
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHexafluoroisobutylation of enolates through a tandem elimination/allylic shift/hydrofluorination reaction†
Aline
Delamare‡
a,
Guillaume
Naulet‡
a,
Brice
Kauffmann
 b,
Gilles
Guichard
*a and
Guillaume
Compain
*a
b,
Gilles
Guichard
*a and
Guillaume
Compain
*a
aUniv. Bordeaux, CNRS, Bordeaux INP, CBMN, UMR 5248, IECB, F-33600 Pessac, France. E-mail: g.guichard@iecb.u-bordeaux.fr; g.compain@iecb.u-bordeaux.fr
bUniv. Bordeaux, CNRS, INSERM, IECB, UAR 3033, F-33600 Pessac, France
First published on 20th July 2022
Abstract
The isobutyl side chain is a highly prevalent hydrophobic group in drugs, and it notably constitutes the side chain of leucine. Its replacement by a hexafluorinated version containing two CF3 groups may endow the target compound with new and advantageous properties, yet this modification remains overlooked due to the absence of a general and practical synthetic methodology. Herein, we report the first general method to introduce the hexafluoroisobutyl group into ketoesters, malonates, 1,3-diketones, Schiff base esters and malononitrile. We demonstrated that the reaction occurs through an elimination/allylic shift/hydrofluorination cascade process which efficiently overcomes the usual fluoride β-elimination observed with α-CF3-vinyl groups. We showed that with alkali metal bases, a pentafluorinated alkene is obtained predominantly, whereas the use of tetrabutylammonium fluoride (TBAF) allows hydrofluorination to occur. This tandem process represents a conceptually new pathway to synthesize bis-trifluoromethylated compounds. This methodology was applied to the multigram-scale synthesis of enantiopure (S)-5,5,5,5′,5′,5′-hexafluoroleucine.
Introduction
Fluorine is highly prevalent in pharmaceuticals due to its potential beneficial effects.1 The incorporation of one or several fluorine atoms is a well-established approach to improve the physical properties, stability and/or biological activity of a lead compound.2,3 This approach was highly successful as shown by the large number of approved fluorinated drugs on the market,1c,4 and many of them are polyfluorinated.5 Therefore, there is a growing interest in developing methods to introduce emerging polyfluorinated groups.6 In this context, we were interested in incorporating a hexafluoroisobutyl group, a fluorinated analogue of the leucine side chain. The isobutyl group is found in many peptide therapeutics and numerous other medicinal compounds (Chart 1). Replacing this hydrophobic side chain in such bioactive compounds by its hexafluorinated counterpart could enhance/modulate their biological properties (Fig. 1, top). With a dipole moment of 1.98 D,7 it is more polar than a single CF3 group (1.65 D),8 and could promote dipolar interactions with a biological target. More importantly, the presence of two CF3 groups significantly increases the hydrophobicity of the molecule while preserving the morphology of the parent compound.9 This could favor the affinity for a biological target and/or the membrane permeability. Additionally, polyfluorinated versions of proteinogenic hydrophobic amino acids have proved particularly well suited to studying the structure and function of proteins as they provide additional sensitivity in 19F NMR experiments and can be incorporated into proteins/peptides by either synthetic or biosynthetic methods.10 In these respects, 5,5,5,5′,5′,5′-hexafluoroleucine which bears six fluorine atoms is a key fluorinated amino acid.9b,c,11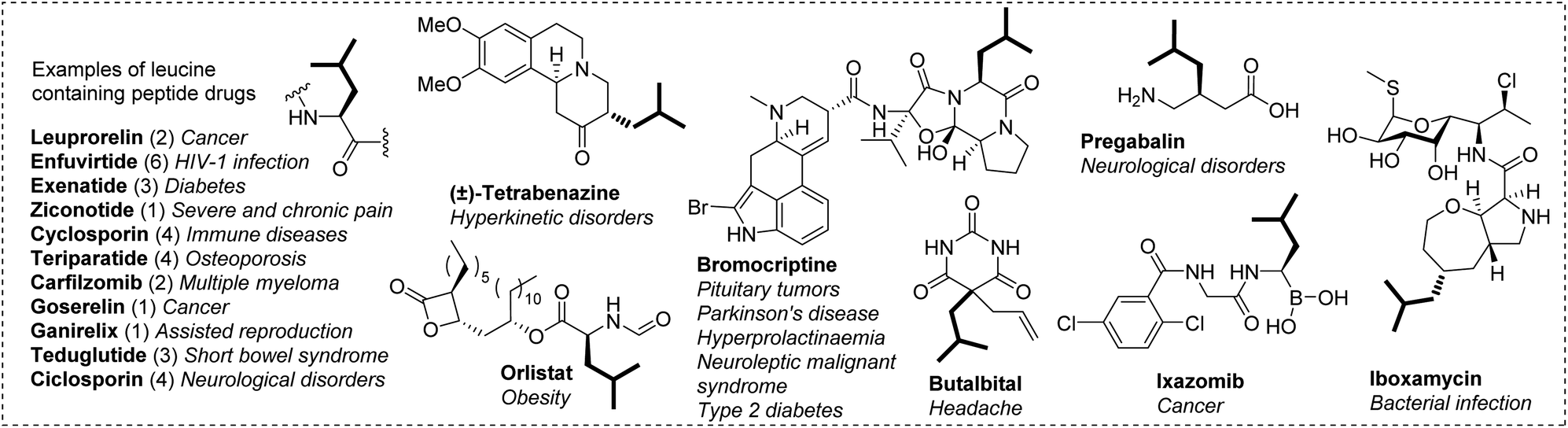 | ||
| Chart 1 Selected drugs bearing the isobutyl side chain and their medical applications. For peptides, the number under brackets refers to the number of leucine residues. | ||
 | ||
| Fig. 1 Top: Changes in physical properties when replacing the isobutyl group by its hexafluorinated counterpart. π corresponds to the hydrophobic substituent constant.9d Middle: SN2′ mechanism previously described on α-CF3-vinyl derivatives.12–17 Bottom: The reaction reported in this work. | ||
However, as there is no general synthetic method reported so far to incorporate this fluoroalkyl group, we seek to work out an efficient and practical protocol. Ideally, the side chain should be introduced in one step. The hexafluoroisobutene reagent would be a suitable reagent to perform such fluoroalkylation on enolates as it is highly electrophilic due to the presence of both CF3 groups. However, α-CF3-vinyl reagents usually react with nucleophiles through the SN2′ mechanism leading to β-fluoride eliminations (Fig. 1, middle). This reaction mechanism has often been considered as an opportunity to synthesize gem-difluoroalkenes for several decades.12–15 Nevertheless, this elimination reaction remains the main obstacle for effective synthesis of trifluoromethylated compounds using α-CF3-vinyl groups or more generally α-CF3 carbanion chemistry (Fig. 1, top).16,17 Another disadvantage is that hexafluoroisobutene is a gas at room temperature18 and applying this method would require specific safety equipment and make the measurement of small quantities inaccurate.
