
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHot-exciton harvesting via through-space single-molecule based white-light emission and optical waveguides†
Debasish
Barman
 a,
Mari
Annadhasan
b,
Rajadurai
Chandrasekar
*b and
Parameswar Krishnan
Iyer
*ac
a,
Mari
Annadhasan
b,
Rajadurai
Chandrasekar
*b and
Parameswar Krishnan
Iyer
*ac
aDepartment of Chemistry, Indian Institute of Technology Guwahati, Guwahati-781039, Assam, India. E-mail: pki@iitg.ac.in
bSchool of Chemistry and Centre for Nanotechnology, University of Hyderabad, Gachibowli, Prof. C. R. Rao Road, Hyderabad-500046, India
cCentre for Nanotechnology, Indian Institute of Technology Guwahati, Guwahati-781039, Assam, India
First published on 4th July 2022
Abstract
Through-space donor–alkyl bridge–acceptor (D–σ–A) luminogens are developed as new organic single-molecule white light emitters (OSMWLEs) involving multiple higher lying singlet (Sn) and triplet (Tm) states (hot-excitons). Experimental and theoretical results confirm the origin of white light emission due to the co-existence of prompt fluorescence from locally excited states, thermally activated delayed fluorescence (TADF), and fast/slow dual phosphorescence color mixing simultaneously. Notably, the fast phosphorescence was observed due to trace amounts of isomeric impurities from commercial carbazole, while H-/J-aggregation resulted in slow phosphorescence. Crystal structure-packing-property analysis revealed that the alkyl chain length induced supramolecular self-assembly greatly influenced the solid-state optical properties. Remarkably, the 1D-microrod crystals of OSMWLEs demonstrated the first examples of triplet harvesting waveguides by self-guiding the generated phosphorescence through light propagation along their longitudinal axis. This work thus highlights an uncommon design strategy to achieve multi-functional OSMWLEs with in-depth mechanistic insights and optical waveguiding applications making them a potentially new class of white emissive materials.
Introduction
Organic single-molecule white light emitters (OSMWLEs)1 have stimulated enormous interest owing to their potential applications in next-generation white light-emitting diodes (WLEDs),2 sensing,3 anti-counterfeiting,4 biological imaging5,6 and optical waveguides (OWGs).7 The unique photophysical processes governing the broad visible spectrum emission of OSMWLEs are fundamentally very intriguing. The majority of WLEs reported, thus far, rely on harnessing singlet excitons either in a single molecule and/or polymers8 or multicomponent fluorescent blends.9 These systems typically include monomer/excimer complexes,10 inter/intramolecular charge transfer (ICT) systems,11 Förster resonance energy transfer (FRET) systems,12 excited-state intramolecular proton transfer (ESIPT) systems,13 and dual (prompt/delayed) fluorescence (DFL) systems.14 In contrast, weakly susceptible triplet excitons in organic molecules adopt non-radiative pathways, and dissipate as heat energy instead of light due to the poor spin–orbit coupling (SOC), thereby exhibiting dim phosphorescence.15 Besides, triplet excitons show high quenching propensities in the presence of moisture and molecular oxygen at room temperature (RT), while phosphorescence from organics generally exists only at cryogenic temperature.16 These limitations impede the effective utilization of both singlet and triplet energy, especially to generate white light in purely organic compounds. Essentially, phosphorescent OSMWLEs have immense prospects of triplet harvesting features under ambient conditions. They offer alternatives to conventional emitters due to the favourable no phase segregation, no colour aging, uniform film-forming ability and simple device fabrication.17,18 However, due to ineffective molecular design and lack of fundamental insights on the excited-state dynamic (ESD) process, the origin of white light emission (WLE) is often overlooked.Recently, multiple radiative decays from both singlet and triplet excitons generating WLE were demonstrated in organic molecules by typical FL or thermally activated delayed FL (TADF) and room temperature phosphorescence (RTP) or two distinct dual-phosphorescence (DP) studies.19–22 Strategically, an engineered TADF emitter possesses a well-separated highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO), comprising small singlet–triplet splitting (ΔEST), that can harvest triplet excitons through an efficient reverse intersystem crossing (RISC) process.23 After the first report of seminal TADF emitter 4CZIPN,24 tremendous efforts have been devoted to exploring new materials based on triazines, oxadiazoles, cyanobenzenes, sulfones and benzothiazole cores.25 These molecules with a common electron donor–acceptor (D–A) dyad/triad structure exhibit narrow ΔEST and robust TADF.26 On the other hand, long-lived RTP from organic luminogens was realized by populating triplet excitons through introducing a heavy atom effect, El-Sayed's rule, and charge transfer (CT) state into the bipolar molecular skeleton involving carbazole, carbonyl, borate, sulfone, and phenothiazine respectively.27–29 Subsequently, crystal packing, polymer matrices and a rigid host enhance the SOC to promote the intersystem crossing (ISC) process by suppressing non-radiative dissipation.30 Nonetheless, recent breakthroughs in ultra-long organic phosphorescence due to the presence of trace amounts of isomeric impurities (in carbazole or boronic acid-based systems) promoted the realization of stable phosphorescence emission under ambient conditions without any protection from moisture or molecular oxygen.31,32 Although a few triplet harvesting mechanistic processes are known, through-space triplet–triplet energy transfer (TTET) remains a key photophysical process, which defines the exchange of both spin and energy between a pair of molecules or intramolecular fragments located at a short distance of <10 Å, and releases free energy (exothermic) via a Dexter-type channel.33 Anisotropic one-dimensional (1D) molecular crystals could harvest excitons and display robust light propagation on account of a long-range ordered molecular π–π stacking, free grain boundary, and efficient light guiding propensity.34 Such types of high refractive index and defect free crystals perform as excellent optical waveguides (OWGs) suitable for next-generation photonic and communication technologies.35,36
In order to address the challenge of constructing multifunctional OSMWLEs, a few promising strategies that rely on simple material design with an inherent triplet harvesting mechanism have been reported37–41 (Fig. S1†). These reports confirm that nearly all the WLEs rely on the conventional knowledge of crystallization and the external/internal heavy atom effect in a directly connected D–A system. Despite the progress in designing WLEs, potential solid state applications at RT using a combination of multiple excited states in purely organic molecules are unexplored. Thus far, optical crystals with hot-exciton harvesting OSMWLEs having DFL and DP demonstrating efficient OWG attributes have not been reported to the best of our knowledge.
In this work, four through-space charge transfer (TSCT) OSMWLE materials have been designed and synthesized, denoted as BT2OxCz (x = 3, 4, 5 and 6), which contain acceptor 2,2′-(1,3-phenylene)bis(benzothiazole) [BT2] and donor carbazole [Cz], connected via a series of odd/even alkyl chains. The synthesized BT2OxCz molecular crystals exhibit highly sought after DFL and DP at RT. Experimental and theoretical results attribute the origin of WLE to substantially high lying manifold singlet and triplet excited states. A key conceptual insight on the emission process in the solid state is prompt fluorescence (locally excited emission) and TADF, accompanied by DP at RT. Further, the enhancement of SOC via fine adjustment of moderate ΔEST (∼0.50–0.55 eV) between S1 and T1 with collateral narrow ΔEST (∼0.02–0.12 eV) between S1 and T3/T4 facilitates efficient ISC and RISC to upsurge the TADF and RTP simultaneously. By varying the chain length between D and A, singlet and triplet states could be manipulated to realize a broadband visible range emission in powder, polymethyl methacrylate (PMMA) doped thin-films and in single molecular crystals. Notably, sterically constrained shorter chain BT2OxCZ (x = 3, 4, 5) molecules display TADF and RTP. In contrast, the longest chain analogue BT2O6Cz exhibits relatively intense RTP, owing to dimer formation and subsequent inter-TSCT. Moreover, sterically strained ‘J-aggregation’ and geometrically relaxed ‘H-aggregation’ in the crystal lattice enable harvesting of singlet and triplet-excitons, simultaneously. Remarkably, 1D crystals of BT2O3Cz and BT2O6Cz efficiently guide triplet-harvesting light along their long axes with a low optical loss coefficient (α′ = 0.3260 dB µm−1, and 0.2158 dB µm−1 respectively).
