
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControlled, one-pot synthesis of recyclable poly(1,3-diene)-polyester block copolymers, catalyzed by yttrium β-diketiminate complexes†
Sophia C.
Kosloski-Oh
 a,
Yvonne
Manjarrez
a,
Taleen J.
Boghossian
a and
Megan E.
Fieser
*ab
a,
Yvonne
Manjarrez
a,
Taleen J.
Boghossian
a and
Megan E.
Fieser
*ab
aDepartment of Chemistry, University of Southern California, Los Angeles, California 90089, USA
bWrigley Institute for Environmental Studies, University of Southern California, Los Angeles, California 90089, USA. E-mail: fieser@usc.edu
First published on 8th August 2022
Abstract
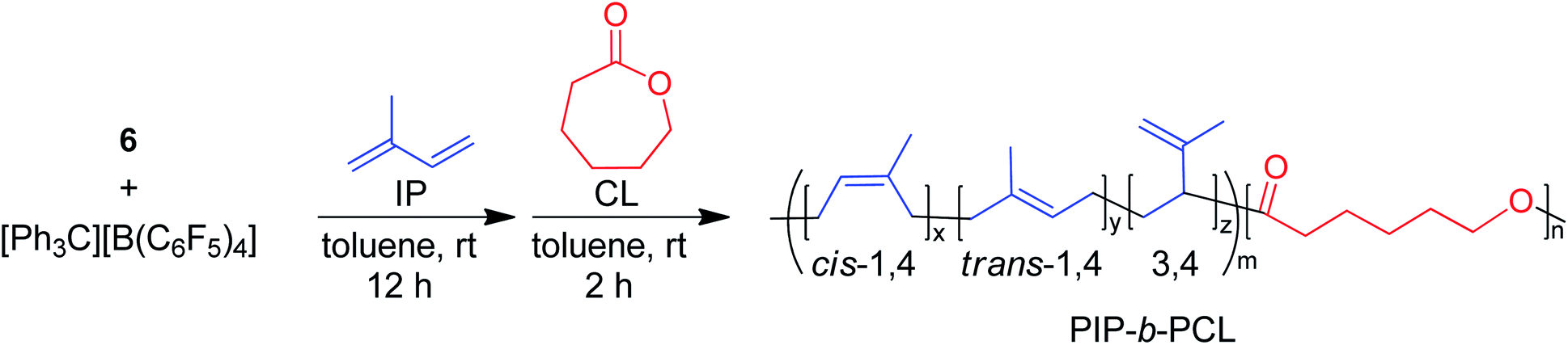
The one-pot synthesis of well-defined block copolymers of olefins/1,3-dienes and polar monomers, such as cyclic esters and acrylates has long been the focus of intense research. Cationic alkyl rare earth metal catalysts, activated by organoborates, have shown to be promising for the polymerization of isoprene or styrene and ε-caprolactone. In this study, we synthesize a series of yttrium bis(alkyl) complexes supported by simple β-diketiminate ancillary ligands. Subtle changes have been made to the β-diketiminate ligand framework to elucidate the effect of ligand structure on the rate and selectivity of olefin/1,3-diene and cyclic ester polymerization, with small ligand changes having a large impact on the resulting polymerizations. Generation of the active cationic species was easily streamlined by identification of appropriate catalyst![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :organoborate ratios, allowing for high catalyst efficiencies. Notably, we demonstrate the first cationic rare earth metal alkyl-initiated polymerization of δ-valerolactone and ε-decalactone as well as introduced five new block copolymer morphologies. In addition, selective degradation of the ester block in poly(isoprene-b-caprolactone) enabled recovery of the polyisoprene block with identical spectroscopic and thermal properties. Significantly, recopolymerization of the recovered poly(1,3-diene) with fresh ε-caprolactone reproduced the desired diblocks with nearly identical thermal and physical properties to those of virgin copolymer, illustrating a plausible recycling scheme for these materials.
:organoborate ratios, allowing for high catalyst efficiencies. Notably, we demonstrate the first cationic rare earth metal alkyl-initiated polymerization of δ-valerolactone and ε-decalactone as well as introduced five new block copolymer morphologies. In addition, selective degradation of the ester block in poly(isoprene-b-caprolactone) enabled recovery of the polyisoprene block with identical spectroscopic and thermal properties. Significantly, recopolymerization of the recovered poly(1,3-diene) with fresh ε-caprolactone reproduced the desired diblocks with nearly identical thermal and physical properties to those of virgin copolymer, illustrating a plausible recycling scheme for these materials.
Introduction
In the last few decades, block copolymers have become an indispensable class of soft materials with an expanding range of applications including drug delivery, adhesives, electronics, and construction.1 Poly(1,3-diene)-based block copolymers have been extensively commercialized due to their low cost, lightweight, durability, wide service temperature range, and resistance to chemical reactivity.2 Block copolymers of poly(1,3-dienes) and polar polymers often exhibit improved adhesive, dyeing, and moisture absorption properties, making them suitable for a broader spectrum of applications.3 Current efforts in synthesizing olefin/polar block copolymers have focused on incorporating olefins with polar functional groups such as alkyl acrylates.4–9 This produces block copolymers with strong aliphatic carbon–carbon bonds in the backbone, making these materials difficult to chemically recycle. In addition, block copolymerization of these two dissimilar monomers is challenging because catalysts that excel at olefin or 1,3-diene polymerization often struggle with polar monomers. Furthermore, these catalysts tend to be highly oxophilic and can be easily poisoned by polar functional groups.6 Alternative polar monomers, such as cyclic esters, could maintain valuable physical properties while providing a degradable polymer block. Additionally, cyclic esters can be sourced from biomass; therefore, incorporation of more sustainable monomers in polymers derived from petroleum sources would alleviate depletion of petroleum feedstocks.10,11 Yet, switching from olefin/1,3-diene polymerization to cyclic ester polymerization can also poison