
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Aldehyde-functional thermoresponsive diblock copolymer worm gels exhibit strong mucoadhesion†
Emma E.
Brotherton‡
a,
Thomas J.
Neal‡
 a,
Daulet B.
Kaldybekov‡
bc,
Mark J.
Smallridge
d,
Vitaliy V.
Khutoryanskiy
*b and
Steven P.
Armes
*a
a,
Daulet B.
Kaldybekov‡
bc,
Mark J.
Smallridge
d,
Vitaliy V.
Khutoryanskiy
*b and
Steven P.
Armes
*a
aDainton Building, Department of Chemistry, University of Sheffield, Brook Hill, Sheffield, South Yorkshire S3 7HF, UK. E-mail: s.p.armes@sheffield.ac.uk
bSchool of Pharmacy, University of Reading, Whiteknights, PO Box 224, Reading, Berkshire RG6 6DX, UK. E-mail: v.khutoryanskiy@reading.ac.uk
cDepartment of Chemistry and Chemical Technology, Al-Farabi Kazakh National University, Almaty 050040, Kazakhstan
dGEO Specialty Chemicals, Hythe, Southampton, Hampshire SO45 3ZG, UK
First published on 26th May 2022
Abstract
A series of thermoresponsive diblock copolymer worm gels is prepared via reversible addition–fragmentation chain transfer (RAFT) aqueous dispersion polymerization of 2-hydroxypropyl methacrylate using a water-soluble methacrylic precursor bearing pendent cis-diol groups. Selective oxidation using an aqueous solution of sodium periodate affords the corresponding aldehyde-functional worm gels. The aldehyde groups are located within the steric stabilizer chains and the aldehyde content can be adjusted by varying the periodate/cis-diol molar ratio. These aldehyde-functional worm gels are evaluated in terms of their mucoadhesion performance with the aid of a fluorescence microscopy-based assay. Using porcine urinary bladder mucosa as a model substrate, we demonstrate that these worm gels offer a comparable degree of mucoadhesion to that afforded by chitosan, which is widely regarded to be a ‘gold standard’ positive control in this context. The optimum degree of aldehyde functionality is approximately 30%: lower degrees of functionalization lead to weaker mucoadhesion, whereas higher values compromise the desirable thermoresponsive behavior of these worm gels.
Introduction
It is well known that amphiphilic diblock copolymers undergo spontaneous self-assembly in aqueous solution to form a wide range of nano-objects, including spheres, worms, vesicles or lamellae.1–7 Typically, such morphologies are accessed via post-polymerization processing via initial copolymer dissolution in a suitable water-miscible solvent such as THF or DMF, followed by dilution via slow addition of water.8 Under such near-equilibrium conditions, the precise copolymer morphology usually depends solely on the relative volume fraction of each block, as indicated by the fractional packing parameter originally introduced for conventional small molecule surfactants.8–10Over the past decade or so, the development of polymerization-induced self-assembly (PISA) has provided convenient access to many copolymer morphologies.11–22 Unlike spheres or vesicles, diblock copolymer worms usually occupy relatively narrow phase space. Nevertheless, we and others have shown that the construction of pseudo-phase diagrams facilitates the reproducible synthesis of worms, which are usually well-defined in terms of their mean cross-sectional area but typically somewhat polydisperse in terms of their length.12,18,20,23–31
Such worms typically form 3D networks in semi-concentrated solution, which leads to macroscopic gelation under zero shear at ambient temperature.25,26 In particular, the reversible addition–fragmentation chain transfer (RAFT) aqueous dispersion polymerization of 2-hydroxypropyl methacrylate (HPMA) provides access to thermoresponsive worms that exhibit a worm-to-sphere transition on cooling to sub-ambient temperature.27,32–35 This morphological transition is accompanied by degelation and is reversible.36 This is important in the context of potential cell biology applications because it allows the media to be sterilized via ultrafiltration and enables the cells to be readily harvested after cell culture studies.34,37–40
Polymeric hydrogels have many applications in biomedical research, ranging from soft contact lenses to gel electrophoresis.41,42 In principle, hydrogels bearing appropriate chemical functionality can adhere to biological surfaces. This is likely to be particularly important for mucosal drug delivery, for which therapeutic efficiency is often substantially reduced by the continuous production and flow of biological fluids.43,44 This can result in drug leakage from the site of administration, which prevents effective localized delivery. For example, poor retention on mucosal surfaces is a common problem in delivering drugs to the eye, where the continuous production of tear fluid causes rapid removal of the active pharmaceutical ingredient from ocular surfaces.45,46 Similar problems are well-documented for the nasal cavity: the generation of mucus and the protective function afforded by mucociliary clearance does not allow drug molecules to be retained on the olfactory epithelium, which would otherwise potentially offers efficient nasal delivery to the brain.47,48 Similarly, drugs administered by catheter to treat bladder cancer also suffer from short residence times owing to the continuous production of urine and the periodic need for organ voiding.49,50
In principle, more effective drug delivery via mucosal surfaces should be feasible by designing mucoadhesive hydrogels. Various strategies to enhance mucoadhesion have been reported, including the design of copolymers containing thiol,51,52 acryloyl,53,54 methacryloyl55,56 or maleimide groups.57,58 These reactive moieties can form covalent bonds with the thiol group in cysteine, which is one of the amino acid building blocks present within mucins. Another strategy is the introduction of phenylboronic acid groups, which can form dynamic covalent bonds with the 1,2-diol-functional sugar groups expressed by mucins.59,60 Alternatively, catechol-based mucoadhesive polymers have been evaluated owing to their ability to form catechol-thiol or catechol-amine adducts with mucins.61,62 More recently, Bernkop-Schnürch and co-workers reported the synthesis of polymers functionalized with N-hydroxy(sulfo)succinimide esters that form amide bonds with mucins.63,64
Recently, we reported the use of RAFT polymerization65–67 for the synthesis of a new water-soluble methacrylic polymer (denoted as PGEO5MA) that contains pendent cis-diol groups, see Scheme S1.†68 This precursor can be oxidized under mild conditions in aqueous solution using sodium periodate to produce the corresponding aldehyde-functionalized water-soluble polymer. Subsequently, we employed a PGEO5MA precursor for the RAFT aqueous dispersion polymerization of HPMA to prepare a series of well-defined diblock copolymer spheres, worms or vesicles.69 In particular, it was shown that a model globular protein could be chemically adsorbed onto periodate-treated PGEO5MA-PHPMA vesicles via Schiff base chemistry (followed by in situ reduction of the initial labile imine linkages to produce hydrolytically stable secondary amine bonds). In the present study, we revisit this aqueous PISA formulation to prepare well-defined aldehyde-functional diblock copolymer worm gels and examine whether such materials offer any potential use in the context of mucoadhesion using porcine urinary bladder mucosa as a model system.