Results and discussion
Materials design
By considering important aspects of OSMWLEs, a through-space rational design strategy was developed to achieve solid-state WLE at RT. The conceived TADF and DP luminogens (BT2OxCz, x = 3, 4, 5 and 6) bear a donor Cz and an acceptor BT2 connected via short (three/four membered) and long (five/six membered) alkyl chains viz. BT2O3Cz, BT2O4Cz, BT2O5Cz, and BT2O6Cz. The key requisites to achieve white-emission in organic molecules depend on the following quantum-mechanical aspects and a few stringent requirements, which include: (1) singlet exciton equilibration with parent and other accessible excited states (e.g., ≥S1), favouring RISC from ≥T2via a small ΔEST producing TADF.42 (2) Utilization of triplet excitons either from the distinct radiative decay of the T1 states (populated via TTET/IC, from ≥T2) or directly from ≥T3 by evolving with ≥S1via a mixed ISC process. (3) Realization of stable phosphorescence from a small amount of impurities that could protect against triplet exciton quenching and result in faster decay from ≥T3.31 (4) Faster decay of ≥T3 to counterbalance the smaller extents with T1 to facilitate radiative decay from ≥T3 and T1, producing DP. (5) By creating excited state charge-separation (ESCS) involving the hot-excitons in highly-flexible molecular systems and controlling the complex excited-state dynamics, solid state white light emission could be generated. Collectively, these aspects directly influence and contribute to the higher-lying singlet and triplet energy (≥S4/S7 and ≥T8/T10) states allowing TADF and RTP simultaneously. The proposed Jablonski diagram illustrates the prominent photophysical processes and relative energy levels involved in fine tuning of the higher-lying excited states for WLE emission for the integrated OSMWLEs (Fig. 1). | ||
| Fig. 1 (A) Proposed Jablonski diagram illustrating the OSMWLE emission mechanism. (B) Chemical structures of OSMWLE BT2OxCz (x = 3, 4, 5 and 6). (C) Pristine powder images of BT2OxCz captured under a 365 nm UV lamp. (D) Illustration of exciton distribution in WLE emission spectra. | ||
Synthesis of BT2OxCZ (x = 3, 4, 5 and 6) OSMWLEs
2,2′-(5-(3-(9H-Carbazol-9-yl)alkoxy)-1,3-phenylene)bis(benzothiazole) compounds (BT2OxCz, x = 3, 4, 5 and 6) were synthesized using a simple two-step substitution reaction with 3,5-bis (benzothiazol-2-yl)phenol (BT2OH) in the presence of 9-(3-bromoalkyl)-9H-carbazole (1, n-CzBr, n = 3, 4, 5 and 6). 3,5-Bis(benzothiazol-2-yl)phenol was synthesized via in situ cross-coupling reaction and 9-(3-bromoalkyl)-9H-carbazole from base substitution reaction (Schemes S1–S3†). Detailed structural characterization using NMR (1H and 13C) and HRMS is shown in Fig. S2–S25.† The electron-accepting (A) (3,5-bis(benzothiazol-2-yl)phenoxy) unit bearing a large number of lone pairs and two sulphur atoms makes the conjugated fragment highly electron deficient. D–A units are connected through-space via various non-conjugated alkyl chains. The electron balance in the molecule happens via through-space charge transfer (TSCT) from electron rich Cz to the electron deficient BT2O unit. Moreover, by changing the length of the alkyl chain (propyl, butyl, pentyl and hexyl), the resultant molecules (BT2OxCz, x = 3, 4, 5 and 6) show an impressive tunable WLE at RT in the solid-state. From their crystal structures and computational optimized geometry it was observed that the molecules containing a shorter chain length adopt large twisted conformations rather than the structurally relaxed longer chain molecules. Geometrically strained BT2O3Cz and BT2O4Cz molecules showed a strong twisted intramolecular charge transfer (TICT) feature rather than the more relaxed BT2O5Cz and BT2O6Cz which formed dimers. However, in the solid state all the emitters exhibited distinct TADF and RTP properties with Commission Internationale de l'éclairage (CIE) coordinate values very close to those of pure WLEs (0.33, 0.33).Solid state optical properties of OSMWLEs
A clear mechanistic insight on the origin of WLE was methodically obtained by studying crystalline powders obtained from the slow evaporation of chloroform/hexane (8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) solution. To decipher the details of intrinsic solid state WLE, PL and TRPL characteristics were investigated for all the through-space BT2OxCz emitters in their crystalline/amorphous powder states. UV-visible spectra for shorter chain BT2O3Cz and BT2O4Cz exhibited absorption bands at 350 and 315 nm, respectively. In contrast, longer chain BT2O5Cz and BT2O6Cz showed comparatively broader absorption bands at 340 and 330 nm, respectively (Fig. 2A–D, solid black lines). The broad absorption for longer chain emitters can be attributed to the dimeric assembly evident from the crystal packings. Further, steady state optical emission spectra recorded (Fig. 2A–D, blue dotted line) for all the BT2OxCzs displayed broad emission spectra with multiple peaks covering the 400 to 700 nm region. Remarkably, an intense peak around ∼430 nm and other broad and weak shoulder peaks at ∼550 and >600 nm were observed for all the emitters, suggesting multimode emission due to the combined existence of fluorescence and phosphorescence. Besides, the intensities of later peaks for BT2O5Cz and BT2O6Cz gradually decrease with increasing chain length due to the weakening of TSCT properties. To confirm the TICT effect on varying chain length between D–A, solvatochromism studies were performed for BT2O3Cz and BT2O6Cz. Notably, a robust TICT feature with 150 nm red-shifted emission was observed for BT2O3Cz containing a highly twisted shorter chain, while the strain free structure of BT2O6Cz showed no such TICT effect (Fig. S26†).
:2) solution. To decipher the details of intrinsic solid state WLE, PL and TRPL characteristics were investigated for all the through-space BT2OxCz emitters in their crystalline/amorphous powder states. UV-visible spectra for shorter chain BT2O3Cz and BT2O4Cz exhibited absorption bands at 350 and 315 nm, respectively. In contrast, longer chain BT2O5Cz and BT2O6Cz showed comparatively broader absorption bands at 340 and 330 nm, respectively (Fig. 2A–D, solid black lines). The broad absorption for longer chain emitters can be attributed to the dimeric assembly evident from the crystal packings. Further, steady state optical emission spectra recorded (Fig. 2A–D, blue dotted line) for all the BT2OxCzs displayed broad emission spectra with multiple peaks covering the 400 to 700 nm region. Remarkably, an intense peak around ∼430 nm and other broad and weak shoulder peaks at ∼550 and >600 nm were observed for all the emitters, suggesting multimode emission due to the combined existence of fluorescence and phosphorescence. Besides, the intensities of later peaks for BT2O5Cz and BT2O6Cz gradually decrease with increasing chain length due to the weakening of TSCT properties. To confirm the TICT effect on varying chain length between D–A, solvatochromism studies were performed for BT2O3Cz and BT2O6Cz. Notably, a robust TICT feature with 150 nm red-shifted emission was observed for BT2O3Cz containing a highly twisted shorter chain, while the strain free structure of BT2O6Cz showed no such TICT effect (Fig. S26†).
 | ||
| Fig. 2 Solid state photophysical properties of the OSMWLEs. (A–D) Ultraviolet-visible absorption, steady state fluorescence and phosphorescence spectra in crystalline powder form for all the OSMWLEs of BT2OxCz (x = 3, 4, 5 and 6) (inset photograph at daylight and 365 nm UV-lamp). (E–H) Time-resolved PL based delayed lifetime recorded under ambient and inert conditions for all the OSMWLEs at RT. (I) Multiple delayed emissions for a rigid PMMA (0.5 wt%) doped spin-coated film for all the OSMWLEs. (J and K) Life-time decay and time resolved emission spectra (TRES) of BT2O3Cz at 545 and 700 nm phosphorescence emission. (L) CIE coordinate diagram for the steady state (blue) and delayed emission (white) for all the OSMWLEs. | ||
Likewise, the solid state phosphorescence spectra of all the WLE BT2OxCz showed multiple peaks distributed over the visible region (Fig. 2A–D, orange dotted line). A weaker fluorescence band (blue shaded) superimposes with steady state fluorescence at ∼430 nm, whereas intense phosphorescence signals (red shaded) appear in the green (∼550 nm) and near-IR region (640, 634, 620 and 635 nm) for BT2O3Cz, BT2O4Cz, BT2O5Cz, and BT2O6Cz. These signals were also found to be identical with a steady state faint shoulder emission band (>600 nm). However, BT2O5Cz exhibited bright fluorescence and showed weak phosphorescence because of its amorphous nature. To confirm the ascertained hypothesis for BT2O5Cz, powder X-ray diffraction (PXRD)/thin film XRD and FETEM studies were conducted with pristine OSMWLEs at RT (Fig. S27†). In both the XRD patterns, broad and weak peaks were observed for BT2O5Cz relating to its amorphous nature, whereas, other emitters of BT2OxCz (x = 3, 4, 6) showed sharp diffracting peak patterns in the low d-spacing region (marked by a red dotted box) depicting a high degree of crystallinity. In the FETEM, despite the micro-rod shaped morphologies of the OSMWLEs, the selected area (electron) diffraction (SAED) images showed a clear amorphous nature of BT2O5Cz powder (Fig. S28†) which does not affect the singlets. Hence, it had bright fluorescence and due to the lack of rigid crystallinity, non-radiative deactivation of triplets has not been fully circumvented, and thereby it showed weak phosphorescence. Notably, with increasing chain length, BT2O5Cz and BT2O6Cz exhibited blue-shifted phosphorescence, probably due to the less geometrical strain reduced thermal relaxation of high lying triplet energy. Hence, we hypothesized that the former blue emission at 430 nm originated from a higher singlet state (>S1) and TADF in nature. However, the latter peaks are assigned to room temperature dual phosphorescence (RTDP) emission, derived from low (T1) and high lying triplet states (>T1). Subsequently, the lifetime recorded at 430 nm showed a bi-exponential decay comprising a ns range lifetime, due to the prompt locally excited emission (τPF = 0.94, 4.09, 5.27 and 3.11 ns for all BT2OxCz WLEs) (Fig. S29†) and a microsecond (µs) range lifetime for delayed components (τDF = 11.5, 11.4, 55.0 and 17.3 µs) (Fig. S30†), which confirms that the shorter wavelength blue emission arises from singlet states. The long average lifetime (5.27 ns and 55 µs) for BT2O5Cz further confirms the prominent fluorescence nature of the emitter compared to the other three emitters. Besides, longer wavelength green/red region peaks are actually derived from the triplet state. To validate this, lifetime studies were carried out under ambient and inert conditions for all the emitters. As expected, the average lifetime calculated from bi-exponential decays showed huge enhancement from 6.77 to 68.07 µs for BT2O3Cz (λem = 640 nm), 10.89 to 18.89 µs for BT2O4Cz (λem = 634 nm), 21.65 to 30.75 µs for BT2O5Cz (λem = 620 nm) and 20.72 to 29.12 µs for BT2O6Cz (λem = 635 nm) (Fig. 2E–H) under inert conditions.
Notably, the higher lifetime for BT2O5Cz and BT2O6Cz was presumably due to the thermally elevated phosphorescence while the comparatively lower lifetime for BT2O3Cz and BT2O4Cz was because of TADF emission under ambient conditions. Further, the temperature-dependent delayed emission and the corresponding lifetime were verified by recording emission at 100, 150, 200 and 300 K respectively for BT2O3CZ and BT2O6Cz (Fig. S31–S36†). Collectively, peaks at ∼430 and ∼600 nm with their delayed lifetime were verified as TADF and thermally elevated RTP in character. In contrast, delayed intensity and lifetime increase with increasing temperature from 100 to 300 K, providing concrete evidence to simultaneously support the thermally activated singlet and triplet exciton release process at RT.