the polymerization catalyst.12 Cationic rare earth metal alkyl catalysts are highly efficient at the homopolymerization of olefins and 1,3-dienes.13 These complexes usually bear an outer-sphere borate anion generated in situ and have shown resistance to poisoning. These complexes can also lead to polymers with different stereoselectivity depending on the monomer and catalyst structure. To date, only five rare earth metal catalysts have been identified as efficient catalysts for the block copolymerization of olefins or 1,3-dienes and cyclic esters (Fig. 1).14–19 Hou and coworkers reported the first block copolymerization of styrene (S) and ε-caprolactone (CL) with a scandium alkyl half-sandwich pre-catalyst (1).14 Cheng and coworkers later extended this work to include a bifunctional initiating alkyl to form triblock copolymers of polystyrene (PS) and polycaprolactone (PCL), with PS in the middle block.18 Hou and coworkers also developed a PNP carbazolide bis(alkyl) rare earth metal catalyst capable of producing block copolymers consisting of cis-1,4-polyisoprene (PIP) and PCL in a living manner (2), Fig. 2.15 In 2014, Cui and coworkers reported an amidino N-heterocyclic carbene-supported lutetium bis(alkyl) complex for the block copolymerization of isoprene (IP) and CL (4).16 In contrast with the Hou pre-catalyst (2), this catalyst produced 3,4-regulated polyisoprene (PIP). The change in selectivity could be due to the reduced ionic radii of the metal as well as the greater steric crowding of the carbene ligand in the latter complex. However, due to the vast differences between the ligands in 2 and 4, it is difficult to designate the exact factors that led to the disparate catalyst selectivity.13 Pan and coworkers synthesized lutetium and yttrium bis(alkyl) complexes supported by an anilido-oxazoline ligand (3) for the block copolymerization of IP with CL.17 More recently, Shi and coworkers altered the pendant carbene arm of the amidinate ligand in 4 to a pyridine donor (5), and tested these new complexes for the block copolymerization of IP and CL.19 This subtle change dramatically altered the selectivity of IP polymerization to trans-1,4 polymerization. Changing the pendant arm length of the pyridine donor also made a large impact on the rate of polymerization and selectivity of IP polymerization.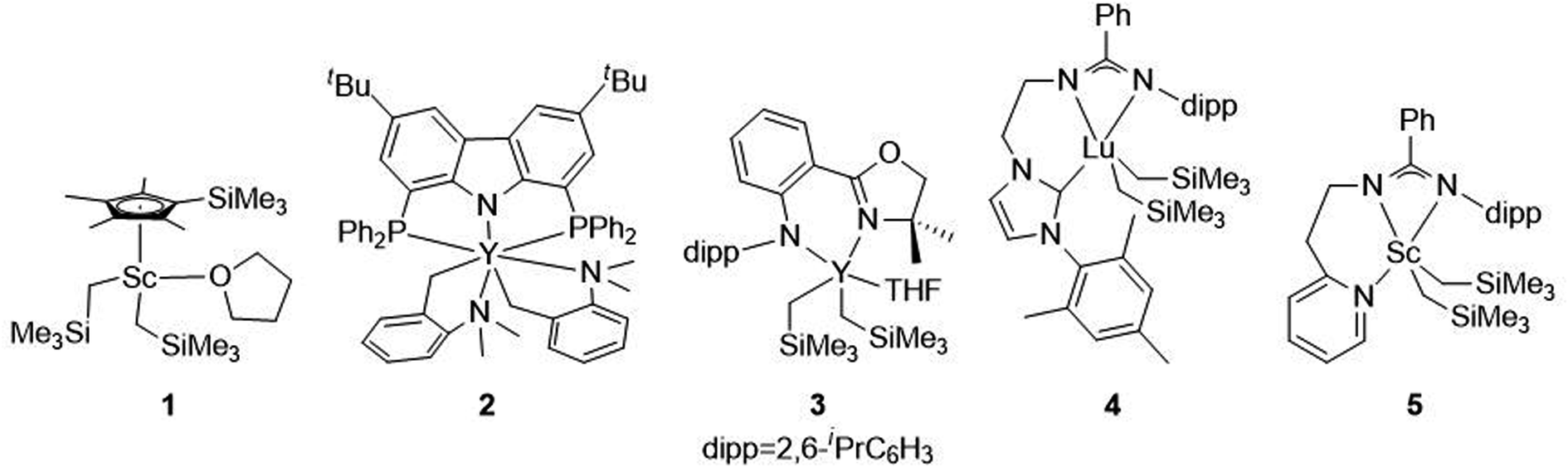 | ||
| Fig. 1 Five pre-catalysts reported for the block copolymerization of CL with S (1),14 IP (2,153,174,16519). | ||
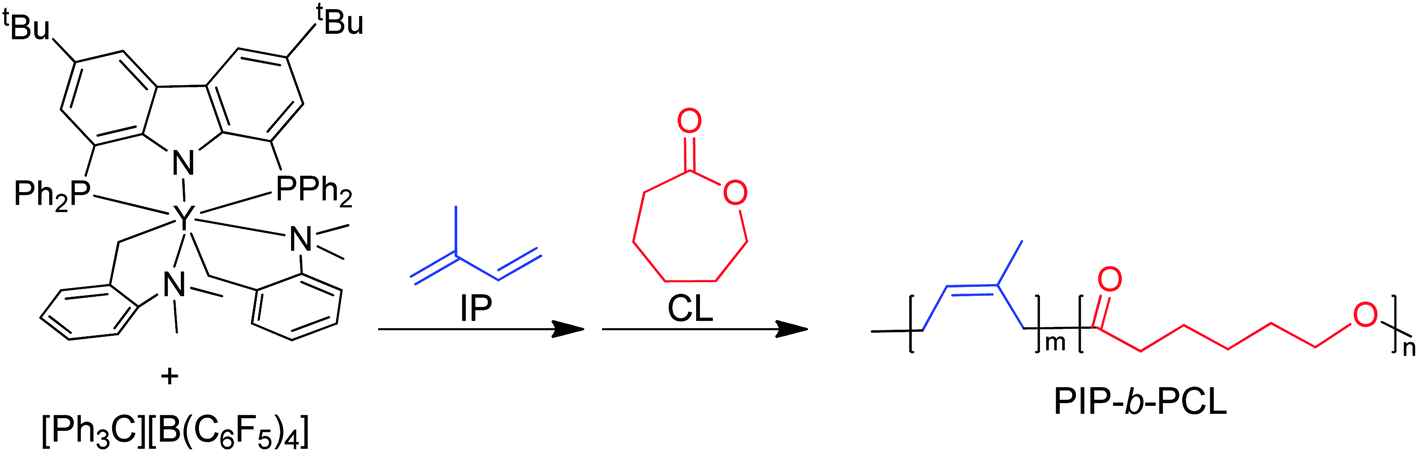 | ||
| Fig. 2 Representative diblock copolymerization of IP and CL with yttrium pre-catalyst 2.15 | ||
Since these previous reports have generally polymerized the monomers to full conversion, there have been no studies identifying how ligand structure impacts the polymerization rate and control for the block copolymerization of olefins/1,3-dienes and cyclic esters. The monomer scope has also been limited to one olefin, one 1,3-diene, and only one cyclic ester. Finally, no efforts have been directed towards addressing the recyclability of these block copolymers.
Herein, we report the use of β-diketiminate (BDI) supported yttrium complexes for the catalytic block copolymerization of several 1,3-dienes and cyclic esters, expanding the literature monomer scope with the addition of one bioderived 1,3-diene (β-myrcene (Myr)) and two cyclic esters (δ-valerolactone (VL), and ε-decalactone (DL)). Subtle changes in pendant neutral donors on the BDI ligands show a large impact on the polymerization of both monomer types. Additionally, we demonstrate that the poly(1,3-diene) block can be recycled to remake the same block copolymers.
Results and discussion
Catalyst selection
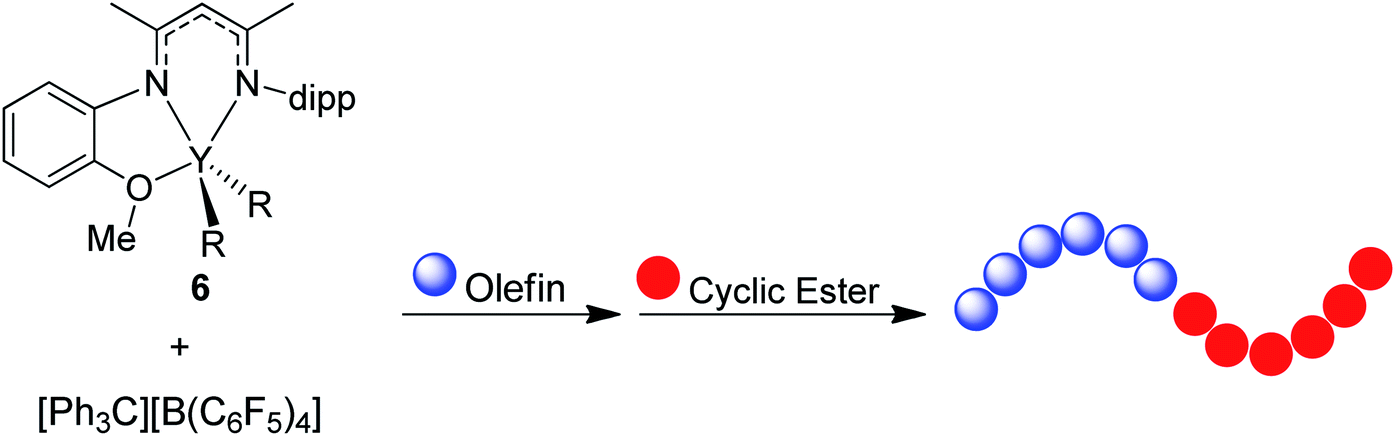
BDI ligands have been utilized to support rare earth metal catalysts for the polymerization of various polyesters.20 In particular, complexes 6–8 were found to be highly active for the ring-opening polymerization (ROP) of cyclic esters; however, reaction conditions did not reveal any trends on how the ligand structure impacts the rate of polymerization. These complexes were extended to the perfectly alternating copolymerization of epoxides and cyclic anhydrides, particularly butylene oxide with phthalic anhydride, where complex 6 was the slowest catalyst and 8 was the fastest.21 This suggested that strong field donors enhanced the rate of polymerization. We sought to use complexes 6–8 to identify if related trends could be discovered for the block copolymerization of IP and CL. All three complexes were synthesized according to literature methods by reacting the protonated BDI ligand with one equivalent of Y(CH2SiMe3)3(THF)2 (Fig. 3).22–24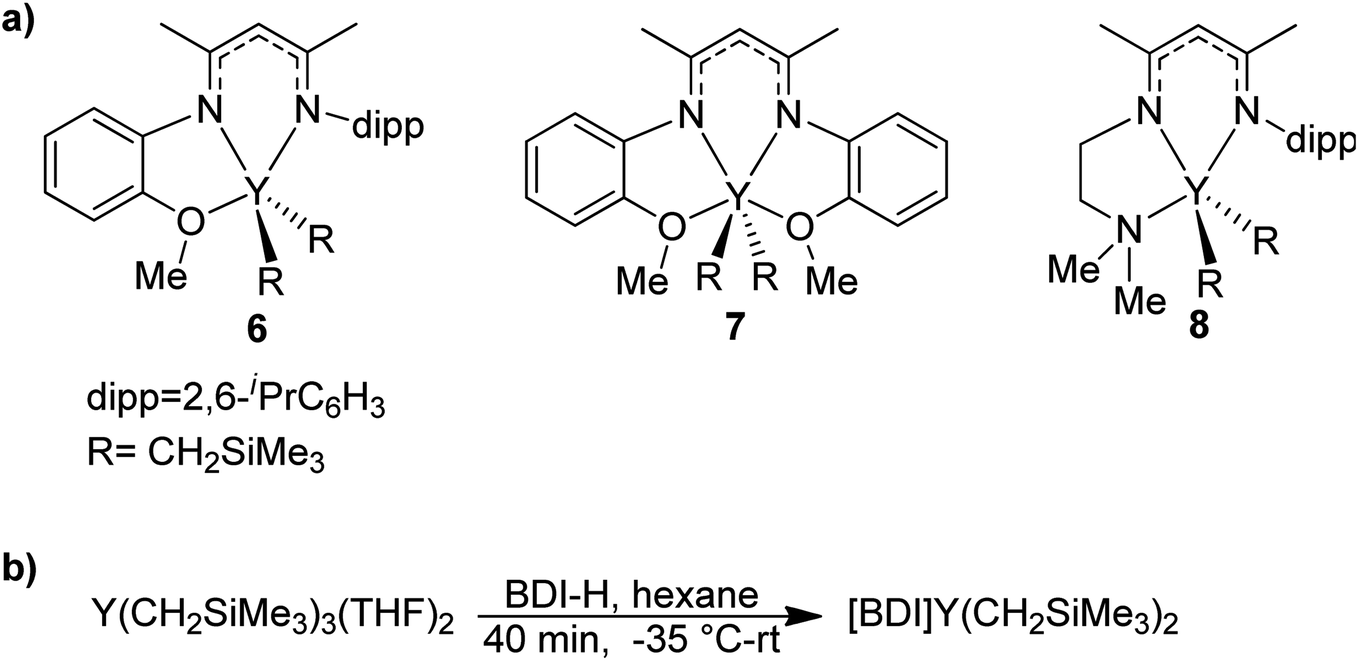 | ||
