
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cu(II)/SPDO complex catalyzed asymmetric Baeyer–Villiger oxidation of 2-arylcyclobutanones and its application for the total synthesis of eupomatilones 5 and 6†
Chang-Sheng
Zhang
a,
Ya-Ping
Shao
a,
Fu-Min
Zhang
 *a,
Xue
Han
a,
Xiao-Ming
Zhang
a,
Kun
Zhang
b and
Yong-Qiang
Tu
*ac
*a,
Xue
Han
a,
Xiao-Ming
Zhang
a,
Kun
Zhang
b and
Yong-Qiang
Tu
*ac
aState Key Laboratory of Applied Organic Chemistry, College of Chemistry and Chemical Engineering, Lanzhou University, Lanzhou 730000, P. R. China. E-mail: tuyq@lzu.edu.cn; zhangfm@lzu.edu.cn
bSchool of Biotechnology and Health Sciences, Wuyi University, Jiangmen, 529020, Guangdong, P. R. China
cSchool of Chemistry and Chemical Engineering, Shanghai Jiao Tong University, Shanghai 200240, P. R. China
First published on 23rd June 2022
Abstract
A novel classical kinetic resolution of 2-aryl-substituted or 2,3-disubstituted cyclobutanones of Baeyer–Villiger oxidation catalyzed by a Cu(II)/SPDO complex is reported for the first time, producing normal lactones in excellent enantioselectivities (up to 96% ee) and regioselectivities (up to >20/1), along with unreacted ketones in excellent enantioselectivities (up to 99% ee). The current transformation features a wide substrate scope. Moreover, catalytic asymmetric total syntheses of natural eupomatilones 5 and 6 are achieved in nine steps from commercially available 3-methylcyclobutan-1-one.
Introduction
Baeyer–Villiger (B–V) oxidation, which was first reported in 1898,1 is an important transformation in organic synthesis because it provides a concise and convenient approach toward esters from ketones or aldehydes, especially lactones from cycloketones.2–5 Asymmetric B–V oxidation using chiral substrates as the starting materials has been widely applied to prepare chiral lactones; however, its catalytic asymmetric version was explored by Bolm in the 1990s.5a Since then, catalytic asymmetric B–V oxidation has received much attention from synthetic chemists, and chiral bio-,3 org-,4 and metal catalytic systems5 have been developed, delivering valuable synthetic building blocks such as γ-, δ-, and ε-lactones with excellent enantioselectivities and/or regioselectivities (rs). Although some achievements have been made in recent decades,2 catalytic asymmetric investigations still need to be fully developed; in particular, the scope of substrates is limited to two types, namely prochiral and cyclic meso-ketones,4a–j,5d–y which were investigated through desymmetric B–V oxidation, and racemic cyclic ketones through the kinetic resolution of B–V oxidation.4i,k,n,5a–d,h–y In the latter, 2-substituted or 3-substituted cyclohexanones and 2-substituted cyclopentanones were explored,4g,i,n,5v–y resulting in ideal selectivity and excellent enantioselectivity. However, for their ring-strained cyclobutanone analogues, just limited bicyclic or tricyclic substrates could produce the desired γ-lactone with good enantiomeric excess (ee) (Scheme 1b),4i,m,n,5h–y while 2-substituted cyclobutanones and more challenging 2,3-chain substituted cyclobutanones have remained an unsolved problem till now. To the best of our knowledge, only one example, i.e., 2-phenylcyclobutanone, was reported by Feng and co-workers in 2014,5w and a moderate ee value and poor rs ratio were observed using their own developed privileged chiral N,N′- dioxide/Sc(III) system that exhibited excellent activities and enantioselectivities for a wide range of cyclohexanones and cyclopentanones in the desymmetric or kinetic resolution of B–V oxidations5v–y and other diverse asymmetric transformations.6 From a synthetic perspective, there are some synthetic challenges in the classical kinetic resolution of 2-substituted cyclobutanones and their derivatives, including the following: (1) easy racemization of the unreacted 2-substituted enantiomer is inevitable due to the effect of the adjacent carbonyl group during its purification at room temperature; (2) both the regioselectivity and enantioselectivity of the reacted enantiomer can be simultaneously controlled in the subsequent transformation of its key Criegee intermediates;7 (3) the chiral catalyst could tolerate the corresponding oxidation conditions; and (4) the lack of appropriate functional groups in 2-substituted cyclobutanones which could interact with the chiral catalysts. However, this classical kinetic resolution through asymmetric B–V oxidation could consistently deliver two synthetically valuable products, namely chiral γ-lactone and 2-substituted cyclobutanone. Therefore, it is essential to develop a general B–V oxidation involving 2-substituted cyclobutanone and its derivatives.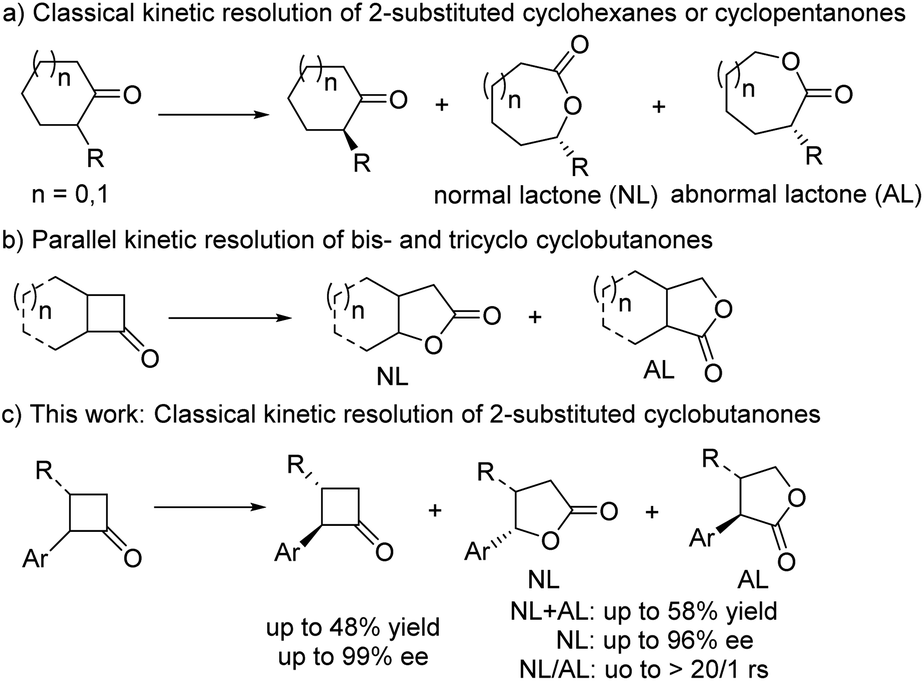 | ||
| Scheme 1 The overview of asymmetric B–V oxidations involving 2-substituted cycloketones. | ||
Over the past few years, our group has developed structurally unique spiro-pyrrolidine oxazoline (SPDO)-derived catalysts,8,9 which exhibit excellent properties in some catalytic asymmetric transformations, especially aerobic oxidative coupling reactions.9a,c Therefore, we wondered whether SPDO catalysts could be used as suitable ligands to catalyse the challenging classical kinetic resolution of 2-substituted cyclobutanone under B–V oxidation conditions.5w If successful, this transformation could not only solve the above-mentioned synthetic challenges but also further expand this rarely reported Cu-catalyzed asymmetric B–V oxidation, which would simultaneously provide two synthetically useful chiral products. Herein, we wish to report our research results as a communication (Scheme 1c).