Herein, we report the first general synthetic method to introduce this fluorinated side chain in enolates based on an α,α-bis-CF3-vinyl electrophile. The reaction requires the use of a non-gaseous simple fluorinated reagent generating in situ the α,α-bis-CF3-vinyl electrophile. Furthermore, instead of avoiding fluoride elimination, the reaction involves a tandem allylic shift/hydrofluorination process (Fig. 1, bottom), overcoming the usual SN2′ undesired mechanism. Remarkably, the reaction occurs through a well-controlled cascade process under optimized conditions. This method was successfully applied to the synthesis of (S)-5,5,5,5′,5′,5′-hexafluoroleucine.
Results and discussion
Optimization of the reaction conditions
To incorporate the hexafluorinated side chain, we thought to use commercially available 2-(bromomethyl)-1,1,1,3,3,3-hexafluoropropane. With a boiling point of 78 °C, this reagent is a liquid at room temperature, thus facilitating handling. We started this study by evaluating the alkylation reaction on ketoester 1a using NaOH powder in THF at −20 °C (Table 1, entry 1). The reaction reached a maximum of conversion after 2 h. Unfortunately, the desired product 1b was formed with a very low yield (12%, entry 1) and the pentafluoroalkenylated compound 1c was obtained as the predominant product (45% yield). Despite the successful alkylation of 1a, an elimination of fluoride is thus taking place at some point in the process. With LiOH, the elimination product was obtained with a better yield (51%) but compound 1b was still the minor compound (14%). As the pentafluoroalkene moiety should be quite electrophilic due to the presence of five electron-withdrawing fluorine atoms, we tested whether the elimination could be reversed by using a source of fluoride as a base.19 To our delight, TBAF alone was found to be effective in promoting the reaction. Disappointingly, with 4 equiv. of tetrabutylammonium fluoride (TBAF), 1c was again obtained as the predominant product and the yield was even higher (69%). Only a trace amount of 1b was observed in the crude mixture. In contrast, compound 1b was successfully isolated with a good 63% yield when a larger quantity of TBAF (10 equiv.) was employed (entry 4). It is noteworthy that under these conditions, no elimination product 1c was formed after 1 h of reaction. Additionally, an unexpected byproduct, compound 1d, was also isolated as a minor compound formed during the reaction (9% yield). This compound is the result of two consecutive fluoroalkylations of 1a with a close polarity to that of 1b. At that stage, it was necessary to further improve the selectivity for the desired mono(hexafluoroalkylated) compound not only to increase the yield, but also to simplify the purification step.|
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Base | y | Solvent | Conc. [mM] | x | t [h] | Yield | 1b/1c/1da | ||
| 1b | 1c | 1d | ||||||||
| a Crude ratio determined by 19F NMR. b Other side products were observed in the crude NMR spectra. c The ratio could not be determined due to signal overlaps with other side products. d Reaction conducted at −50 °C for 1 h. e Reaction conducted at −50 °C for 1 h and then at RT for 1 h. f Reaction conducted at −20 °C for 1 h and then at RT for 4 h; no evolution of the reaction mixture after 4 h. g Reaction conducted at 0 °C. | ||||||||||
| 1 | NaOH | 4 | THF | 33 | 2.0 | 2.0 | 12% | 45% | — | 1/4.3/0b |
| 2 | LiOH | 4 | THF | 33 | 2.0 | 2.0 | 14% | 51% | — | 1/4.8/0b |
| 3 | TBAF | 4 | THF | 33 | 2.0 | 1.0 | Trace | 69% | — | 1/15.1/0 |
| 4 | TBAF | 10 | THF | 33 | 2.0 | 1.0 | 63% | — | 9% | 1/0/0.23 |
| 5 | TBAF | 10 | THF | 17 | 2.0 | 1.0 | 65% | — | 8% | —c |
| 6d | TBAF | 10 | THF | 17 | 2.0 | 1.0 | Trace | 16% | Trace | 1/7.2/0.28 |
| 7e | TBAF | 10 | THF | 17 | 2.0 | 2.0 | 17% | 19% | 11% | 1/1.8/0.67 |
| 8 | TBAF | 10 | Toluene | 33 | 2.0 | 5.0 | 2% | — | — | 1/0/0 |
| 9f | TBAF | 10 | CH2Cl2 | 33 | 2.0 | 5.0 | 27% | 11% | — | 1/0.44/0 |
| 10 | TBAF | 10 | AcOEt | 33 | 2.0 | 1.5 | 32% | — | — | 1/0/0 |
| 11 | TBAF | 10 | CH3CN | 33 | 2.0 | 1.5 | 61% | — | — | 1/0/0 |
| 12 | TBAF | 10 | DMF | 33 | 2.0 | 1.5 | 8% | — | — | —c |