| Fig. 3 (a) Targeted yttrium BDI complexes for the block copolymerization of 1,3-dienes with polar monomers. (b) Reported synthetic pathway to targeted yttrium BDI complexes.22–24 | ||
Isoprene and caprolactone
Initially, 6–8 were tested as pre-catalysts for the homopolymerization of IP at room temperature for 12 hours, with 10 μmol (0.125 mol%) of catalyst and 10 μmol of [Ph3C][B(C6F5)4] (Table 1, entries 1–3). Complex 7 showed no polymerization of IP within 12 hours and was not pursued further (Table 1, entry 2). This inactivity is likely due to inhibiting steric or coordinative saturation from the two pendant donors. Complex 8 showed slow polymerization of IP, achieving only 85% conversion after 12 hours (Table 1, entry 3), and had a preferred selectivity (62%) for trans-1,4 polymerization. Surprisingly, complex 6 showed full conversion to PIP with 98% selectivity for 1,4 polymerization, with a slight preference for cis-1,4 over trans-1,4 selectivity (Table 1, entry 1). Even when reactions are run past full conversion, a dispersity below 1.10 is maintained, suggestive of excellent polymerization control (Table 1, entry 4).|
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Cat. | Monomer | Time | Conv.b (%) | M n (kDa) | Đ | MicrostructuredCis-1,4/Trans-1,4/3,4 | T g (°C) | T m (°C) | Efff (%) |
| a Conditions: [cat.], 10 μmol; [Ph3C][B(C6F5)4], 10 μmol; IP 0.80 M; CL 0.30 M; [IP]/Y = 800; [CL]/Y = 300; toluene, 10 mL; room temperature; entries 1, 4 and 9 are done in triplicate. b Determined by 1H NMR spectroscopy of crude reaction mixtures, comparing monomer peaks to polymer. c Determined by gel permeation chromatography (GPC) in THF using a Wyatt DAWN HELEOS II MALS detector. d 1,4 and 3,4 selectivity determined by 1H NMR. Cis-1,4 and trans-1,4 selectivity determined by 13C NMR. e Determined by low temperature differential scanning calorimetry (DSC). f Catalyst efficiency, calculated by Mn(theor.)/Mn(exp.). g Average of the triplicate runs. h Data for one of the individual runs. | ||||||||||
| 1 | 6 | IP | 12 h | >99 | 84(9) | 1.04(1) | 52/46/2g | −67h | — | 67(6) |
| 2 | 7 | IP | 12 h | 0 | 0 | 0 | — | — | — | — |
| 3 | 8 | IP | 12 h | 85 | 51 | 1.10 | 0/62/38 | −53 | — | 91 |
| 4 | 6 | IP | 24 h | >99 | 82(11) | 1.03(1) | 58/40/2g | −65h | — | 67(9) |
| 5 | 6 | IP | 30 min | 64 | 76 | 1.03 | 67/32/1 | −65 | — | 54 |
| 6 | 8 | IP | 30 min | 22 | 15 | 1.10 | 8/65/27 | −53 | — | 80 |
| 7 | 6 | CL | 10 min | 89 | 40 | 1.13 | — | — | 55 | 75 |
| 8 | 8 | CL | 10 min | >99 | 37 | 1.34 | — | — | 55 | 93 |
| 9 | 6 | CL | 2 h | >99 | 66(5) | 1.2(1) | — | — | 55h | 52(3) |
Contrary to the perfectly alternating copolymerization of epoxides and cyclic anhydrides,21 the superior rate of complex 6 suggests that a weak field donor leads to the fastest polymerization of IP. We attribute this to the more electron-deficient yttrium center in complex 6 compared to complex 8, which would likely lead to better activation of IP towards polymerization. The change in selectivity is interesting, as the slightly bulkier –NMe2 donor in 8 shows a higher preference for trans-1,4 and 3,4-polymerization, while the –OMe donor in 6 shows almost exclusive preference for 1,4-polymerization. This is consistent with what is found in the literature where a bulkier ligand leads to increased 3,4-selectivity.16,25,26 While it is also true that bulkier ligands have been shown to promote cis-1,4 over trans-1,4 polymerization, we speculate 8's selectivity for trans-1,4 might arise from the decreased Lewis acidity of its yttrium center.27
Shortening the reaction time revealed that 64% conversion of IP is already achieved within 30 minutes for complex 6, while 8 shows only 22% conversion within this timeframe (Table 1, entries 5 and 6, respectively). Interestingly, both shortened reactions show more cis-1,4 selectivity than their respective 12 hours reactions. Previous reports have indicated that higher concentrations of IP can lead to a preference for cis-1,4, which explains the higher cis-1,4 selectivity at shorter reaction times.28 It is interesting that the longer reaction time with 8 (Table 1, entry 3) has no presence of cis-1,4 selectivity, while the shorter time has 8% cis-1,4 selectivity (Table 1, entry 6). Since cis-1,4 selectivity seems to drop with IP concentration, this could be due to the cis-1,4 getting buried in the baseline of the NMR spectrum for the long reaction times. Alternatively, this could be due to variability in selectivity between separate reactions.
