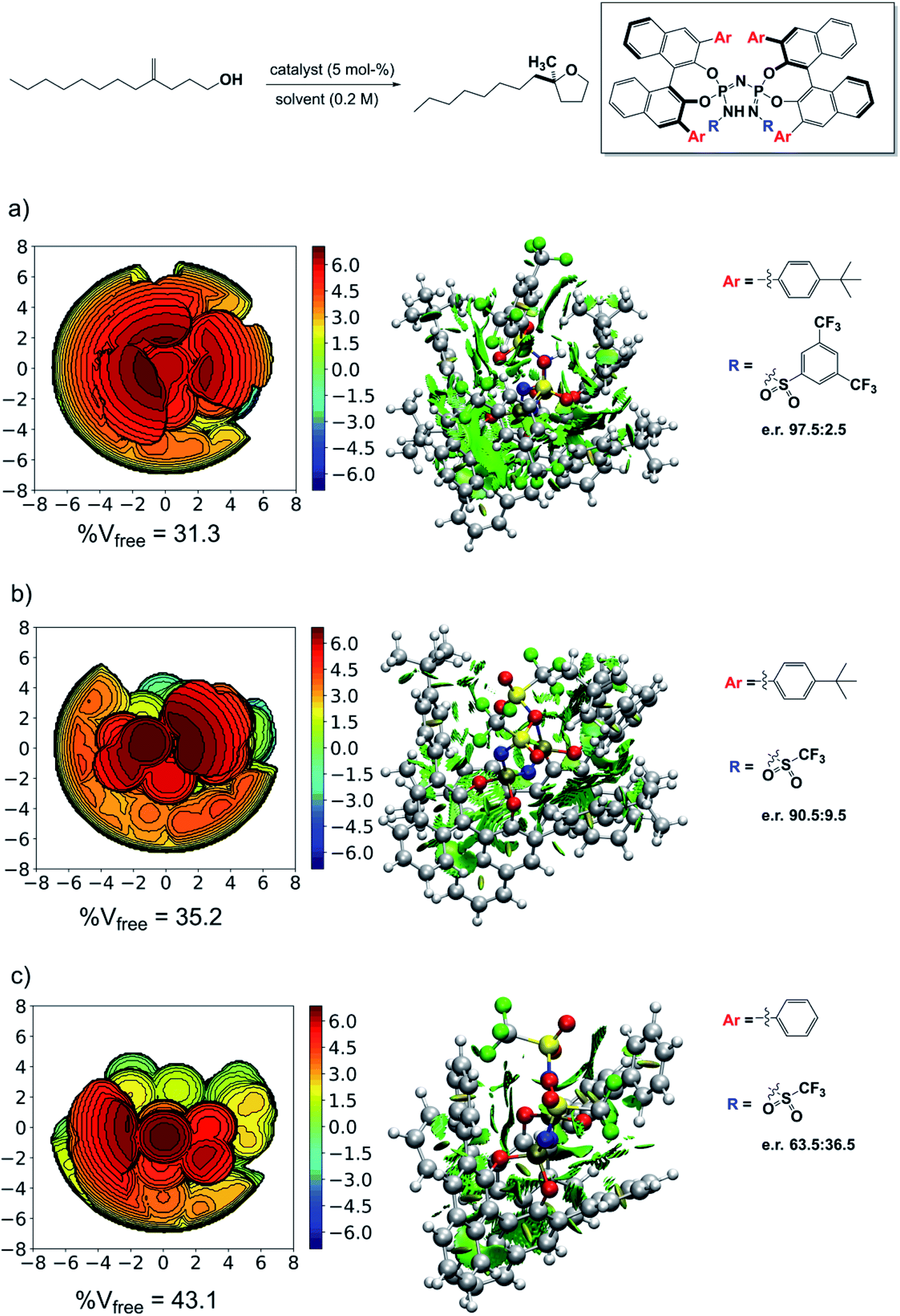 | ||
| Fig. 7 Steric maps, NCI plots and experimental enantiomeric ratios for different catalysts in their geometric ground state. (a) Catalyst 3b. (b) Catalyst 3a. (c) Catalyst 3d. The isovalue for the density in the NCI plots is 0.5 a.u. | ||
The experimental results were taken from ref. 38. Catalyst 3d showed almost no stereoselectivity (e.r. 63.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :36.5), catalyst 3a is significantly more selective (e.r. 90.5:9.5), and catalyst 3b is the most selective (e.r. 97.5:2.5). Remarkably enough, analysis of the steric maps reveal that the catalyst selectivity does correlate with catalyst confinement as quantified by % Vfree. As expected, based on our previous analysis, the NCI plots also reveal an increase of stabilizing noncovalent interactions with increasing confinement, which fully supports our dispersion-based induced-fit model for these systems.