| 13 | TBAF | 10 | CH 3 CN | 33 | 1.1 | 1.0 | 71% | — | — | 1/0/0 |
| 14 | TBAF | 10 | THF | 33 | 1.1 | 1.0 | 48% | — | — | —c |
| 15g | TBAF | 10 | CH3CN | 33 | 1.1 | 1.0 | 16% | — | — | —c |
| 16 | TBAF | 10 | CH3CN | 100 | 1.1 | 1.0 | 47% | — | — | 1/0/0 |

Therefore, we next focused our effort on the optimization of the reaction conditions by changing the concentration, the temperature and the solvent. The concentration was reduced two-fold to see whether it could improve the selectivity, but similar yield and selectivity were observed (entry 5). Then, when running the reaction at −50 °C for 1 h, the conversion was dramatically reduced and the compound distribution was unsatisfactory (entry 6), even if the mixture was run at RT for one more hour (entry 7). Next, we tested different solvents (entries 8–12). When using toluene, a less polar solvent than THF, we observed almost no conversion after 5 hours of reaction (entry 8). In dichloromethane, the reaction provided only 27% of the desired compound 1b and 11% of 1c was also recovered. Pleasingly, the use of ethyl acetate and acetonitrile favored the selective formation of 1b with no trace of 1c or 1d observed in the NMR spectra of the crude product (entries 10 and 11 respectively). Notably, the yield was twice as high in acetonitrile as it was in ethyl acetate, respectively at 61% versus 32%. Finally, the use of the more polar solvent DMF resulted in a complex mixture and compound 1b was isolated with only 8% yield (entry 12). We thus selected acetonitrile to pursue our investigation. To our delight, when using only 1.1 equiv. of the fluorinated electrophile instead of 2.0 equiv. (entry 13 versus 11), the yield of compound 1b was increased substantially (71%). However, changing back acetonitrile to THF (entry 14), increasing the temperature to 0 °C (entry 15) or increasing the concentration (entry 16) drastically reduced the yield of 1b.
Scope of the hexafluoroisobutylation reaction
With our optimized conditions in hand (Table 1, entry 13), we examined the substrate scope of this reaction for a series of ketoesters (Fig. 2) with various substitutions at R1, EWG1 and EWG2 positions, i.e. alkyl, cycloalkyl and aromatic groups. Overall, all substrates tested provided the desired compounds with high selectivity, i.e. no other side product was observed in the NMR spectra of the crude product. The reaction was found to be very effective with ethyl and benzyl groups positioned at the central carbon (R1 group) leading to compounds 2b and 3b with 84% and 95% yield respectively. However, a lower yield was obtained when using a more hindered substrate such as 4a bearing an isopropyl group which gave 4b with a moderate yield (44%). Replacing the CH3 group at the EWG1 position by an aromatic substituent was found to be well tolerated (compounds 5–7b) leading to the desired hexafluorinated products with good yields (60 to 78%). Then cyclic substrates were tested (compounds 8–11a). The size of the ring was found to be critical. While with cyclopentanone 8a, the resulting product 8b was isolated with only 15% yield, cyclohexanone 9a and cycloheptanone 10a provided the hexafluorinated products with high yields, 84% and 87% respectively. | ||
| Fig. 2 Scope of the hexafluoroisobutylation reaction. a1.5 h of reaction time; bTHF was used instead of CH3CN; c2 h of reaction time; dTHF was used instead of CH3CN, 1.5 h of reaction time; eTHF was used instead of CH3CN with 2 equiv. of the electrophile; fthe deacetylated product 11e was obtained with 39% yield (see Scheme 1); g2 h of reaction time; h2.5 h of reaction time, 35% of the starting materials were recovered; i2 h of reaction time; jthis compound was found unstable on silica gel and could not be separated from benzophenone, its decomposition product. The yield was estimated using the NMR ratio of the two products. | ||
Finally, the lactone 11a was found to be a special case. The expected compound 11b was successfully isolated but with a moderate yield of 38%, and the reaction provided the deacetylated product 11e as well with 39% yield. The structure of 11e was confirmed by X-ray structure analysis. Notably, we thought that promoting the deacetylation reaction would be of special interest since it could provide the hexafluoroalkylated ester in one step. The reaction was carried out for a longer time and at higher temperature to see whether 11b could be converted to 11e. Pleasantly, this cascade reaction exclusively afforded 11e, isolated with 63% yield which is a remarkable yield for such a multi-reaction process, and this reaction allows direct access to a substituted ester (Scheme 1).