Results and discussion
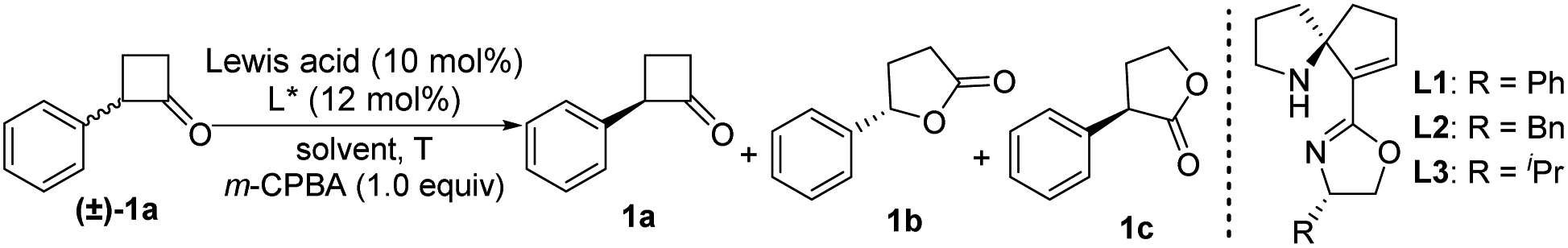
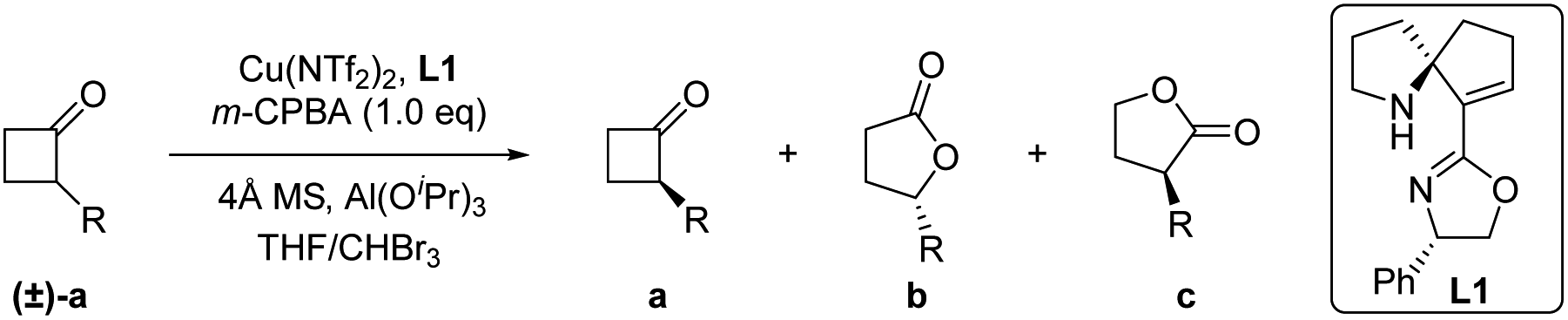
Initially, we selected 2-phenylcyclobutan-1-one5w,10 as a model substrate and 3-chloroperoxybenzoic acid (m-CPBA)11 as a stoichiometric oxidant in the presence of the Cu(OTf)2 complex and ligand L1 to evaluate our desired classical kinetic resolution procedure. The preliminary results indicate that unreacted 2-phenylcyclobutan-1-one 1a was isolated with moderate ee, along with normal lactone 1b and its regioisomer 1c in an almost equal ratio (entry 1, Table 1). Compared to other tested copper salts (entries 2–4, Table 1), Cu(NTf2)2 exhibited excellent enantioselectivity with unreacted 2-phenylcyclobutan-1-one 1a and better regioselectivity with lactones 1b and 1c (4.7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Subsequently, other chiral SPDO ligands L2 and L3 were used to replace L1, but no better result was obtained (entries 5 and 6, Table 1).11 To our delight, the improved regioselectivity of lactones b and c was obtained in different halogenated solvents (entries 7–9, Table 1). For comprehensive consideration of the enantioselectivity of the two desired products 1a and 1b and regioselectivity of 1b and 1c, different ratios of mixed solvent of tetrahydrofuran (THF) and haloalkane were further screened. Better results were observed when the mixed solvent (THF:CHBr3 = 1:1) was used (entries 10–12, Table 1).11 When the model reaction was performed at −40 °C, improvements in the ee value of lactone 1b to 92% and regioselectivity ratio of 1b/1c to 15/1 were obtained, although the ee value of unreacted ketone 1a was only 72% (entry 13, Table 1). Inspired by Feng's excellent work in the B–V oxidations of a variety of substrates,5v–y various additives were introduced into the reaction mixture to further improve the enantioselectivity of ketone 1a, and the combination of Al(OiPr)3 and 4 Å molecular sieves (MS) showed the best reaction results.11 In this case, the enantioselectivity of unreacted ketone 1a increased to 91% ee, and the enantioselectivity of lactone 1b and regioselectivity of 1b/1c were also less influenced (entry 15, Table 1). On further decreasing the reaction temperature to −50 °C, no better result was obtained (entry 16, Table 1). Notably, the ligand ent-L1 exhibited similar reaction results, providing the enantiomers ent-1a and ent-1b, respectively (entry 17, Table 1). Therefore, the reaction parameters listed in entry 15 (Table 1) were selected as the optimal reaction conditions for subsequent investigations.