In the temperature dependency results we observed anomalous behaviour for the OSMWLEs, which can be explained by considering the following aspects: (1) isomeric impurities act as charge traps and generate charge-separated excited states which are responsible for fast phosphorescence decay (at 550 nm emission with µs lifetime), (2) at elevated temperature hotter excitons are produced that may prefer a direct transition from higher-lying states (T3/T4 → S0) or go via TTET for favourable relaxation to feed T1 thereby resulting in 600 nm red phosphorescence and (3) communications among these emitting states at local minima by thermal motions (e.g., vibrations) may be weakened at low temperatures so that hot excitons remain concealed (experimental results related to these properties are discussed in the “Mechanism for multi-emission section”).
Moreover, the longest phosphorescence lifetime (68.07 µs) for BT2O3Cz can be attributed to the high crystallinity, densely packed supramolecular assembly (ρcal = 1.400 mg mm−3), intermolecular electronic coupling and through-space TICT effect in sterically strained molecular structure. Phosphorescence QY for all the BT2OxCz powders was calculated to be 4.5%, 4.4%, 5.3% and 13.3%. The radiative rate constant (kr) was calculated to be 6.72 × 107 s−1, 4.08 × 107 s−1, 2.55 × 107 s−1, and 6.14 × 107 s−1 and non-radiative rate constants (knr) were found to be 1.40 × 104 s−1, 0.87 × 104 s−1, 0.40 × 104 s−1, and 0.42 × 104 s−1, for all BT2OxCz WLEs. These results signify that excitons were utilized to the maximum extent for radiative decay (×107) under photo-illumination.
Further, ISC and RISC rate constants calculated from PLQY and lifetime values of the prompt and delayed components were estimated to be kISC = 7.5 × 106 s−1, 1.53 × 106 s−1, 1.25 × 106 s−1, 0.75 × 106 s−1 and kRISC = 1.47 × 105 s−1, 0.24 × 105 s−1, 0.46 × 105 s−1, 0.48 × 105 s−1, for BT2OxCz (x = 3–6) respectively (summarized in Table S1†).43 To obtain clear insights into phosphorescence emission at the molecular level, including elimination of the molecular aggregation effect and to restrict non-radiative motion, a set of control experiments were conducted on spin-coated films after doping all the BT2OxCz WLEs at 0.5 wt% and 5 wt% concentration into PMMA under inert conditions using dry toluene. The phosphorescence spectrum in low concentrated films (0.5%) exhibited three distinct peaks located at 450 nm (TADF), 545 nm (fast RTP, τav = ∼80 µs) and 700 nm (slow RTP, τav = ∼2.5 ms) covering the entire visible range (Fig. 2I and J), clearly suggesting exclusion of molecular oxygen in the rigid polymer matrix, allowing significant RTP contribution (triplet excitons are dominant over singlet excitons). Notably, in films, there was an ∼20 nm bathochromic shift compared to the powder form with no change in peak positions for all four emitters, which could be due to the restriction in molecular motion (RIM), and strong intramolecular interactions rigidifying the structural conformation and resulting in suppression of non-radiative deactivation.44 The colour purity and uniform single molecular surface morphologies were established from the CIE diagram and atomic force microscopy (AFM) topography, respectively (Fig. S37 and S38†). However, molecular aggregation caused the quenching effect reducing the RTP efficiency of the highly concentrated (5.0 wt%) PMMA-doped film. As expected, the film displayed only a broad fluorescence and no phosphorescence at RT (Fig. S39 and S40†). This result suggests that conformational rigidity in the proximity of phosphors is more important than the environmental matrix rigidity. Moreover, the two distinct triplet emission peaks from the 0.5 wt% PMMA-doped film due to boosted ISC allowed triplet exciton migration from the high lying Tm state to the high lying T1 state via the intramolecular TTET process33 which was further elucidated by recording time-resolved emission spectra (TRES) at RT. The TRES spectra of model PMMA-doped BT2O3Cz exhibited two persistent delayed emission bands in the millisecond (ms) lifetime region within the 400 to 800 nm wavelength region (Fig. 2K). Here the emission band centered at 545 nm was inherited from the high energy level of T3/T4 states, corresponding to fast RTP, whereas the emission at 700 nm stems through the TTET process from T3/T4 to the low energy stable T1 state revealing slow RTP. It has also been observed that these compounds adopted broad and switchable emission colours by changing their excitation source. Upon steady state excitation the emission colour for crystalline powder had CIE coordinates near the blue region (0.24, 0.24) for BT2O3Cz, (0.26, 0.27) for BT2O4Cz, (0.18, 0.11) for BT2O5Cz and (0.19, 0.13) for BT2O6Cz. These CIE values revealed that OSMWLEs turn bluer with increasing chain length. In contrast, a very close pure white CIE (0.33, 0.33) was obtained upon excitation with a microsecond pulse as a source. The obtained phosphorescence CIE values were (0.35, 0.30) for BT2O3Cz, (0.39, 0.33) for BT2O4Cz, (0.40, 0.31) for BT2O5Cz and (0.36, 0.31) for BT2O6Cz (Fig. 2L). The emitters containing shorter and longer chains emit close to pure white, likely due to the strong packing and wider overlaps of dual fluorescence and phosphorescence in the crystalline state that enabled the suppression of triplet non-radiative decays and promoted equilibrium in their states. Furthermore, phosphorescence spectra and decays were recorded for the neat film of model compound BT2O3Cz under heating conditions (298 K, 323 K, 353 K and 373 K). Considering the photostability of OSMWLEs, both TADF and thermally elevated phosphorescence were involved. The phosphorescence spectra of the heated films of BT2O3Cz exhibited distinct TADF and phosphorescence properties with increasing temperatures and relatively higher intensity was observed at 353 K. Notably, at RT (298 K) no distinguishable peaks at 550 and 600 nm were observed which may be due to the quenching effect in neat films. However, peaks were distinguished at elevated temperatures due to the contributions of hot-excitons in their respective emission sources. Moreover, no significant difference in life-time was observed in the film, signifying good photo-stability of BT2O3Cz (Fig. S41†). The overall photophysical properties are summarized in Table 1, which identifies multimode emissions and their vital difference in various states.
| OSMWLEs | λ UV (nm) | λ PL (nm) | λ Phos (nm) | τ avP (ns) | τ avD (µs) | ϕ P | k r (×107 s−1) | k nr (×104 s−1) | K ISC (×106 s−1) | K RISC (×105 s−1) | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Solid | Solid | Solid | Film | Solid | Solid air/inert | Film | Phos. RT | Solid air/RT | Solid air/RT | Solid air/RT | Solid air/RT | |
| a Radiative decay rate [kr = ϕ/τ] calculated from QY and delayed lifetime of crystals at RT in air. b Non-radiative decay rate [knr = (1 − ϕ)/τ] calculated from QY and delayed lifetime of crystals at RT in air. | ||||||||||||
| BT2O3Cz | 350 | 415 | 430 | 430 | 0.94 | 6.77 | 71.73 | 4.5 | 6.72 | 1.40 | 7.5 | 1.47 |
| 530 | 540 | 535 | 68.07 | 2200 | ||||||||
| 567 | 640 | 700 | ||||||||||
| BT2O4Cz | 320 | 430 | 445 | 430 | 4.09 | 10.89 | 76.85 | 4.4 | 4.08 | 0.87 | 1.53 | 0.24 |
| 540 | 546 | 535 | 18.89 | 2059 | ||||||||
| 610 | 633 | 700 | ||||||||||
| BT2O5Cz | 340 | 420 | 420 | 430 | 5.27 | 21.65 | 85.57 | 5.3 | 2.44 | 0.40 | 1.25 | 0.46 |
| 540 | 615 | 535 | 30.76 | 2200 | ||||||||
| 700 | ||||||||||||
| BT2O6Cz | 330 | 415 | 450 | 430 | 3.11 | 20.72 | 74.24 | 13.3 | 6.14 | 0.42 | 0.75 | 0.48 |
| 538 | 630 | 535 | 29.12 | 2270 | ||||||||
| 700 | ||||||||||||
Mechanism for multi-emission
Furthermore, to understand the actual origin of all the independent emissions observed for solids and thin films of BT2OxCZ emitters, steady state PL spectra, PLQY and TRPL emission of neat films of each of the moieties Cz, BT2OH and a mixture (Cz:BT2OH = 1:1) were recorded. This experiment also facilitates discrimination of all independent excited states involved in the fluorescence and phosphorescence (Fig. S42†). Steady state PL was observed at 375, 420 and 400 nm with PLQY values of 7.79%, 4.61% and 9.54% for Cz, BT2OH and mixtures (Cz:BT2OH = 1:1), respectively. These results clearly depict that every individual moiety contributed towards 1π–π*, 1n–π* and 1ICT transitions independent of the singlet emission. Furthermore, average lifetime values of 3.0, 0.26 and 1.53 ns with biexponential fit results were observed for Cz, BT2OH and the mixture (Cz:BT2OH = 1:1), from prompt and delayed species, respectively.