Results and discussion
Water-soluble PGEO5MA13 and PGEO5MA16 precursors were prepared via RAFT solution polymerization of GEO5MA in ethanol (Scheme S2†). DMF gel permeation chromatography (GPC) analysis (using a series of poly(methyl methacrylate) calibration standards) indicated that these homopolymers had Mn values of 9.3 and 11.2 kg mol−1, respectively, and relatively narrow molecular weight distributions (Đ = 1.19 and 1.18, respectively; Fig. S1†). Each PGEO5MA precursor was then chain-extended via RAFT aqueous dispersion polymerization of HPMA at 10% w/w solids (Scheme 1). A series of PGEO5MA13-PHPMAy (y = 120–200) and PGEO5MA16-PHPMAy (y = 140–220) diblock copolymer nanoparticles were prepared in order to identify a pure worm phase. All polymerizations had high HPMA conversions (>99%) as determined by 1H NMR spectroscopy while DMF GPC analysis indicated reasonably good RAFT control (Đ ≤ 1.25; Fig. S2†). A high molecular weight shoulder can be observed in each chromatogram, which has been previously attributed to dimethacrylate impurities in the HPMA monomer (<0.30 mol%).70,71 In particular, PGEO5MA13-PHPMA150-190 formed soft, thermoresponsive free-standing gels and a pure worm morphology was confirmed by transmission electron microscopy (TEM) studies (Fig. S3†). Similarly, a pure worm phase was obtained for PGEO5MA16-PHPMA170–200 as judged by TEM studies (Fig. S4†).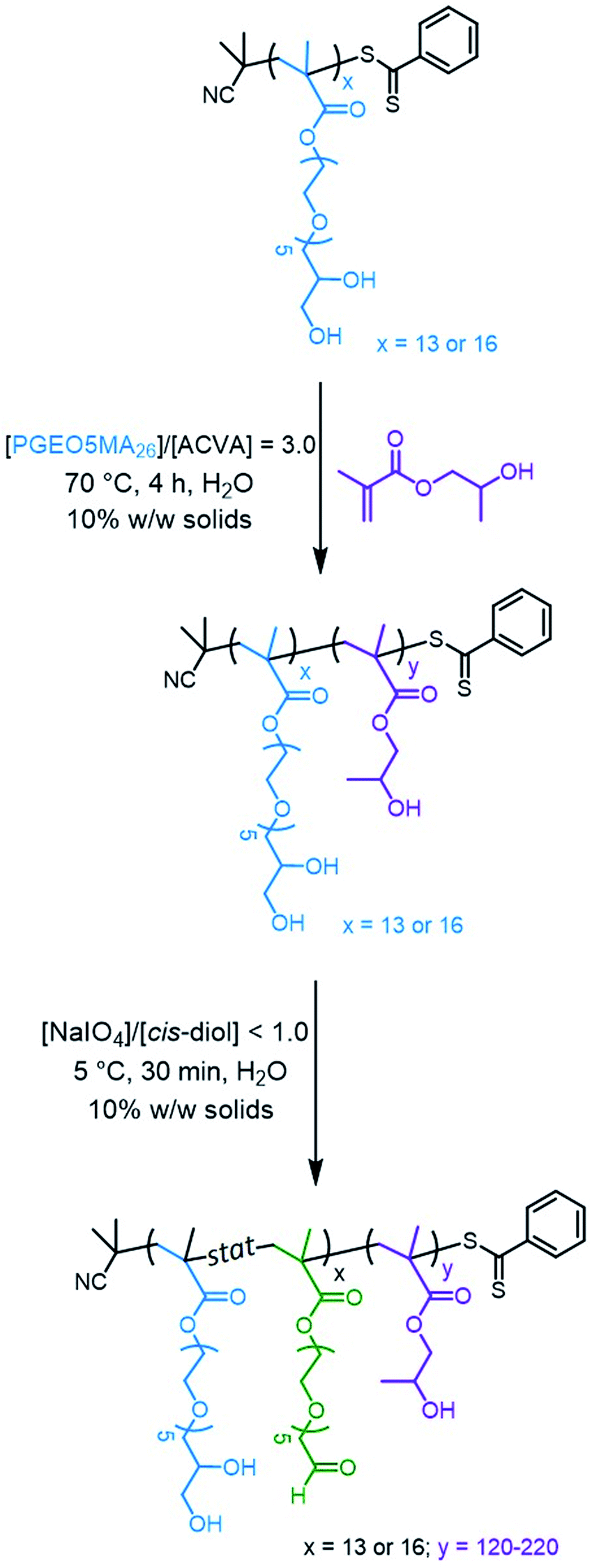 | ||
| Scheme 1 Two-step synthesis of aldehyde-functionalized PAGEO5MAx–PHPMAy diblock copolymer worms starting from the cis-diol groups in the PGEO5MAx homopolymer. In the first step, this water-soluble precursor is chain-extended via RAFT aqueous dispersion polymerization of HPMA. The second step involves partial selective oxidation of the PGEO5MAx stabilizer block using aqueous sodium periodate at 22 °C. | ||
The thermoresponsive nature of a PGEO5MA13-PHPMA155 and a PGEO5MA16-PHPMA200 worm gel was initially confirmed by visual inspection. A 10% w/w aqueous copolymer dispersion of each sample formed a soft, free-standing gel at 22 °C, see Fig. 1 and S5.† On cooling to 5 °C, degelation occurred to afford free-flowing liquids in both cases, with TEM analysis indicating a concomitant worm-to-sphere transition.
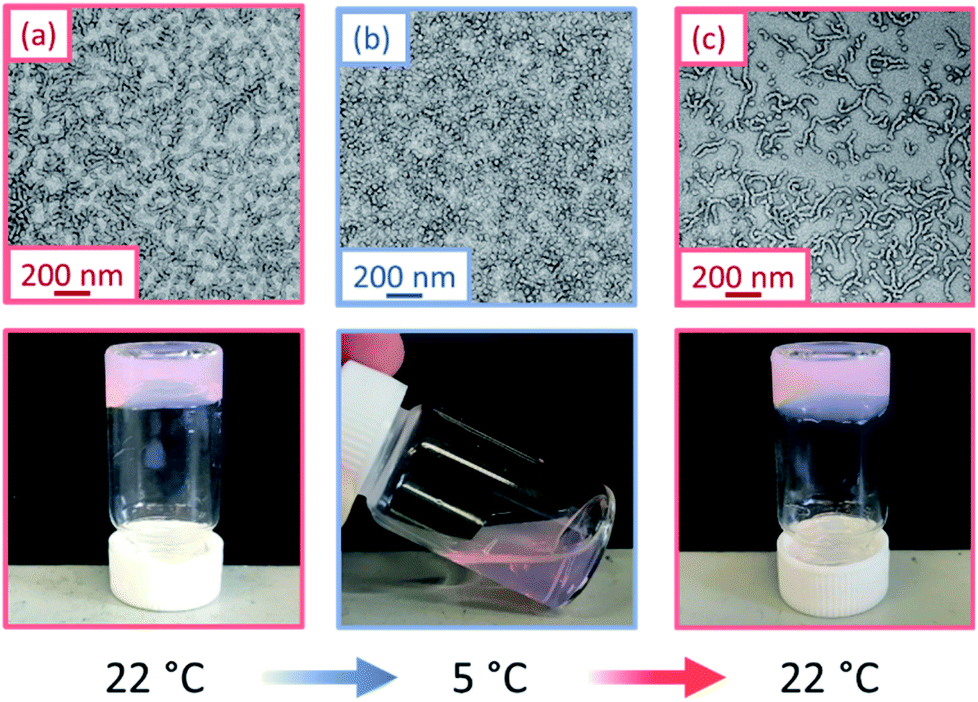 | ||
| Fig. 1 TEM images and corresponding digital photographs recorded for a 10% w/w aqueous dispersion of PGEO5MA13-PHPMA155 nano-objects: (a) soft, free-standing worm gel formed at 22 °C, (b) free-flowing fluid obtained on cooling to 5 °C and (c) the reconstituted worm gel formed after returning to 22 °C. [N.B. The pink coloration is conferred by the dithiobenzoate-based RAFT chain-ends]. | ||
On returning to 22 °C, regelation was observed for PGEO5MA13-PHPMA155 owing to a sphere-to-worm transition. However, no regelation was observed for PGEO5MA16-PHPMA200, and TEM analysis indicated the presence of kinetically-trapped spheres and short worms in this case (Fig. S6†). Fully reversible thermoresponsive behavior is highly desirable for biomedical applications since this enables facile sterilization via cold ultrafiltration.38 Thus, only the PGEO5MA13-PHPMA155 worm gel was selected for the subsequent mucoadhesion studies.