:1). Subsequently, other chiral SPDO ligands L2 and L3 were used to replace L1, but no better result was obtained (entries 5 and 6, Table 1).11 To our delight, the improved regioselectivity of lactones b and c was obtained in different halogenated solvents (entries 7–9, Table 1). For comprehensive consideration of the enantioselectivity of the two desired products 1a and 1b and regioselectivity of 1b and 1c, different ratios of mixed solvent of tetrahydrofuran (THF) and haloalkane were further screened. Better results were observed when the mixed solvent (THF:CHBr3 = 1:1) was used (entries 10–12, Table 1).11 When the model reaction was performed at −40 °C, improvements in the ee value of lactone 1b to 92% and regioselectivity ratio of 1b/1c to 15/1 were obtained, although the ee value of unreacted ketone 1a was only 72% (entry 13, Table 1). Inspired by Feng's excellent work in the B–V oxidations of a variety of substrates,5v–y various additives were introduced into the reaction mixture to further improve the enantioselectivity of ketone 1a, and the combination of Al(OiPr)3 and 4 Å molecular sieves (MS) showed the best reaction results.11 In this case, the enantioselectivity of unreacted ketone 1a increased to 91% ee, and the enantioselectivity of lactone 1b and regioselectivity of 1b/1c were also less influenced (entry 15, Table 1). On further decreasing the reaction temperature to −50 °C, no better result was obtained (entry 16, Table 1). Notably, the ligand ent-L1 exhibited similar reaction results, providing the enantiomers ent-1a and ent-1b, respectively (entry 17, Table 1). Therefore, the reaction parameters listed in entry 15 (Table 1) were selected as the optimal reaction conditions for subsequent investigations.
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Ligand | Lewis acid | Solvent (mL) | T (°C) | Time (h) | 1a: yieldb (%) | 1b + 1c: yieldb (%) | rsd |
| eec (%) | 1b: eec (%) | 1b/1c | ||||||
| a Reaction conditions: unless otherwise noted, the reactions were performed with 1a (0.2 mmol), Cu(NTf2)2 (10 mol%), ligand (12 mol%) and m-CPBA (1.0 equiv.) in THF (2.0 mL) at 0 °C. b Isolated yield. c Determined by UPC2 analysis. d The regioselectivity (rs) of b/c was determined by 1H NMR of crude products. e 2 mL THF and 2 mL haloalkane were used. f 4 Å MS (60 mg) was added. g Al(OiPr)3 (50 mol%) was used. | ||||||||
| 1 | L1 | Cu(OTf)2 | THF | 0 | 18 | 43/56 | 55/87 | 1.1/1 |
| 2 | L1 | Cu(NTf2)2 | THF | 0 | 10 | 45/70 | 52/74 | 4.7/1 |
| 3 | L1 | Cu(BF4)2·6H2O | THF | 0 | 10 | 50/34 | 45/86 | 1.7/1 |
| 4 | L1 | Cu(ClO4)2·6H2O | THF | 0 | 10 | 52/30 | 40/84 | 1.2/1 |
| 5 | L2 | Cu(NTf2)2 | THF | 0 | 18 | 48/35 | 50/45 | 3.8/1 |
| 6 | L3 | Cu(NTf2)2 | THF | 0 | 18 | 43/41 | 54/40 | 3.9/1 |
| 7 | L1 | Cu(NTf2)2 | DCM | 0 | 6 | 46/40 | 48/47 | 15.0/1 |
| 8 | L1 | Cu(NTf2)2 | DCE | 0 | 5 | 49/30 | 48/46 | 14.1/1 |
| 9 | L1 | Cu(NTf2)2 | CHCl3 | 0 | 5 | 46/57 | 50/70 | 11.2/1 |
| 10e | L1 | Cu(NTf2)2 | THF/DCM | 0 | 10 | 43/80 | 52/69 | 9.0/1 |
| 11e | L1 | Cu(NTf2)2 | THF/CHCl3 | 0 | 10 | 46/77 | 51/80 | 8.5/1 |
| 12e | L1 | Cu(NTf2)2 | THF/CHBr3 | 0 | 18 | 48/75 | 45/88 | 8.0/1 |
| 13e | L1 | Cu(NTf2)2 | THF/CHBr3 | −40 | 36 | 54/72 | 45/92 | 15.0/1 |
| 14e,f | L1 | Cu(NTf2)2 | THF/CHBr3 | −40 | 36 | 51/66 | 46/94 | 14.8/1 |
| 15e,f,g | L1 | Cu(NTf2)2 | THF/CHBr3 | −40 | 36 | 43/91 | 52/92 | 12.5/1 |
| 16e,f,g | L1 | Cu(NTf2)2 | THF/CHBr3 | −50 | 60 | 48/85 | 48/90 | 13.0/1 |
| 17e,f,g | ent-L1 | Cu(NTf2)2 | THF/CHBr3 | −40 | 36 | 44/−91 | 52/−92 | 12.5/1 |
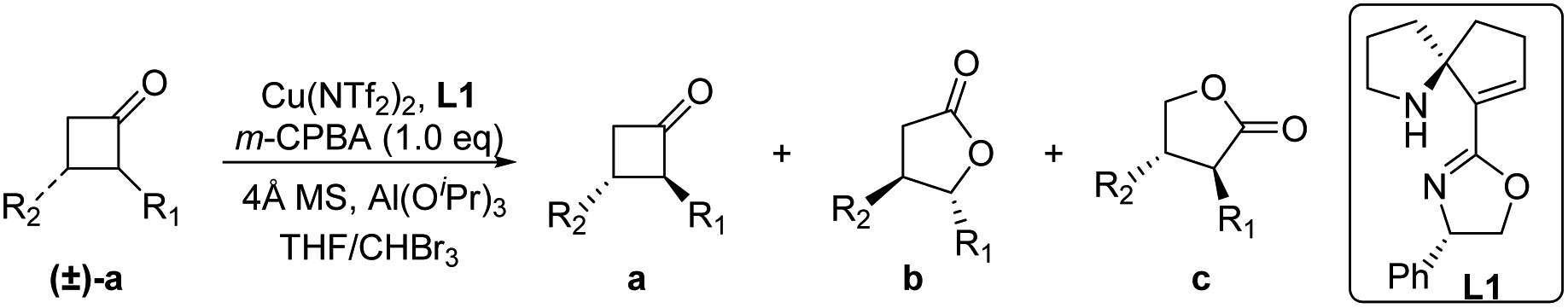
With the optimal reaction conditions in hand, the substrate scope of 2-aryl cyclobutanone was investigated (Table 2). In most cases, high ee of γ-lactone b (82–94%) and unreacted ketone a (78–99%), decent rs of γ-lactones b/c, and good selectivity factor (s-factor) were obtained.5v,12 For the substrates bearing mono-substituents on the aryl ring, some experimental phenomena were observed from the reaction results, including the following: (1) The substituents on the aromatic ring at the para-position affected the reaction results, that is, the steric hindrance of substituents on the aromatic ring was greater, reaction time was longer, and ratio of γ-lactones b/c was lower (entries 1–5, Table 2). (2) The substituents at the ortho-position dramatically affected the desired B–V oxidation, and poor results