:36.5), catalyst 3a is significantly more selective (e.r. 90.5:9.5), and catalyst 3b is the most selective (e.r. 97.5:2.5). Remarkably enough, analysis of the steric maps reveal that the catalyst selectivity does correlate with catalyst confinement as quantified by % Vfree. As expected, based on our previous analysis, the NCI plots also reveal an increase of stabilizing noncovalent interactions with increasing confinement, which fully supports our dispersion-based induced-fit model for these systems.
Conclusions
We elucidated the mechanism and stereo-controlling factors of the stereoselective intramolecular hydroalkoxylation of terminal olefins catalyzed by confined IDPi Brønsted acids using state of the art computational techniques. It was found that the catalyst–substrate adduct formation is better described using an “induced-fit” model rather than a “key-and-lock” mechanism, in which both the substrate and the catalyst adopt the conformation and orientation that is optimal for the interaction. Thus, the major pathway minimizes the energy penalty associated with the distortion of the catalyst, while maximizing at the same time the attractive catalyst–substrate interactions. Remarkably enough, the energy penalty associated with the catalyst distortion is dominated by the change in the intramolecular dispersion interactions within the catalyst, while intermolecular catalyst–substrate dispersion interactions are the major stabilizing contribution in the transition state. Thus, a simple possible path towards rational catalyst design is the identification of confined catalysts that are largely stabilized by dispersion forces, e.g., via the use of appropriate dispersion energy donors.Data availability
The Cartesian coordinates of all structures, the results of benchmark calculations on transition state geometries, energies, and thermochemical corrections, local energy decomposition analyses for the low-energy transition states and a detailed analysis of the key noncovalent interactions are provided in the ESI.†Author contributions
I. H. performed all the calculations. G. B. conceived and directed the project. I. H. and G. B. wrote the original draft of the manuscript and prepared the figures. G. B., F. N. and I. H. contributed to the data analysis and to the writing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Max Planck Society for financial support.References
- G. Rothenberg, Catalysis: concepts and green applications. John Wiley & Sons: 2017 Search PubMed.
- W. P. Jencks, Catalysis in chemistry and enzymology. Courier Corporation: 1987 Search PubMed.
- M. Yoshizawa, M. Tamura and M. Fujita, Diels-alder in aqueous molecular hosts: unusual regioselectivity and efficient catalysis, Science, 2006, 312(5771), 251–254 CrossRef CAS PubMed.
- C. J. Hastings, M. D. Pluth, R. G. Bergman and K. N. Raymond, Enzymelike Catalysis of the Nazarov Cyclization by Supramolecular Encapsulation, J. Am. Chem. Soc., 2010, 132(20), 6938–6940 CrossRef CAS PubMed.
- M. Dusselier, P. Van Wouwe, A. Dewaele, A. Jacobs Pierre and F. Sels Bert, Shape-selective zeolite catalysis for bioplastics production, Science, 2015, 349(6243), 78–80 CrossRef CAS PubMed.