 | ||
| Scheme 1 Hexafluoroisobutylation/deacetylation domino reaction.20 | ||
Next, we explored the reactivity of malonate derivatives (Fig. 2, right) bearing saturated, unsaturated and aromatic substituents. Gratifyingly, this reaction is quite compatible with these substrates. Good to excellent yields were obtained for compounds 12–18b, having methyl, allyl, benzyl, homobenzyl, phenyl, p-nitrophenyl and isobutyl groups. Interestingly, compound 13b could be used as a potential precursor of the fluorinated analogue of butalbital (see Chart 1). Nonetheless, the reaction afforded a lower yield with an isopropyl group (substrate 19a), even lower than that for substrate 4a in the ketoester series, which seems to confirm the sensitivity of the reaction to steric effects. A low yield was also obtained for the compound 20b bearing a nitrile functional group.
We finally tested a set of other pronucleophiles including 1,3-diketones, iminoester, and malononitrile (Fig. 2, bottom). The reaction was found to be compatible with 1,3-diketones 21a and 22a. Notably, a quantitative yield was obtained for compound 21b. The reaction on iminoester 23a provided the desired fluorinated compound with 33% yield. It is worth noting that the presence of a second acidic proton on the molecule did not provide a dialkylated compound. Finally, only 12% yield was obtained for the compound 24b starting from malononitrile.
Synthesis of (S)-5,5,5,5′,5′,5′-hexafluoroleucine
Then, we sought to apply this methodology to the synthesis of (S)-5,5,5,5′,5′,5′-hexafluoroleucine whose potential use is still limited by its synthetic accessibility. Although several enantiopure syntheses of this fluorinated amino acid have been reported,21 the incorporation of the hexafluoroisobutyl group essentially relies on the use of either hexafluoroacetone, a highly toxic gas requiring specific safety equipment, or the expensive [(CF3)2C]2S2 reagent. Moreover, several steps are still necessary after the installation of the fluorinated moiety to access the desired fluoroalkyl amino acid and cognate amino acid. We tested our methodology starting from the Ni(II) chiral complex of the glycine Schiff base (S)-25a (Table 2).22–24 The stereoselective homologation of Ni(II) chiral complexes is a robust synthesis approach to access non-canonical amino acids,25 which has been efficiently employed for the synthesis of various fluorinated amino acids.26 To our delight, the use of our optimized procedure provided the desired mono(hexafluoroalkylated) compound 25b with 63% yield (Table 2, entry 1). The compound was obtained with a very good diastereoselectivity ((S,S)-25b/(S,R)-25b: 92/8). The major diastereoisomer was readily crystallizing and its structure was confirmed by X-ray analysis.|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Reaction timea | Yield 25bb | (S,S)-25b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :(S,R)-25bc :(S,R)-25bc |
| a The reaction time corresponds to 1 + x. b The isolated yield refers to the isolation of the mixture of diastereoisomers (S,S)-25 and (S,R)-25. c Determined by 19F NMR. d 5 equiv. of TBAF were used. e 19% of 25a was recovered after flash chromatography. | ||||
| 1 | CH3CN | 21 h | 63% | 92:8 |
| 2 | CH2Cl2 | 21 h | 39% | 93:7 |
| 3 | DMF | 1.5 h | 36% | 92:8 |
| 4 | THF | 3 h | 66% | 90:10 |
| 5d,e | THF | 21 h | 53% | n.d. |

However, the reaction was found to be much slower compared to the previous substrates, requiring 1 h at −20 °C and then 20 h at room temperature to reach a full conversion. This is probably due to the increased steric hindrance of the nucleophile. Less polar solvents CH2Cl2 gave 25b in 39% yield (entries 2). In DMF, 36% yield was obtained (entry 3) which is significantly higher than the yield attained with 1a (8%). In contrast, when using THF (entry 4), the yield was improved (66%) and the reaction was found to proceed much faster than in acetonitrile (3 h versus 21 h). If only 5 equiv. of TBAF were used, the reaction was slowed down and provided 25b with a lower yield confirming that 10 equiv. of TBAF are necessary (entry 5).
This methodology is compatible with a multi-gram scale procedure as shown in Scheme 2. The two diastereoisomers (S,S)-25b and (S,R)-25b were successfully separated by flash chromatography affording pure (S,S)-25b with a diastereomeric ratio of 99:1. The hydrolysis of the alkylated complex (S,S)-25b afforded hexafluoroleucine (S)-26 with an almost quantitative yield. To confirm the high enantiopurity of the resulting fluorinated amino acid, the enantiomeric excess was determined using the Marfey's derivatization method (see ESI†),27 and an enantiomeric ratio of 99:1 ((S)-26:(R)-26) was obtained.