This thermally-induced morphological transition was further characterized using small-angle X-ray scattering (SAXS). A 1.0% w/w aqueous dispersion of PGEO5MA13-PHPMA155 worms was studied at 37 °C and 5 °C. At 37 °C, a gradient of −1 was observed in the Guinier region (low q) of the SAXS pattern (Fig. 2a), which is consistent with the highly anisotropic worms observed by TEM (Fig. 1). However, a gradient of zero is observed in the same low q region on cooling to 5 °C (Fig. 2a). This indicates the presence of spherical nanoparticles, which agrees with the TEM image recorded at the same temperature (Fig. 1). Finally, the 1.0% w/w dispersion was warmed to 37 °C and allowed to equilibrate at this temperature for 1 h. The SAXS pattern recorded after equilibration is almost identical to that original pattern acquired at 37 °C (Fig. 2a). This demonstrates that these PGEO5MA13-PHPMA155 nano-objects exhibit thermoreversible behavior with minimal hysteresis. Moreover, the core radii for the worms (rw) and the spheres (rs) can be estimated using rw = 3.83/q and rs = 4.49/q respectively, where q corresponds to the intensity minimum. The approximate core radii for the initial worm gel, the cold spheres, and the reconstituted worm gel are calculated to be 11, 10, and 12 nm, respectively. These values are comparable with core radii estimated by TEM analysis (counting at least 100 nanoparticles per sample).
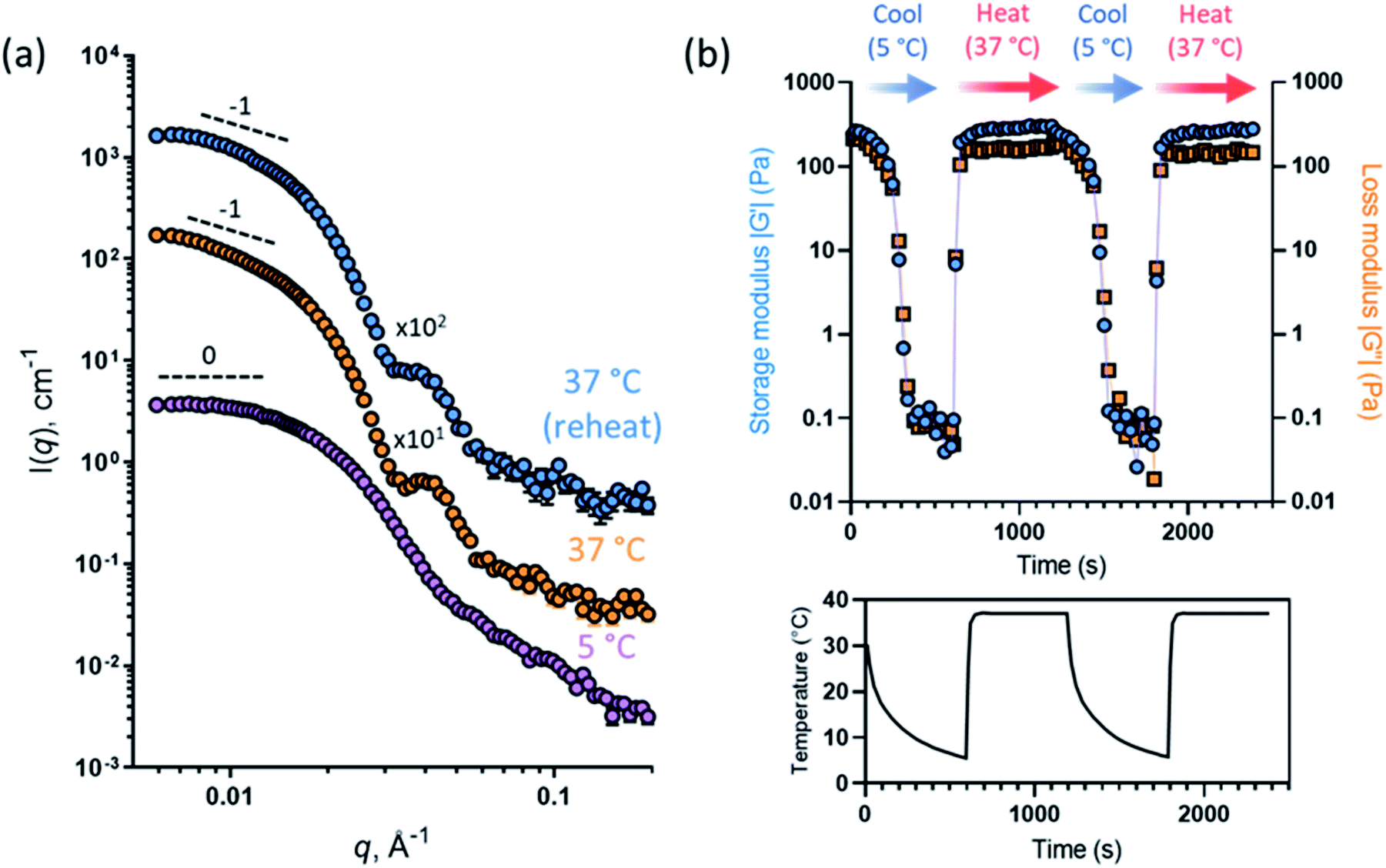 | ||
| Fig. 2 (a) SAXS patterns obtained for a 1.0% w/w aqueous dispersion of PGEO5MA13-PHPMA155 nano-objects initially at 37 °C (orange circles), after cooling to 5 °C (purple circles), and after returning to 37 °C (blue circles) [N.B. the two upper patterns are offset by the stated numerical factors to aid clarity]. Dashed lines indicate low q gradients of 0 and −1 as guidance for the eye, where such gradients indicate the presence of spheres and worms, respectively. (b) Storage and loss moduli (G′ and G′′, respectively) recorded for a 12% w/w aqueous dispersion of PGEO5MA13-PHPMA155 nano-objects over two 37 °C to 5 °C to 37 °C thermal cycles using oscillatory rheology. A temperature–time profile for such experiments is also displayed. | ||
The PGEO5MA13-PHPMA155 worm gel was lyophilized to produce a freeze-dried powder. Redispersion of this copolymer powder in ice-cold deionized water (which ensures near-molecular dissolution of the amphiphilic copolymer chains72) followed by warming to 22 °C produced a soft, free-standing worm gel. Empirically, it was found that redispersion at 12% w/w solids produced longer, more linear worms than redispersion at 10% w/w solids. Thus, all subsequent experiments were conducted at 12% w/w solids.