- A. Corma, L. T. Nemeth, M. Renz and S. Valencia, Sn-zeolite beta as a heterogeneous chemoselective catalyst for Baeyer-Villiger oxidations, Nature, 2001, 412(6845), 423–425 CrossRef CAS PubMed.
- I. Čorić and B. List, Asymmetric spiroacetalization catalysed by confined Brønsted acids, Nature, 2012, 483(7389), 315–319 CrossRef PubMed.
- S. Das, L. Liu, Y. Zheng, M. W. Alachraf, W. Thiel, C. K. De and B. List, Nitrated Confined Imidodiphosphates Enable a Catalytic Asymmetric Oxa-Pictet–Spengler Reaction, J. Am. Chem. Soc., 2016, 138(30), 9429–9432 CrossRef CAS PubMed.
- J. H. Kim, I. Čorić, C. Palumbo and B. List, Resolution of Diols via Catalytic Asymmetric Acetalization, J. Am. Chem. Soc., 2015, 137(5), 1778–1781 CrossRef CAS PubMed.
- J. H. Kim, I. Čorić, S. Vellalath and B. List, The Catalytic Asymmetric Acetalization, Angew. Chem., Int. Ed., 2013, 52(16), 4474–4477 CrossRef CAS PubMed.
- L. Liu, P. S. J. Kaib, A. Tap and B. List, A General Catalytic Asymmetric Prins Cyclization, J. Am. Chem. Soc., 2016, 138(34), 10822–10825 CrossRef CAS PubMed.
- P. S. J. Kaib, L. Schreyer, S. Lee, R. Properzi and B. List, Extremely Active Organocatalysts Enable a Highly Enantioselective Addition of Allyltrimethylsilane to Aldehydes, Angew. Chem., Int. Ed., 2016, 55(42), 13200–13203 CrossRef CAS PubMed.
- L. Schreyer, R. Properzi and B. List, IDPi Catalysis, Angew. Chem., Int. Ed., 2019, 58(37), 12761–12777 CrossRef CAS PubMed.
- S. Ghosh, S. Das, C. K. De, D. Yepes, F. Neese, G. Bistoni, M. Leutzsch and B. List, Strong and Confined Acids Control Five Stereogenic Centers in Catalytic Asymmetric Diels–Alder Reactions of Cyclohexadienones with Cyclopentadiene, Angew. Chem., Int. Ed., 2020, 59(30), 12347–12351 CrossRef CAS PubMed.
- J. Ouyang, J. L. Kennemur, C. K. De, C. Farès and B. List, Strong and Confined Acids Enable a Catalytic Asymmetric Nazarov Cyclization of Simple Divinyl Ketones, J. Am. Chem. Soc., 2019, 141(8), 3414–3418 CrossRef CAS PubMed.
- T. Amatov, N. Tsuji, R. Maji, L. Schreyer, H. Zhou, M. Leutzsch and B. List, Confinement-Controlled, Either syn- or anti-Selective Catalytic Asymmetric Mukaiyama Aldolizations of Propionaldehyde Enolsilanes, J. Am. Chem. Soc., 2021, 143(36), 14475–14481 CrossRef CAS PubMed.
- B. Mitschke, M. Turberg and B. List, Confinement as a Unifying Element in Selective Catalysis, Chem, 2020, 6(10), 2515–2532 CAS.
- A. J. Sterling, S. Zavitsanou, J. Ford and F. Duarte, Selectivity in organocatalysis—From qualitative to quantitative predictive models, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2021, 11(5), e1518 CAS.
- E. H. Krenske, K. N. Houk and M. Harmata, Origin of Stereoselectivity in the (4 + 3) Cycloadditions of Chiral Alkoxy Siloxyallyl Cations with Furan, Org. Lett., 2010, 12(3), 444–447 CrossRef CAS PubMed.