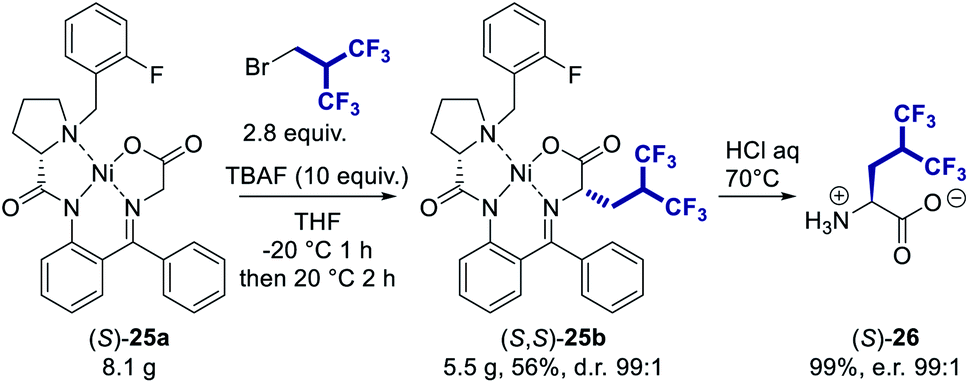 | ||
| Scheme 2 Hexafluoroisobutylation scale-up procedure on nickel complex (S)-25a and hydrolysis of alkylated complexes (S,S)-25b to provide enantiomerically pure amino acid (S)-26. | ||
Mechanistic study
To get insights into the reaction mechanism, the reactivity of the fluorinated electrophile was studied in the presence of TBAF and the reaction was followed by 19F NMR (Fig. 3A) and 1H NMR (Fig. S1†). After only 2 minutes, the brominated reagent undergoes an elimination of HBr to produce HFIB, and the latter one is relatively stable in the reaction medium beyond 3 h. Indeed, this reaction is favored due to the presence of two CF3 groups which extensively contribute to enhancing the acidity of the central C–H bond.28 Consequently, the alkylating reagent in the reaction is unlikely to be the bromo derivative but rather the alkene instead. Interestingly, we did not observe HF addition to the alkene by NMR despite the high content of TBAF. To get additional insights on the reaction mechanism, we performed several experiments with 25a, with which the reaction was found to be slower than with other substrates. As observed with 1a, the pentafluoroalkene product 25c was formed predominantly when using NaOH as the base (55% yield). The structure of compound 25c was confirmed by X-ray diffraction analysis (Fig. 4). Aside from 25c, compound 25b was recovered only in 6% yield together with 20% of the starting material (see the ESI†). Interestingly, when monitoring the reaction performed with TBAF by TLC (Table 2, entry 5), the pentafluorinated elimination product 25c was observed first and then converted into the hexafluorinated compound 25b (Fig. S2†). We also performed in situ NMR experiments to monitor the reaction of 25a at 0 °C (Fig. 3B). The formation of both compounds 25b and 25c was rapidly observed (after only 7 minutes). Then, the proportion of compound 25c started to decrease progressively over time with a concomitant increase in the intensity of the signal corresponding to compound 25b. These observations indicate that compound 25c is formed first and then converted into compound 25b, thus suggesting that TBAF promotes the addition of HF to the alkene. To confirm this mechanism, alkene 25c was treated with 10 equiv. of TBAF, and under these conditions, the compound was fully converted into 25b within 1 h. To see whether the hexafluorinated compound is in equilibrium with the alkene, 25b was treated with both TBAF (10 equiv.) and NaOH (4 equiv.) for 1 h at 0 °C. Under these conditions, 25b was found to be very stable with no trace of 25c being observed (Fig. 4 and S3†). | ||
| Fig. 3 (A) In situ19F NMR spectra of the reaction of 2-(bromomethyl)-1,1,1,3,3,3-hexafluoropropane and TBAF in THF-d8. (B) In situ19F NMR spectra of the reaction of 25a, 2-(bromomethyl)-1,1,1,3,3,3-hexafluoropropane and TBAF (10 equiv.) in THF-d8. | ||
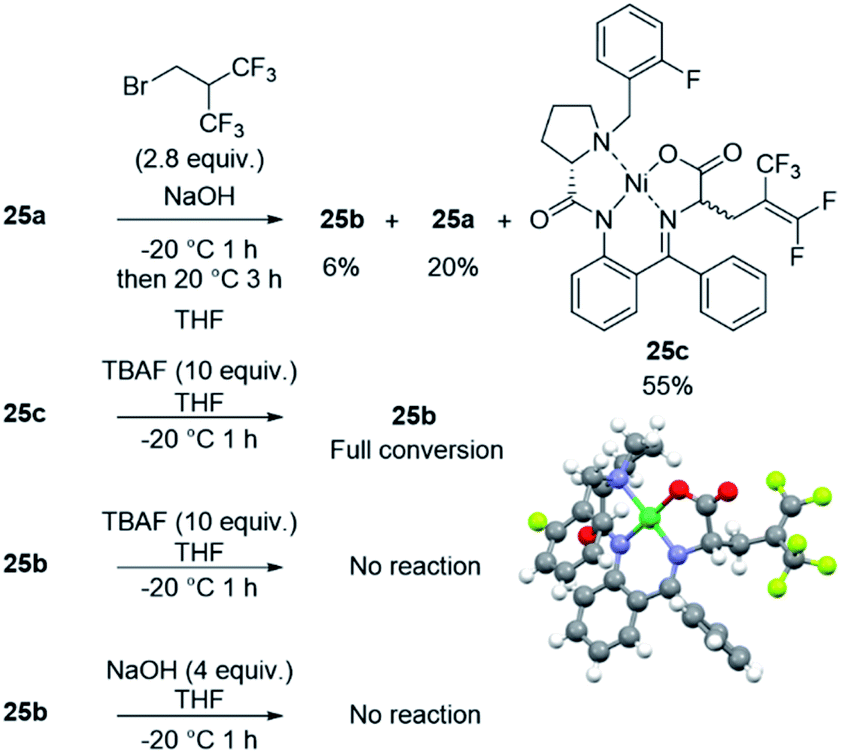 | ||
| Fig. 4 Reactions of nickel complex 25a with NaOH, 25c with TBAF, 25b with TBAF and NaOH. The X-ray structure of compound (S,S)-25c. | ||
Based on the overall results, we propose the mechanisms shown in Scheme 3. The brominated reagent rapidly undergoes an elimination of HBr under basic conditions to provide HFIB. In parallel, the deprotonation of the substrate a leads to the formation of enolate, which then reacts with HFIB through an SN2′ mechanism. This provides the elimination product c. Then, the difluoroalkene undergoes a fluoride addition to give an anionic intermediate. The latter one can either react with a proton to provide b or with HFIB leading to d. It is noteworthy that despite the complexity of the multiple cascade processes, the optimized procedure allows the selective and efficient formation of compound b.