Oscillatory rheology was used to characterize the thermoresponsive behavior of a 12% w/w aqueous dispersion of PGEO5MA13-PHPMA155 nano-objects. This sample was subjected to shear (1.0% strain at an angular frequency of 1 rad s−1; Fig. 2b) over two thermal cycles (from 5 °C to 37 °C to 5 °C). On cooling to 5 °C the initial worms are converted into spheres, which causes in situ degelation (G′′ > G′; Fig. 2b) and a significant reduction in the complex viscosity (from 345 Pa s at 37 °C to 0.09 Pa s at 5 °C). On warming to 37 °C, worms are reformed and regelation occurs (G′′ < G′; Fig. 2b), with the complex viscosity increasing to 347 Pa s. Essentially the same (de)gelation behavior was observed during the second thermal cycle, which indicates excellent thermoreversibility.
Importantly, the pendent cis-diol units on these copolymer worms can be selectively oxidized using sodium periodate in aqueous solution under mild conditions to introduce aldehyde groups within the steric stabilizer chains (Scheme 1).68,69 In principle, such derivatization might be expected to produce mucoadhesive worm gels since it is well-known that aldehydes can react readily with amines via Schiff base chemistry.73 However, fluorescence labelling is normally required for mucoadhesion flow-through assays.74,75 Therefore, HPMA and fluorescein methacrylate (FMA) were statistically copolymerized to produce fluorescein-tagged worms (Scheme S3†). A FMA content of 0.1 mol% was targeted and the overall comonomer conversion was more than 99% as indicated by 1H NMR spectroscopy. The initial pink worm gel formed a bright yellow worm gel on adjusting the solution from pH 5 to pH 9 with NaOH, indicating that the FMA was incorporated within the core-forming block (Fig. S7†). Moreover, UV GPC analysis performed at λ = 495 nm (which corresponds to the maximum absorbance for the FMA repeat units when they are in their anionic carboxylate form) produced a very similar molecular weight distribution curve to that recorded using a refractive index detector (Fig. S8†).76 TEM analysis confirmed that there was no discernible difference between the fluorescently-labeled PGEO5MA13-P(HPMA155-stat-FMA0.15) worms and the non-fluorescent PGEO5MA13-PHPMA155 worms (Fig. S9†).
The former worms were subsequently oxidized with sodium periodate targeting periodate/cis-diol molar ratios of 0.10, 0.20, 0.30 or 0.50 using a previously reported protocol developed for crosslinked PGEO5MA26-PHPMA350-PEGDMA20 vesicles.69 In each case, the extent of oxidation was determined by 1H NMR spectroscopy (Fig. S10†). We chose to conduct periodate oxidation on the final diblock copolymer nanoparticles rather than on the PGEO5MA precursor. This approach means that the same cis-diol-functional worm gel precursor was used to produce each aldehyde-functional worm gel examined in this study, which eliminates batch-to-batch variability. Moreover, 1H NMR spectroscopy studies (data not shown) confirmed that a sample of 100% aldehyde-functional PAGEO5MA26-PHPMA250 worms reported in our prior study69 remained stable with respect to aerial oxidation for at least one year when stored at ambient temperature. TEM studies confirmed that there was no discernible change in morphology after oxidation (Fig. S9 and S11†). Furthermore, oscillatory strain sweeps (from 0.1% to 20%) were performed on these partially oxidized worm gels to assess how the degree of aldehyde functionality affected the gel properties (Fig. S12†). It was found that a higher degree of oxidation led to a lower strain being required for degelation (G′′ < G′). For example, degelation of the cis-diol functional precursor worm gel required an applied strain of 16.8%, whereas the 50% aldehyde-functionalized worm gel underwent degelation at just 1.1% strain (Fig. S12†). Moreover, higher degrees of aldehyde functionalization led to higher gel viscosities (Fig. S13†). For example, the gel viscosity of the cis-diol functional precursor worm gel was 72 Pa s at an applied strain of 1.0% when equilibrated at ambient temperature, whereas the 50% aldehyde-functionalized worm gel exhibited a gel viscosity of 263 Pa s under the same conditions (Fig. S13†). This indicates that the introduction of aldehyde groups produces stronger (but more fragile/brittle) gels. There are multiple two possible for why the gels become stronger with increased aldehyde content. In principle, the pendent aldehyde groups can react with the remaining cis-diols to form hemiacetal bonds between neighboring worms, thus leading to stronger gels. However, GPC analysis (see Fig. S14†) of the aldehyde-functional diblock copolymer chains provides no evidence for inter-chain crosslinking, which would produce a high molecular weight shoulder. Alternatively, the higher storage moduli observed for the aldehyde-functional worm gels may be related to the formation of stronger hydrogen bonds between the hydroxyl (or ester carbonyl) groups on the remaining cis-diol repeat units and the aldehyde groups (or geminal diol) groups. Further studies (perhaps with model compounds) are required to answer this question but this is beyond the scope of the present study.
Further variable temperature oscillatory rheology experiments were performed on the oxidized worm gels, whereby samples were first cooled to 5 °C and subsequently heated to 37 °C under an applied shear (Fig. 3a and S15†). Thermoreversible degelation was observed in most cases but the rate of degelation was notably slower for gels with higher degrees of aldehyde functionality. Such thermoresponsive behavior was confirmed by visual inspection: free-standing gels became free-flowing liquids after cooling from 22 °C to 5 °C for 50 min and reformed free-standing gels on returning to ambient temperature (Fig. S16†). These observations were consistent with TEM studies, which indicated the presence of spheres at 5 °C and worms at 25 °C (Fig. 3b). However, the 50% aldehyde-functional worm gel did not undergo degelation at all on the timescale (20 min) of the rheology experiments (Fig. S15d†). Thus, the highest degree of aldehyde functionality that can be incorporated into the PGEO5MA13-P(HPMA155-stat-FMA0.15) worm gel precursor without significantly affecting its thermoresponsive behavior is 30%.Interestingly, the worm gels remained pink after oxidation, suggesting retention of the dithiobenzoate end-groups (Fig. S16†). This was confirmed by UV GPC studies (λ = 298 nm), which indicated that this RAFT end-group is retained after periodate oxidation (Fig. S14†).
The retention of such worm gels on mucosal surfaces was studied using a porcine urinary bladder mucosa model under a constant flow of artificial urine (AU). This model mimics the physiologically relevant conditions within the urinary bladder following the intravesical administration of therapeutic agents for the treatment of bladder cancer or interstitial cystitis. Fig. 4 shows fluorescence images recorded for urinary bladder tissue when using a series of fluorescently-labeled worm gels plus two control samples after washing with varying volumes of AU. Fluorescein isothiocyanate (FITC)-chitosan and FITC-dextran were used as positive and negative controls owing to their strong and weak adhesion to mucosal tissues, respectively.77 Worm gels bearing 0, 10, 20, 30 or 50 mol% aldehyde functionality were evaluated in these experiments. Visual inspection of these images indicates that the incorporation of aldehyde groups within such worm gels clearly improves their retention on mucosal tissue. All images were analyzed using ImageJ software to determine fluorescence intensities, which were then converted into % mucosal retention (Fig. 5). This approach enables quantitative interpretation of the wash-off experiments. The worm gel containing no aldehyde groups exhibited relatively weak adhesion to the mucosa, which is initially comparable to that for FITC-dextran. However, unlike this negative control, this worm gel are still retained on mucosal surface to some extent even after washing with up to 120 mL of AU. This may be related to their favorable rheological characteristics relative to non-gelling FITC-dextran. The worm gel containing 10% aldehyde groups exhibited substantially improved retention with around 20% remaining on the bladder mucosa after washing with 120 mL of AU. Further increasing the aldehyde content in the worm gels up to either 20 or 30% led to progressively stronger mucoadhesion. Most notably, the worm gel bearing 50% aldehyde groups exhibited comparable mucoadhesion to that of chitosan, which is widely considered to be a ‘gold standard’ mucoadhesive polymer.78 It is perhaps worth emphasizing that such polyelectrolytes usually exhibit superior mucoadhesive properties compared to non-ionic polymers.78 In contrast, the hydroxyl-rich worm gels examined in this study possess solely non-ionic character.