- F. R. Clemente and K. N. Houk, Theoretical Studies of Stereoselectivities of Intramolecular Aldol Cyclizations Catalyzed by Amino Acids, J. Am. Chem. Soc., 2005, 127(32), 11294–11302 CrossRef CAS PubMed.
- R. R. Fraser, T. B. Grindley and S. Passannanti, Stereoselectivity in the Reactions of Nitrosopiperidine Carbanions. Steric vs. Stereoelectronic Control, Can. J. Chem., 1975, 53(16), 2473–2480 CrossRef CAS.
- S. Ichikawa, S. Shuto and A. Matsuda, The First Synthesis of Herbicidin B. Stereoselective Construction of the Tricyclic Undecose Moiety by a Conformational Restriction Strategy Using Steric Repulsion between Adjacent Bulky Silyl Protecting Groups on a Pyranose Ring, J. Am. Chem. Soc., 1999, 121(44), 10270–10280 CrossRef CAS.
- D. J. Cram and K. R. Kopecky, Studies in Stereochemistry. XXX. Models for Steric Control of Asymmetric Induction1, J. Am. Chem. Soc., 1959, 81(11), 2748–2755 CrossRef CAS.
- J. P. Wagner and P. R. Schreiner, London Dispersion in Molecular Chemistry—Reconsidering Steric Effects, Angew. Chem., Int. Ed., 2015, 54(42), 12274–12296 CrossRef CAS PubMed.
- R. S. J. Proctor, A. C. Colgan and R. J. Phipps, Exploiting attractive non-covalent interactions for the enantioselective catalysis of reactions involving radical intermediates, Nat. Chem., 2020, 12(11), 990–1004 CrossRef PubMed.
- Y. E. Türkmen, Y. Zhu and V. H. Rawal, Brønsted Acids, in Comprehensive Enantioselective Organocatalysis, 2013, pp. 239–288 Search PubMed.
- Y. Xi, B. Su, X. Qi, S. Pedram, P. Liu and J. F. Hartwig, Application of Trimethylgermanyl-Substituted Bisphosphine Ligands with Enhanced Dispersion Interactions to Copper-Catalyzed Hydroboration of Disubstituted Alkenes, J. Am. Chem. Soc., 2020, 142(42), 18213–18222 CrossRef CAS PubMed.
- A. A. Thomas, K. Speck, I. Kevlishvili, Z. Lu, P. Liu and S. L. Buchwald, Mechanistically Guided Design of Ligands That Significantly Improve the Efficiency of CuH-Catalyzed Hydroamination Reactions, J. Am. Chem. Soc., 2018, 140(42), 13976–13984 CrossRef CAS PubMed.
- G. Lu, R. Y. Liu, Y. Yang, C. Fang, D. S. Lambrecht, S. L. Buchwald and P. Liu, Ligand–Substrate Dispersion Facilitates the Copper-Catalyzed Hydroamination of Unactivated Olefins, J. Am. Chem. Soc., 2017, 139(46), 16548–16555 CrossRef CAS PubMed.
- N. I. Saper, A. Ohgi, D. W. Small, K. Semba, Y. Nakao and J. F. Hartwig, Nickel-catalysed anti-Markovnikov hydroarylation of unactivated alkenes with unactivated arenes facilitated by non-covalent interactions, Nat. Chem., 2020, 12(3), 276–283 CrossRef CAS PubMed.
- E. Lyngvi, I. A. Sanhueza and F. Schoenebeck, Dispersion Makes the Difference: Bisligated Transition States Found for the Oxidative Addition of Pd(PtBu3)2 to Ar-OSO2R and Dispersion-Controlled Chemoselectivity in Reactions with Pd[P(iPr)(tBu2)]2, Organometallics, 2015, 34(5), 805–812 CrossRef CAS.
- R. C. Wende, A. Seitz, D. Niedek, S. M. M. Schuler, C. Hofmann, J. Becker and P. R. Schreiner, The Enantioselective Dakin–West Reaction, Angew. Chem., Int. Ed., 2016, 55(8), 2719–2723 CrossRef CAS PubMed.