were obtained even at room temperature (entry 3, Table 2). (3) An electron-donating group (EDG) on the arenes accelerated the reaction and improved the ratio of b/c (entries 7–10, Table 2), while electron-withdrawing groups (EWGs) exhibited the opposite effect on the reaction results (entries 11–15, Table 2), especially regarding the rs of γ-lactones (entries 7–10 & 11–14, Table 2). For bis-substituted aryl substrates, ketones 16a–19a or lactones 16b–19b were concisely isolated with excellent selectivity (entries 16–19, Table 2), and substituents at the 3,4-positions had less effect on the results. More importantly, the 3,4,5-tri-OMe-substituted substrate reacted well in terms of the resulting enantioselectivity and rs (entries 17 & 20, Table 2). Although 1-naphthyl cyclobutanone showed moderate rs with an excellent ee (entry 22, Table 2), the greater steric hindrance of the 2-naphthyl and 1-pyrenyl analogues improved the rs, enantioselectivities, and reactivities, and just 0.6 equiv. of m-CPBA was needed (21a, 23a). Notably, the absolute configuration of lactones 6b, 16b, and 21b was further determined by X-ray analysis. Additionally, substrates with heteroaromatic benzothiophenyl (24a), thienyl (25a, 26a), and benzofuranyl (27a) substituents were well tolerated, resulting in ketones 24a–27a and lactones 24b–27b with slightly decreased enantioselectivities; unsatisfactorily, substrate 24a showed moderate regioselectivity (4.0/1). Overall, whether for aryl rings with a mono-substituent or multi-substituents or for heteroaryl rings, the desired B–V oxidation reactions proceeded well in most cases, providing two valuable synthetic building blocks (2-aryl-substituted cycloketones and γ-lactones) with excellent enantioselectivities.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Substituents | Time | a: yield/ee | b + c: yield/b: ee | b/c rs | s-factor |
| a Reaction conditions: unless otherwise noted, the reactions were performed with a (0.2 mmol), Cu(NTf2)2 (10 mol%), L1 (12 mol%), 60 mg 4 Å MS and m-CPBA (1.0 equiv.) in THF/CHBr3 (2.0/2.0 mL) for the indicated time at −40 °C. Isolated yield. ee was determined by UPC2 analysis. The rs of b/c was determined by crude 1H NMR. b The reaction was performed at 20 °C, and the ee value of 3c was 44.8%. c THF/Et2O/CHBr3 (1.0/1.0/2.0 mL) was used. d 0.6 equiv. of m-CPBA was used. e The ee value of 22c was 95%. f The ee value of 24c was 85%. g The absolute configuration of the products was confirmed by X-ray analysis. | ||||||
| 1 | 1a: R = Ph | 36 h | 43%/91% | 52%/93% | 12.5/1 | 88 |
| 2 | 2a: R = 4-Me-Ph | 48 h | 43%/98% | 57%/90% | 10.0/1 | 87 |
| 3 | 3a: R = 2-Me-Ph | 72 h | 52%/7% | 44%/61% | 1.5/1b,c | 4.4 |
| 4 | 4a: R = 4-iPr-Ph | 50 h | 44%/90% | 52%/91% | 9.2/1c | 65 |
| 5 | 5a: R = 4-tBu-Ph | 50 h | 53%/78% | 43%/92% | 10.0/1c | 57 |
| 6 | 6a: R = 4-Ph-Ph | 42 h | 45%/90% | 54%/91% | 12.5/1g | 65 |
| 7 | 7a: R = 4-MeO-Ph | 28 h | 44%/95% | 54%/91% | 16.6/1 | 79 |
| 8 | 8a: R = 3-MeO-Ph | 38 h | 47%/95% | 51%/91% | 12.4/1 | 79 |
| 9 | 9a: R = 4-PhO-Ph | 36 h | 45%/99% | 48%/93% | 12.5/1 | 145 |
| 10 | 10a: R = 4-BnO-Ph | 39 h | 43%/99% | 52%/88% | 13.0/1 | 82 |
| 11 | 11a: R = 4-F-Ph | 39 h | 49%/82% | 50%/92% | 6.3/1c | 61 |
| 12 | 12a: R = 3-F-Ph | 39 h | 48%/88% | 50%/91% | 5.0/1c | 62 |
| 13 | 13a: R = 4-Cl-Ph | 36 h | 43%/87% | 56%/92% | 5.9/1c | 68 |
| 14 | 14a: R = 4-Br-Ph | 38 h | 45%/85% | 50%/92% | 6.3/1c | 65 |
| 15 | 15a:R = 3-MeCO2Ph | 38 h | 45%/92% | 53%/91% | 6.1/1 | 70 |
| 16 | 16a: R = 2,3-dihydrobenzofuranyl | 38 h | 40%/95% | 56%/86% | 16.7/1g | 49 |
| 17 | 17a: R = 3,4-diMeO-Ph | 39 h | 40%/99% | 58%/87% | 14.3/1 | 75 |
| 18 | 18a: R = 3-F-4-MeO-Ph | 38 h | 47%/94% | 49%/89% | 12.5/1 | 61 |
| 19 | 19a: R = 3-Cl-4-MeO-Ph | 38 h | 46%/97% | 52%/90% | 11.1/1 | 80 |
| 20 | 20a: R = 3,4,5-triMeO-Ph | 48 h | 48%/91% | 48%/85% | >20/1 | 39 |
| 21 | 21a: R = 2-naphthyl | 42 h | 48%/91% | 51%/92% | >20/1d,g | 76 |
| 22 | 22a: R = 1-naphthyl | 38 h | 41%/98% | 56%/94% | 6.0/1e | 149 |
| 23 | 23a: R = 1-pyrenyl | 42 h | 44%/96% | 55%/89% | >20/1d | 67 |
| 24 | 24a: R = 3-benzothiophenyl | 48 h | 42%/84% | 57%/90% | 4.0/1c,f | 50 |
| 25 | 25a: R = 3-thienyl | 36 h | 45%/90% | 51%/90% | 17.1/1c | 58 |
| 26 | 26a: R = 2-thienyl | 48 h | 40%/98% | 54%/86% | 12.0/1 | 60 |
| 27 | 27a: R = 2-benzofuranyl | 36 h | 42%/96% | 53%/82% | 10.0/1 | 39 |