To precisely obtain insights on the multi-emission sources at 430, 550 and 700 nm, we hypothesized first that the 430 nm emission resulted from higher lying singlets. To verify this, excitation wavelength-dependent emission studies were performed for BT2O3Cz and BT2O6Cz. A series of excitation wavelengths were used to excite the luminogens, and the PL spectra illustrated the bright state emission (λem = 430 nm) which disappears when excitation shifted to a longer wavelength (λex = 300 to 500 nm) from normal excitation (λex = ∼320 nm) (Fig. S43 and S44†). Moreover, on excitation at λex = 500 nm a red-shifted dark emission feature emerges at ∼530 and ∼570 nm, indicating that the molecule relaxes to the lower energy dark states viz. S1/S2. By fixing the excitation wavelength to a lower energy (500 nm), weak emission was observed from a lower-lying excited state (the dark state, which in this scenario is S1). This experiment proved that the main emission is from an energy level higher than S1, while S1 is a weakly emissive one and corroborates the existence of dark states via SOKR.45 Further, viscosity-induced enhanced fluorescence of BT2OxCz (x = 3 and 6) was also studied in various micro-viscous environments where a binary mixture, glycerol (Gly) and methanol, was used to regulate the viscosity of the medium (Fig. S45 and S46†). The emission maxima initially shifted to longer wavelengths with increasing Gly fraction (fGly) due to ICT. However, tenfold enhanced emission intensity was observed at 99% fGly, compared to the methanol solution. Since, in the low viscous medium, excitation to the emissive excited state followed by rotation about the rotor axis significantly decreases the energy gap between the emissive and dark excited states, it resulted in a rapid internal conversion (IC) to S1, while in a highly viscous medium or in the aggregated state, the non-radiative deactivation of the twisted complex is circumvented by suppressing IC from the >S1 state to the dark S1 state, due to restricted rotation thereby resulting in huge fluorescence enhancement.
Further, to shed light on the 550 nm emission, carbazole units containing alkyl chains were taken independently, resulting in ultra-long room temperature phosphorescence emission (URTP)4,31 of up to several seconds afterglow (removing UV-excitation) for carbazole moieties containing four, five and six carbon chains, respectively (Fig. S47 including live Videos SIV1-URTP-1,3CzBr, SIV2-URTP-1,4CzBr, SIV3-URTP-1,5CzBr and SIV4-URTP-1,6CzBr respectively†). Moreover, steady state PL spectra of 1, nCzBr exhibited a sharp prompt fluorescence band near 375 nm and a shoulder phosphorescence peak at 550 nm. Additionally, phosphorescence spectra recorded in air showed a very distinct band centered at ∼550 nm with lifetime values of 95.1 µs, 187.5 ms, 47.57 ms and 126.2 ms for 1,3CzBr, 1,4CzBr, 1,5CzBr and 1,6CzBr, respectively. More interestingly, the fast RTP at ∼550 nm emission of BT2OxCz exactly matched with the URTP emission of bromo alkylated carbazole derivatives.
Notably, it is reported that URTP emission can be observed due to the presence of trace amounts of isomeric impurities [1H-benzo[f]indole (Bd)] of commercial carbazole derivatives.31 Therefore, the fast phosphorescence emission from the present BT2OxCz emitter is also inherited because of the impurity effect confirmed by HPLC analysis (Fig. 3A, B and S48†).
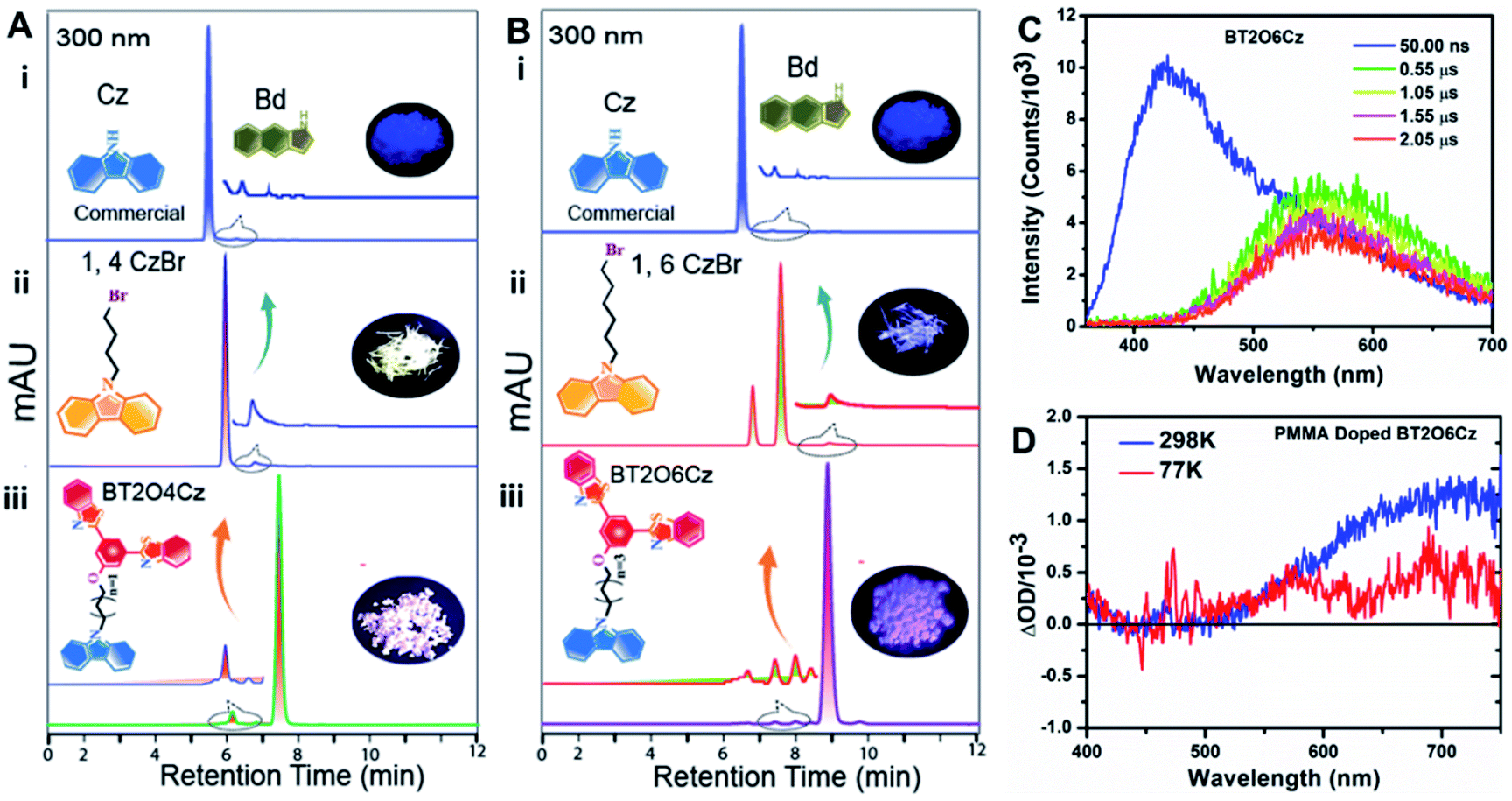 | ||
| Fig. 3 Isomeric impurity effect on the solid-state emission mechanism: (A and B) High-performance liquid chromatography (HPLC) spectra of commercial Cz (i) 1,4CzBr and 1,6CzBr crystals (ii) and BT2O4Cz and BT2O6Cz crystals (iii) monitored at 300 nm with acetonitrile as eluent. All the crystals up to the final compound inherit an impurity peak of BD compounds. Chemical structures of all the compounds are shown in the inset photograph of each crystal captured under a 365 nm UV-lamp. (C) Laser induced fluorescence (LIF) spectra of BT2O6Cz acquired at RT in LP980 at varying delays after the pump pulse (color coding in the graph). (D) Transient absorption (TA) spectra of the PMMA doped BT2O6Cz spin-coated film (0.5%) acquired with the ICCD at 298 K (blue) and 77 K (red). | ||
The hypothesis was further confirmed by laser-induced fluorescence (LIF) spectra and transient absorption (TA) study. We recorded LIF spectra for BT2O6Cz at 77 K and RT from LP980 at varying delays after the pump pulse (Fig. 3C and S49–S51†). The LIF spectra demonstrated that on moving from ns to µs delay, distinctive phosphorescence was observed in the range of 500–700 nm. Further, TA study was conducted with the PMMA doped spin-coated film (0.5%) of BT2O6Cz, and two distinct positive transient absorption signals appeared at ∼550 and ∼700 nm at 77 K while at RT both peaks increased and the 700 nm TA signal dominated (Fig. 3D). Besides, kinetic TA trace also exhibited an exponential decay profile (Fig. S53B†). These results clearly depicted that the trace amounts of alkylated carbazole isomeric impurities generate charge-separated states (hot-excitons) and result in fast and stable phosphorescence at 550 nm. Consecutively, the hotter excitons (emissive higher-lying triplet excited states T3/T4) are likely feeding the lowest T1 state via TTET at RT. Substantially, the communication is cut off at 77 K, indicative of such higher triplet excited states that are being separated from the lowest T1.46 At the same time, ∼700 nm emission has also been rationalized as due to due to aggregation-induced supramolecular non-covalent interactions evolving alkyl chains furnishing tight molecular packing via dimerization and circumventing non-radiative decays in the solid state. To investigate the intrinsic insights and support the experimentally obtained multimode emission results, quantum-mechanical calculations were performed for BT2O3Cz and BT2O6Cz from their optimized single crystal structure geometry using the Gaussian 16 program.47 Density functional theory (DFT) and time dependent density functional theory (TD-DFT) were performed at the B3LYP/6-31G(d,p) level up to the 10th state at their different spin multiplicities (Fig. 4A, B and S54†). Frontier molecular orbital (FMO) results displayed well separated electron density distributions in their respective HOMO and LUMO with a wider band gap profile (3.61/3.48 eV) depicting TSCT behaviour of these emitters (Fig. 4A(i) and B(i)). Nonetheless, zero energy splitting (0.02 and 0.12 eV) between S1 and T3/T4 strongly favours an efficient RISC process, where, relatively larger differences of S1 (3.24, 3.03 eV) and T1 (2.74, 2.39 eV) lead to moderate ΔEST values of 0.50 and 0.71 eV for BT2O3Cz and BT2O6Cz (Fig. 4A(ii) and B(ii)), which corresponds to the basic prerequisite for displaying a simultaneous TADF and RTP emission.19,20
 | ||
| Fig. 4 Computational calculations: (A(i) and B(i)) HOMO and LUMO density distributions with the corresponding band gap profiles, (A(ii) and B(ii)) adiabatic energy transitions dependent oscillator strength (f) and possible manifold active ISC channels and energy splitting (ΔEST) for BT2O3Cz and BT2O6Cz from Gaussian 16 package at the B3LYP-6-31G(d,p) level of theory. (C and D) The functions of the reduced density gradient (RDG) and sign (λ2)ρ isosurface map with an isovalue of 0.5 for BT2O3Cz and BT2O6Cz respectively. | ||