- C. Eschmann, L. Song and P. R. Schreiner, London Dispersion Interactions Rather than Steric Hindrance Determine the Enantioselectivity of the Corey–Bakshi–Shibata Reduction, Angew. Chem., Int. Ed., 2021, 60(9), 4823–4832 CrossRef CAS PubMed.
- Y.-h. Lam and K. N. Houk, Origins of Stereoselectivity in Intramolecular Aldol Reactions Catalyzed by Cinchona Amines, J. Am. Chem. Soc., 2015, 137(5), 2116–2127 CrossRef CAS PubMed.
- B. Li, H. Xu, Y. Dang and K. N. Houk, Dispersion and Steric Effects on Enantio-/Diastereoselectivities in Synergistic Dual Transition-Metal Catalysis, J. Am. Chem. Soc., 2022, 144(4), 1971–1985 CrossRef CAS PubMed.
- S. Singha, M. Buchsteiner, G. Bistoni, R. Goddard and A. Fürstner, A New Ligand Design Based on London Dispersion Empowers Chiral Bismuth–Rhodium Paddlewheel Catalysts, J. Am. Chem. Soc., 2021, 143(15), 5666–5673 CrossRef CAS PubMed.
- D. Yepes, F. Neese, B. List and G. Bistoni, Unveiling the Delicate Balance of Steric and Dispersion Interactions in Organocatalysis Using High-Level Computational Methods, J. Am. Chem. Soc., 2020, 142(7), 3613–3625 CrossRef CAS PubMed.
- N. Tsuji, J. L. Kennemur, T. Buyck, S. Lee, S. Prévost, P. S. J. Kaib, D. Bykov, C. Farès and B. List, Activation of olefins via asymmetric Brønsted acid catalysis, Science, 2018, 359(6383), 1501–1505 CrossRef CAS PubMed.
- A. R. Rosales, J. Wahlers, E. Limé, R. E. Meadows, K. W. Leslie, R. Savin, F. Bell, E. Hansen, P. Helquist, R. H. Munday, O. Wiest and P.-O. Norrby, Rapid virtual screening of enantioselective catalysts using CatVS, Nat. Catal., 2019, 2(1), 41–45 CrossRef CAS.
- A. C. Doney, B. J. Rooks, T. Lu and S. E. Wheeler, Design of organocatalysts for asymmetric propargylations through computational screening, ACS Catal., 2016, 6(11), 7948–7955 CrossRef CAS.
- Y. Guan, V. M. Ingman, B. J. Rooks and S. E. Wheeler, AARON: An Automated Reaction Optimizer for New Catalysts, J. Chem. Theory Comput., 2018, 14(10), 5249–5261 CrossRef CAS PubMed.
- C. R. Corbeil, S. Thielges, J. A. Schwartzentruber and N. Moitessier, Toward a computational tool predicting the stereochemical outcome of asymmetric reactions: development and application of a rapid and accurate program based on organic principles, Angew. Chem., 2008, 120(14), 2675–2678 CrossRef.
- G. dos Passos Gomes, R. Pollice and A. Aspuru-Guzik, Navigating through the Maze of Homogeneous Catalyst Design with Machine Learning, Trends Chem., 2021, 3(2), 96–110 CrossRef CAS.
- F. Neese, Software update: the ORCA program system, version 4.0, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2018, 8(1), e1327 Search PubMed.
- S. Grimme, Exploration of Chemical Compound, Conformer, and Reaction Space with Meta-Dynamics Simulations Based on Tight-Binding Quantum Chemical Calculations, J. Chem. Theory Comput., 2019, 15(5), 2847–2862 CrossRef CAS PubMed.