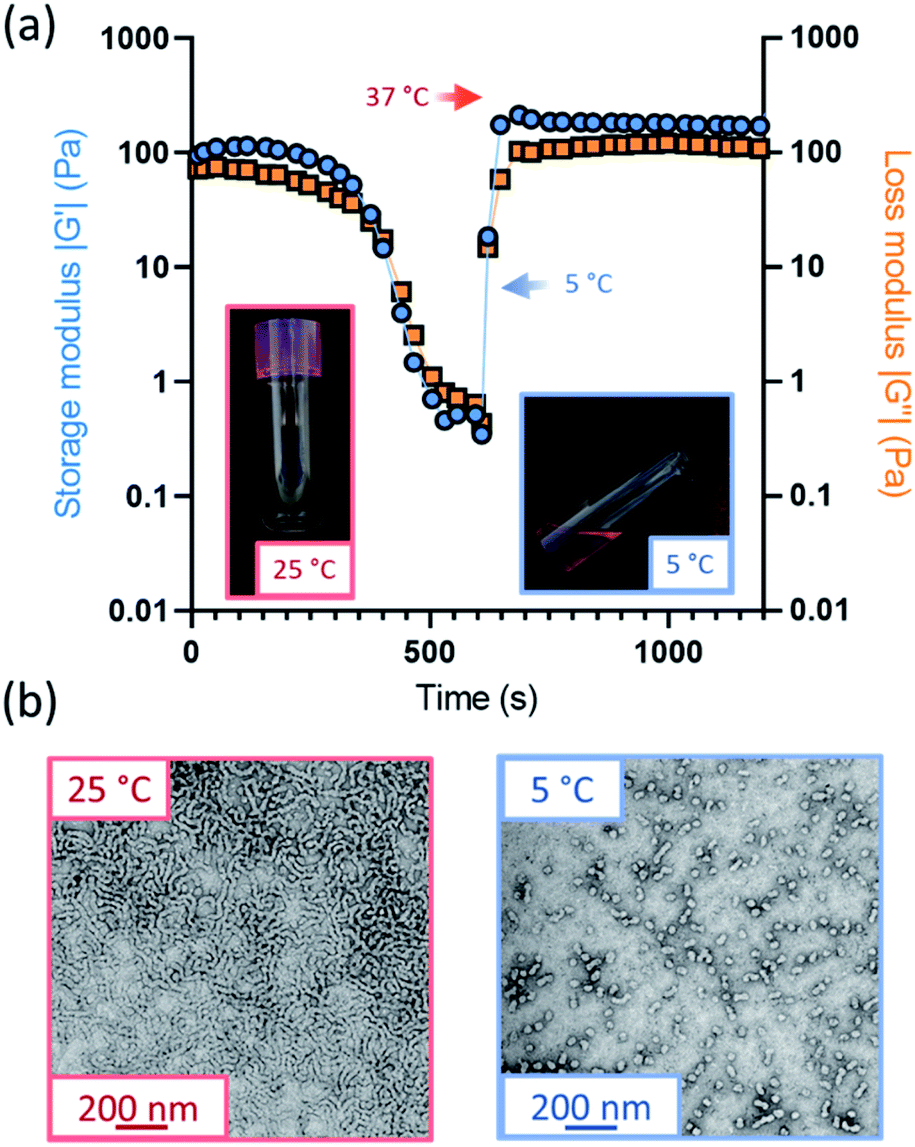 | ||
| Fig. 3 (a) Variable temperature oscillatory rheology monitoring the storage and loss modulus (G′ and G′′, respectively) at both 5 °C and 37 °C for PGEO5MA13-P(HPMA155-stat-FMA0.15) worms with 30% aldehyde functionality. (b) TEM images for PGEO5MA13-P(HPMA155-stat-FMA0.15) worms with 30% aldehyde at 25 °C and the spherical nanoparticles at 5 °C. | ||
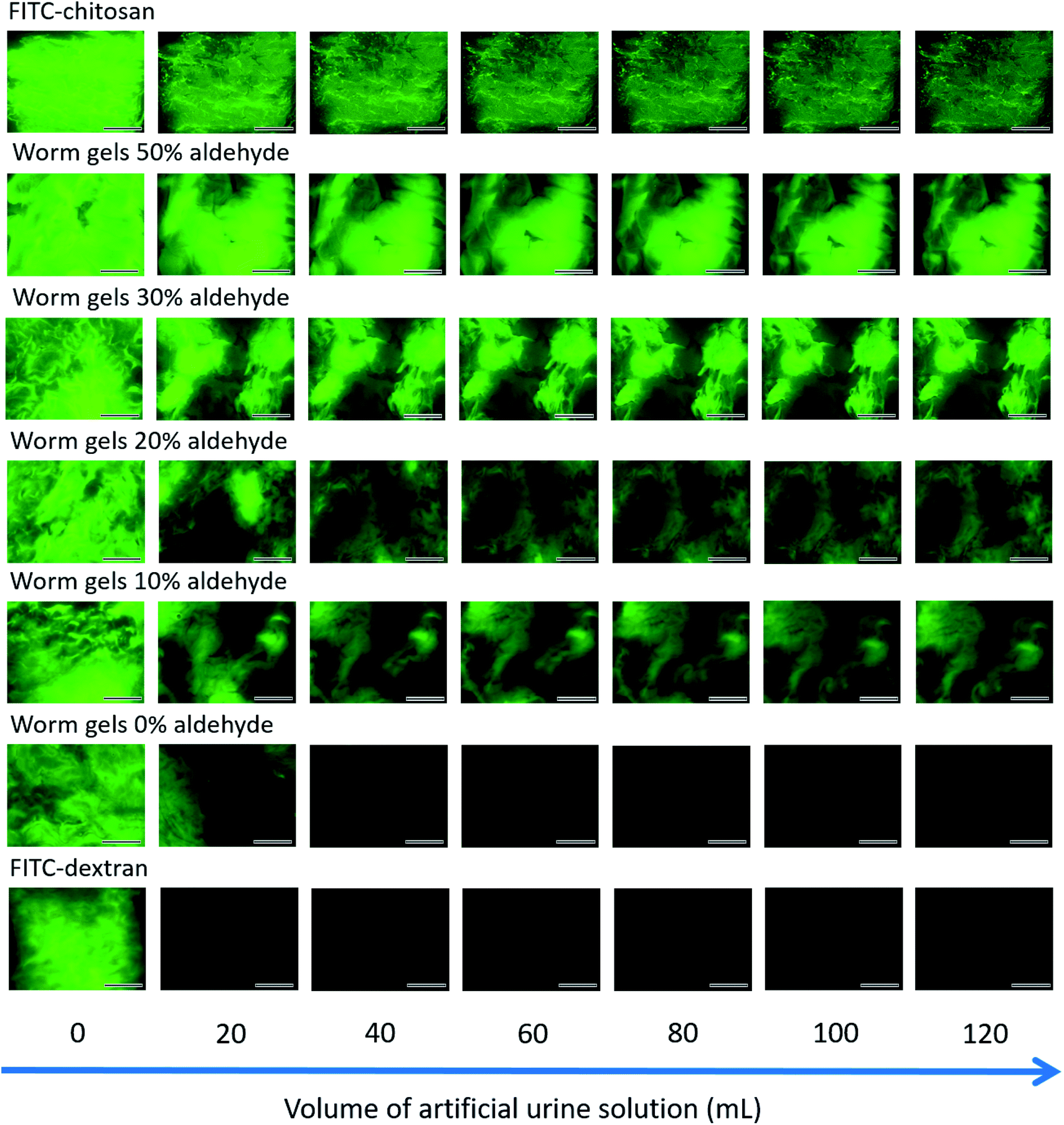 | ||
| Fig. 4 Representative fluorescence images of freshly-dissected porcine urinary bladder mucosa illustrating the retention of fluorescently-labeled PGEO5MA13-P(HPMA155-stat-FMA0.15) worm gels bearing varying degrees of aldehyde functionality after irrigation with varying volumes of AU solution at a flow rate of 2.0 mL min−1, plus positive and negative controls (FITC-chitosan and FITC-dextran, respectively). Scale bars correspond to 6 mm. | ||
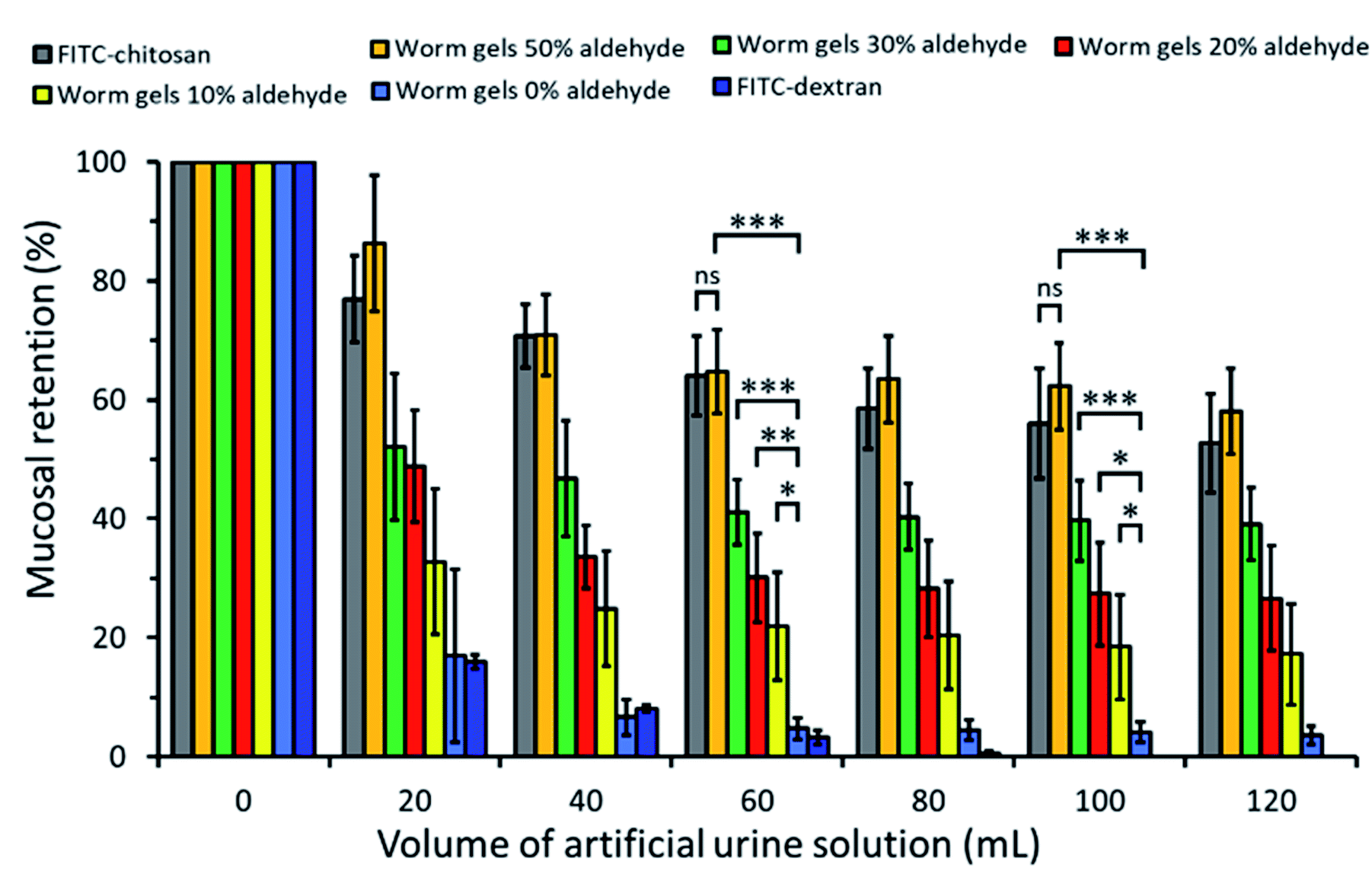 | ||
| Fig. 5 Percentage retention for fluorescently-labeled PGEO5MA13-P(HPMA155-stat-FMA0.15) worm gels with differing degrees of aldehyde functionality on freshly dissected porcine urinary bladder mucosa after irrigating with varying volumes of AU solution at a flow rate of 2.0 mL min−1, plus positive and negative controls. Data are expressed as mean values ± standard deviations (n = 3). Statistically significant differences are given as: * = p < 0.05; ** = p < 0.01; *** = p < 0.001; ns denotes no significance. | ||