Energy band gap and ΔEST values suggest a strong susceptibility of TADF for shorter chain and intense RTP for longer chain emitters respectively. Further, active manifold ISC channels were also investigated using sequential adiabatic energies related to the ground state for S1, >S1 and T1 to higher triplet states of up to the 10th state and their corresponding major frontier orbital contributions are summarized in Tables S2–S9.† Noticeably, the oscillator strengths (f) at S1–S3 states for both the emitters were found to be zero (f = 0), indicating that the lowest S1 state is a non-emissive state. Large f values were observed beyond S4/S5 depicting electronic relaxation from higher-lying states (>S4) contributing an emissive state which is unusual and reverse of conventional and many experimental results. Moreover, ambient delayed fluorescence or phosphorescence emission by harvesting the triplet state is a very difficult process, as most of the organic molecules undergo S1 (π–π*) → T1 (π–π*), a forbidden ISC transition. However, in the present molecules, natural transition orbital (NTO) calculations suggested that S1 and >S1 have pure 1ICT and 1LE (π–π*) characters while T1 and T3 have a 3LE (n–π*) character (Fig. S55†). Therefore, the multimode emissions by the involved NTOs corresponding to higher excited states producing 1ICT → 3LE and 1LE → 3LE/3ICT transitions (allowed transition) within singlet and triplet (Sn and Tm) manifolds result in substantially enhanced SOC thereby leading to TADF and RTP simultaneously. These theoretical findings are able to rationalize the simultaneous process of TADF and RTP that occurred from higher-lying states (hot-excitons).48 Furthermore, to reveal the dissimilar intramolecular non-covalent interactions which induced emission from excited states, the functions of the reduced density gradient (RDG) and sign (λ2)ρ were calculated for BT2O3Cz and BT2O6Cz using the TD-DFT geometry with Multiwfn software.49 RDG analysis (Fig. 4C and D) confirms the presence of obvious intramolecular attractive interactions (green region) and larger steric hindrance (brown region) between D–A and long alkyl chain segments in BT2O6Cz which could effectively restrict the intramolecular vibrations and prevent energy loss of the excited molecules, revealing more RTP susceptibility. In contrast, the short alkyl chain linked conformation of BT2O3CZ exhibits relatively weak intramolecular interactions, which is a common phenomenon found in general TADF molecules.50
Supramolecular structures and luminescence
To gain a deeper understanding of supramolecular interactions and molecular packing arrangement induced TADF and RTP emission in the solid state, single-crystal X-ray structures were analyzed carefully. Crystal structures of BT2O3Cz (CCDC 1969742) and BT2O6Cz (CCDC 1969743) were obtained by very slow evaporation of chloroform/hexane (8:2) solvents. Unfortunately, the crystal structures of BT2O4Cz and BT2O5Cz could not be solved. Two needle-like crystals of BT2O3Cz and BT2O6Cz exhibited a monoclinic and orthorhombic crystal system with P21/c and Pca21 space groups (crystal structure information summarized in Table S10†). Both the molecules exhibited multiple intra- and intermolecular interactions via hydrogen bonding due to the presence of N/O/S atoms and other non-covalent interactions such as weak CH–π, π–π, and through-space CT (Fig. 5A and B). These interactions enable formation of a rigid and dense 3D network which further assists to restrict vibrational relaxation and prevents exciton quenching.51 The intramolecular hydrogen bonds involving S and N atoms were close to active hydrogen on the phenyl in the acceptor unit (C–S⋯H: 2.74 Å; C–N⋯H: 2.55 Å in BT2O3Cz and C–S⋯H: 2.64 Å; C–N⋯H: 2.57 Å in BT2O6Cz), and enhance the co-planarity of the three units [two benzothiazole (BT) and central phenyl core], evidenced by their short contact distances and torsions angles of 170.60° and 173.50°. Other intramolecular interactions involved the active hydrogen present in alkyl chain with the π-cloud of the donor carbazole (Cz) unit (C–H⋯π: 2.83 Å; π⋯C–H: 2.72 Å in BT2O3Cz and C–H⋯π: 2.80 Å; π⋯C–H: 2.75 Å in BT2O6Cz). Additionally, multiple intermolecular interactions were also apparent in both the crystal structures. Intermolecular hydrogen bonding by a N atom present in BT and the O atom present in the alkoxy chain in BT2O3Cz (C–H⋯N: 2.66 Å, C–H⋯O: 2.70 Å) also interacts non-covalently with the nearby set of molecules creating a rigid 3D molecular framework, thereby reducing non-radiative deactivation. However, BT2O6Cz exhibited a single intermolecular hydrogen bond, involving the O atom in the alkoxy chain (C–H⋯O: 2.64 Å). Unlike hydrogen bonds, many other intermolecular strong interactions were also observed due to the presence of active hydrogen atoms, while the BT2O6Cz showed more intermolecular interactions involving carbazole (Cz), alkyl chain hydrogens and the S atom in BT (C–H⋯π: 2.85 Å; 2.86 Å and π⋯S: 3.31 Å) than BT2O3Cz which displayed only C–H⋯π: 2.79 Å interaction. Besides, very weak π–π stacking between the two BT units in BT2O6Cz (3.35 Å, 3.36 Å) leads to a dimeric structure, whereas, BT2O3Cz exhibits a J-aggregated structure (slip angle 43.71° between BT cores, 37.56° between Cz cores). BT2O6Cz exhibits an H-aggregated structure (slip angle 82.61° between BT cores, 78.48° between Cz cores) (Fig. 5C and D), and this dimeric form is stabilized by multiple non-covalent interactions and separated electron clouds of individual D–A units (detailed SC-structural analysis summarized in Table S11†). These key factors enabled considerably weak π–π stacking, leading to strengthening of TSCT interactions. Additionally, their bulk crystal tight packing shows that H-aggregated BT2O6Cz leads to overall denser molecular packing rather than J-aggregated BT2O3Cz in their crystalline state, which suppressed vibrational deactivation and activated the radiative channels52 (Fig. 5E and F). Simulation studies by electrostatic potential (ESP) mapping and their corresponding two-dimensional (2D) fingerprint plots (Crystal Explorer 3.1 software) substantiated the packing and interactions (π–π stacking and intermolecular HB interactions). The ESP mapped on the Hirshfeld surface of BT2O3Cz/BT2O6Cz exhibited bright red spots on the strongly linked hydrogen bonds which clearly demonstrated effective interactions, influencing their emission properties53 (Fig. 5G and H). The 2D fingerprint plot suggested that the overall interactions for BT2O6Cz were higher (71%) than those for BT2O3Cz (65%) (Fig. 5I and J). To clarify their distinct packing motifs, schematic illustrations of J- and H-aggregation with their corresponding energy profile as a function of slip angles in the dimeric structure are presented (Fig. 5K). Therefore, the rigid interlocked BT2O3Cz and the involvement of the donor (Cz) group and longer alkyl chain interactions in BT2O6Cz inhibited internal conversions and photochemical reactions which facilitated emissions from higher lying excited states, resulting in enhanced delayed fluorescence and phosphorescence at RT. SC-XRD analysis Fig. S56–S58.† The X-ray powder diffraction (XPRD) peaks of crystalline powder obtained from as-synthesized pristine compounds exactly matched with the simulated patterns obtained from SCXRD-structure analysis (Fig. 5L). The BT2O3Cz crystal grew preferentially along [011], whereas BT2O6Cz grew along [200] with planar layered structures. The BT2O6Cz exhibited multiple diffraction peaks in the low d-space region (θ = 20°–30°) (highlighted by the orange dotted line) compared to BT2O3Cz confirming more non-covalent interactions and high packing density due to alkyl chain and dimer formations. The calculated attachment energy54 suggests a rod like growth morphology at different crystal facets (001), (002), (100) and (11![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) in BT2O3Cz with high attachment energy and (200), (201), (20) and (101) in BT2O6Cz displayed relatively less attachment energy for the binary growth model. Moreover, (001) and (002) are the primary crystal growth facet for BT2O3Cz while (200) is for BT2O6Cz (Fig. 5M and N and Tables S12 and S13†). Further, both crystals were thermally stable up to ∼400 °C (Fig. S59†). Overall, these studies reveal the contribution of denser supramolecular non-covalent interactions, self-assembly induced solid-state packing, growth morphology and thermal stability profoundly to their ESD and multi-emissions. Further, the H-aggregated molecular packing greatly impedes higher triplet state stabilization thereby favouring phosphorescence for longer chain BT2O6Cz.
) in BT2O3Cz with high attachment energy and (200), (201), (20) and (101) in BT2O6Cz displayed relatively less attachment energy for the binary growth model. Moreover, (001) and (002) are the primary crystal growth facet for BT2O3Cz while (200) is for BT2O6Cz (Fig. 5M and N and Tables S12 and S13†). Further, both crystals were thermally stable up to ∼400 °C (Fig. S59†). Overall, these studies reveal the contribution of denser supramolecular non-covalent interactions, self-assembly induced solid-state packing, growth morphology and thermal stability profoundly to their ESD and multi-emissions. Further, the H-aggregated molecular packing greatly impedes higher triplet state stabilization thereby favouring phosphorescence for longer chain BT2O6Cz.