After expansion of the substrate scope of 2-substituted cyclobutanones, we turned our attention to more synthetically challenging 2,3-disubstituted cyclobutanones with an additional functional group at the ketone ring (Table 3). The reaction of 2,3-disubstituted cyclobutanones also proceeded well (28a–32a). With respect to 3-aryl-substituted substrates, excellent enantioselectivities (94–98% ee) and good rs (8.3/1–12.5/1 rs) were obtained with either EDG (OMe) or EWG (CF3) substituents at the para-position of aryl substrates. Notably, this reaction was performed on the gram scale, and products 28a and 28b were isolated without a decrease in the yield and selectivity (entry 2, Table 3). For the 3-methyl-2-aryl-substituted substrate, the reaction also proceeded well under the optimal reaction conditions; both an excellent ee of ketone 32a (91% ee) and excellent rs of 32b and 32c (>20/1) were observed, although with a moderate ee of γ-lactone 32b. Further improvement in the ee value of lactone ent-32b will be discussed in the asymmetric total synthesis of natural products.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Substituents | Time | a: yield/ee | b + c: yield/b: ee | b/c rs | s-factor |
| a Reaction conditions: unless otherwise noted, the reactions were performed with a (0.2 mmol), Cu(NTf2)2 (10 mol%), L1 (12 mol%), 60 mg 4 Å MS. and m-CPBA (1.0 equiv.) in THF/CHBr3 (2.0/2.0 mL) for the time indicated at −40 °C. Isolated yield. ee was determined by UPC2 analysis. The rs of b/c was determined by crude 1H NMR. b 1.0 g 28a was used. c ent-L1 (12% mol) was applied. d Cu(OTf)2 (10 mol%) was applied to replace Cu(NTf2)2. e The absolute configuration of 28b was confirmed by X-ray analysis. | ||||||
| 1 | 28a: R1 = Ph, R2 = Ph | 36 h | 46%/96% | 52%/94% | 12.5/1e | 127 |
| 2 | 28a: R1 = Ph, R2 = Ph | 38 h | 48%/95% | 52%/94% | 12.5/1b | 121 |
| 3 | 29a: R1 = Ph, R2 = 4-F-Ph | 37 h | 49%/96% | 50%/94% | 8.3/1 | 127 |
| 4 | 30a: R1 = Ph, R2 = 4-CF3-Ph | 37 h | 48%/95% | 50%/93% | 9.1/1 | 103 |
| 5 | 31a: R1 = Ph, R2 = 4-MeO-Ph | 37 h | 48%/98% | 51%/96% | 12.5/1 | 226 |
| 6 | 32a: R1 = 3,4,5-triMeO-Ph, R2 = Me | 26 h | 40%/91% | 50%/86% | >20/1c | 42 |
| 7 | 32a: R1 = 3,4,5-triMeO-Ph, R2 = Me | 32 h | 48%/91% | 48%/92% | 19.0/1c,d | 121 |
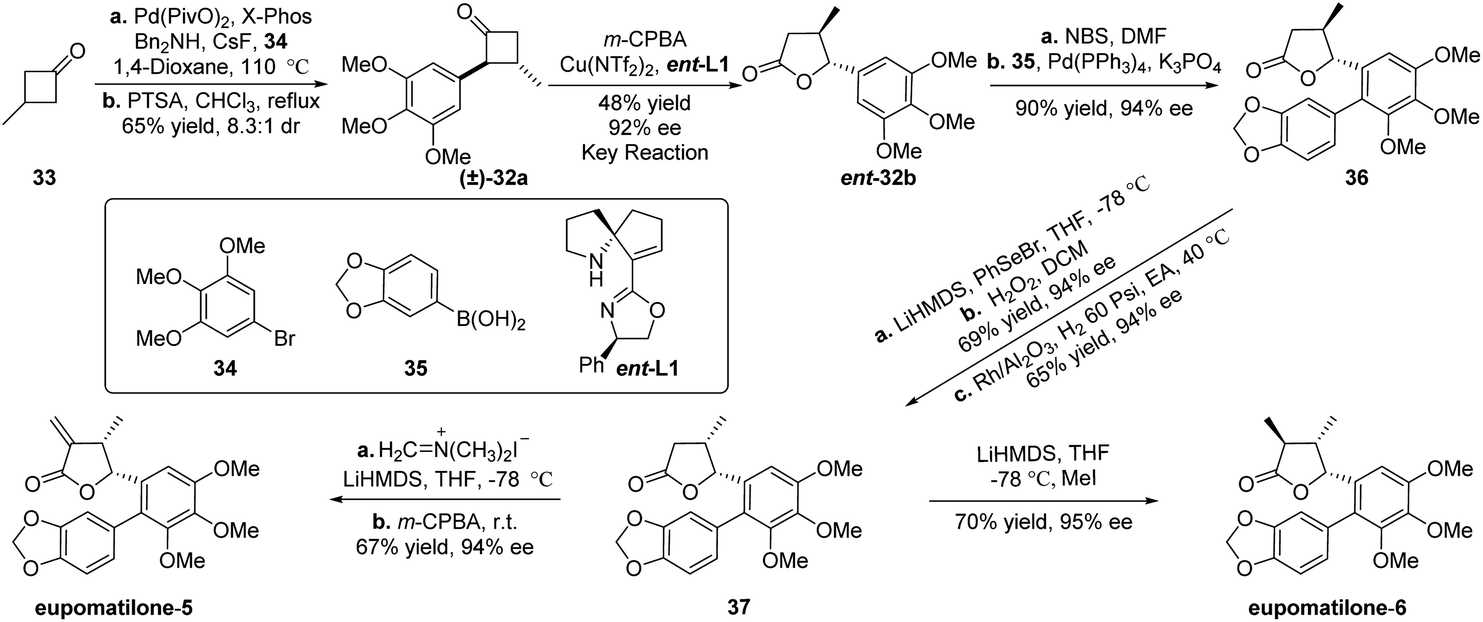
After completing the study of generality of this classical kinetic resolution of 2-aryl-substituted and 2,3-disubstituted cyclobutanones, we focused our efforts on exploring the synthetic application of this unexplored methodology and targeted eupomatilones 5 and 6 isolated from the Australian shrub Eupomatia (Scheme 2).13,14 Starting from commercially available 3-methylcyclobutan-1-one (33), Pd-catalyzed α-arylation of cyclobutanone with 1-bromo-3,4,5-trimethoxybenzene (34) was carried out to prepare racemic precursor 32a for the B–V reaction. However, poor yield and/or diastereoisomeric ratio (dr) was observed under the tested reaction conditions.11 To our delight, after extensive investigation, the desired racemate 32a was obtained in 5.6:1 dr using a more sterically hindered ligand (X-Phos), and the dr of (±)-32a was further increased to 8.3:1 by treating with p-toluenesulfonic acid in refluxing chloroform. To improve the dissatisfactory results of the desired lactone 32b achieved under the optimal reaction conditions (entry 6, Table 3), slightly adjusted reaction conditions (Cu(OTf)2 and ent-L1 were applied)11 were used to furnish the expected lactone ent-32b in 48% yield with an excellent ee of 92% (entry 7, Table 3). With chiral lactone ent-32b in hand, mono-bromination of the aryl ring with N-bromosuccinimide (NBS) and the subsequent Pd-catalyzed Suzuki–Miyaura reaction with 1,3-benzodioxole-5-boronic acid (35) gave biaryl compound 3613b,15in 90% yield and 94% ee (two steps). To reverse the configuration of the methyl group at the β-position of γ-lactone, compound 36 was treated with phenylselenyl bromide (PhSeBr) and lithium bis(trimethylsilyl)amide (LiHMDS) at −78 °C and then oxidized with 30% H2O2 to yield an unsaturated lactone. The subsequent catalytic hydrogenation of the resulting lactone with Rh/Al2O3 in ethyl acetate (EA) at 40 °C afforded γ-butyrolactone 37 with high diastereoselectivity.16 Finally, the total synthesis of eupomatilones 5 and 6 was concisely completed in just a one-step transformation. Treatment of 37 with Eschenmoser's salt in THF at −78 °C and subsequent elimination produced eupomatilone-5 in 67% yield and 94% ee, while eupomatilone-6 was obtained through stereoselective methylation using LiHMDS and MeI at −78 °C (70% yield, 95% ee).13e The spectral data of two synthetic natural products were consistent with the reported literature.13b,d–g