 | ||
| Scheme 3 Proposed mechanism for the formation of compounds b, c and d through cascade reactions. | ||
Conclusions
In summary, we report the first general method to incorporate the hexafluoroisobutyl group into enolates, including ketoesters, malonates, Schiff base esters, diketones, and malononitrile. The reaction is based on the nucleophilic attack on HFIB, rapidly formed under basic conditions from 2-(bromomethyl)-1,1,1,3,3,3-hexafluoropropane, as revealed by NMR. This method is highly practical since the brominated reagent is liquid at room temperature, unlike HFIB. The reaction first promotes the β-elimination of a fluoride through an SN2′ mechanism affording the corresponding pentafluorinated alkene. Unfortunately, when using alkali metal bases, the reaction predominantly provides this undesired alkene. However, we found that the use of TBAF as a base allows the efficient and selective formation of the hexafluoroisobutylated compounds by promoting the addition of HF to the alkene. In situ NMR data and other experiments support the tandem elimination/allylic shift/hydrofluorination mechanism. This methodology was successfully applied to the synthesis of (S)-5,5,5,5′,5′,5′-hexafluoroleucine thanks to diastereoselective fluoroalkylation of a Schiff base chiral nickel complex. Hydrolysis of the nickel complex readily affords the fluorinated amino acid in one step with high enantiopurity. The ease to manipulate the brominated reagent, liquid at room temperature, makes the synthesis of (S)-5,5,5,5′,5′,5′-hexafluoroleucine much more simple and practical than the previously reported syntheses allowing its preparation on a multi-gram scale. This hexafluoroisobutylation reaction is the first example of a tandem allylic shift/hydrofluorination process and provides a conceptually new pathway to perform fluoroalkylation reactions. This methodology affords an easy protocol to incorporate hexafluoroisobutyl groups to engineer bioactive compounds, for applications in medicinal chemistry and chemical biology.Data availability
The data that support the findings of this study are available in the ESI† and in the CIF files for crystallographic data.Author contributions
A. D., G. N., and G. C. performed the experiments, optimized the reaction conditions, developed substrate scope, synthesized (S)-5,5,5,5′,5′,5′-hexafluoroleucine and conducted detailed mechanistic studies. B. K. performed the X-ray diffraction analysis of the compounds. G. C. and G. G. conceived the idea, designed the research, and wrote the manuscript. All the authors commented on the draft of the manuscript and contributed to the analysis and interpretation of the data.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We gratefully thank the Ministère de l'Enseignement Supérieur de la Recherche et de l'Innovation (MESRI) for the PhD funding of Aline Delamare and the Univ. Bordeaux for the temporary teaching and research assistant position of Guillaume Naulet. We thank ANR for its financial support (Project ANR-20-CE06-0008). Pierre Waffo Teguo, from the Institut des Sciences de la Vigne et du Vin, is gratefully acknowledged for his help with specific rotation measurements. We thank Estelle Morvan from the IECB Biophysical and Structural Chemistry Platform (BPCS), Univ. Bordeaux, CNRS UAR3033, Inserm US001, for her assistance with the NMR experiments.Notes and references
- (a) N. A. Meanwell, J. Med. Chem., 2018, 61, 5822–5880 CrossRef CAS PubMed; (b) E. P. Gillis, K. J. Eastman, M. D. Hill and D. J. Donnelly, J. Med. Chem., 2015, 58, 8315–8359 CrossRef CAS PubMed; (c) A. C. Flick, C. A. Leverett, H. X. Ding, E. McInturff, S. J. Fink, C. J. Helal and C. J. O'Donnell, J. Med. Chem., 2019, 62, 7340–7382 CrossRef CAS PubMed.
- Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Aceña, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422–518 CrossRef CAS PubMed.
- S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC.
- (a) H. Mei, J. Han, S. Fustero, M. Medio-Simon, D. M. Sedgwick, C. Santi, R. Ruzziconi and V. A. Soloshonok, Chem.–Eur. J., 2019, 25, 11797–11819 CrossRef CAS PubMed; (b) M. Inoue, Y. Sumii and N. Shibata, ACS Omega, 2020, 5, 10633–10640 CrossRef CAS PubMed.