- P. Pracht, F. Bohle and S. Grimme, Automated exploration of the low-energy chemical space with fast quantum chemical methods, Phys. Chem. Chem. Phys., 2020, 22(14), 7169–7192 RSC.
- P. Pracht and S. Grimme, Calculation of absolute molecular entropies and heat capacities made simple, Chem. Sci., 2021, 12(19), 6551–6568 RSC.
- C. Bannwarth, E. Caldeweyher, S. Ehlert, A. Hansen, P. Pracht, J. Seibert, S. Spicher and S. Grimme, Extended tight-binding quantum chemistry methods, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2021, 11(2), e1493 CAS.
- C. Bannwarth, S. Ehlert and S. Grimme, GFN2-xTB—An Accurate and Broadly Parametrized Self-Consistent Tight-Binding Quantum Chemical Method with Multipole Electrostatics and Density-Dependent Dispersion Contributions, J. Chem. Theory Comput., 2019, 15(3), 1652–1671 CrossRef CAS PubMed.
- S. Grimme, C. Bannwarth and P. Shushkov, A Robust and Accurate Tight-Binding Quantum Chemical Method for Structures, Vibrational Frequencies, and Noncovalent Interactions of Large Molecular Systems Parametrized for All spd-Block Elements (Z = 1–86), J. Chem. Theory Comput., 2017, 13(5), 1989–2009 CrossRef CAS PubMed.
- A. D. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98(7), 5648–5652 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7(18), 3297–3305 RSC.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132(15), 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 2011, 32(7), 1456–1465 CrossRef CAS PubMed.
- K. Eichkorn, O. Treutler, H. Öhm, M. Häser and R. Ahlrichs, Auxiliary basis sets to approximate Coulomb potentials, Chem. Phys. Lett., 1995, 240(4), 283–290 CrossRef CAS.
- F. Neese, An improvement of the resolution of the identity approximation for the formation of the Coulomb matrix, J. Comput. Chem., 2003, 24(14), 1740–1747 CrossRef CAS PubMed.
- F. Weigend, Accurate Coulomb-fitting basis sets for H to Rn, Phys. Chem. Chem. Phys., 2006, 8(9), 1057–1065 RSC.
- A. Hansen, D. G. Liakos and F. Neese, Efficient and accurate local single reference correlation methods for high-spin open-shell molecules using pair natural orbitals, J. Chem. Phys., 2011, 135(21), 214102 CrossRef PubMed.
- D. G. Liakos, A. Hansen and F. Neese, Weak Molecular Interactions Studied with Parallel Implementations of the Local Pair Natural Orbital Coupled Pair and Coupled Cluster Methods, J. Chem. Theory Comput., 2011, 7(1), 76–87 CrossRef CAS PubMed.
- F. Neese, A. Hansen and D. G. Liakos, Efficient and accurate approximations to the local coupled cluster singles doubles method using a truncated pair natural orbital basis, J. Chem. Phys., 2009, 131(6), 064103 CrossRef PubMed.
- C. Riplinger and F. Neese, An efficient and near linear scaling pair natural orbital based local coupled cluster method, J. Chem. Phys., 2013, 138(3), 034106 CrossRef PubMed.
- C. Riplinger, P. Pinski, U. Becker, E. F. Valeev and F. Neese, Sparse maps—A systematic infrastructure for reduced-scaling electronic structure methods. II. Linear scaling domain based pair natural orbital coupled cluster theory, J. Chem. Phys., 2016, 144(2), 024109 CrossRef PubMed.
- C. Riplinger, B. Sandhoefer, A. Hansen and F. Neese, Natural triple excitations in local coupled cluster calculations with pair natural orbitals, J. Chem. Phys., 2013, 139(13), 134101 CrossRef PubMed.
- F. Neese, F. Wennmohs, A. Hansen and U. Becker, Efficient, approximate and parallel Hartree-Fock and hybrid DFT calculations. A ‘chain-of-spheres’ algorithm for the Hartree-Fock exchange, Chem. Phys., 2009, 356(1), 98–109 CrossRef CAS.