 | ||
| Scheme 2 Asymmetric total syntheses of eupomatilones 5 and 6. | ||
Conclusions
In summary, we developed an efficient Cu–SPDO catalytic system that exhibits excellent activity in the classical B–V oxidation of 2-aryl-substituted or 2,3-disubstituted cyclobutanones for the first time. The current transformation features a wide substrate scope and excellent enantioselectivity and regioselectivity, providing an alternative and concise approach for the simultaneous preparation of chiral γ-lactones and chiral aryl-substituted cyclobutanones. Additionally, asymmetric total syntheses of natural eupomatilones 5 and 6 were completed using this newly developed methodology as a key step. Other asymmetric reactions catalyzed using metal/SPDO complexes, and their synthetic applications are underway in our group.Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
C.-S. Zhang, F.-M. Zhang and Y.-Q. Tu designed this project. C.-S. Zhang performed the main experiments and prepared the ESI; Y.-P. Shao performed part of the experiments. X. Han synthesised some substrates. X.-M. Zhang, F.-M. Zhang and Y.-Q. Tu supervised and directed the project. C.-S. Zhang, F.-M. Zhang and Y.-Q. Tu wrote the manuscript. All authors discussed the finalized manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the NSFC (No. 21502080, 21772019, 21772076, 21871117, 91956203, and 21971095), the ‘111’ Program of MOE, the Major Project (2018ZX09711001-005-002) of MOST, and the STCSM (19JC1430100) and the lzujbky-2020-ct01 for the financial support.Notes and references
- (a) A. Baeyer and V. Villiger, Ber. Dtsch. Chem. Ges., 1899, 32, 3625–3633 CrossRef; (b) A. Baeyer and V. Villiger, Ber. Dtsch. Chem. Ges., 1900, 33, 858–864 CrossRef CAS.
- For selected reviews, see: (a) G. R. Krow, Org. React., 1993, 43, 251–809 CAS; (b) G. Strukul, Angew. Chem., Int. Ed., 1998, 37, 1198–1209 CrossRef; (c) S. M. Roberts and P. W. H. Wan, J. Mol. Catal. B: Enzym., 1998, 4, 111–136 CrossRef CAS; (d) M. Renz and B. Meunier, Eur. J. Org. Chem., 1999, 737–750 CrossRef CAS; (e) G.-J. t. Brink, I. W. C. E. Arends and R. A. Sheldon, Chem. Rev., 2004, 104, 4105–4123 CrossRef CAS PubMed; (f) M. D. Mihovilovic, Curr. Org. Chem., 2006, 10, 1265–1287 CrossRef CAS; (g) R. A. Michelin, P. Sgarbossa, A. Scarso and G. Strukul, Coord. Chem. Rev., 2010, 254, 646–660 CrossRef CAS; (h) I. A. Yaremenko, V. A. Vil’, D. V. Demchuk and A. O. Terent’ev, Beilstein J. Org. Chem., 2016, 12, 1647–1748 CrossRef CAS PubMed; (i) B. Mao, M. Fañanás-Mastral and B. L. Feringa, Chem. Rev., 2017, 117, 10502–10566 CrossRef CAS PubMed; (j) K. P. Bryliakov, Chem. Rev., 2017, 117, 11406–11459 CrossRef CAS PubMed; (k) C. Liu, K.-G. Wen, X.-P. Zeng and Y.-Y. Peng, Adv. Synth. Catal., 2020, 362, 1015–1031 CrossRef CAS.
- (a) J. D. Stewart, K. W. Reed, C. A. Martinez, J. Zhu, G. Chen and M. M. Kayser, J. Am. Chem. Soc., 1998, 120, 3541–3548 CrossRef CAS; (b) M. J. Taschner and D. J. Black, J. Am. Chem. Soc., 1988, 110, 6892–6893 CrossRef CAS; (c) M. M. Kayser, G. Chen and J. D. Stewart, J. Org. Chem., 1998, 63, 7103–7106 CrossRef CAS PubMed; (d) M. T. Reetz, B. Brunner, T. Schneider, F. Schulz, C. M. Clouthier and M. M. Kayser, Angew. Chem., Int. Ed., 2004, 43, 4075–4078 CrossRef CAS PubMed; (e) M. D. Mihovilovic, F. Rudroff, B. Grötzl, P. Kapitan, R. Snajdrova, J. Rydz and R. Mach, Angew. Chem., Int. Ed., 2005, 44, 3609–3613 CrossRef CAS PubMed; (f) F. Hollmann, A. Taglieber, F. Schulz and M. T. Reetz, Angew. Chem., Int. Ed., 2007, 46, 2903–2906 CrossRef CAS PubMed; (g) M. T. Reetz and S. Wu, J. Am. Chem. Soc., 2009, 131, 15424–15432 CrossRef CAS PubMed; (h) J. Rehdorf, M. D. Mihovilovic and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2010, 49, 4506–4508 CrossRef CAS PubMed; (i) H. Leisch, K. Morley and P. C. K. Lau, Chem. Rev., 2011, 111, 4165–4222 CrossRef CAS PubMed; (j) J. Liu and Z. Li, ACS Catal., 2013, 3, 908–911 CrossRef CAS; (k) Y. Hu, J. Wang, Y. Cen, H. Zheng, M. Huang, X. Lin and Q. Wu, Chem. Commun., 2019, 55, 2198–2201 RSC.