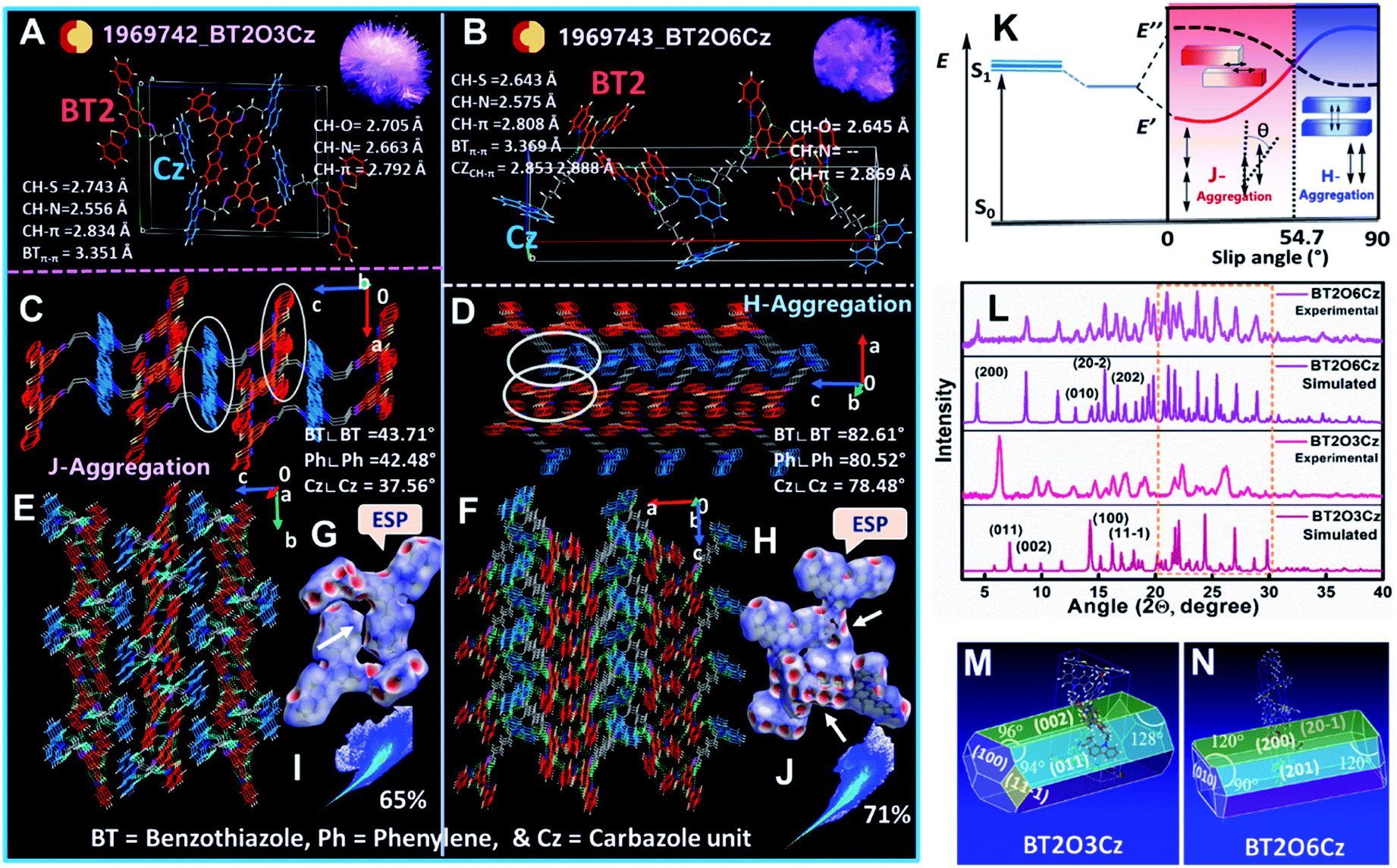 | ||
| Fig. 5 SC-XRD and PXRD analysis. (A and B) Single-crystal XRD structure of BT2O3Cz and BT2O6Cz OSMWLEs in the unit cell (inset: single crystals images captured under a 365 nm UV lamp). (C and D) Side views of bulk stacked architectures, relative distances and angles from each constituent are shown to exhibit distinct J-type and H-type aggregation patterns for BT2O3Cz and BT2O6Cz OSMWLEs. (E and F) Top view of the bulk intramolecular packing of rigid BT2O3Cz and comparatively highly dense packed intermolecularly rigid BT2O6Cz OSMWLE. (G–J) Electrostatic potential (ESP) and 2D finger plot mapped on the Hirshfeld surface in the BT2O3Cz and BT2O6Cz OSMWLEs. (K) Schematic illustration of J- and H-aggregation with their energy profile as a function of slip angles in dimeric structures. (L) Powder X-ray diffraction (PXRD) characterization of BT2O3Cz and BT2O6Cz. (M and N) Simulated crystal growth. | ||
The densely packed crystalline state is a key strategy to realize triplet emission for most organic phosphorescent materials.55 The solid state features of BT2O3Cz and BT2O6Cz motivated us to probe the triplet-harvesting optical wave-guiding properties in their single crystal states. We performed single-crystal micro-spectroscopy studies on crystals of BT2O3Cz and BT2O6Cz of length ≈202.9 µm and ≈75.6 µm.
Optical waveguide study
A 405 nm diode laser (continuous wave) was used as an excitation source. Excitation at the left terminal of the microcrystal generated bright greenish-white luminescence which subsequently propagated and out-coupled at the opposite terminal (Fig. 6A and B). The microcrystals were excited at different points along their long axes (202.9, 163.2, 132.3, 101.4, 67.6, 32.3 and 0 µm for BT2O3Cz and 75.6, 63.1, 46.5, 39, 27, 15 and 0 µm for BT2O6Cz). The recorded broad band PL spectra covered the 500–750 nm region and notably, two distinct phosphorescence peaks appeared at 550 and 600 nm. These peaks exactly match with the triplet emission of crystalline powders of OSMWLEs. Noticeably, the spectral profiles did not shift with the excitation position. However, with an increase of light propagation distance, the PL intensity gradually decreased due to propagation-induced optical loss (Fig. 6C and D).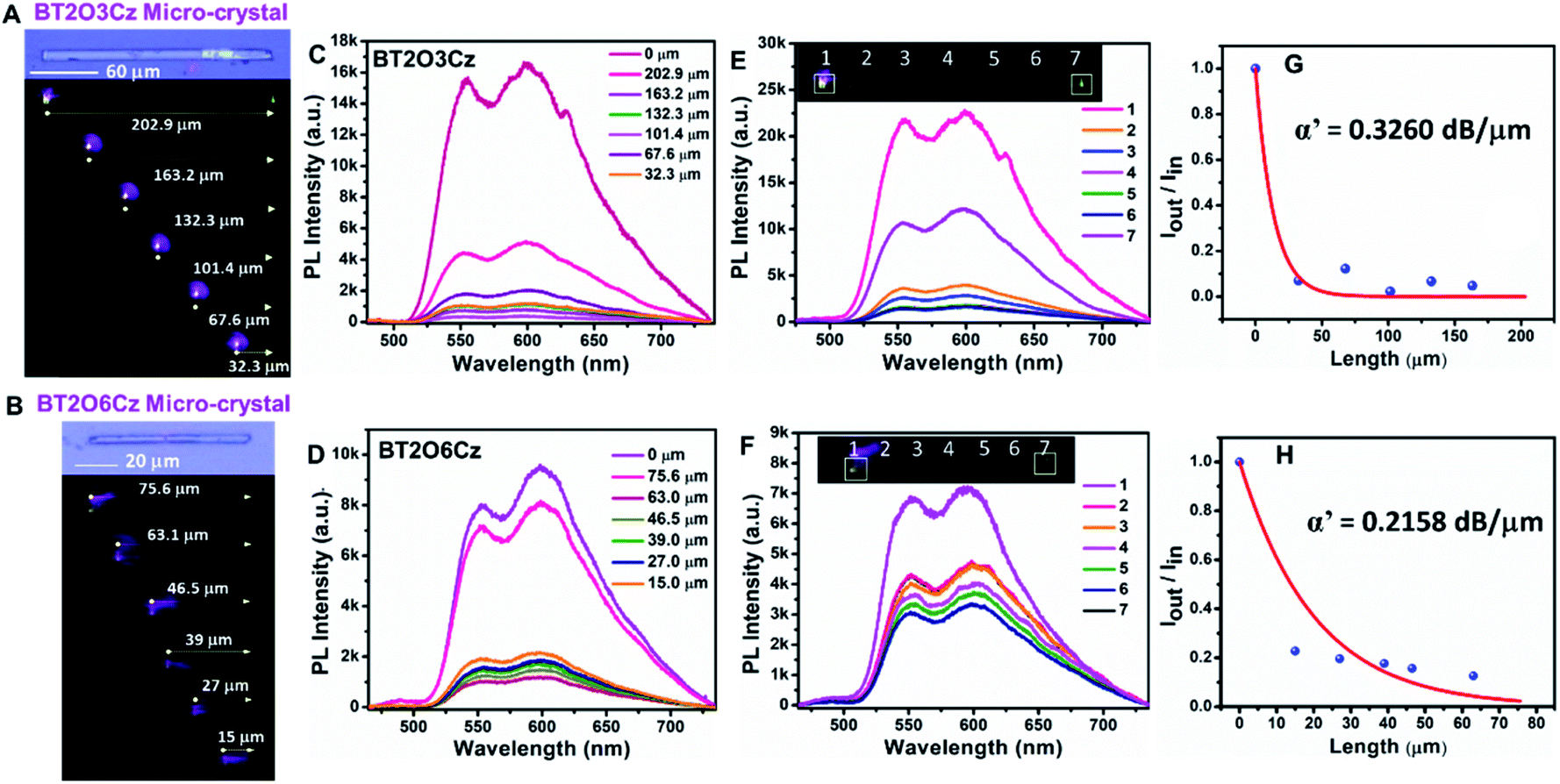 | ||
| Fig. 6 Optical waveguide study. (A and B) Optical and PL images of microcrystals of BT2O3Cz and BT2O6Cz showing excitation at different positions with a 405 nm laser. (C and D) PL spectra recorded at the end of the BT2O3Cz and BT2O6Cz microcrystals. (E and F) Spatially resolved PL spectra obtained at each point keeping the excitation position constant. (G and H) Estimation of the optical loss coefficient (α′) from the plot of Iout/Iinversus the light propagation distance. | ||
By fitting of an exponential decay function, Iout/Iin = exp(−αD), where D is the distance between the illuminated position and the signal collection point of the crystal, the loss coefficient α′ (where α′ = 4.34α) was determined to be as low as 0.3260 dB µm−1 and 0.2158 dB µm−1 for BT2O3Cz and BT2O6Cz respectively (Fig. 6E and F).56 These data suggest good optical wave-guiding performance of these crystalline organic phosphorescent micro-rods (Fig. 6G and H). Notably, these α′ values are lower than those of most of the state-of-the art 1D optical waveguide materials such as solvate crystals, metal–organic frameworks and hybrid perovskite crystals.57,58 These results confirm the triplet-harvesting light propagation ability of microcrystals suitable for photonic device applications.