- S. H. Vosko, L. Wilk and M. Nusair, Accurate spin-dependent electron liquid correlation energies for local spin density calculations: a critical analysis, Can. J. Phys., 1980, 58(8), 1200–1211 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields, J. Phys. Chem., 1994, 98(45), 11623–11627 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B, 1988, 37(2), 785–789 CrossRef CAS PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions, J. Phys. Chem. B, 2009, 113(18), 6378–6396 CrossRef CAS PubMed.
- A. Altun, F. Neese and G. Bistoni, Effect of Electron Correlation on Intermolecular Interactions: A Pair Natural Orbitals Coupled Cluster Based Local Energy Decomposition Study, J. Chem. Theory Comput., 2019, 15(1), 215–228 CrossRef CAS PubMed.
- A. Altun, M. Saitow, F. Neese and G. Bistoni, Local Energy Decomposition of Open-Shell Molecular Systems in the Domain-Based Local Pair Natural Orbital Coupled Cluster Framework, J. Chem. Theory Comput., 2019, 15(3), 1616–1632 CrossRef CAS PubMed.
- G. Bistoni, Finding chemical concepts in the Hilbert space: Coupled cluster analyses of noncovalent interactions, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2020, 10(3), e1442 CAS.
- W. B. Schneider, G. Bistoni, M. Sparta, M. Saitow, C. Riplinger, A. A. Auer and F. Neese, Decomposition of Intermolecular Interaction Energies within the Local Pair Natural Orbital Coupled Cluster Framework, J. Chem. Theory Comput., 2016, 12(10), 4778–4792 CrossRef CAS PubMed.
- G. Bistoni, A. A. Auer and F. Neese, Understanding the Role of Dispersion in Frustrated Lewis Pairs and Classical Lewis Adducts: A Domain-Based Local Pair Natural Orbital Coupled Cluster Study, Chem.–Eur. J., 2017, 23(4), 865–873 CrossRef CAS PubMed.
- Q. Lu, F. Neese and G. Bistoni, London dispersion effects in the coordination and activation of alkanes in σ-complexes: a local energy decomposition study, Phys. Chem. Chem. Phys., 2019, 21(22), 11569–11577 RSC.
- L. Falivene, Z. Cao, A. Petta, L. Serra, A. Poater, R. Oliva, V. Scarano and L. Cavallo, Towards the online computer-aided design of catalytic pockets, Nat. Chem., 2019, 11(10), 872–879 CrossRef CAS PubMed.
- J. Contreras-García, E. R. Johnson, S. Keinan, R. Chaudret, J. P. Piquemal, D. N. Beratan and W. Yang, NCIPLOT: a program for plotting non-covalent interaction regions, J. Chem. Theory Comput., 2011, 7(3), 625–632 CrossRef PubMed.
- R. A. Boto, F. Peccati, R. Laplaza, C. Quan, A. Carbone, J.-P. Piquemal, Y. Maday and J. Contreras-García, NCIPLOT4: Fast, Robust, and Quantitative Analysis of Noncovalent Interactions, J. Chem. Theory Comput., 2020, 16(7), 4150–4158 CrossRef CAS PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, Revealing Noncovalent Interactions, J. Am. Chem. Soc., 2010, 132(18), 6498–6506 CrossRef CAS PubMed.
- S. Grimme, R. Huenerbein and S. Ehrlich, On the Importance of the Dispersion Energy for the Thermodynamic Stability of Molecules, Chemphyschem, 2011, 12(7), 1258–1261 CrossRef CAS PubMed.
- A. Wuttke and R. A. Mata, Visualizing dispersion interactions through the use of local orbital spaces, J. Comput. Chem., 2017, 38(1), 15–23 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc02274e |
| This journal is © The Royal Society of Chemistry 2022 |