- (a) M. Bassetto, S. Ferla and F. Pertusati, Future Med. Chem., 2015, 7, 527–546 CrossRef CAS PubMed; (b) F. Pertusati, M. Serpi and E. Pileggi, Polyfluorinated Scaffolds in Drug Discovery, in Fluorine in Life Sciences: Pharmaceuticals, Medicinal Diagnostics, and Agrochemicals, Elsevier, 2019, pp. 141–180 Search PubMed.
- (a) D. Cahard and J. Ma, in Emerging Fluorinated Motifs: Synthesis, Properties, and Applications, Wiley, 1st edn, 2020 Search PubMed; (b) S. Meyer, J. Häfliger and R. Gilmour, Chem. Sci., 2021, 12, 10686–10695 RSC; (c) Q. Wang, Q. Tao, H. Dong, C. Ni, X. Xie and J. Hu, Angew. Chem. Int. Ed., 2021, 60, 27318–27323 ( Angew. Chem. , 2021 , 133 , 27524–27529 ) CrossRef CAS PubMed.
- A. R. H. Goodwin and J. B. Mehl, Int. J. Thermophys., 1997, 18, 795–806 CrossRef CAS.
- D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308–319 RSC.
- (a) B. Linclau, Z. Wang, G. Compain, V. Paumelle, C. Q. Fontenelle, N. Wells and A. Weymouth-Wilson, Angew. Chem. Int. Ed., 2016, 55, 674–678 ( Angew. Chem. , 2016 , 128 , 684–688 ) CrossRef CAS PubMed; (b) S. Huhmann, A.-K. Stegemann, K. Folmert, D. Klemczak, J. Moschner, M. Kube and B. Koksch, Beilstein J. Org. Chem., 2017, 13, 2869–2882 CrossRef CAS PubMed; (c) E. N. G. Marsh, Acc. Chem. Res., 2014, 47, 2878–2886 CrossRef CAS PubMed; (d) C. Hansch, A. Leo, S. H. Unger, K. H. Kim, D. Nikaitani and E. J. Lien, J. Med. Chem., 1973, 16, 1207–1216 CrossRef CAS PubMed.
- (a) M. A. Miller and E. M. Sletten, ChemBioChem, 2020, 21, 3451–3462 CrossRef CAS PubMed; (b) E. N. G. Marsh and Y. Suzuki, ACS Chem. Biol., 2014, 9, 1242–1250 CrossRef CAS PubMed.
- (a) H. Mei, J. Han, K. D. Klika, K. Izawa, T. Sato, N. A. Meanwell and V. A. Soloshonok, Eur. J. Med. Chem., 2020, 186, 111826 CrossRef CAS PubMed; (b) H. Meng and K. Kumar, J. Am. Chem. Soc., 2007, 129, 15615–15622 CrossRef CAS PubMed; (c) L. M. Gottler, H.-Y. Lee, C. E. Shelburne, A. Ramamoorthy and E. N. G. Marsh, ChemBioChem, 2008, 9, 370–373 CrossRef CAS PubMed; (d) H. Meng, S. T. Krishnaji, M. Beinborn and K. Kumar, J. Med. Chem., 2008, 51, 7303–7307 CrossRef CAS PubMed; (e) S. Huhmann and B. Koksch, Eur. J. Org. Chem., 2018, 2018, 3667–3679 CrossRef CAS.
- (a) X. Zhang and S. Cao, Tetrahedron Lett., 2017, 58, 375–392 CrossRef CAS; (b) G. Chelucci, Chem. Rev., 2012, 112, 1344–1462 CrossRef CAS PubMed; (c) F. Tian, G. Yan and J. Yu, Chem. Commun., 2019, 55, 13486–13505 RSC.
- (a) T. Fuchikami, Y. Shibata and Y. Suzuki, Tetrahedron Lett., 1986, 27, 3173–3176 CrossRef CAS; (b) T. Kitazume and T. Ohnogi, Synthesis, 1988, 1988, 614–615 CrossRef; (c) T. Kitazume, T. Ohnogi, H. Miyauchi, T. Yamazaki and S. Watanabe, J. Org. Chem., 1989, 54, 5630–5632 CrossRef CAS; (d) S. Watanabe, K. Sugahara, T. Fujita, M. Sakamoto and T. Kitazume, J. Fluorine Chem., 1993, 62, 201–206 CrossRef CAS; (e) H. M. Park, T. Uegaki, T. Konno, T. Ishihara and H. Yamanaka, Tetrahedron Lett., 1999, 40, 2985–2988 CrossRef CAS; (f) K. Funabiki, K. Sawa, K. Shibata and M. Matsui, Synlett, 2002, 2002, 1134–1136 CrossRef; (g) W. Dai, Y. Lin, Y. Wan and S. Cao, Org. Chem. Front., 2018, 5, 55–58 RSC.
- (a) V. Martin, H. Molines and C. Wakselman, J. Org. Chem., 1992, 57, 5530–5532 CrossRef CAS; (b) J. Ichikawa, H. Fukui and Y. Ishibashi, J. Org. Chem., 2003, 68, 7800–7805 CrossRef CAS PubMed; (c) J.-P. Bégué, D. Bonnet-Delpon and M. H. Rock, Tetrahedron Lett., 1995, 36, 5003–5006 Search PubMed; (d) J.-P. Bégué, B.-D. Delpon and M. H. Rock, J. Chem. Soc., Perkin Trans. 1, 1996, 1409–1413 RSC; (e) J. Ichikawa, Y. Ishibashi and H. Fukui, Tetrahedron Lett., 2003, 44, 707–710 CrossRef CAS; (f) J. Ichikawa, H. Miyazaki, K. Sakoda and Y. Wada, J. Fluorine Chem., 2004, 125, 585–593 CrossRef CAS; (g) J. Ichikawa, M. Yokota, T. Kudo and S. Umezaki, Angew. Chem. Int. Ed., 2008, 47, 4870–4873 ( Angew. Chem. , 2008 , 120 , 4948–4951 ) CrossRef CAS PubMed; (h) K. Fuchibe, M. Takahashi and J. Ichikawa, Angew. Chem. Int. Ed., 2012, 51, 12059–12062 ( Angew. Chem. , 2012 , 24 , 12225–12228 ) CrossRef CAS PubMed.