Experimental
Materials
GEO5MA monomer was synthesized at GEO Specialty Chemicals (Hythe, UK) by Dr C. P. Jesson as previously reported.68 2-Hydroxypropyl methacrylate (HPMA, 97%) was provided by GEO Specialty Chemicals (Hythe, UK). 4,4′-Azobis(4-cyanopentanoic acid) (ACVA; >98%), sodium periodate (NaIO4, ≥99.8%), chitosan (low molecular weight), fluorescein methacrylate (FMA; 95%), fluorescein isothiocyanate (FITC) (isomer l) and FITC-dextran (MW = 3000–5000 Da) were purchased from Sigma-Aldrich (UK). 2-Cyano-2-propyl dithiobenzoate (CPDB, >97%) was purchased from Strem Chemicals Ltd (Cambridge, UK). Ethanol and diethyl ether were purchased from Fisher Scientific (UK). d7-Dimethylformamide (DMF) was purchased from Goss Scientific Instruments Ltd (Cheshire, UK). All reagents were used as received unless otherwise stated. Dialysis tubing with a molecular weight cut-off of 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–14000 Da was purchased from Medicell Membranes Ltd. (UK). Deionized water was used for all experiments involving aqueous solutions.
000–14000 Da was purchased from Medicell Membranes Ltd. (UK). Deionized water was used for all experiments involving aqueous solutions.
Methods
000 g mol−1. Chromatograms were analyzed using Agilent GPC/SEC software.