Conclusions
In summary, four through-space charge transfer (TSCT) OSMWLE materials, BT2OxCz (alkyl spacer x = 3, 4, 5 and 6) were reported, which displayed multi-functional properties involving hot-exciton contributions i.e. high lying Sn (singlet) and Tm (triplet) manifolds via charge-separated states. The bridging alkyl chain-length between D–A and a trace amount of carbazole isomer plays a crucial role in controlling the excited state relaxation processes and fine tunes the emission behaviour in the solid state and PMMA doped film states, respectively. The integrated heavy atom free and amorphous OSMWLEs with shorter chain BT2O3Cz, BT2O4Cz and BT2O5Cz displayed TADF-RTP while longer chain BT2O6Cz was more susceptible to exhibiting intense RTP under ambient conditions. Notably, two distinct fast/slow phosphorescence emissions were realized where the fast phosphorescence was observed due to trace amounts of isomeric impurities of commercial carbazole, while H-/J-aggregation results in slow phosphorescence. Theoretical studies provided valuable information on the effective manifold ISC channel and narrow ΔEST values (0.02/0.50 and 0.12/0.71 eV) strongly supporting the experimental results in the isolated and dimeric form of the emitters from their single crystal geometry. Transient PL studies revealed the co-existence of thermally elevated fluorescence-phosphoresce at the same time. Remarkably, the single-crystal structure illustrates that aggregation induced densely packed supramolecular crystals of BT2O3Cz and BT2O6Cz exhibited self-guided triplet-harvesting light propagation capability along the primary growth direction with a low optical loss coefficient. Overall, this new approach presents a unique strategy to obtain efficient photonic materials by stressing the importance of modulating and optimizing the bridging groups in achieving multi-emissions in a single molecule. The multifunctional OSMWLEs from a single component system demonstrate the first example of simultaneous observation of locally excited emission, TADF, RTDP and optical wave-guiding properties.Data availability
All experimental, computational and crystallographic data are available in the ESI.†Author contributions
D. B. designed, synthesized and conducted all the experiments, analysis, computational calculations, and prepared the manuscript. M. A. and R. C. performed the optical waveguide study and were involved in manuscript preparation. P. K. I. supervised the project, and was involved in planning, analysis and manuscript preparation.Conflicts of interest
There are no conflicts to declare.Note added after first publication
This version replaces the manuscript published on 18th July 2022 which contained errors in Table 1. The RSC apologises for any confusion.Acknowledgements
Financial grants from the Department of Science and Technology, New Delhi, India project no. DST/CRG/2019/002614, Deity, India no. 5(1)/2022-NANO, ICMR, Grant no. 5/3/8/20/2019-ITR and Max-Planck-Gesellschaft IGSTC/MPG/PG(PKI)/2011A/48 are acknowledged. R. C. acknowledges ERB (CRG-2018/001551) and the UoH-IoE/RC/RC1-003 project [MHRD F11/9/2019-U3(A)] for financial support. We are thankful to Prof. Aditya Narayan Panda for valuable discussions on the theoretical calculations. We also thank the Param-Ishan supercomputing facility, Centre for Nanotechnology, Department of Chemistry and CIF-IITG for computing and instrument facilities.Notes and references
- Y. Liu, M. Nishiura, Y. Wang and Z. Hou, J. Am. Chem. Soc., 2006, 128, 5592–5593 CrossRef CAS PubMed.
- (a) G. M. Farinola and R. Ragni, Chem. Soc. Rev., 2011, 40, 3467–3482 RSC; (b) D. Das, P. Gopikrishna, D. Barman, R. B. Yathirajula and P. K. Iyer, Nano Convergence, 2019, 6, 31 CrossRef PubMed.
- P. Lehner, C. Staudinger, S. M. Borisov and I. Klimant, Nat. Commun., 2014, 5, 4460 CrossRef CAS PubMed.
- L. Bian, H. Shi, X. Wang, K. Ling, H. Ma, M. Li, Z. Cheng, C. Ma, S. Cai, Q. Wu, N. Gan, X. Xu and Z. An, J. Am. Chem. Soc., 2018, 140, 10734–10739 CrossRef CAS PubMed.
- T. Maldiney, A. Bessière, J. Seguin, E. Teston, S. K. Sharma, B. Viana, A. J. J. Bos, P. Dorenbos, M. Bessodes, D. Gourier, D. Scherman and C. Richard, Nat. Mater., 2014, 13, 418–426 CrossRef CAS PubMed.
- W.-L. Zhou, Y. Chen, Q. Yu, H. Zhang, Z.-X. Liu, X.-Y. Dai, J.-J. Li and Y. Liu, Nat. Commun., 2020, 11, 4655 CrossRef CAS PubMed.
- (a) H. Liu, Z. Lu, Z. Zhang, Y. Wang and H. Zhang, Angew. Chem., Int. Ed., 2018, 57, 8448–8452 CrossRef CAS PubMed; (b) Z. Lu, Y. Zhang, H. Liu, K. Ye, W. Liu and H. Zhang, Angew. Chem., Int. Ed., 2020, 59, 4299–4303 CrossRef CAS PubMed; (c) S. Hayashi and T. Koizumi, Angew. Chem., Int. Ed., 2016, 55, 2701–2704 CrossRef CAS PubMed; (d) L. Catalano, D. P. Karothu, S. Schramm, E. Ahmed, R. Rezgui, T. J. Barber, A. Famulari and P. Naumov, Angew. Chem., Int. Ed., 2018, 57, 17254–17258 CrossRef CAS PubMed; (e) M. Annadhasan, D. P. Karothu, R. Chinnasamy, L. Catalano, E. Ahmed, S. Ghosh, P. Naumov and R. Chandrasekar, Angew. Chem., Int. Ed., 2020, 59, 13821–13830 CrossRef CAS PubMed; (f) D. P. Karothu, G. Dushaq, E. Ahmed, L. Catalano, S. Polavaram, R. Ferreira, L. Li, S. Mohamed, M. Rasras and P. Naumov, Nat. Commun., 2021, 12, 1326 CrossRef CAS PubMed; (g) M. P. Zhuo, J. J. Wu, X. D. Wang, Y. C. Tao, Y. Yuan and L. S. Liao, Nat. Commun., 2019, 10, 9 CrossRef PubMed; (h) J. Ravi, M. Annadhasan, A. V. Kumar and R. Chandrasekar, Adv. Funct. Mater., 2021, 31, 2100642 CrossRef CAS; (i) J. Ravi and R. Chandrasekar, Adv. Opt. Mater., 2021, 9, 2100550 CrossRef CAS; (j) J. Ravi, A. Vinod Kumar, D. P. Karothu, M. Annadhasan, P. Naumov and R. Chandrasekar, Adv. Funct. Mater., 2021, 31, 2105415 CrossRef CAS.
- (a) S. Shao, J. Ding, L. Wang, X. Jing and F. Wang, J. Am. Chem. Soc., 2012, 134, 20290–20293 CrossRef CAS PubMed; (b) P. Gopikrishna, P. D. Das, L. R. Adil and P. K. Iyer, J. Phys. Chem. C, 2017, 121, 18137–18143 CrossRef CAS.
- (a) Y. Yang, M. Lowry, C. M. Schowalter, S. O. Fakayode, J. O. Escobedo, X. Xu, H. Zhang, T. J. Jensen, F. R. Fronczek, I. M. Warner and R. M. Strongin, J. Am. Chem. Soc., 2006, 128, 14081–14092 CrossRef CAS PubMed; (b) S. Basak, Y. S. L. V. Narayana, M. Baumgarten, K. Müllen and R. Chandrasekar, Macromolecules, 2013, 46, 362–369 CrossRef CAS; (c) Y. S. L. V. Narayana, M. Baumgarten, K. Müllen and R. Chandrasekar, Macromolecules, 2015, 48, 4801–4812 CrossRef CAS.
- Y.-H. Chen, K.-C. Tang, Y.-T. Chen, J.-Y. Shen, Y.-S. Wu, S.-H. Liu, C.-S. Lee, C.-H. Chen, T.-Y. Lai, S.-H. Tung, R.-J. Jeng, W.-Y. Hung, M. Jiao, C.-C. Wu and P.-T. Chou, Chem. Sci., 2016, 7, 3556–3563 RSC.
- H. Naito, K. Nishino, Y. Morisaki, K. Tanaka and Y. Chujo, Angew. Chem., Int. Ed., 2017, 56, 254–259 CrossRef CAS PubMed.
- D. K. Maiti, R. Bhattacharjee, A. Datta and A. Banerjee, J. Phys. Chem. C, 2013, 117, 23178–23189 CrossRef CAS.