- (a) B. M. Kraft and W. D. Jones, J. Am. Chem. Soc., 2002, 124, 8681–8689 CrossRef CAS PubMed; (b) Y. Huang and T. Hayashi, J. Am. Chem. Soc., 2016, 138, 12340–12343 CrossRef CAS PubMed; (c) Y. Liu, Y. Zhou, Y. Zhao and J. Qu, Org. Lett., 2017, 19, 946–949 CrossRef CAS PubMed; (d) M. Wang, X. Pu, Y. Zhao, P. Wang, Z. Li, C. Zhu and Z. Shi, J. Am. Chem. Soc., 2018, 140, 9061–9065 CrossRef CAS PubMed.
- C. Ni and J. Hu, Chem. Soc. Rev., 2016, 45, 5441–5454 RSC.
- K. Burger and B. Helmreich, J. Chem. Soc., Chem. Commun., 1992, 348–349 RSC.
- P. M. Murphy, J. Fluorine Chem., 2013, 156, 345–362 CrossRef CAS.
- For some examples of the use of TBAF as a base, see: (a) S. Liu, X. Chen, Y. Hu, L. Yuan, S. Chen, P. Wu, W. Wang, S. Zhang and W. Zhang, Adv. Synth. Catal., 2015, 357, 553–560 CrossRef CAS; (b) G. V. M. Sharma, V. G. Chander, A. S. Reddy and K. R. Reddy, Tetrahedron: Asymmetry, 2002, 13, 21–24 CrossRef CAS; (c) J. H. Clark, Chem. Rev., 1980, 80, 429–452 CrossRef CAS.
- J. J. Ritter and T. J. Kaniecki, J. Org. Chem., 1962, 27, 622–623 CrossRef CAS.
- (a) Y. Tang and D. A. Tirrell, J. Am. Chem. Soc., 2001, 123, 11089–11090 CrossRef CAS PubMed; (b) X. Xing, A. Fichera and K. Kumar, Org. Lett., 2001, 3, 1285–1286 CrossRef CAS PubMed; (c) J. T. Anderson, P. L. Toogood and E. N. G. Marsh, Org. Lett., 2002, 4, 4281–4283 CrossRef CAS PubMed; (d) H.-P. Chiu and R. P. Cheng, Org. Lett., 2007, 9, 5517–5520 CrossRef CAS PubMed; (e) H.-P. Chiu, Y. Suzuki, D. Gullickson, R. Ahmad, B. Kokona, R. Fairman and R. P. Cheng, J. Am. Chem. Soc., 2006, 128, 15556–15557 CrossRef CAS PubMed.
- H. Mei, T. Hiramatsu, R. Takeda, H. Moriwaki, H. Abe, J. Han and V. A. Soloshonok, Org. Process Res. Dev., 2019, 23, 629–634 CrossRef CAS.
- F. Drouet, A. F. M. Noisier, C. S. Harris, D. P. Furkert and M. A. A Brimble, Eur. J. Org. Chem., 2014, 2014, 1195–1201 CrossRef CAS.
- (a) A. S. Saghyan, A. S. Dadayan, S. A. Dadayan, A. F. Mkrtchyan, A. V. Geolchanyan, L. L. Manasyan, H. R. Ajvazyan, V. N. Khrustalev, H. H. Hambardzumyan and V. I. Maleev, Tetrahedron: Asymmetry, 2010, 21, 2956–2965 CrossRef CAS; (b) J. Wang, D. Lin, S. Zhou, X. Ding, V. A. Soloshonok and H. Liu, J. Org. Chem., 2011, 76, 684–687 CrossRef CAS PubMed; (c) The Ni(II) complex can be easily synthesized in four steps. Please see the ESI.†.
- Y. Wang, X. Song, J. Wang, H. Moriwaki, V. A. Soloshonok and H. Liu, Amino Acids, 2017, 49, 1487–1520 CrossRef CAS PubMed.
- J. L. Aceña, A. E. Sorochinsky, H. Moriwaki, T. Sato and V. A. Soloshonok, J. Fluorine Chem., 2013, 155, 21–38 CrossRef.
- R. Bhushan and H. Brückner, Amino Acids, 2004, 27, 231–247 CrossRef CAS PubMed.
- R. N. Haszeldine, J. Chem. Soc., 1953, 3565–3572 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and analytical data (1H, 13C, and 19F NMR and HRMS) for all compounds; additional experiments to support the proposed mechanism including in situ NMR spectra; crystallographic data for compounds 11e, 24b, (S,S)-25c and (S,S)-25b. CCDC 2144790 (11e), 2144789 (24b), 1998317 ((S,S)-25b), 1998318 ((S,S)-25c). For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc02871a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |