
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanistic dichotomy in the solvent dependent access to E vs. Z-allylic amines via decarboxylative vinylation of amino acids†
Samir
Manna
a,
Shunta
Kakumachi
bc,
Kanak Kanti
Das
a,
Youichi
Tsuchiya
 *c,
Chihaya
Adachi
bc and
Santanu
Panda
*a
*c,
Chihaya
Adachi
bc and
Santanu
Panda
*a
aDepartment of Chemistry, Indian Institute of Technology Kharagpur, Kharagpur 721302, India. E-mail: spanda@chem.iitkgp.ac.in
bDepartment of Chemistry and Biochemistry, Kyushu University, 744 Motooka, Nishi-ku, Fukuoka 819-0395, Japan
cCenter for Organic Photonics and Electronics Research (OPERA), Kyushu University, 744 Motooka, Nishi-ku, Fukuoka 819-0395, Japan
First published on 21st July 2022
Abstract
The solvent plays an important role in the photophysical properties of donor–acceptor based photocatalysts. The solvent-dependent access to E vs. Z-allylic amines was achieved via decarboxylative vinylation of amino acids with vinyl sulfones. Detailed experimental studies have been conducted to understand the role of the solvent in the reactivity and stereoselectivity of the vinylation reactions.
Introduction
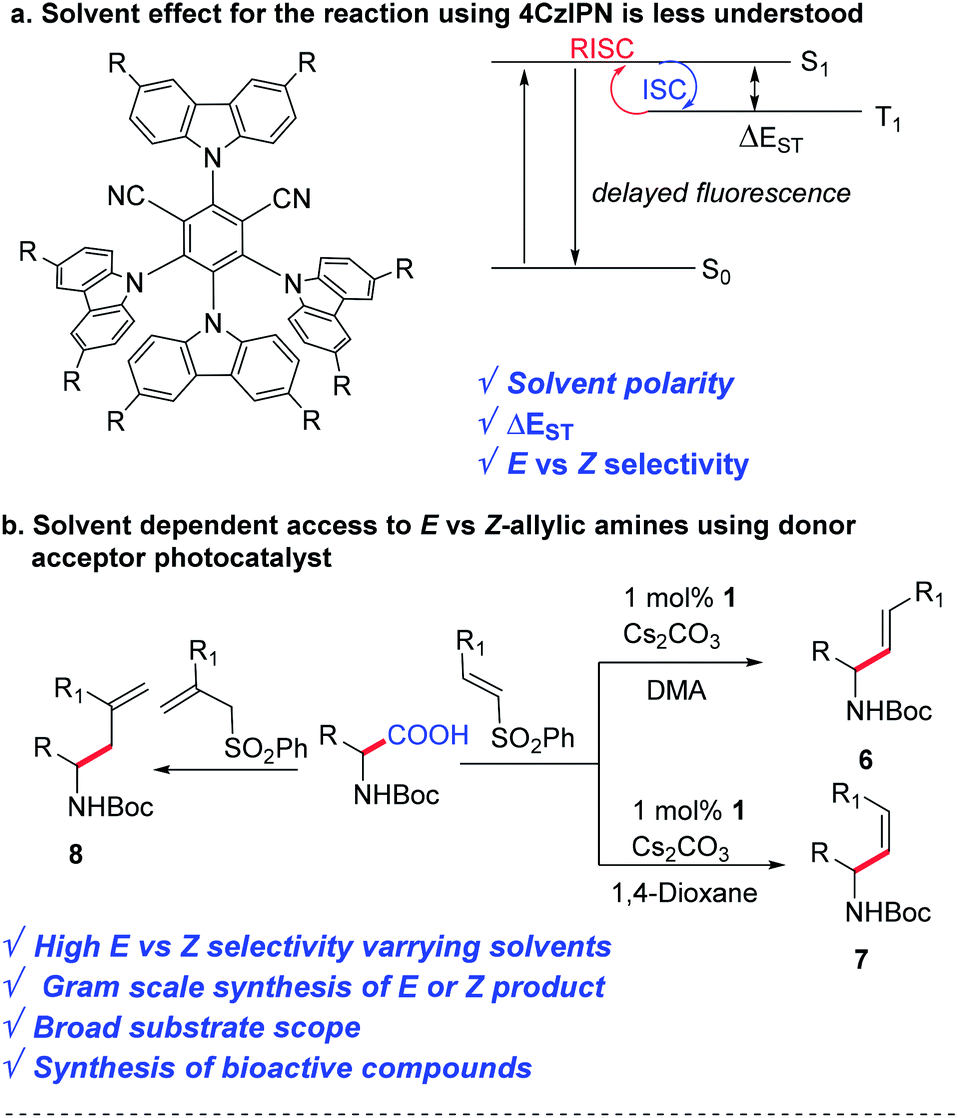
Since the last decade, visible light photoredox catalysis has played an important role in activating small molecules for C–C and C–heteroatom bond formation.1 Iridium- and ruthenium-based inorganic complexes have been commonly used as photocatalysts for many unique and valuable chemical transformations due to their superb photophysical properties.2 However, there has been a paradigm shift in the pharmaceutical industry in conducting high-value organic transformations using transition metal-free catalysts.3 This year the Nobel prize in chemistry was awarded for exemplary work in organocatalysis, suggesting the importance of organic-based catalysts for valuable synthetic transformations.4 Several organic photocatalysts have been developed as a sustainable and cost-effective solution to this problem, which include organic dyes, donor–acceptor materials, carbonyl-based photosensitizers, and many more.5,6In 2012, Uoyama et al. reported a carbazolyl-dicyanobenzene (CDCB) based thermally activated delayed fluorescence (TADF) emitter 4CzIPN, which is comparable to the highly efficient organic light-emitting diodes.7 Such donor–acceptor-based TADF materials have been reported as an alternative to the Ru- or Ir-based photocatalysts due to the similarity of their redox window as well as excited state lifetimes (Scheme 1b).8 Besides the low cost, the easy access to various CDCB derivatives with a wide range of redox potentials, by varying the steric and electronic nature of the donor and acceptor units, has created a toolbox for reaction discovery.9
 | ||
| Scheme 1 Solvent dependent access to E vs. Z-allylic amines. | ||
The solvent plays an important role in photochemical reactions.10 However, the effect of the solvent on the reactivity of 4CzIPN remains poorly understood. Although other groups have observed the difference in reactivities using 4CzIPN in the presence of different solvents,11 a detailed understanding is missing in the literature. While developing a one-pot method for the synthesis of E vs. Z-allylic amines from feedstock amino acids and vinyl sulphone, we observed a huge difference in reactivity and stereo-selectivity in the presence of various polar and non-polar solvents. A detailed experimental study has been conducted to understand their effect, which can be helpful for future reaction discovery using 4CzIPN (Scheme 1).
Allyl amines represent an important and highly versatile class of building blocks for organic synthesis.12 They are essential pharmacophores in numerous marketed drugs. Therefore, various methods have been implemented over the years to access allylic amines.13 Recently, Weaver's group reported uphill catalysis to access Z-allylic amines from E-allylic amines using an Ir(ppy)3 photocatalyst.14 MacMillan's group reported the synthesis of E-allylic amines by the α-vinylation of N-aryl amines and α-amino acids using an Ir-based photocatalyst.15 However, decarboxylative vinylation of amino acids to access either E or Z selective allylic amines using an organophotocatalyst is yet to be discovered. There are other reports on decarboxylative vinylation, via hydroalkylation of alkynes in the presence of transition metals,16 cross-coupling with vinyl precursors,17 and other reports18 using expensive ligand and transition metal-based catalysts.
Here we disclose a decarboxylative E or Z selective vinylation of amino acids using an affordable donor–acceptor organophotocatalyst. Our initial hypothesis was to find an organophotocatalyst that can allow decarboxylation through single electron transfer and E to Z isomerization of olefins through energy transfer. Finding a suitable combination of a solvent and a photocatalyst would be the key to the success of this project.
Results and discussion
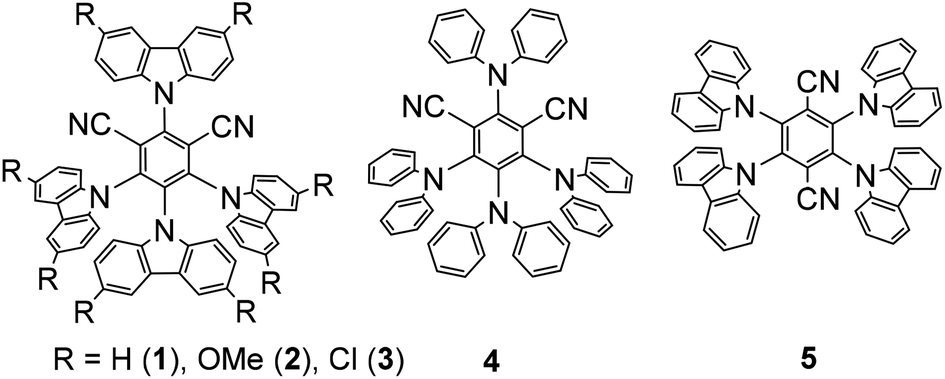
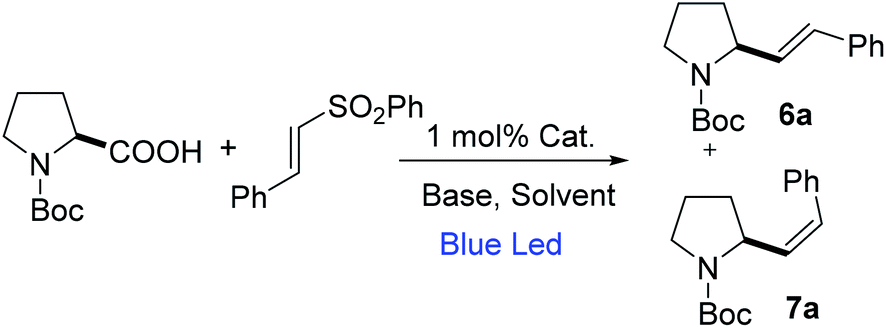
Hence, we initiated our study using N-Boc-proline and phenyl vinyl sulphone in the presence of 4CzIPN as a photocatalyst. We envisioned that absorption of visible light by 4CzIPN will generate excited state 4CzIPN* (E1/2(4CzIPN*/4CzIPN˙−) = +1.4 V vs. SCE in acetonitrile).9a,c As the oxidative potential of an amino acid (Boc-Pro-OCs) is +1.0 V (vs. SCE), it should be feasible for 4CzIPN* to participate in a reductive quenching cycle by removing one electron from Boc-Pro-OCs to generate a carboxyl radical, which would deliver an α-amino radical after expelling CO2 (Scheme 2). The addition of an amino radical to the vinyl sulphone would generate a β-sulfonyl radical, which would afford the desired allylic amine after the expulsion of the sulfinyl radical. The reduced photocatalyst would transfer an electron to the sulfinyl radical (E1/2(PhSO2˙/PhSO2Na) = +0.5 V vs. SCE compared to E1/2(4CzIPN/4CzIPN˙−) = −1.2 V vs. SCE) to generate a sulfinate anion restoring the photocatalyst. Based on this hypothesis we decided to screen this reaction using various solvents and at different reaction temperatures. The reaction was carried out at room temperature using N-Boc-proline, phenyl vinyl sulfone, and Cs2CO3, with 4CzIPN as a photocatalyst under blue light. Gratifyingly, we observed the formation of the desired product with good yield but moderate E-selectivity using acetonitrile as a solvent (Table 1, entry 1). However, the reaction did not proceed with a polar protic solvent like methanol. More than 87% of E-selectivity was achieved using DMA as a solvent (Table 1, entry 5). Moving from polar solvents to non-polar solvents like toluene, benzene, or chlorobenzene, we observed good Z-selectivity. Furthermore, to improve the E-selectivity we decided to screen other catalysts for this reaction based on their redox potential and triplet energy transfer. It is well known that introduction of the electron-donating 6-methoxy group on the carbazole ring of 4CzIPN results in a negative shift of the excited state redox potentials, and introduction of an electron-withdrawing group, e.g. halogen atom, leads to a positive shift.9a,c Also, we have synthesized catalyst 4 by replacing carbazole with diphenylamine. However, we did not observe any improvement using these modified catalysts and bases (Table 1, entry 13–17). We suspect that the E-isomer is produced kinetically (the geometry is likely set in the beta-elimination step), then the increase in reaction temperature might improve the E-selectivity. The decrease of olefin isomerization from the E to Z-isomer at higher temperatures in DMA solvent could be the reason behind the exclusive formation of the E-isomer at higher temperatures. Warmer temperatures will broaden the Boltzman distribution of alkene conformers; those that are more than a few degrees out of planarity will become impossible to excite. Hence, at warmer temperatures the rate of isomerization is expected to slow down due to a decrease in the concentration of excitable E-alkene conformers. Indeed, we achieved 100% E-selectivity by conducting the reaction at 50 °C (Table 1, entry 18).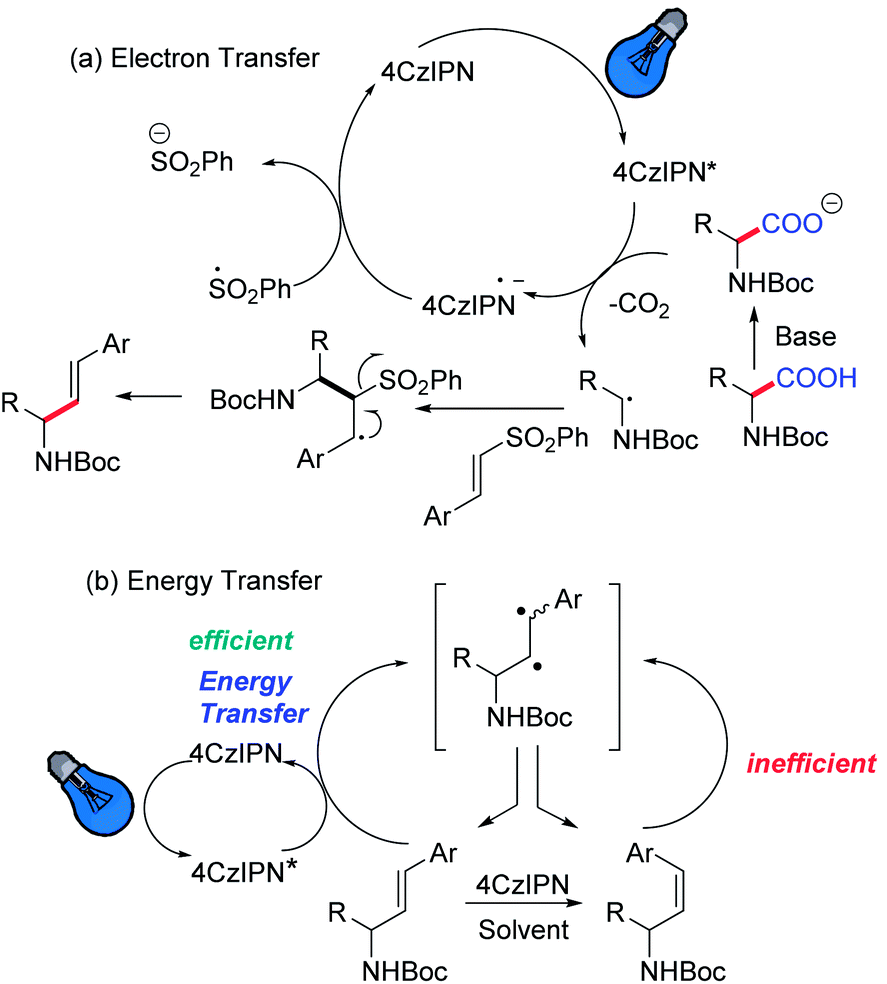 | ||
| Scheme 2 Plausible reaction mechanism. | ||
|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Catalyst | Base | dr (6a/7a) | Yieldb (%) |
| a Vinyl sulfone (1 equiv.), Boc-amino acids (2 equiv.), Cs2CO3 (2 equiv.), 4-CzIPN (1 mol%) at RT. b Isolated yield. c Reaction at 50 °C. | |||||
| 1 | Acetonitrile | 1 | Cs2CO3 | 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30 :30 |
69 |
| 2 | DMSO | 1 | Cs2CO3 | 45:55 |
70 |
| 3 | DMF | 1 | Cs2CO3 | 66:33 |
67 |
| 4 | MeOH | 1 | Cs2CO3 | NA | 0 |
| 5 | DMA | 1 | Cs2CO3 | 83:17 |
70 |
| 6 | Benzene | 1 | Cs2CO3 | 40:60 |
20 |
| 7 | Chlorobenzene | 1 | Cs2CO3 | 25:75 |
20 |
| 8 | Ethyl acetate | 1 | Cs2CO3 | 53:47 |
85 |
| 9 | DCE | 1 | Cs2CO3 | 50:50 |
20 |
| 10 | Toluene | 1 | Cs2CO3 | 20:80 |
20 |
| 11 | o-Xylene | 1 | Cs2CO3 | NA | 0 |
| 12 | DMA | 2 | Cs2CO3 | NA | 0 |
| 13 | DMA | 3 | Cs2CO3 | NA | 0 |
| 14 | DMA | 4 | Cs2CO3 | NA | 0 |
| 15 | DMA | 5 | Cs2CO3 | 80:20 |
65 |
| 16 | DMA | 1 | Cs2CO3 | 76:24 |
70 |
| 17 | DMA | 1 | Cs2CO3 | 60:40 |
20 |
| 18 | DMA | 1 | Cs 2 CO 3 | 100:0 | 75 |
| 19c | DMF | 1 | Cs2CO3 | 90:10 |
70 |
|
|||||
After obtaining the optimal conditions, we started exploring the substrate scope for this reaction (Scheme 3). A variety of natural and unnatural α-amino acids with both acid- and base-sensitive protective groups could be applied very well. Good yield and 100% E-selectivity were obtained using N-Cbz-Pro-OH. Reactions with Boc-protected glycine, alanine, valine, leucine, and isoleucine afforded the desired product in great yields and up to 100% E-selectivity (6c–6g). Most importantly, the reaction tolerated the free methyl sulfide (6k), unprotected amines (6l), and unprotected alcohols (6o) with excellent yields and exclusively E-selectivity, which supports the mildness of the reaction conditions. We have not found references where N-protected amino acids with an unprotected amine and alcohol were reported for oxidative decarboxylative transformations.19 Unprotected indoles are also well tolerated under these conditions affording the products in good yield and selectivity. Further examination of the substrate scope revealed a general tolerance for electron deficient (6q–6t; up to 100% E-selectivity) and electron-rich (6u; 100% E-selective) aryl vinyl sulfones. Good yields and selectivity were observed irrespective of the vinyl sulfone-carrying substituents at the different positions of the aryl ring (6w, 6x). However, we did not observe appreciable yields with aliphatic carboxylic acids.
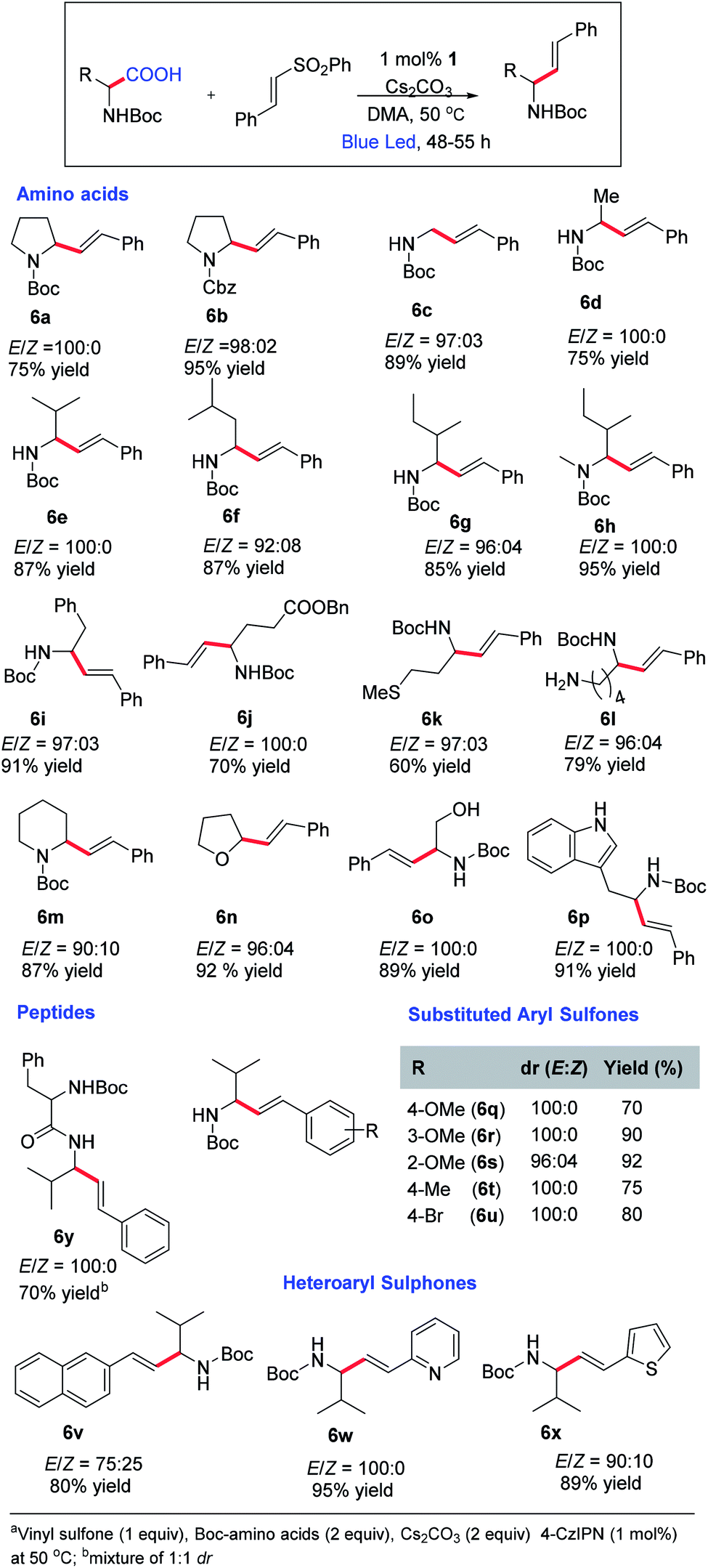 | ||
| Scheme 3 Substrate scope for E-selective vinylationa. | ||
At this point, we decided to optimize the conditions to access Z-selective allylic amines owing to their presence in bioactive compounds and natural products (please see the ESI† for detailed optimization).20 Initial screening indicates that the reaction using toluene afforded the product with Z-selectivity in poor yield. We have screened several bases and catalysts to improve the yield and selectivity, keeping toluene as a solvent. Also, we have tried a mixed solvent system to improve the yield and the selectivity. However, we did not observe any substantial improvement either in yield or Z-selectivity by varying the mixed solvent system, bases and catalysts. Further screening of the reaction in the presence of other solvents revealed that the reaction using 1,4-dioxane afforded 79% yield and 92/8, Z/E ratio. No further improvement was observed by varying the base by keeping 1,4-dioxane as a solvent.
At this point, we decided to explore the substrate scope for Z-selective vinylation (Scheme 4). Various aryl sulfones with both electron-donating and withdrawing groups furnish the desired Z-selective allylic amines in good yields (7a–7e, 7o, 7p). The reaction tolerates various amino acids like Boc-protected proline, glycine, alanine, phenylalanine, and valine, and furnishes the desired products in good yield and high Z-selectivity (7h–7n). The reaction with N-Boc-2-piperidine carboxylic acid and tetrahydrofuran-2-carboxylic acid was well tolerated (7g and 7h). The reaction also tolerates N-Boc–N-Me-protected amino acids, affording the Z-product in good yield and high Z-selectivity.
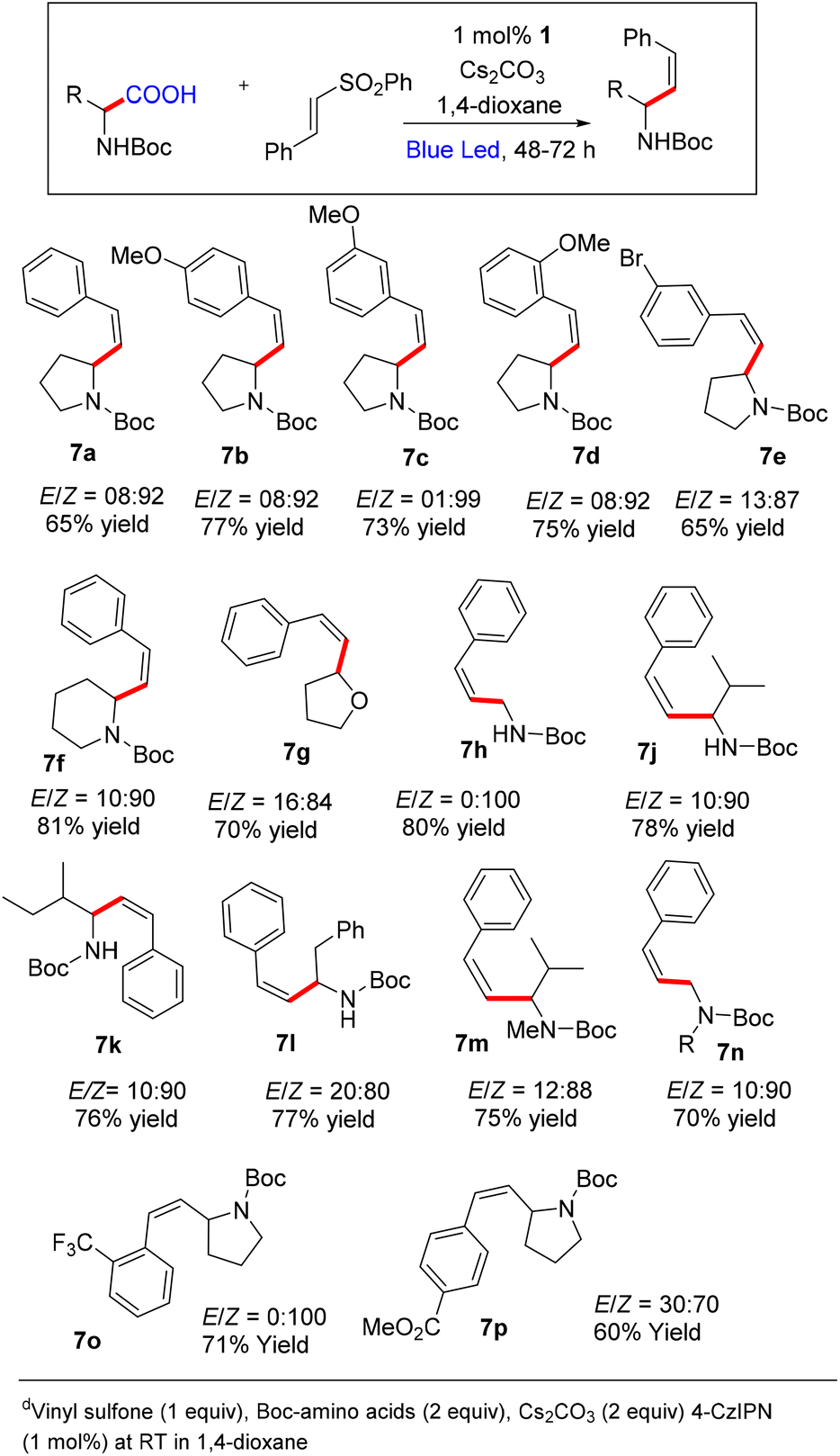 | ||
| Scheme 4 Substrate scope for Z-selective vinylationa. | ||
Next, considering the importance of homoallylic amines in organic synthesis,17 we focused on the synthesis of homoallylic amines via the amino acid decarboxylation strategy (Scheme 5). We could achieve this by replacing the vinyl sulfone with an allyl sulfone (please see the ESI† for optimization studies). A good number of amino acids were well tolerated, affording the homoallylic amines in good yields. To further extend the utility of this decarboxylative vinylation reaction we investigated the applicability of this method for gram-scale synthesis of (±)-norruspoline21 and the synthesis of a bioactive compound for hepatitis C,22 with an over 85% yield and 95/5 E/Z selectivity. These results indicate the potential application of TADF based organic photocatalysts for the large-scale synthesis of value-added materials (Scheme 6).
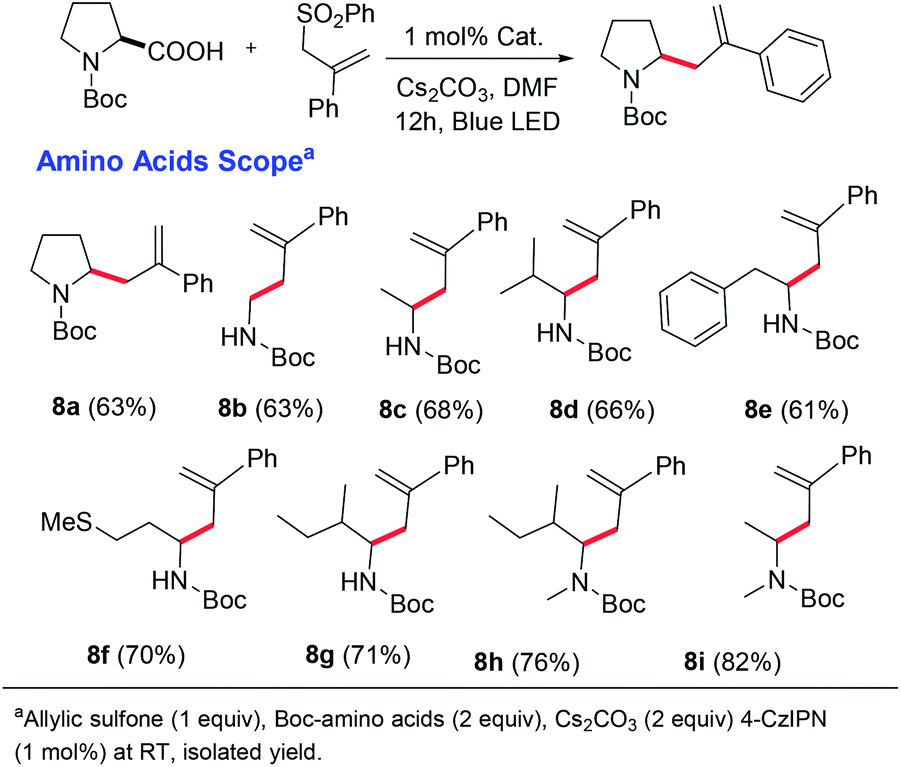 | ||
| Scheme 5 Decarboxylative synthesis of homoallylic amines. | ||
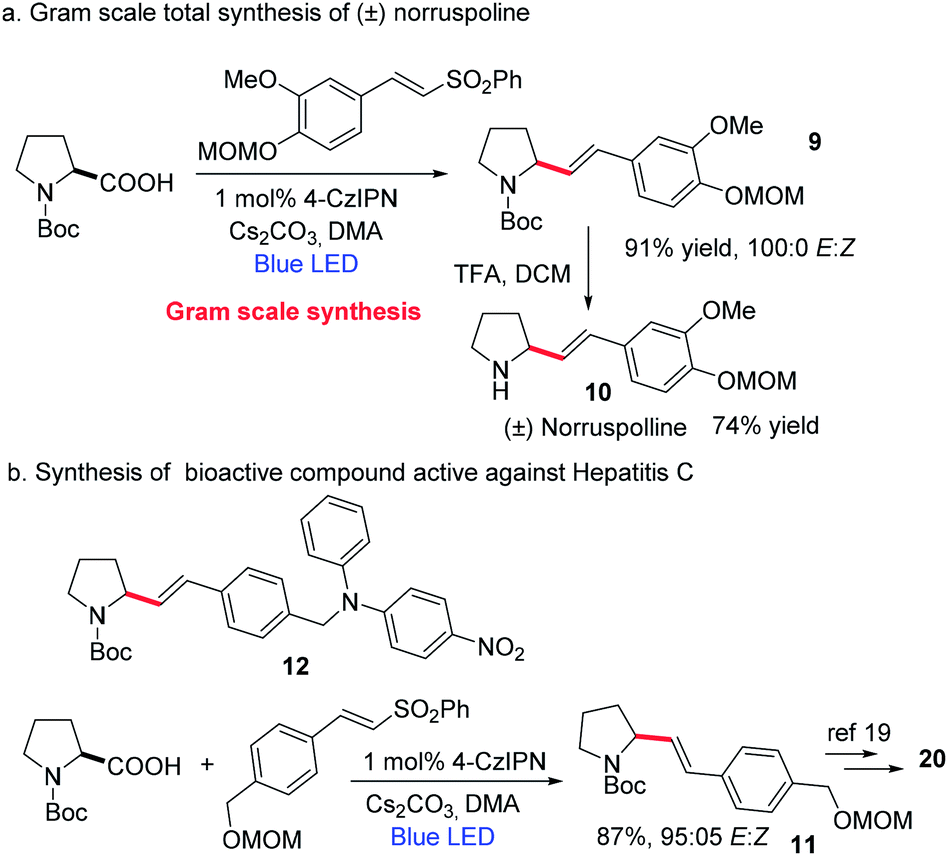 | ||
| Scheme 6 Synthetic applicability. | ||
Finally, we investigated the mechanism of E vs. Z selectivity of this reaction system. As explained above, the E-selectivity can be achieved at a higher temperature because of kinetic selectivity. Therefore, we focused on finding out the reason behind the high Z-selectivity. We did not observe any isomerization of the pure E-product in the absence of a photocatalyst. Also, stopping the reaction in 1,4-dioxane after 12 h indicates the formation of E, Z-mixtures, which isomerized to majorly the Z-isomer after the indicated time. Here, both the solvent and the photocatalyst play an important role. Table 2 shows the physical parameters of the reaction solvent with the resulting E/Z selectivity and reaction yield for the reaction explained in Table 1. It seems that the reaction selectivity has a correlation with ET(30) which is well known as a practical solvent polarity rather than the solvent polarity parameter (εr), but this correlation provides little predictive values. The apolar solvent having ET(30) less than 36.8 provided the Z-isomer as the major product. Also, a trend could be observed wherein high polarity solvents ET(30) above 40 provided high E-selectivity with a good yield. The poor solubility of the amino acid and the base in apolar solvents might result from the poor yield.
| Solvent | n D | ε r | E T(30) | E/Z (% yield) |
|---|---|---|---|---|
| MeOH | 1.328 | 33.6 | 55.4 | NA |
| Acetonitrile | 1.344 | 36 | 45.6 | 70:30 (69) |
| DMSO | 1.479 | 46.71 | 45.1 | 45:55 (70) |
| DMF | 1.43 | 37.06 | 43.2 | 66:33 (67) |
| DMA | 1.438 | 38.3 | 42.9 | 83:17 (70) |
| DCE | 1.417 | 10.1 | 39.4 | 50:50 (20) |
| Ethyl acetate | 1.372 | 6.03 | 38 | 53:47 (85) |
| Chlorobenzene | 1.524 | 5.74 | 36.8 | 25:75 (20) |
| 1,4-Dioxane | 1.422 | 2.27 | 36 | 08:92 (79) |
| Benzene | 1.501 | 2.4 | 34.3 | 40:60 (20) |
| Toluene | 1.497 | 2.43 | 33.9 | 20:80 (20) |
To clarify the high Z selectivity in 1,4-dioxane, the Stern–Volmer plot of 4CzIPN to the various concentration of E- and Z-isomers, 6a, and 7a, was investigated (Fig. 1). The plots clearly indicate that the E-isomer quenched the 4CzIPN emission compared to the Z-isomer with two-order magnitude higher constants; the quenching constant KSV was 14.0 and 0.06 M−1, respectively. Therefore, the Z-isomer has no interaction with 4CzIPN in 1,4-dioxane.
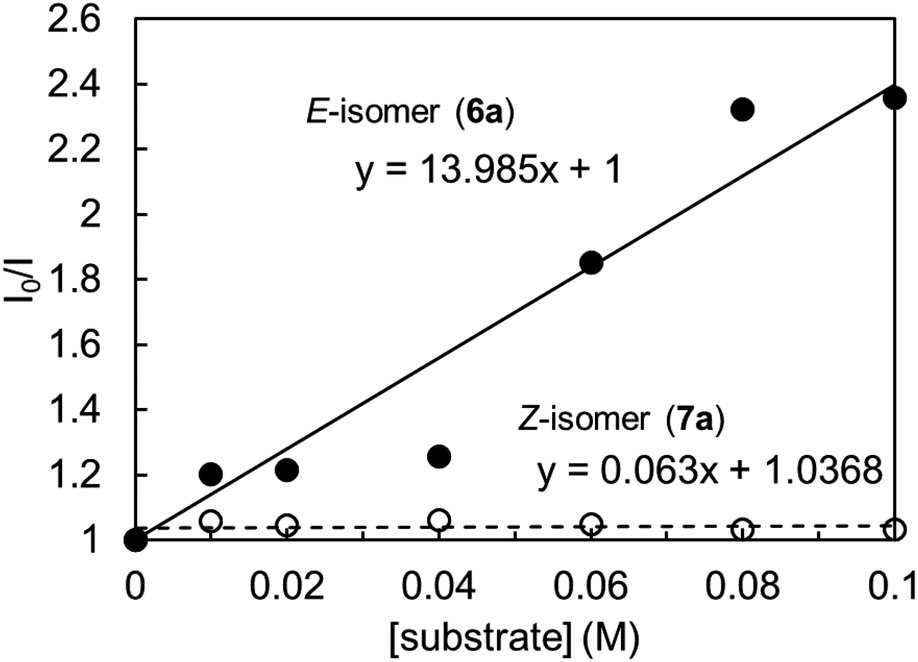 | ||
| Fig. 1 Stern–Volmer plot of 4CzIPN emission quenching with substrates 6a and 7a in 1,4-dioxane, [4CzIPN] = 1.0 × 10−5 M, λex = 365 nm, λem = 510 nm. | ||
Similar to the previous reports,24 4CzIPN showed red-shifted emission in polar solvents (Fig. 2). The lowest singlet excited state (S1) energy drop of 4CzIPN was only 0.1 eV between toluene and DMF (S1 energy: 2.7–2.8 eV) in spite of its emission peak shift of 50 nm (0.2 eV). The fluorescence of 6a (E-isomer) and 7a (Z-isomer) showed a very weak emission peak around 300 nm (see the ESI, S25†) in 1,4-dioxane at room temperature, but they showed additional fluorescence emission at a longer wavelength at 77 K (Fig. 3). This means, the S1 exciton quickly decayed non-radiatively to the dark state (S0, T1, or biradical species) at high temperature, and emissions from higher lying local excited state (Sn) were only observed. Therefore, S1 energy would be ca. 3.1 and 3.5 eV, for E- and Z-isomers, respectively; those were estimated by the onset of the lowest energy emission band on total emission at 77 K. This clearly indicates that both isomers do not have a chance to receive the exciton energy from 4CzIPN via the energy resonation of S1.
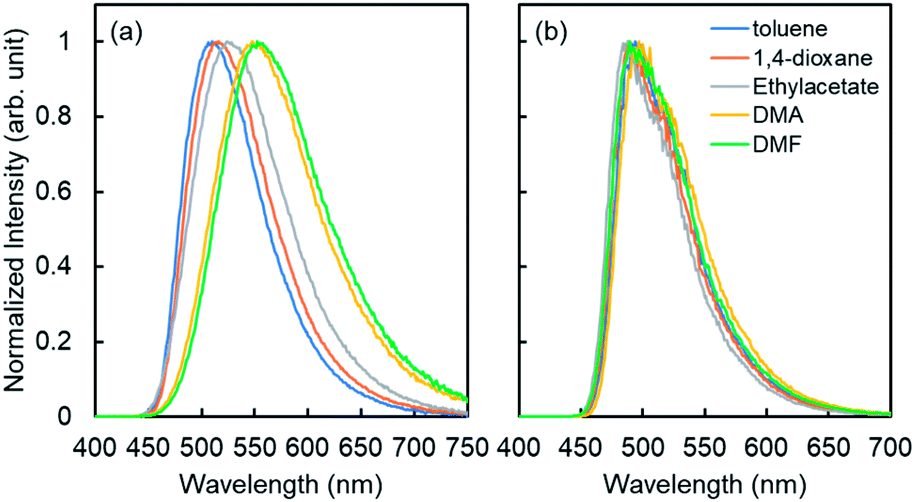 | ||
| Fig. 2 Fluorescence (a) and phosphorescence spectra (b) of 4CzIPN in various solvents; [4CzIPN] = 1.0 × 10−5 M, λex = 330 nm. | ||
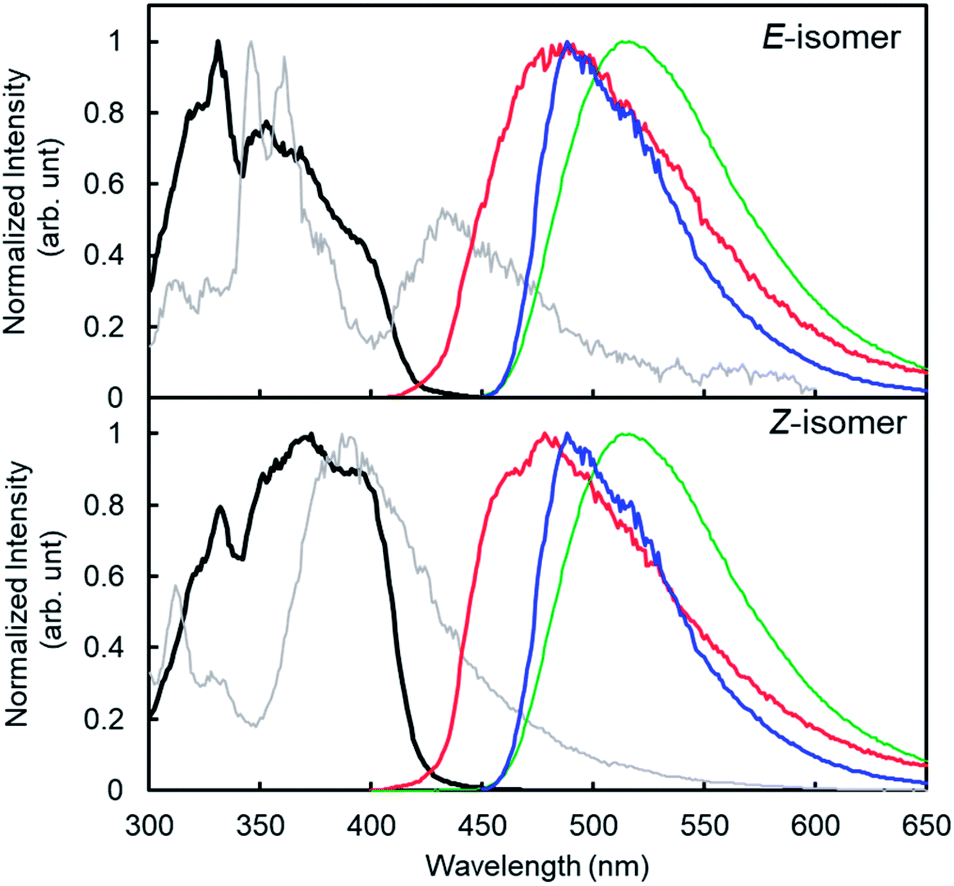 | ||
| Fig. 3 Excitation (for phosphorescence, λem = 500 nm; thick black line), total emission (at 77 K, λex = 280 nm; thin gray line) and phosphorescence spectra (at 77 K, λex = 330 nm; red line) of 6a (E-isomer) and 7a (Z-isomer) with fluorescence (at r.t., λex = 330 nm; green line) and phosphorescence spectra (at 77 K, λex = 330 nm; blue line) of 4CzIPN in 1,4-dioxane; [M] = 1.0 × 10−5 M. Phosphorescence and excitation for phosphorescence were measured with a PL spectrometer (FP-8600, Jasco), and the emission lifetime was measured with a QuantaurusTau (C16361, Hamamatsu Photonics K. K.) | ||
The phosphorescence spectra of 4CzIPN at 77 K in various solvents showed similar spectra and the estimated lowest triplet state (T1) energies were almost the same (ca. 2.5 eV) for all solvents (Fig. 2b). It is difficult to explain the difference between E- and Z-selectivity in the various solvents by using only the photo properties of 4CzIPN. The phosphorescence spectra of E- and Z-isomers were measured in 1,4-dioxane. However, the T1 energies of both isomers were larger than 2.8 eV (Fig. 3). This relationship between the T1 energies of E- and Z-isomers, and 4CzIPN was also similar in DMA and toluene (see the ESI, S24†). In addition, the excitation spectra for phosphorescence, which means the emission was detected at 500 nm at 77 K with the delay time and excitation frequency settings being the same as those for phosphorescence measurement, showed no spectral overlap with 4CzIPN emission. This strongly suggests that the exciton energy of 4CzIPN is too low to achieve the direct excitation of both E- and Z-isomers by energy transfer from the T1 state of 4CzIPN. However, the E-isomer quenched the 4CzIPN emission in fact (Fig. 1). Therefore, the Z-isomerization would be achieved not via the electron exchange Dexter energy transfer process, which also requires the spectral overlap, but via the radical pair formation as an intermediate CT state. The difference between the E- and Z-selectivity would be explained by the LUMO energy difference or interaction strength with 4CzIPN of these isomers.
Furthermore, it was suggested that the Z-selectivity is related to the T1 generation efficiency and length of its lifetime. Mechanistically, initiation of the photoredox reaction would occur through excitation of 4CzIPN using blue light, which moves the electron to the singlet excited state (S1); the lifetime is 28 ns; this decay lifetime would be too fast for the photo-induced electron transfer from the reactant to happen. Because the decay rate from S1 to the ground state (S0) via the radiative and non-radiative process is 2.2 × 107 s−1 and that for intersystem crossing (ISC) is 1.3 × 107 s−1,25 it means 37% of the generated exciton would provide the T1 exciton via the ISC process. 4CzIPN also showed delayed emission with a lifetime of 1.6 μs; the reverse ISC (RISC) rate was 1.0 × 106 s−1. On the other hand, 4CzIPN showed 19 ns prompt and 4.6 μs delayed fluorescence with 70% T1 generation in 1,4-dioxane (1.6 × 107, 3.7 × 107 and 7.4 × 106 s−1 for S1–S0 decay, ISC and RISC rates, respectively). Also, the lifetimes were 11 ns and 4.6 μs for prompt and delayed fluorescence in toluene; the T1 generation ratio was 75% (2.2 × 107, 6.6 × 107, and 1.0 × 106 s−1 for S1–S0 decay, ISC and RISC rates, respectively). The low Z-selectivity in DMA solvent could be due to half of T1 generation efficiency and three times shorter T1 lifetime compared to apolar solvents like 1,4-dioxane and toluene giving high Z-selectivity. The E-products in 1,4-dioxane should be exchanged to Z-products by efficient electron transfer to form the radical ion pair via the long-lived T1 state and back-electron transfer to 4CzIPN. The E-isomer reproduces both E- and Z-isomers but the number of E-isomers should reduce with each cycle. As a result, the Z-isomer will enrich over time and end up as the major product. In addition, the fluorescence of 4CzIPN was also quenched by the primary reactant, N-Boc-proline carboxylate both in DMF and in 1,4-dioxane. Most interestingly, the fluorescence quenching rate is low in 1,4-dioxane compared to DMF. These data suggest that the main effect of solvent change is limiting the solubility of the carboxylate, hence limiting its ability to quench and as a result accelerating the isomerization process.
Conclusions
In summary, a highly efficient visible-light-induced decarboxylative E and Z-selective vinylation of amino acids and peptides using an organophotoredox catalyst was reported by varying the solvents. The reaction tolerated a broad range of amino acids. It is mild, operationally simple, and transition metal-free, and can be performed in the gram scale. The reaction methodology was applied in the gram-scale synthesis of natural products and bioactive compounds. We demonstrated how to control the reaction rate and E vs. Z selectivity by changing the reaction conditions. The reaction mechanism was also suggested to be the photoinduced electron transfer and electron exchange energy transfer from T1via the CT state. Finally, considering the mildness of this methodology, we believe that it is promising to be applied to a wide variety of reactions, e.g., site-selective modification of peptide chains, which has huge importance in chemical biology.Data availability
All experimental and characterisation data in this article are available in the ESI.†Author contributions
SM has carried out optimization, substrate scope for both E and Z-selective allylic amines, applications in total synthesis and fluorescence quenching, SV plot, photostationary state studies; KKD completed synthesis of homoallylic amines; SK, YT & CA helped us with mechanistic studies, manuscript writing and completed phosphorescence and fluorescence experiments; SP designed the project, supervised and prepared the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by DST-Ramanujan fellowship (SB/S2/RJN-171/2017). SM thanks CSIR India for the fellowship & KKD thanks IIT Kharagpur for the fellowship. We are also grateful for financial support from Kyulux Inc.Notes and references
- (a) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC; (b) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed; (c) K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035 CrossRef CAS PubMed; (d) Y. Xi, H. Yi and A. Lei, Org. Biomol. Chem., 2013, 11, 2387 RSC; (e) W. Liu and C.-J. Li, Synlett, 2017, 28, 2714 CrossRef CAS.
- M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2006, 81, 6898 CrossRef PubMed.
- (a) C. Li, Y. Xu, W. Tu, G. Chen and R. Xu, Green Chem., 2017, 19, 882–899 RSC; (b) M. A. Graham, G. Noonan, J. H. Cherryman, J. J. Douglas, M. Gonzalez, L. V. Jackson, K. Leslie, Z.-Q. Liu, D. McKinney, R. H. Munday, C. D. Parsons, D. T. E. Whittaker, E.-X. Zhang and J.-W. Zhang, Org. Process Res. Dev., 2021, 25, 57 CrossRef CAS.
- https://www.nobelprize.org/prizes/chemistry/2021/press-release/ .
- (a) D. P. Hari and B. König, Chem. Commun., 2014, 50, 6688–6699 RSC; (b) T.-Y. Shang, L.-H. Lu, Z. Cao, Y. Liu, W.-M. He and B. Yu, Chem. Commun., 2019, 55, 540 Search PubMed; (c) M. V. Bobo, J. J. Kuchta and A. K. Vannucci, Org. Biomol. Chem., 2021, 19, 4816–4834 RSC; (d) P. P. Singh and V. Srivastava, Org. Biomol. Chem., 2021, 19, 313–321 RSC.
- (a) C. Chen, Org. Biomol. Chem., 2016, 14, 8641–8647 RSC; (b) J.-B. Xia, C. Zhu and C. Chen, J. Am. Chem. Soc., 2013, 135, 17494–17500 CrossRef CAS PubMed; (c) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234 CrossRef CAS PubMed.
- M. A. Bryden and E. Z-Colman, Chem. Soc. Rev., 2021, 50, 7587 RSC.
- (a) J. Luo and J. Zhang, ACS Catal., 2016, 6, 873 CrossRef CAS; (b) J. Lu, B. Pattengale, Q. Liu, S. Yang, W. Shi, S. Li, J. Huang and J. Zhang, J. Am. Chem. Soc., 2018, 140, 13719 CrossRef CAS PubMed; (c) E. Speckmeier, T. G. Fischer and K. Zeitler, J. Am. Chem. Soc., 2018, 140, 15353 CrossRef CAS PubMed.
- (a) R. H. Young, K. Wehrly and R. L. Martin, J. Am. Chem. Soc., 1971, 93, 5774 CrossRef CAS; (b) J. Saltiel and E. D. Megarity, J. Am. Chem. Soc., 1972, 94, 2742 CrossRef CAS; (c) J. Xu, N. Liu, H. Lv, C. He, Z. Liu, X. Shen, F. Cheng and B. Fan, Green Chem., 2020, 22, 2739 RSC.
- (a) G. G. Pawar, F. Robert, E. Grau, H. Cramail and Y. Landais, Chem. Commun., 2018, 54, 9337 RSC; (b) H.-L. Zhu, F.-L. Zeng, X.-L. Chen, K. Sun, H.-C. Li, X.-Y. Yuan, L.-B. Qu and B. Yu, Org. Lett., 2021, 23, 2976 CrossRef CAS PubMed; (c) Y. Liu, X.-L. Chen, X.-Y. Li, S.-S. Zhu, S.-J. Li, Y. Song, L.-B. Qu and B. Yu, J. Am. Chem. Soc., 2021, 143, 964 CrossRef CAS PubMed; (d) C. Shu, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2019, 58, 3870 CrossRef CAS PubMed.
- (a) M. Johannsen and K. A. Jørgensen, Chem. Rev., 1998, 98, 1689 CrossRef CAS PubMed; (b) E. M. Skoda, G. C. Davis and P. Wipf, Org. Process Res. Dev., 2012, 16, 26 CrossRef CAS PubMed; (c) A. Stütz, Angew. Chem., Int. Ed. Engl., 1987, 26, 320 CrossRef.
- (a) C. E. Anderson and L. E. Overman, J. Am. Chem. Soc., 2003, 125, 12412 CrossRef CAS PubMed; (b) Y. K. Chen, A. E. Lurain and P. J. Walsh, J. Am. Chem. Soc., 2002, 124, 12225 CrossRef CAS PubMed; (c) E. Skucas, M.-Y. Ngai, V. Komanduri and M. J. Krische, Acc. Chem. Res., 2007, 40, 1394 CrossRef CAS PubMed; (d) N. R. Candeias, F. Montalbano, P. M. S. D. Cal and P. M. P. Gois, Chem. Rev., 2010, 110, 6169 CrossRef CAS PubMed.
- (a) K. Singh, S. Staig and J. D. Weaver, J. Am. Chem. Soc., 2014, 136, 5275 CrossRef CAS PubMed; (b) A. Singh, C. J. Fennell and J. D. Weaver, Chem. Sci., 2016, 7, 6796 RSC.
- A. Noble and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 11602 CrossRef CAS PubMed.
- (a) N. A. Till, R. T. Smith and D. W. C. MacMillan, J. Am. Chem. Soc., 2018, 140, 5701 CrossRef CAS PubMed; (b) H. Yue, C. Zhu, R. Kancherla, F. Liu and M. Rueping, Angew. Chem., Int. Ed., 2020, 59, 5738 CrossRef CAS PubMed; (c) M. M. Mastandrea, S. Cañellas, X. Caldentey and M. A. Pericàs, ACS Catal., 2020, 10, 6402 CrossRef CAS; (d) J. Li, Q. Lefebvre, H. Yang, Y. Zhao and H. Fu, Chem. Commun., 2017, 53, 1029 RSC; (e) S. Karmakar, A. Silamkoti, N. A. Meanwell, A. Mathur and A. K. Gupta, Adv. Synth. Catal., 2021, 363, 3693 CrossRef CAS.
- (a) J. T. Edwards, R. R. Merchant, K. S. McClymont, K. W. Knouse, T. Qin, L. R. Malins, B. Vokits, S. A. Shaw, D.-H. Bao, F.-L. Wei, T. Zhou, M. D. Eastgate and P. S. Baran, Nature, 2017, 545, 213 CrossRef CAS PubMed; (b) T. Qin, J. Cornella, C. Li, L. R. Malins, J. T. Edwards, S. Kawamura, B. D. Maxwell, M. D. Eastgate and P. S. Baran, Science, 2016, 352, 801 CrossRef CAS PubMed; (c) T. Patra and D. Maiti, Chem.–Eur. J., 2017, 23, 7382 CrossRef CAS PubMed.
- (a) C. Zheng, W.-M. Cheng, H.-L. Li, R.-S. Na and R. Shang, Org. Lett., 2018, 20, 2559 CrossRef CAS PubMed; (b) H. Cao, H. Jiang, H. Feng, J. M. C. Kwan, X. Liu and J. Wu, J. Am. Chem. Soc., 2018, 140, 163 CrossRef PubMed.
- (a) R. Takeuchi and N. Shiga, Org. Lett., 1999, 1, 265 CrossRef CAS; (b) A. Gerpe, M. Bollini, M. González and H. Cerecetto, Synth. Commun., 2008, 39, 29 CrossRef; (c) A. M. Martínez-Gualda, R. Cano, L. Marzo, R. Pérez-Ruiz, J. Luis-Barrera, R. Mas-Ballesté, A. Fraile, V. A. de la Peña O'Shea and J. Alemán, Nat. Commun., 2019, 10, 2634 CrossRef PubMed.
- (a) T. R. Ramadhar and R. A. Batey, Synthesis, 2011, 9, 1321 Search PubMed; (b) S. J. Meek, R. V. O'Brien, J. Llaveria, R. R. Schrock and A. H. Hoveyda, Nature, 2011, 471, 461 CrossRef CAS PubMed; (c) R. Jiang, L. Ding, C. Zheng and S.-L. You, Science, 2021, 371, 380 CrossRef CAS PubMed.
- F. Roessler, D. Ganzinger, S. Johne, E. Schöpp and M. Hesse, Helv. Chim. Acta, 1978, 61, 1200 CrossRef CAS.
- D. A. DeGoey et al. , Preparation of 4,4′-(phenylazanediyl)bisbenzyls and analogs end capped with amino acids or peptides as Hepatitis C virus replication inhibitors for treating HCV infections, WO 2010/120935 A1, 2010 Search PubMed.
- J. P. Cerón-Carrasco, D. Jacquemin, C. Laurence, A. Planchat, C. Reichardt and K. Sraïdi, J. Phys. Org. Chem., 2014, 27, 512 CrossRef.
- (a) R. Ishimatsu, S. Matsunami, K. Shizu, C. Adachi, K. Nakano and T. Imato, J. Phys. Chem. A, 2013, 117, 5607 CrossRef CAS PubMed; (b) T. Hosokai, H. Noda, H. Nakanotani, T. Nawata, Y. Nakayama, H. Matsuzaki and C. Adachi, J. Photonics Energy, 2018, 8, 032102 Search PubMed; (c) X. Zhang, Y. Shi, L. Cai, Y. Zhou, C.-K. Wang and L. Lin, Spectrochim. Acta, Part A, 2020, 225, 117473 CrossRef CAS PubMed.
- Y. Tsuchiya, S. Diesing, F. Bencheikh, Y. Wada, P. L. dos Santos, H. Kaji, E. Z-Colman, I. D. W. Samuel and C. Adachi, J. Phys. Chem. A, 2021, 125, 8074 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc02090d |
| This journal is © The Royal Society of Chemistry 2022 |