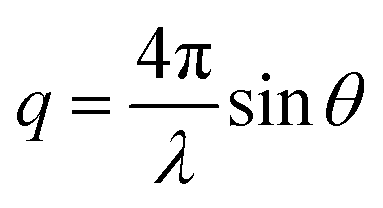 and θ is half of the scattering angle. Glass capillaries of 2.0 mm diameter were used as a sample holder and the sample temperature was controlled using a HFSX350-CAP heating/cooling stage (Linkam Scientific Instruments Ltd, Tadworth, UK), with 10 min being allowed for thermal equilibration of each capillary prior to data collection. 2D X-ray scattering patterns were reduced using software supplied by the SAXS instrument manufacturer. Background subtraction and further data analysis were performed using Irena SAS macro (version 2.61) for Igor Pro.79 The scattering of pure water was used for absolute intensity calibration of the SAXS patterns.
and θ is half of the scattering angle. Glass capillaries of 2.0 mm diameter were used as a sample holder and the sample temperature was controlled using a HFSX350-CAP heating/cooling stage (Linkam Scientific Instruments Ltd, Tadworth, UK), with 10 min being allowed for thermal equilibration of each capillary prior to data collection. 2D X-ray scattering patterns were reduced using software supplied by the SAXS instrument manufacturer. Background subtraction and further data analysis were performed using Irena SAS macro (version 2.61) for Igor Pro.79 The scattering of pure water was used for absolute intensity calibration of the SAXS patterns.
Ex vivo mucoadhesion studies on porcine urinary bladder tissues
Aqueous solutions of FITC-chitosan (1.0 mg mL−1 in 0.5% acetic acid) and FITC-dextran (1.0 mg mL−1 in deionized water) were prepared for use as positive and negative controls, respectively. The FITC-chitosan solution was adjusted to pH 6.0 using 0.1 M NaOH. Then a 200 μL aliquot of either a fluorescently-labeled 12% w/w PGEO5MA13-P(HPMA155-stat-FMA0.15) worm gel prepared in deionized water or a control sample was pipetted onto a mucosal surface and repeatedly washed with AU solution at a flow rate of 2.0 mL min−1 using a syringe pump (total washing time was 60 min). A microscopy image of the mucosal surface of each bladder sample was collected at predetermined time points and then analyzed with ImageJ software by measuring the pixel intensity after each wash. The pixel intensity of the control samples was subtracted from each measurement to obtain normalized intensities. Images from control samples were collected using an exposure time of 20 ms at 1.0× gain. All measurements were conducted in triplicate.
Synthesis
Conclusions
We report the synthesis of thermoresponsive diblock copolymer worm gels via RAFT aqueous dispersion polymerization of HPMA using a water-soluble methacrylic precursor bearing pendent cis-diol groups. Selective oxidation using an aqueous solution of sodium periodate introduces aldehyde groups within the steric stabilizer chains and the aldehyde content can be readily adjusted by varying the NaIO4/cis-diol molar ratio. A series of such aldehyde-functional worm gels are evaluated in the context of mucoadhesion using porcine urinary bladder as a model substrate. A bespoke fluorescence microscopy assay demonstrates that such worm gels can offer similar performance as that afforded by chitosan, which is widely employed as a ‘gold standard’ positive control in this field. One potentially important advantage of these worm gels over chitosan is their non-ionic character, which should enable potential compatibility problems (e.g., complexation with anionic drugs) to be avoided. The optimum degree of aldehyde functionality is approximately 30%: lower degrees of functionalization lead to significantly weaker mucoadhesion, whereas higher values compromise the desirable thermoresponsive behavior of these worm gels. In summary, aldehyde-functionalized worm gels represent a new family of strongly mucoadhesive polymers that can form dynamic covalent imine bonds with mucosal membranes under physiological conditions.Data availability
Relevant additional data are provided in the ESI.†Author contributions
E. E. B. and T. J. N. prepared all the diblock copolymer worms and performed NMR and GPC analyses. E. E. B conducted the periodate oxidation experiments and analysed the nanoparticles by TEM. T. J. N. characterized the worms by SAXS and performed the gel rheology experiments. D. B. K. performed all the mucoadhesion studies. S. P. A. and V. V. K. conceived the project, M. J. S. provided partial funding and all authors contributed to writing this manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. P. A. acknowledges a four-year EPSRC Established Career Particle Technology Fellowship (EP/R003009) which provided postdoctoral support for T. J. N. We thank EPSRC for a CDT PhD studentship for E. E. B. and GEO Specialty Chemicals (Hythe, UK) for CASE support. V. V. K. thanks University of Reading and Royal Berkshire NHS Foundation Trust for postdoctoral support of D. B. K.References
- L. Zhang and A. Eisenberg, Science, 1995, 268, 1728–1731 CrossRef CAS PubMed
.
- L. Zhang, K. Yu and A. Eisenberg, Science, 1996, 272, 1777–1779 CrossRef CAS PubMed
- G. Riess, Prog. Polym. Sci., 2003, 28, 1107–1170 CrossRef CAS
- X. Wang, G. Guerin, H. Wang, Y. Wang, I. Manners and M. A. Winnik, Science, 2007, 317, 644–647 CrossRef CAS PubMed
- L. Zhang and A. Eisenberg, Macromolecules, 1996, 29, 8805–8815 CrossRef CAS
- H. Shen and A. Eisenberg, Angew. Chem., Int. Ed., 2000, 39, 3310–3312 CrossRef CAS PubMed
- Z. Gao, S. K. Varshney, S. Wong and A. Eisenberg, Macromolecules, 1994, 27, 7923–7927 CrossRef CAS
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC
- J. N. Israelachvili, D. J. Mitchell and B. W. Ninham, J. Chem. Soc. Faraday Trans. 2, 1976, 72, 1525–1568 RSC
- M. Antonietti and S. Förster, Adv. Mater., 2003, 15, 1323–1333 CrossRef CAS
- N. J. Warren and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 10174–10185 CrossRef CAS PubMed