- (a) T. Sugimura, Y. Fujiwara and A. Tai, Tetrahedron Lett., 1997, 38, 6019–6022 CrossRef CAS; (b) S.-I. Murahashi, S. Ono and Y. Imada, Angew. Chem., Int. Ed., 2002, 41, 2366–2368 CrossRef CAS; (c) B. Wang, Y.-M. Shen and Y. Shi, J. Org. Chem., 2006, 71, 9519–9521 CrossRef CAS PubMed; (d) S. Xu, Z. Wang, X. Zhang, X. Zhang and K. Ding, Angew. Chem., Int. Ed., 2008, 47, 2840–2843 CrossRef CAS PubMed; (e) S. Xu, Z. Wang, Y. Li, X. Zhang, H. Wang and K. Ding, Chem.–Eur. J., 2010, 16, 3021–3035 CrossRef CAS PubMed; (f) S. Xu, Z. Wang, X. Zhang and K. Ding, Chin. J. Chem., 2010, 28, 1731–1735 CrossRef CAS; (g) S. Xu, Z. Wang, X. Zhang and K. Ding, Eur. J. Org. Chem., 2011, 2011, 110–116 CrossRef; (h) N. Sasakura, K. Nakano, Y. Ichikawa and H. Kotsuki, RSC Adv., 2012, 2, 6135–6139 RSC; (i) M. W. Giuliano, C.-Y. Lin, D. K. Romney, S. J. Miller and E. V. Anslyn, Adv. Synth. Catal., 2015, 357, 2301–2309 CrossRef CAS PubMed; (j) A. L. Featherston, C. R. Shugrue, B. Q. Mercado and S. J. Miller, ACS Catal., 2019, 9, 242–252 CrossRef CAS PubMed; (k) D. K. Romney, S. M. Colvin and S. J. Miller, J. Am. Chem. Soc., 2014, 136, 14019–14022 CrossRef CAS PubMed; (l) N. C. Abascal and S. J. Miller, Org. Lett., 2016, 18, 4646–4649 CrossRef CAS PubMed; (m) P. P. Poudel, K. Arimitsu and K. Yamamoto, Chem. Commun., 2016, 52, 4163–4166 RSC; (n) K. Zhu, S. Hu, M. Liu, H. Peng and F.-E. Chen, Angew. Chem., Int. Ed., 2019, 58, 9923–9927 CrossRef CAS PubMed.
- (a) C. Bolm, G. Schlingloff and K. Weickhardt, Angew. Chem., Int. Ed. Engl., 1994, 33, 1848–1849 CrossRef; (b) A. Gusso, C. Baccin, F. Pinna and G. Strukul, Organometallics, 1994, 13, 3442–3451 CrossRef CAS; (c) M. Lopp, A. Paju, T. Kanger and T. Pehk, Tetrahedron Lett., 1996, 37, 7583–7586 CrossRef CAS; (d) C. Bolm, T. K. K. Luong and G. Schlingloff, Synlett, 1997, 1151–1152 CrossRef CAS; (e) T. Kanger, K. Kriis, A. Paju, T. Pehk and M. Lopp, Tetrahedron: Asymmetry, 1998, 9, 4475–4482 CrossRef CAS; (f) T. Uchida and T. Katsuki, Tetrahedron Lett., 2001, 42, 6911–6914 CrossRef CAS; (g) Y. Peng, X. Feng, K. Yu, Z. Li, Y. Jiang and C.-H. Yeung, J. Organomet. Chem., 2001, 619, 204–208 CrossRef CAS; (h) C. Bolm, O. Beckmann, T. Kühn, C. Palazzi, W. Adam, P. B. Rao and C. R. Saha-Möller, Tetrahedron: Asymmetry, 2001, 12, 2441–2446 CrossRef CAS; (i) A. Watanabe, T. Uchida, K. Ito and T. Katsuki, Tetrahedron Lett., 2002, 43, 4481–4485 CrossRef CAS; (j) Y. Miyake, Y. Nishibayashi and S. Uemura, Bull. Chem. Soc. Jpn., 2002, 75, 2233–2237 CrossRef CAS; (k) A. Watanabe, T. Uchida, R. Irie and T. Katsuki, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5737–5742 CrossRef CAS PubMed; (l) J.-C. Frison, C. Palazzi and C. Bolm, Tetrahedron, 2006, 62, 6700–6706 CrossRef CAS; (m) M. Colladon, A. Scarso and G. Strukul, Synlett, 2006, 3515–3520 CAS; (n) K. Matsumoto, B. Saito and T. Katsuki, Chem. Commun., 2007, 3619–3627 RSC; (o) A. V. Malkov, F. Friscourt, M. Bell, M. E. Swarbrick and P. Kočovský, J. Org. Chem., 2008, 73, 3996–4003 CrossRef CAS PubMed; (p) G. Bianchini, A. Cavarzan, A. Scarso and G. Strukul, Green Chem., 2009, 11, 1517–1520 RSC; (q) C. Paneghetti, R. Gavagnin, F. Pinna and G. Strukul, Organometallics, 1999, 18, 5057–5065 CrossRef CAS; (r) A. Cavarzan, G. Bianchini, P. Sgarbossa, L. Lefort, S. Gladiali, A. Scarso and G. Strukul, Chem.–Eur. J., 2009, 15, 7930–7939 CrossRef CAS PubMed; (s) K. Ito, A. Ishii, T. Kuroda and T. Katsuki, Synlett, 2003, 643–646 CrossRef CAS; (t) K. S. Petersen and B. M. Stoltz, Tetrahedron, 2011, 67, 4352–4357 CrossRef CAS; (u) R. Sandaroos, M. T. Goldani, S. Damavandi and A. Mohammadi, J. Chem. Sci., 2012, 124, 871–876 CrossRef CAS; (v) L. Zhou, X. Liu, J. Ji, Y. Zhang, X. Hu, L. Lin and X. Feng, J. Am. Chem. Soc., 2012, 134, 17023–17026 CrossRef CAS PubMed; (w) L. Zhou, X. Liu, J. Ji, Y. Zhang, W. Wu, Y. Liu, L. Lin and X. Feng, Org. Lett., 2014, 16, 3938–3941 CrossRef CAS PubMed; (x) N. Yang, Z. Su, X. Feng and C. Hu, Chem.–Eur. J., 2015, 21, 7264–7277 CrossRef CAS PubMed; (y) W. Wu, W. Cao, L. Hu, Z. Su, X. Liu and X. Feng, Chem. Sci., 2019, 10, 7003–7008 RSC.