Complexes 6 and 8, activated with [Ph3C][B(C6F5)4], were also used for the homopolymerization of CL at room temperature (Table 1, entries 7 and 8, respectively). While complex 8 showed faster polymerization, the higher dispersity of the resulting polymer (1.34) suggested either lack of polymerization control or the presence of transesterification reactions when polymerization is complete. Activated complex 6 showed high conversion (89%) of CL after just 10 min while maintaining an excellent dispersity (1.13). Leaving the reaction well past full conversion showed no evidence of transesterification, with dispersity remaining low (Table 1, entry 9). It is worth noting that the activated complex 6 also showed better molecular weight control and/or less transesterification than the non-activated complex 6 (Table S2,† entry 5). Considering the necessity of a cationic catalyst species for olefin or 1,3-diene polymerization, this result presents the activated complex as better suited for further study. While several reports have demonstrated the efficacy of neutral non-activated catalysts with cyclic ester polymerizations, using the activated cationic complex would provide a better representative understanding of the transition between the IP and CL polymerizations.29,30
The results found herein suggest that although 8 was the fastest catalyst for CL polymerization, its rate for IP polymerization was significantly lower in comparison to 6. Since the target block copolymerization requires an efficient catalyst that can provide control for both olefin or 1,3-diene and cyclic ester monomers, 6 was used as the ideal candidate for further studies in synthesizing block copolymers.
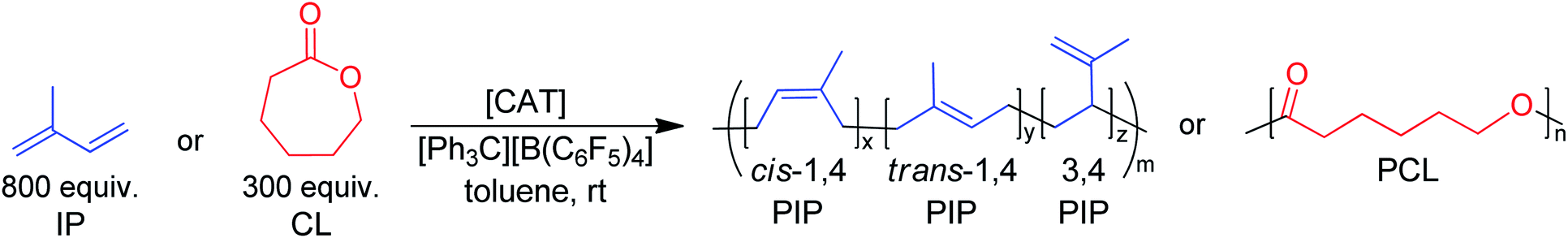
PIP:PCL block copolymers
Stepwise block copolymerization of IP and CL was conducted in triplicate using organoborate activated complex 6 (Table 2). Longer reaction times (12 h and 2 h, respectively) were chosen to identify any side reactions present after full conversion of each monomer. Different ratios of IP and CL could be polymerized effectively and reproducibly, in which the polymerization control of both steps was maintained well past full conversion, as evidenced by the low dispersities of the resulting block copolymers. Selectivity for cis-1,4 PIP was highest for the 800:300 IP:CL combination (Table 2, entry 1). As the IP amount was lowered, the ratio of trans-1,4 to cis-1,4 selectivity increased, as was discussed previously in IP homopolymerizations. For all feed ratio combinations, narrow dispersities were seen. The molecular weight of the 800:300 IP:CL combination (Table 2, entry 1) indicates catalyst activation to be much higher (81–88%) than that of other catalysts in the literature for the block copolymerization of both monomers, which has been reported to be between 24 and 56%.19
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Feed ratio (IP:CL) |
Conv.b (%) | M n (kDa) | Đ | Poly(1,3-diene):polyesterd,e (%) |
Microstructuree,fCis-1,4/Trans-1,4/3,4 | T g , (°C) | T m , (°C) |
| a Conditions: 6, 10 μmol; [Ph3C][B(C6F5)4], 10 μmol; toluene, 10 mL; room temperature; IP 12 h; CL 2 h; all entries are done in triplicate. b Determined by 1H NMR spectroscopy of crude reaction mixtures, comparing monomer peaks to polymer. c Determined by gel permeation chromatography (GPC) in THF using a Wyatt DAWN HELEOS II MALS detector. d Determined by 1H NMR spectroscopy of the isolated polymer. e Average of the triplicate runs. f 1,4 and 3,4 selectivity determined by 1H NMR. Cis-1,4 and trans-1,4 selectivity determined by 13C NMR. g Determined by low temperature differential scanning calorimetry (DSC). h Data for one of the individual runs. | ||||||||
| 1 | 800:300 |
>99 | 110(21) | 1.10(2) | 68:32 |
51/48/1 | −66 | 51 |
| 2 | 550:550 |
>99 | 90.9(17) | 1.12(1) | 47:53 |
44/54/2 | −67 | 55 |
| 3 | 300:800 |
>99 | 101(20) | 1.23(3) | 26:74 |
28/70/2 | −64 | 54 |
Catalyst efficiency
It was identified that the molecular weights of most polymerizations with activated 6 were often inconsistent and higher than expected. While the literature often describes this as catalyst efficiency, i.e., the degree of catalyst activation by the organoborate, we aimed to probe this further.It is presumed that the reaction of complex 6 with one equivalent of [Ph3C][B(C6F5)4] leads to the abstraction of one alkyl to form 6a, which serves as the active catalyst (Fig. 4). Overactivation of 6 with two equivalents of [Ph3C][B(C6F5)4] would lead to the abstraction of both alkyls, generating 6b, a species with no bound initiators which is likely inactive for polymerization of IP. The higher-than-expected molecular weights would indicate that there is less active catalyst in solution than anticipated, implying that there is incomplete activation to 6a.
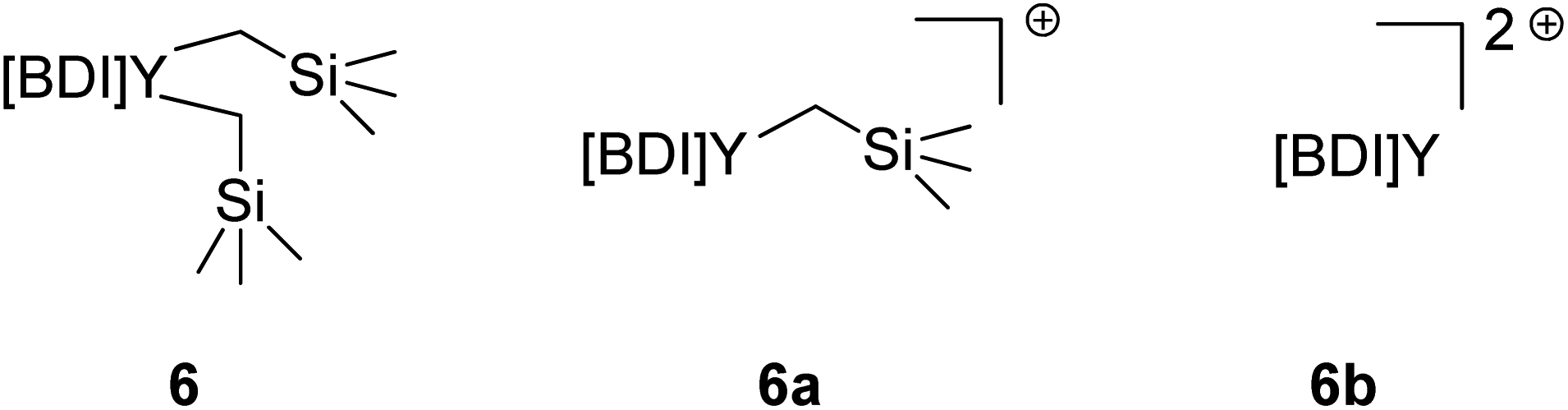 | ||
| Fig. 4 Comparison of activated complex 6 where BDI = {MeC(NDIPP)CHC(Me)[N(2-OMeC6H4)]}Y(CH2SiMe3)2 (DIPP = 2,6-iPr2C6H3) and counter anions are [B(C6F5)4]. | ||
We aimed to execute control reactions in triplicate to allow us to better understand this activation (Table 3). First, the polymerization activity of unactivated 6 (with no [Ph3C][B(C6F5)4]) and 6b (with 3 equivalents of [Ph3C][B(C6F5)4]) were tested. Neither condition showed any polymerization of IP, further validating the active catalyst as 6a.
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Activator equiv. | IP addition | Monomer (M1:M2) |
Conv.b (%) | M n (kDa) | Đ | MicrostructuredCis-1,4/Trans-1,4/3,4 | Effe (%) |
| a Conditions: 6, 10 μmol; [Ph3C][B(C6F5)4], 5–30 μmol; [IP]/6 = 800; [CL]/6 = 300; toluene, 10 mL; room temperature; IP 12 h; CL 2 h; all entries are done in triplicate; at full conversion, PIP:PCL is 800:300 for entries 3 and 4. b Determined by 1H NMR spectroscopy of crude reaction mixtures, comparing monomer peaks to polymer. c Determined by gel permeation chromatography (GPC) in THF using a Wyatt DAWN HELEOS II MALS detector. d 1,4 and 3,4 selectivity determined by 1H NMR. Cis-1,4 and trans-1,4 selectivity determined by 13C NMR. e Catalyst efficiency, calculated by Mn(theor.)/Mn(exp.). | ||||||||
| 1 | 1 | 0 | IP | >99 | 83.5(4) | 1.04(2) | 57/40/3 | 65(3) |
| 2 | 1 | 30 min | IP | >99 | 82.6(3) | 1.04(2) | 58/40/2 | 66(2) |
| 3 | 0.5 | 10 min | IP:CL | >99 | 188(30) | 1.41(3) | 43/55/2 | 48(8) |
| 4 | 1.5 | 10 min | IP:CL | >99 | 93.1(13) | 1.15(1) | 57/41/2 | 96(14) |
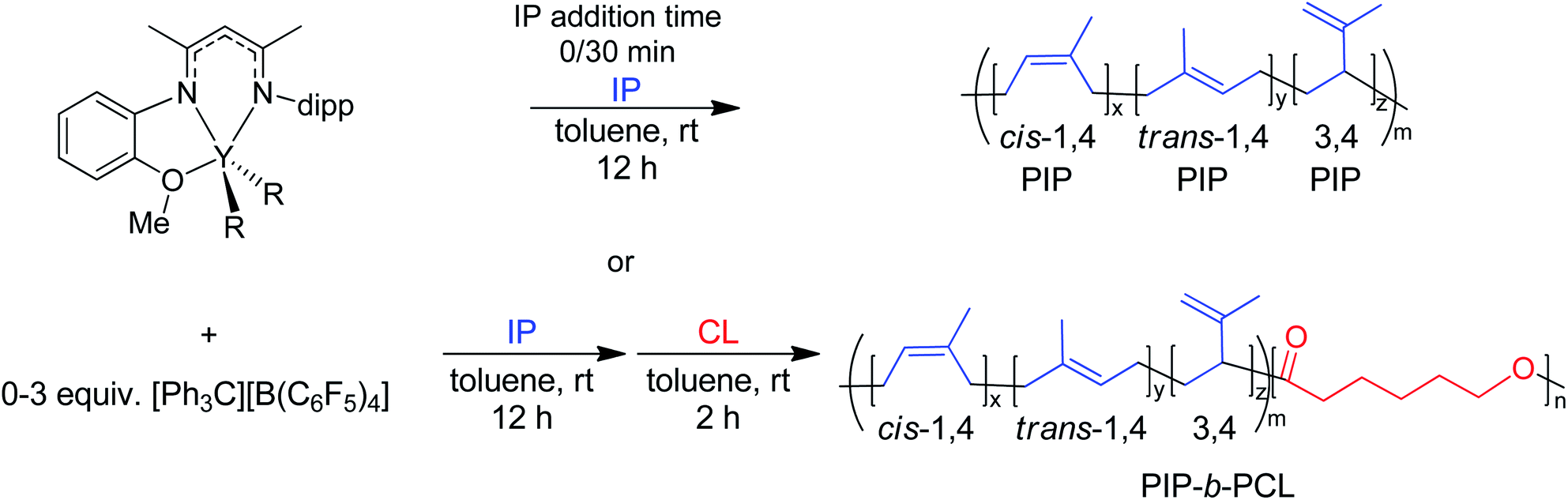
To test whether catalyst activation is being disrupted by preemptive monomer addition, we adjusted the time between the addition of [Ph3C][B(C6F5)4] and monomer. In prior experiments, 6 and the [Ph3C][B(C6F5)4] are mixed for 10 minutes before exposure to monomers. A reaction in which 6 and the [Ph3C][B(C6F5)4] are mixed quickly, followed by immediate IP addition gave a similar molecular weight (Mn) of 84 kDa to that shown in Table 1, entry 1 (Table 3, entry 1). Additionally, mixing 6 and the [Ph3C][B(C6F5)4] for 30 minutes prior to IP addition also led to indistinguishable Mn of 83 kDa (Table 3, entry 2). These studies indicate that catalyst activation is not being interrupted by monomer addition. 1H NMR spectra of 6 activated with one equivalent of [Ph3C][B(C6F5)4] maintains a clean ligand environment, with no evidence of protonated ligand. This result is in contrast to a report by Li and coworkers, where a BDIYCl2(THF)2 complex was activated with [PhNMe2H][B(C6F5)4].31 This reaction led to the protonation of the ligand and the formation of a proposed ion pair [YCl2(THF)2][B(C6F5)4]. We rationalize the absence of an analogous protonation reaction here, as our activating agent does not have an available proton, and the ligand has an added chelate that would likely make dissociation of the ligand more difficult.
A19F NMR spectrum (Fig. S1†) of [Ph3C][B(C6F5)4] revealed the presence of a minor impurity (98% purity). This suggests the amount of [Ph3C][B(C6F5)4] added would need to be tuned to maximize the catalyst efficiency. Thus, the addition of 1.5 equivalents of [Ph3C][B(C6F5)4], relative to 6, showed a drop in molecular weight of the 800:300 PIP:PCL block copolymers to 93 kDa (Table 3, entry 4), in good agreement with the theoretical Mn of 89 kDa, thereby increasing the catalyst efficiency to 96%. In contrast, the use of only 0.5 equivalents of [Ph3C][B(C6F5)4], relative to 6, showed a large increase in the block copolymer molecular weight to 188 kDa (Table 3, entry 3) with a much lower catalyst efficiency of 48%. Additionally, while DOSY NMR experiments do not identify a mixture of two polymers, GPC analysis of the resulting polymer showed a slightly bimodal appearance. With only 0.5 equivalents of [Ph3C][B(C6F5)4], we expect a mixture of 6 and 6a. While only 6 does not initiate IP, it can initiate CL polymerization, suggesting a possible small impurity of PCL homopolymer in the isolated sample. This highlights the importance of high catalyst efficiencies. Notably, molecular weight control was maintained for both reactions, with dispersities remaining below 1.4. Higher ratios of [Ph3C][B(C6F5)4] slightly increased selectivity for cis-1,4 vs. trans-1,4 IP polymerization, as would be expected for higher active catalyst concentrations.29 Since the reactions were done in triplicate, we identified that activation of the catalyst was variable under the same conditions. This suggests that individual runs may not be entirely representative of the average efficiency for a particular condition.