- K.-C. Tang, M.-J. Chang, T.-Y. Lin, H.-A. Pan, T.-C. Fang, K.-Y. Chen, W.-Y. Hung, Y.-H. Hsu and P.-T. Chou, J. Am. Chem. Soc., 2011, 133, 17738–17745 CrossRef CAS PubMed.
- Z. Xie, C. Chen, S. Xu, J. Li, Y. Zhang, S. Liu, J. Xu and Z. Chi, Angew. Chem., Int. Ed., 2015, 54, 7181–7184 CrossRef CAS PubMed.
- C. A. Masmanidis, H. H. Jaffe and R. L. Ellis, J. Phys. Chem., 1975, 79, 2052–2061 CrossRef CAS.
- M. A. El-Sayed, Acc. Chem. Res., 1971, 4, 23–31 CrossRef CAS.
- D. Lee, O. Bolton, B. C. Kim, J. H. Youk, S. Takayama and J. Kim, J. Am. Chem. Soc., 2013, 135, 6325–6329 CrossRef CAS PubMed.
- H. A-Attar and A. Monkman, Adv. Funct. Mater., 2012, 22, 3824–3832 CrossRef.
- L. Zhan, Z. Chen, S. Gong, Y. Xiang, F. Ni, X. Zeng, G. Xie and C. Yang, Angew. Chem., Int. Ed., 2019, 58, 17651–17655 CrossRef CAS PubMed.
- H.-T. Feng, J. Zeng, P.-A. Yin, X.-D. Wang, Q. Peng, Z. Zhao, J. W. Y. Lam and B. Z. Tang, Nat. Commun., 2020, 11, 2617 CrossRef CAS PubMed.
- Z. Mao, Z. Yang, Y. Mu, Y. Zhang, Y.-F. Wang, Z. Chi, C.-C. Lo, S. Liu, A. Lien and J. Xu, Angew. Chem., Int. Ed., 2015, 54, 6270–6273 CrossRef CAS PubMed.
- L. Huang, L. Liu, X. Li, H. Hu, M. Chen, Q. Yang, Z. Ma and X. Jia, Angew. Chem., Int. Ed., 2019, 58, 16445–16450 CrossRef CAS PubMed.
- X.-K. Chen, D. Kim and J.-L. Brédas, Acc. Chem. Res., 2018, 51, 2215–2224 CrossRef CAS PubMed.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- Y. Im, M. Kim, Y. J. Cho, J.-A. Seo, K. S. Yook and J. Y. Lee, Chem. Mater., 2017, 29, 1946–1963 CrossRef CAS.
- A. Pershin, D. Hall, V. Lemaur, J.-C. Sancho-Garcia, L. Muccioli, E. Zysman-Colman, D. Beljonne and Y. Olivier, Nat. Commun., 2019, 10, 597 CrossRef CAS PubMed.
- O. Bolton, K. Lee, H.-J. Kim, K. Y. Lin and J. Kim, Nat. Chem., 2011, 3, 205 CrossRef CAS PubMed.
- W. Zhao, Z. He, J. W. Y. Lam, Q. Peng, H. Ma, Z. Shuai, G. Bai, J. Hao and B. Z. Tang, Chem, 2016, 1, 592 CAS.
- H. Han and E.-G. Kim, Chem. Mater., 2019, 31, 6925–6935 CrossRef CAS.
- (a) Y. Xie, Y. Ge, Q. Peng, C. Li, Q. Li and Z. Li, Adv. Mater., 2017, 29, 1606829 CrossRef PubMed; (b) J. Yang, X. Zhen, B. Wang, X. Gao, Z. Ren, J. Wang, Y. Xie, J. Li, Q. Peng, K. Pu and Z. Li, Nat. Commun., 2018, 9, 840 CrossRef PubMed.
- C. Chen, Z. Chi, K. C. Chong, A. S. Batsanov, Z. Yang, Z. Mao, Z. Yang and B. Liu, Nat. Mater., 2020, 20, 175 CrossRef PubMed.
- C. Chen, K. C. Chong, Y. Pan, G. Qi, S. Xu and B. Liu, ACS Mater. Lett., 2021, 3, 1081–1087 CrossRef CAS.
- W. Zhao, T. S. Cheung, N. Jiang, W. Huang, J. W. Y. Lam, X. Zhang, Z. He and B. Z. Tang, Nat. Commun., 2019, 10, 1595 CrossRef PubMed.
- S. Hayashi, S.-Y. Yamamoto, D. Takeuchi, Y. Ie and K. Takagi, Angew. Chem., Int. Ed., 2018, 57, 17002–17008 CrossRef CAS PubMed.
- F. Di Benedetto, A. Camposeo, S. Pagliara, E. Mele, L. Persano, R. Stabile, R. Cingolani and D. Pisignano, Nat. Nanotechnol., 2008, 3, 614–619 CrossRef CAS PubMed.
- J. M. Halabi, E. Ahmed, L. Catalano, D. P. Karothu, R. Rezgui and P. Naumov, J. Am. Chem. Soc., 2019, 141, 14966–14970 CrossRef CAS PubMed.
- B. Xu, Y. Mu, Z. Mao, Z. Xie, H. Wu, Y. Zhang, C. Jin, Z. Chi, S. Liu, J. Xu, Y.-C. Wu, P.-Y. Lu, A. Lien and M. R. Bryce, Chem. Sci., 2016, 7, 2201–2206 RSC.
- Y. Zhang, Y. Miao, X. Song, Y. Gao, Z. Zhang, K. Ye and Y. Wang, J. Phys. Chem. Lett., 2017, 8, 4808–4813 CrossRef CAS PubMed.
- B. Xu, H. Wu, J. Chen, Z. Yang, Z. Yang, Y.-C. Wu, Y. Zhang, C. Jin, P.-Y. Lu, Z. PChi, S. Liu, J. Xu and M. Aldred, Chem. Sci., 2017, 8, 1909–1914 RSC.
- Z. He, W. Zhao, J. W. Y. Lam, Q. Peng, H. Ma, G. Liang, Z. Shuai and B. Z. Tang, Nat. Commun., 2017, 8, 416 CrossRef PubMed.
- J. Wang, X. Gu, H. Ma, Q. Peng, X. Huang, X. Zheng, S. H. P. Sung, G. Shan, J. W. Y. Lam, Z. Shuai and B. Z. Tang, Nat. Commun., 2018, 9, 2963 CrossRef PubMed.
- I. Bhattacharjee, N. Acharya, H. Bhatia and D. Ray, J. Phys. Chem. Lett., 2018, 9, 2733–2738 CrossRef CAS PubMed.
- D. Barman, K. Narang, R. Gogoi, D. Barman and P. K. Iyer, J. Mater. Chem. C, 2022, 10, 8536–8583 RSC.
- X.-K. Chen, Y. Tsuchiya, Y. Ishikawa, C. Zhong, C. Adachi and J.-L. Brédas, Adv. Mater., 2017, 29, 1702767 CrossRef PubMed.
- H. Qian, M. E. Cousins, E. H. Horak, A. Wakefield, M. D. Liptak and I. Aprahamian, Nat. Chem., 2017, 9, 83–87 CrossRef CAS PubMed.
- T. Wang, Z. Hu, X. Nie, L. Huang, M. Hui, X. Sun and G. Zhang, Nat. Commun., 2021, 12, 1364 CrossRef CAS PubMed.
- M. J. Frisch, et al., Gaussian 16 Revision C.01, Gaussian, Inc., Wallingford CT, USA, 2016 Search PubMed.
- Y. Xu, P. Xu, D. Hu and Y. Ma, Chem. Soc. Rev., 2021, 50, 1030–1069 RSC.
- T. Lu and F. W. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- Z. Yang, Z. Mao, C. Xu, X. Chen, J. Zhao, Z. Yang, Y. Zhang, W. Wu, S. Jiao, Y. Liu, M. P. Aldred and Z. Chi, Chem. Sci., 2019, 10, 8129–8134 RSC.
- X.-N. Li, M. Yang, X.-L. Chen, J.-H. Jia, W.-W. Zhao, X.-Y. Wu, S.-S. Wang, L. Meng and C.-Z. Lu, Small, 2019, 15, 1903270 CrossRef CAS PubMed.
- Z. An, C. Zheng, Y. Tao, R. Chen, H. Shi, T. Chen, Z. Wang, H. Li, R. Deng, X. Liu and W. Huang, Nat. Mater., 2015, 14, 685–690 CrossRef CAS PubMed.
- Z. Yang, C. Xu, W. Li, Z. Mao, X. Ge, Q. Huang, H. Deng, J. Zhao, F. L. Gu, Y. Zhang and Z. Chi, Angew. Chem., Int. Ed., 2020, 59, 17451–17455 CrossRef CAS PubMed.
- D. Winn and M. F. Doherty, AIChE J., 2000, 46, 1348–1367 CrossRef CAS.
- (a) Y. Li, K. Wang, Q. Liao, L. Fu, C. Gu, Z. Yu and H. Fu, Nano Lett., 2021, 21(7), 3287–3294 CrossRef CAS PubMed; (b) M. Godumala, A. V. Kumar and R. Chandrasekar, J. Mater. Chem. C, 2021, 9, 14115–14132 RSC; (c) H. Liu, Z. Bian, Q. Cheng, L. Lan, Y. Wang and H. Zhang, Chem. Sci., 2019, 10, 227–232 RSC.
- W. Yuan, J. Cheng, X. Li, M. Wu, Y. Han, C. Yan, G. Zou, K. Müllen and Y. Chen, Angew. Chem., Int. Ed., 2020, 59, 9940–9945 CrossRef CAS PubMed.
- B. Lu, X. Fang and D. Yan, ACS Appl. Mater. Interfaces, 2020, 12, 31940–31951 CrossRef CAS PubMed.
- S. Du, S. Ma, B. Xu and W. Tian, J. Phys. Chem. Lett., 2021, 12, 9233–9238 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1969742 and 1969743. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc02172b |
| This journal is © The Royal Society of Chemistry 2022 |