- (a) X. Liu, L. Lin and X. Feng, Acc. Chem. Res., 2011, 44, 574–587 CrossRef CAS PubMed; (b) X. Liu, H. Zheng, Y. Xia, L. Lin and X. Feng, Acc. Chem. Res., 2017, 50, 2621–2631 CrossRef CAS PubMed.
- V. R. Criegee, Justus Liebigs Ann. Chem., 1948, 560, 127–135 CrossRef.
- (a) J.-M. Tian, Y.-H. Yuan, Y.-Y. Xie, S.-Y. Zhang, W.-Q. Ma, F.-M. Zhang, S.-H. Wang, X.-M. Zhang and Y.-Q. Tu, Org. Lett., 2017, 19, 6618–6621 CrossRef CAS PubMed; (b) S.-K. Chen, W.-Q. Ma, Z.-B. Yan, F.-M. Zhang, S.-H. Wang, Y.-Q. Tu, X.-M. Zhang and J.-M. Tian, J. Am. Chem. Soc., 2018, 140, 10099–10103 CrossRef CAS PubMed; (c) Q. Zhang, F.-M. Zhang, C.-S. Zhang, S.-Z. Liu, J.-M. Tian, S.-H. Wang, X.-M. Zhang and Y.-Q. Tu, Nat. Commun., 2019, 10, 2507 CrossRef PubMed; (d) Q. Zhang, F.-M. Zhang, C.-S. Zhang, S.-Z. Liu, J.-M. Tian, S.-H. Wang, X.-M. Zhang and Y.-Q. Tu, J. Org. Chem., 2019, 84, 12664–12671 CrossRef CAS PubMed; (e) Y.-H. Yuan, X. Han, F.-P. Zhu, J.-M. Tian, F.-M. Zhang, X.-M. Zhang, Y.-Q. Tu, S.-H. Wang and X. Guo, Nat. Commun., 2019, 10, 3394 CrossRef PubMed; (f) M.-H. Xu, Y.-H. Yuan, D.-D. Liang, X.-M. Zhang, F.-M. Zhang, Y.-Q. Tu, A.-J. Ma, K. Zhang and J.-B. Peng, Org. Chem. Front., 2021, 8, 3292–3297 RSC; (g) X.-Y. Zhang, Y.-P. Shao, B.-K. Guo, K. Zhang, F.-M. Zhang, X.-M. Zhang and Y.-Q. Tu, Chem. Commun., 2021, 57, 11233–11235 RSC; (h) J.-S. Yang, K. Lu, C.-X. Li, Z.-H. Zhao, X.-M. Zhang, F.-M. Zhang and Y.-Q. Tu, Angew. Chem., Int. Ed., 2022, 61, e202114129 CAS.
- (a) J.-M. Tian, A.-F. Wang, J.-S. Yang, X.-J. Zhao, Y.-Q. Tu, S.-Y. Zhang and Z.-M. Chen, Angew. Chem., Int. Ed., 2019, 58, 11023–11027 CrossRef CAS PubMed; (b) C.-C. Xi, X.-J. Zhao, J.-M. Tian, Z.-M. Chen, K. Zhang, F.-M. Zhang, Y.-Q. Tu and J.-W. Dong, Org. Lett., 2020, 22, 4995–5000 CrossRef CAS PubMed; (c) X.-J. Zhao, Z.-H. Li, T.-M. Ding, J.-M. Tian, Y.-Q. Tu, A.-F. Wang and Y.-Y. Xie, Angew. Chem., Int. Ed., 2021, 60, 7061–7065 CrossRef CAS PubMed; (d) Z.-R. Jing, D.-D. Liang, J.-M. Tian, F.-M. Zhang and Y.-Q. Tu, Org. Lett., 2021, 23, 1258–1262 CrossRef CAS PubMed.
- (a) W. Ai, Y. Liu, Q. Wang, Z. Lu and Q. Liu, Org. Lett., 2018, 20, 409–412 CrossRef CAS PubMed; (b) T. Wang, Y.-N. Wang, R. Wang, B.-C. Zhang, C. Yang, Y.-L. Li and X.-S. Wang, Nat. Commun., 2019, 10, 5373 CrossRef PubMed.
- For details, please see the ESI.†.
- The values were calculated using the equation S = ln[(1 − c) (1 + eea)]/ln[(1 − c) (1 − eea)], where ee is the enantiomeric excess of the product and c is the conversion. The conversion was set to equal the ratio of (eea)/(eea + eeb).
- (a) R. S. Coleman and S. R. Gurrala, Org. Lett., 2004, 6, 4025–4028 CrossRef CAS PubMed; (b) S. H. Yu, M. J. Ferguson, R. McDonald and D. G. Hall, J. Am. Chem. Soc., 2005, 127, 12808–12809 CrossRef CAS PubMed; (c) G. W. Kabalka and B. Venkataiah, Tetrahedron Lett., 2005, 46, 7325–7328 CrossRef CAS; (d) M. K. Gurjar, B. Karumudi and C. V. Ramana, J. Org. Chem., 2005, 70, 9658–9661 CrossRef CAS PubMed; (e) S. Mitra, S. R. Gurrala and R. S. Coleman, J. Org. Chem., 2007, 72, 8724–8736 CrossRef CAS PubMed; (f) Y. Hirokawa, M. Kitamura, M. Mizubayashi, R. Nakatsuka, Y. Kobori, C. Kato, Y. Kurata and N. Maezaki, Eur. J. Org. Chem., 2013, 721–727 CrossRef CAS; (g) H. Choi, H. Jang, H. Kim and K. Lee, Org. Lett., 2019, 21, 7857–7862 CrossRef CAS PubMed; (h) H. Choi, J. Choi, J. Han and K. Lee, J. Org. Chem., 2022, 87, 4316–4322 CrossRef CAS PubMed.
- (a) A. R. Carroll and W. C. Taylor, Aust. J. Chem., 1991, 44, 1615–1626 CrossRef CAS; (b) A. R. Carroll and W. C. Taylor, Aust. J. Chem., 1991, 44, 1705–1714 CrossRef CAS.
- J. B. Johnson, E. A. Bercot, C. M. Williams and T. Rovis, Angew. Chem., Int. Ed., 2007, 46, 4514–4518 CrossRef CAS PubMed.
- M. K. Gurjar, J. Cherian and C. V. Ramana, Org. Lett., 2004, 6, 317–319 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2164218–2164220, 2169290, and 2169293. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc02079c |
| This journal is © The Royal Society of Chemistry 2022 |