Monomer scope
After complex 6 was identified as an active and controlled pre-catalyst for the block copolymerization of IP and CL, extension to other monomers of interest was pursued. First, 6 (activated with one equivalent of [Ph3C][B(C6F5)4]) was tested for the homopolymerization of several olefin, 1,3-diene, and cyclic ester monomers (Table 4). Since 8 showed higher activity for CL polymerization, it was also evaluated for these different monomers. However, there were no cases in which 8 outperformed 6 (Table S1†).|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Monomer | Time | Temp. (°C) | Conv.b (%) | M n (kDa) | Đ | MicrostructuredCis-1,4/Trans-1,4/3,4 | T g (°C) | T m (°C) |
| a Conditions: 6, 10 μmol; [Ph3C][B(C6F5)4], 10 μmol; toluene, 10 mL; [olefin or 1,3-diene]/6 = 800; [cyclic ester]/6 = 300. b Determined by 1H NMR spectroscopy of crude reaction mixtures, comparing monomer peaks to polymer. c Determined by gel permeation chromatography (GPC) in THF using a Wyatt DAWN HELEOS II MALS detector. d 1,4 and 3,4 selectivity determined by 1H NMR. Cis-1,4 and trans-1,4 selectivity determined by 13C NMR. e Determined by low temperature differential scanning calorimetry (DSC). | |||||||||
| 1 | Myr | 30 min | rt | 44 | 121 | 1.13 | 97/2/1 | −64 | — |
| 2 | Myr | 90 min | rt | 72 | 293 | 1.15 | 97/2/1 | −64 | — |
| 3 | Myr | 3 h | rt | >99 | 388 | 1.59 | 85/14/1 | −64 | — |
| 4 | S | 30 min | rt | 18 | 9.4 | 2.03 | — | 96 | — |
| 5 | S | 20 h | rt | 23 | 21.2 | 2.17 | — | 99 | — |
| 6 | VL | 10 min | rt | 81 | 26.2 | 1.24 | — | — | 53 |
| 7 | DL | 6 h | 60 | 84 | 24.9 | 1.14 | — | −50 | — |
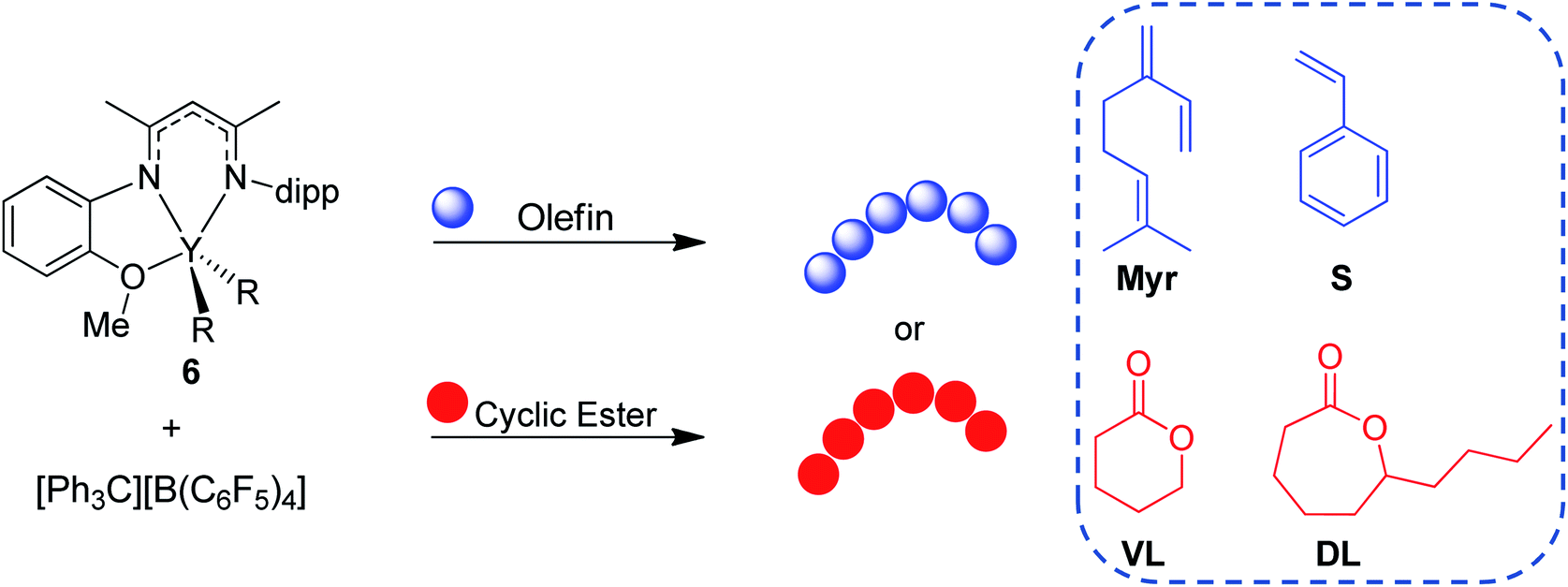
Activated 6 was found to be active for the homopolymerization of Myr at room temperature, in which full conversion of 800 equivalents of monomer could be achieved within 3 hours (Table 4, entries 1–3). While dispersity remained low at conversions of 44% (1.13) and 72% (1.15), upon reaching full conversion the dispersity broadened slightly (1.59). Complex 6 showed excellent selectivity towards cis-1,4 over trans-1,4 or 3,4 Myr polymerization.32 Interestingly, Myr polymerization with complex 8 (Table S1,† entry 1) demonstrated a preference for trans-1,4 selectivity resembling the selectivity found for its polymerization of IP. Additionally, activated 6 was able to polymerize S (Table 4, entry 4) albeit at slow rates. Even at a long reaction time of 20 hours (Table 4, entry 5) only 23% conversion of S was reached. 13C NMR analysis (Fig. S65†) showed only atactic PS was synthesized using 6.33 Activated 6 was also able to polymerize cyclic esters that are often difficult to ring open, such as VL and DL. In particular, high conversion of VL (81%) could be achieved within 10 minutes at room temperature, with a low dispersity of 1.24 (Table 4, entry 6). Polymerization of DL to high conversions could also be achieved with low dispersities, but a higher reaction temperature (60 °C) and longer reaction times (6 h) were needed (Table 4, entry 7). It is worth noting that complex 8 also demonstrated polymerization of VL and DL with comparable rates and marginally broader dispersities (Table S1†, entries 3 and 4, respectively).
Extensions of these studies to the synthesis of block copolymers was conducted for all monomers except S due to the incomplete conversion with 6. Combinations of 1,3-dienes (IP, Myr) with cyclic esters (CL, VL, and DL) has led to five more block copolymerization morphologies, all of which are new polymers never reported in prior literature (Table 5). A consistent 800:300 1,3-diene:cyclic ester ratio was used for all combinations. Block copolymerization of IP with either VL or DL (Table 5, entries 1 and 2, respectively) reached full conversion for both monomers, producing high molecular weight polymers with narrow dispersities comparable to those of their respective homopolymers. IP selectivity was akin to the IP:CL combination (Table 2, entry 1). Myr block copolymerization was next explored with CL, VL, and, DL (Table 5, entries 3, 4, and 5, respectively). In all three cases, full conversion of Myr was achieved, while incomplete conversion of the cyclic ester was observed. Incomplete enchainment of the cyclic ester could be a result of increased viscosity in the reaction medium or due to the bulky high molecular weight polymyrcene (PMyr) blocking access to the active metal center. Additionally, high dispersities (2.07–2.41) and low solubility were seen, indicating the presence of side reactions, such as transesterification and/or cross-linking.34 The lower conversions and broader dispersities of the Myr copolymers highlight the need for a better understanding of catalyst design principles to encourage efficient and controlled polymerization of a range of olefin/1,3-dienes and cyclic ester monomers, as well as seamless transfer from one monomer to the next.
|
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Monomer (M1:M2) |
Time (h) | Temp. (°C) | Conv.b (%) | M n (kDa) | Đ | Poly(1,3-diene):polyesterd (%) |
MicrostructureeCis-1,4/Trans-1,4/3,4 | T g (°C) (1st/2nd) | T m (°C) |
| a Conditions: 6,10 μmol; [Ph3C][B(C6F5)4], 10 μmol; toluene, 10 mL; [1,3-diene]/6 = 800; [cyclic ester]/6 = 300. b Determined by 1H NMR spectroscopy of crude reaction mixtures, comparing monomer peaks to polymer. c Determined by gel permeation chromatography (GPC) in THF using a Wyatt DAWN HELEOS II MALS detector. d Determined by 1H NMR spectroscopy of the isolated polymer. e 1,4 and 3,4 selectivity determined by 1H NMR. Cis-1,4 and trans-1,4 selectivity determined by 13C NMR. f Determined by low temperature differential scanning calorimetry (DSC). | ||||||||||
| 1 | IP:VL | 12:2 |
rt:rt | >99: >99 | 141 | 1.04 | 66:34 |
58/40/2 | −64/– | 50 |
| 2 | IP:DL | 12:12 |
rt:60 | >99: >99 | 75.1 | 1.27 | 60:30 |
46/52/2 | −65/−48 | — |
| 3 | Myr:CL | 3:2 |
rt:rt | >99:58 | 482 | 2.41 | 89:11 |
93/5/2 | −69/– | 51 |
| 4 | Myr:VL | 3:2 |
rt:rt | >99:67 | 277 | 2.27 | 43:57 |
90/8/2 | −63/– | 48 |
| 5 | Myr:DL | 3:12 |
rt:60 | >99:56 | 387 | 2.07 | 88:12 |
87/12/1 | −64/−53 | — |
These results show the versatility of pre-catalyst 6 and introduce two new cyclic esters (VL and DL) and a bio-derived 1,3-diene (Myr) to the literature monomer scope for this block copolymerization. Additionally, these polymerizations represent the first examples of block copolymerization of Myr with cyclic esters. With new polymers now available, the testing and further understanding of polymer physical properties due to the variations of monomers and ratios of each block are currently underway.
Recyclability
One of the main motivations for the block copolymerization of 1,3-dienes with cyclic esters is the ability to recycle the poly(1,3-diene) block. Therefore, it was of interest to show proof of concept that the polyester block from poly(isoprene-block-caprolactone) (PIP-b-PCL) copolymers could be selectively degraded, leaving the PIP block intact to be used again to reform the desired block copolymer (Fig. 5). Towards this end, a 50:50 PIP:PCL block copolymer was synthesized in a stepwise fashion using activated 6. This reaction was done using 20 μmol of catalyst, in which half of the reaction mixture after the first step was used to synthesize the PIP-b-PCL copolymer with 50 equivalents of CL added, while the other half was used to characterize the original PIP block (Fig. 6). The synthesized 50:50 PIP-b-PCL were fully characterized by NMR and IR spectroscopy, GPC, and TGA/DSC (Figs. S42, S84, S147, S215, S282, and S321,† respectively). The molecular weight of the block copolymer was 55 kDa, which was much higher than expected with the catalyst efficiency only being 17%. We anticipated this was due to the low concentration of IP in the solution since the same volume of solvent was used for this reaction as for all other reactions. A reaction done at a much higher concentration lowered the molecular weight of the PIP-b-PCL block copolymer to 21 kDa, improving the overall catalyst efficiency (43%) and getting much closer to the expected molecular weight for the ratio of monomers. Selective degradation of the CL block in the 50:50 PIP-b-PCL copolymer was achieved through alkaline hydrolysis. The copolymer was solubilized by the addition of minimal THF and heated in a 2 M aqueous NaOH solution at 100 °C.35 The degraded polymer was characterized by NMR spectroscopy, GPC, and FT-IR and compared to the original PIP block, which showed nearly identical molecular weights and dispersities, as well as similar NMR, and FT-IR spectral features (Fig. 6, S44 and S149,† respectively).
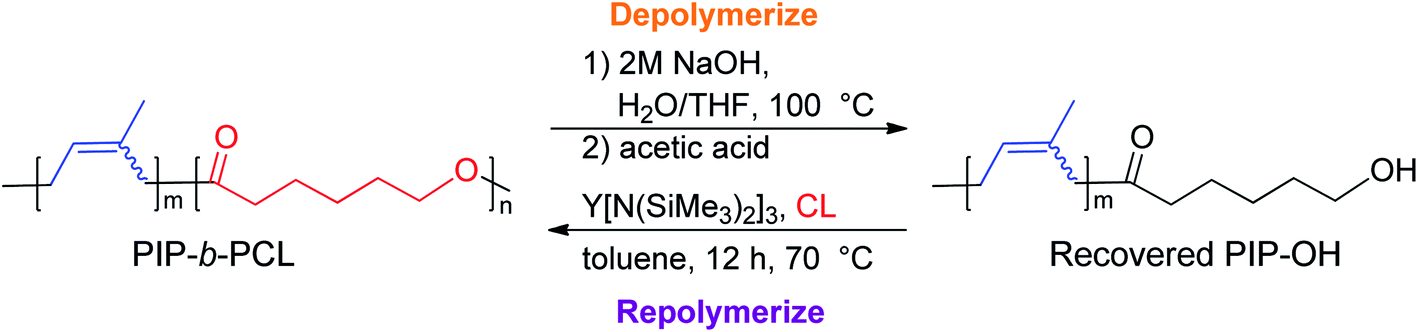 | ||
| Fig. 5 Proposed selective hydrolysis of CL block and repolymerization to IP:CL diblock copolymer. | ||
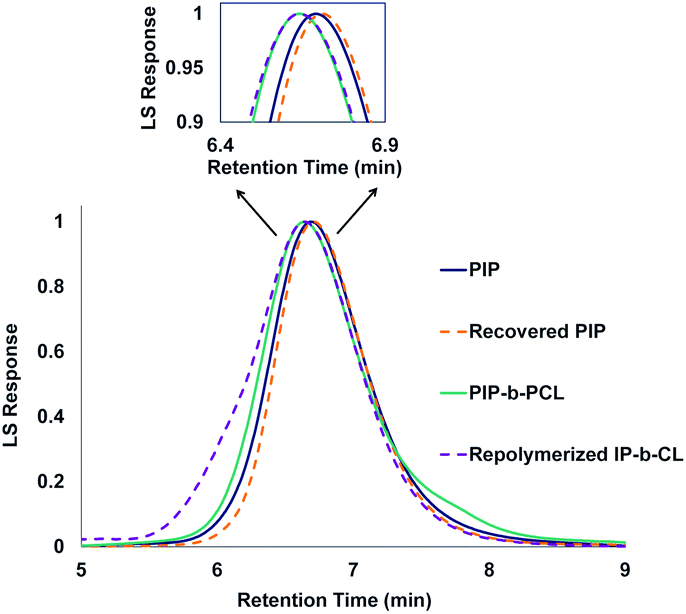 | ||
| Fig. 6 GPC traces of PIP (Mn = 42 kDa, Đ = 1.15), recovered PIP (Mn = 43 kDa, Đ = 1.15), PIP-b-PCL (Mn = 55 kDa, Đ = 1.09), and repolymerized PIP-b-PCL (Mn = 56 kDa, Đ = 1.16). | ||
Yttrium tris[N,N-bis(trimethylsilyl)amide] (Y[N(SiMe3)2]3) was chosen as the repolymerization catalyst as it is commercially available. It is also well known to readily exchange with an alcohol to form an yttrium tris-alkoxide species that is active in the polymerization of CL.36,37 It was reasoned that the recovered 50 PIP block would terminate with an alcohol if complete hydrolysis of the ester bonds was achieved, and could exchange with Y[N(SiMe3)2]3 to form a macroinitiator that could polymerize CL. Indeed, NMR and FT-IR spectroscopy of recovered 50 PIP confirmed the presence of a hydroxyl functional group (Figs. S43 and S148,† respectively). Thus, the recovered 50 PIP block (2.5 equiv.) was combined with Y[N(SiMe3)2]3 (1 equiv.) and 1H NMR monitoring revealed the growth of a hexamethyldisilazane (HMDS) peak (Fig. S2†) consistent with an alkoxide exchange. CL (125 equiv.) was subsequently added, and complete consumption of CL was achieved within 12 hours. The dispersity of the repolymerized PIP-b-PCL was comparable to the virgin PIP-b-PCL (1.09 and 1.16, respectively), while their respective Mn values were essentially identical. These findings demonstrate the potential of 1,3-diene and cyclic ester block copolymers to be efficiently recycled, warranting further studies into diversifying the polymer structure and exploring their future applications.
Conclusions
Herein, we identified cationic alkyl yttrium β-diketiminate complexes as active catalysts for the homopolymerization and block copolymerization of two 1,3-dienes and three cyclic esters. This study demonstrated that the number of pendant donors on the ancillary ligand had a dramatic impact on the target polymerization. Ancillary BDI ligands bearing two donors shut down 1,3-diene polymerization, while ligands with a single pendant donor could be tuned to affect the selectivity and rate of 1,3-diene and cyclic ester polymerization. Overall, the rate of 1,3-diene polymerization was faster with a weaker field donor. The –OMe weak field donor promoted 1,4 selectivity over 3,4 selectivity in IP polymerization with a slight preference for cis-1,4 over trans-1,4, while the strong field –NMe2 donor produced 3,4 and trans–1,4 selectivity. Comparable with results observed with IP, the catalyst with the weak field donor favored 1,4 over 3,4 Myr polymerization, with a strong preference for cis-1,4 over trans-1,4 selectivity. High molecular weight polymers could be achieved with moderate dispersities of 1.13–1.59. Overall, for cyclic ester polymerization, both catalysts with one neutral donor demonstrated fast polymerization and narrow dispersities. One-pot block copolymerizations led to a total of 6 diblock morphologies, 5 of which are entirely new materials. Rigorous inquiry into the thermal and mechanical properties of these new materials is currently underway.Investigating the activation of the pre-catalyst demonstrated that monomer addition did not inhibit the formation of the active catalyst. Also, super stoichiometric ratios of [Ph3C][B(C6F5)4] to pre-catalyst (1.5 equiv.) led to experimental molecular weights in better agreement with theoretical molecular weights, suggesting that stoichiometric addition of [Ph3C][B(C6F5)4] to pre-catalyst is insufficient for complete catalyst activation. On the other hand, a vast excess of [Ph3C][B(C6F5)4] (3 equiv.) completely shuts down catalyst activity. For PIP-b-PCL copolymers, selective degradation of the PCL block can be achieved through simple alkaline catalyzed hydrolysis of ester bonds recovering the PIP block with identical molecular weight and dispersities to virgin PIP. Subsequent repolymerization with CL using a commercially available yttrium catalyst reproduced the PIP-b-PCL copolymers with high molecular weights and narrow dispersities, both of which are analogous to those of virgin block copolymers. For the first time, this study introduces a possible recycling scheme for 1,3-diene and cyclic ester block copolymers.
Data availability
All relevant datasets supporting this article including general considerations for chemicals, metal complexes, monomers and solvents, polymerization conditions and results, characterization data (including 1H and 13C NMR spectra, DOSY NMR specta, FT-IR spectra, GPC spectra, TGA curves, and DSC curves) are available in the ESI.†Author contributions
M. E. F. supervised this project and contributed to the manuscript; S. C. K.-O. performed all experimental studies and data analysis and wrote the manuscript. Y. M. was the first to synthesize the pre-catalysts in our group. T. J. B. aided in the ligand synthesis. All authors approved the final version of this manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding for this project was provided by the American Chemical Society Petroleum Research Fund (PRF# 61544-DNI3) and University of Southern California (USC) start-up funds. We would also like to thank Chris Kosloski-Oh for his help with the Table of Contents graphic. Thank you, also, to Lanja Karadaghi for assistance with a few TGA samples while our instrument was down.References
- R. Shanks and I. Kong, Thermoplastic Elastomers, ed. A. Z. El-Sonbati, IntechOpen, London, 2012, ch. 8, pp. 136–154 Search PubMed.
- H. D. Tabba, Y. M. Hijji and A. S. Abu-Surrah, in Polyolefin Compounds and Materials: Fundamentals and Industrial Applications, ed. M. Al-Ali AlMa'adeed and I. Krupa, Cham Springer, Switzerland, 1, 2016, vol. 3, pp. 51–77 Search PubMed.
- A. R. Padwa, Prog. Polym. Sci., 1989, 14, 811–833 CrossRef CAS.
- C. Moineau, M. Minet, P. Teyssié and R. Jérôme, Macromolecules, 1999, 32, 8277–8282 CrossRef CAS.
- J. D. Tong and R. Jerôme, Polymer, 2000, 41, 2499–2510 CrossRef CAS.
- D. J. Haloi, S. Ata, N. K. Singha, D. Jehnichen and B. Voit, ACS Appl. Mater. Interfaces, 2012, 4, 4200–4207 CrossRef CAS PubMed.
- B. Dufour, K. Koynov, T. Pakula and K. Matyjaszewski, Macromol. Chem. Phys., 2008, 209, 1686–1693 CrossRef CAS.
- S. Oshita, B. K. Chapman and K. Hirata, Acrylic Block Copolymer for Adhesive Application, in PSTC Tape Summit 2012, Boston, 2012 Search PubMed.
- K. Hamada, K. Ishiura, T. Takahashi, S. Yaginuma, M. Akai, T. Ono and K. Shachi, US Pat., 6767976B2, 2004.
- H. Wang, G. Ding, X. Li, H. She, Y. Zhu and Y. Li, Sustainable Energy Fuels, 2021, 5, 930–934 RSC.
- Y. Huang, R. Chang, L. Han, G. Shan, Y. Bao and P. Pan, ACS Sustainable Chem. Eng., 2016, 4, 121–128 CrossRef CAS.
- A. Keyes, H. E. Basbug Alhan, E. Ordonez, U. Ha, D. B. Beezer, H. Dau, Y.-S. Liu, E. Tsogtgerel, G. R. Jones and E. Harth, Angew. Chem., Int. Ed., 2019, 58, 12370–12391 CrossRef CAS PubMed.
- A. A. Trifonov and D. M. Lyubov, Coord. Chem. Rev., 2017, 340, 10–61 CrossRef CAS.
- L. Pan, K. Zhang, M. Nishiura and Z. Hou, Macromolecules, 2010, 43, 9591–9593 CrossRef CAS.
- L. Wang, D. Cui, Z. Hou, W. Li and Y. Li, Organometallics, 2011, 30, 760–767 CrossRef CAS.
- C. Yao, D. Liu, P. Li, C. Wu, S. Li, B. Liu and D. Cui, Organometallics, 2014, 33, 684–691 CrossRef CAS.
- W. Li, X. Jiang, Y.-M. So, G. He and Y. Pan, New J. Chem., 2020, 44, 121–128 RSC.
- Y. Wang, C. Zhou and J. Cheng, Macromolecules, 2020, 53, 3332–3338 CrossRef CAS.
- J. Wang, S. Xu, X. Hu, Y. Huo and X. Shi, Organometallics, 2022, 41, 115–123 CrossRef CAS.
- D. M. Lyubov, A. O. Tolpygin and A. A. Trifonov, Coord. Chem. Rev., 2019, 392, 83–145 CrossRef CAS.
- Y. Manjarrez, M. D. C. L. Cheng-Tan and M. E. Fieser, Inorg. Chem., 2022, 61, 7088–7094 CrossRef CAS PubMed.
- X. Shang, X. Liu and D. Cui, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 5662–5672 CrossRef CAS.
- X. Xu, X. Xu, Y. Chen and J. Sun, Organometallics, 2008, 27, 758–763 CrossRef CAS.
- X. Liu, X. Shang, T. Tang, N. Hu, F. Pei, D. Cui, X. Chen and X. Jing, Organometallics, 2007, 26, 2747–2757 CrossRef CAS.
- G. Du, Y. Wei, L. Ai, Y. Chen, Q. Xu, X. Liu, S. Zhang, Z. Hou and X. Li, Organometallics, 2011, 30, 160–170 CrossRef CAS.
- B. Liu, L. Li, G. Sun, J. Liu, M. Wang, S. Li and D. Cui, Macromolecules, 2014, 47, 4971–4978 CrossRef CAS.
- X. Kang, Y. Luo and Z. Hou, in Computational Quantum Chemistry: Insights Into Polymerization Reactions, ed. M. Soroush, Elsevier, 2019, vol. 10, pp. 327–356 Search PubMed.
- Z. Huang, S. Wang, X. Zhu, Y. Wei, Q. Yuan, S. Zhou, X. Mu and H. Wang, Chin. J. Chem., 2021, 39, 3360–3368 CrossRef CAS.
- J.-F. Carpentier, Organometallics, 2015, 34, 4175–4189 CrossRef CAS.
- I. Cota, Phys. Sci. Rev., 2017, 2, 20160129 Search PubMed.
- D. Li, S. Li, D. Cui and X. Zhang, Organometallics, 2010, 29, 2186–2193 CrossRef CAS.
- B. Liu, L. Li, G. Sun, D. Liu, S. Li and D. Cui, Chem. Commun., 2015, 51, 1039–1041 RSC.
- T. Kawamura, N. Toshima and K. Matsuzaki, Macromol. Rapid Commun., 1994, 15, 479–486 CrossRef CAS.
- S. Loughmari, A. Hafid, A. Bouazza, A. El Bouadili, P. Zinck and M. Visseaux, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2898–2905 CrossRef CAS.
- M. Bartnikowski, T. R. Dargaville, S. Ivanovski and D. W. Hutmacher, Prog. Polym. Sci., 2019, 96, 1–20 CrossRef CAS.
- E. Martin, P. Dubois and R. Jérôme, Macromolecules, 2000, 33, 1530–1535 CrossRef CAS.
- E. Martin, P. Dubois and R. Jérôme, Macromolecules, 2003, 36, 5934–5941 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc02265f |
| This journal is © The Royal Society of Chemistry 2022 |