- S. Boissé, J. Rieger, K. Belal, A. Di-Cicco, P. Beaunier, M.-H. H. Li and B. Charleux, Chem. Commun., 2010, 46, 1950–1952 RSC
- C. J. Ferguson, R. J. Hughes, B. T. T. Pham, B. S. Hawkett, R. G. Gilbert, A. K. Serelis and C. H. Such, Macromolecules, 2002, 35, 9243–9245 CrossRef CAS
- C. J. Ferguson, R. J. Hughes, D. Nguyen, B. T. T. Pham, R. G. Gilbert, A. K. Serelis, C. H. Such and B. S. Hawkett, Macromolecules, 2005, 38, 2191–2204 CrossRef CAS
- P. Yang, Y. Ning, T. J. Neal, E. R. Jones, B. R. Parker and S. P. Armes, Chem. Sci., 2019, 10, 4200–4208 RSC
- J. Rieger, Macromol. Rapid Commun., 2015, 36, 1458–1471 CrossRef CAS PubMed
- C. E. Boott, J. Gwyther, R. L. Harniman, D. W. Hayward and I. Manners, Nat. Chem., 2017, 9, 785–792 CrossRef CAS PubMed
- G. Mellot, J. M. Guigner, L. Bouteiller, F. Stoffelbach and J. Rieger, Angew. Chem., Int. Ed., 2019, 58, 3173–3177 CrossRef CAS PubMed
- E. Vergari, J. G. Knudsen, R. Ramracheya, A. Salehi, Q. Zhang, J. Adam, I. W. Asterholm, A. Benrick, L. J. B. Briant, M. V. Chibalina, F. M. Gribble, A. Hamilton, B. Hastoy, F. Reimann, N. J. G. Rorsman, I. I. Spiliotis, A. Tarasov, Y. Wu, F. M. Ashcroft and P. Rorsman, Nat. Commun., 2019, 10, 139 CrossRef PubMed
- A. Blanazs, A. J. Ryan and S. P. Armes, Macromolecules, 2012, 45, 5099–5107 CrossRef CAS
- C. J. Mable, N. J. Warren, K. L. Thompson, O. O. Mykhaylyk and S. P. Armes, Chem. Sci., 2015, 6, 6179–6188 RSC
- C. J. Mable, I. Canton, O. O. Mykhaylyk, B. Ustbas Gul, P. Chambon, E. Themistou and S. P. Armes, Chem. Sci., 2019, 10, 4811–4821 RSC
- O. J. Deane, J. Jennings, T. J. Neal, O. M. Musa, A. Fernyhough and S. P. Armes, Chem. Mater., 2021, 33, 7767–7779 CrossRef CAS
- O. J. Deane, J. Jennings and S. P. Armes, Chem. Sci., 2021, 12, 13719–13729 RSC
- W. Zhang, B. Charleux and P. Cassagnau, Macromolecules, 2012, 45, 5273–5280 CrossRef CAS
- J. R. Lovett, M. J. Derry, P. Yang, F. L. Hatton, N. J. Warren, P. W. Fowler and S. P. Armes, Chem. Sci., 2018, 9, 1–7 RSC
- A. Blanazs, R. Verber, O. O. Mykhaylyk, A. J. Ryan, J. Z. Heath, C. W. I. Douglas and S. P. Armes, J. Am. Chem. Soc., 2012, 134, 9741–9748 CrossRef CAS PubMed
- X. Zhang, S. Boissé, W. Zhang, P. Beaunier, F. D'Agosto, J. Rieger and B. Charleux, Macromolecules, 2011, 44, 4149–4158 CrossRef CAS
- W. Zhang, F. D'Agosto, O. Boyron, J. Rieger and B. Charleux, Macromolecules, 2012, 45, 4075–4084 CrossRef CAS
- N. J. Warren, M. J. Derry, O. O. Mykhaylyk, J. R. Lovett, L. P. D. Ratcliffe, V. Ladmiral, A. Blanazs, L. A. Fielding and S. P. Armes, Macromolecules, 2018, 51, 8357–8371 CrossRef CAS PubMed
- A. Czajka and S. P. Armes, Chem. Sci., 2020, 11, 11443–11454 RSC
- N. J. Warren, O. O. Mykhaylyk, D. Mahmood, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 1023–1033 CrossRef CAS PubMed
- L. P. D. Ratcliffe, M. J. Derry, A. Ianiro, R. Tuinier and S. P. Armes, Angew. Chem., Int. Ed., 2019, 58, 18964–18970 CrossRef CAS PubMed
- M. Sponchioni, C. T. O'Brien, C. Borchers, E. Wang, M. N. Rivolta, N. J. W. Penfold, I. Canton and S. P. Armes, Chem. Sci., 2020, 11, 232–240 RSC
- D. L. Beattie, O. O. Mykhaylyk, A. J. Ryan and S. P. Armes, Soft Matter, 2021, 17, 5602–5612 RSC
- R. Verber, A. Blanazs and S. P. Armes, Soft Matter, 2012, 8, 9915–9922 RSC
- K. A. Simon, N. J. Warren, B. Mosadegh, M. R. Mohammady, G. M. Whitesides and S. P. Armes, Biomacromolecules, 2015, 16, 3952–3958 CrossRef CAS PubMed
- I. Canton, N. J. Warren, A. Chahal, K. Amps, A. Wood, R. Weightman, E. Wang, H. Moore and S. P. Armes, ACS Cent. Sci., 2016, 2, 65–74 CrossRef CAS PubMed
- R. Spelat, F. Ferro, P. Contessotto, N. J. Warren, G. Marsico, S. P. Armes and A. Pandit, Mater. Today Bio, 2020, 5, 100040 CrossRef PubMed
- A. L. A. Binch, L. P. D. Ratcliffe, A. H. Milani, B. R. Saunders, S. P. Armes and J. A. Hoyland, Biomacromolecules, 2021, 22, 837–845 CrossRef CAS PubMed
- O. Wichterle, D. Lím, 1960, 185, 117–118 Search PubMed
- P. H. O'Farrell, J. Biol. Chem., 1975, 250, 4007–4021 CrossRef
- B. Vigani, S. Rossi, G. Sandri, M. C. Bonferoni, C. M. Caramella and F. Ferrari, Pharmaceutics, 2020, 12, 859 CrossRef CAS PubMed
- F. Zahir-Jouzdani, J. D. Wolf, F. Atyabi and A. Bernkop-Schnürch, Expert Opin. Drug Deliv., 2018, 15, 1007–1019 CrossRef CAS PubMed
- A. Ludwig, Adv. Drug Deliv. Rev., 2005, 57, 1595–1639 CrossRef CAS PubMed
- J. L. Greaves and C. G. Wilson, Adv. Drug Deliv. Rev., 1993, 11, 349–383 CrossRef CAS
- V. Bourganis, O. Kammona, A. Alexopoulos and C. Kiparissides, Eur. J. Pharm. Biopharm., 2018, 128, 337–362 CrossRef CAS PubMed
- M. I. Ugwoke, R. U. Agu, N. Verbeke and R. Kinget, Adv. Drug Deliv. Rev., 2005, 57, 1640–1665 CrossRef CAS PubMed
- S. GuhaSarkar and R. Banerjee, J. Control. Release, 2010, 148, 147–159 CrossRef CAS PubMed
- O. M. Kolawole, W. M. Lau, H. Mostafid and V. V. Khutoryanskiy, Int. J. Pharm., 2017, 532, 105–117 CrossRef CAS PubMed
- A. Bernkop-Schnürch, Adv. Drug Deliv. Rev., 2005, 57, 1569–1582 CrossRef PubMed
- F. Laffleur and A. Bernkop-Schnürch, Future Med. Chem., 2012, 4, 2205–2216 CrossRef CAS PubMed
- J. Lenoir, I. Claerhout, P. Kestelyn, A. Klomp, J. P. Remon and E. Adriaens, Toxicol. Vitr., 2011, 25, 1919–1925 CrossRef CAS PubMed
- T. Eshel-Green and H. Bianco-Peled, Colloids Surf., B, 2016, 139, 42–51 CrossRef CAS PubMed
- X. Shan, S. Aspinall, D. B. Kaldybekov, F. Buang, A. C. Williams and V. V. Khutoryanskiy, ACS Appl. Polym. Mater., 2021, 3, 5882–5892 CrossRef CAS
- O. M. Kolawole, W. M. Lau and V. V. Khutoryanskiy, Int. J. Pharm., 2018, 550, 123–129 CrossRef CAS PubMed
- P. Tonglairoum, R. P. Brannigan, P. Opanasopit and V. V. Khutoryanskiy, J. Mater. Chem. B, 2016, 4, 6581–6587 RSC
- Y. Shtenberg, M. Goldfeder, A. Schroeder and H. Bianco-Peled, Carbohydr. Polym., 2017, 175, 337–346 CrossRef CAS PubMed
- O. M. Kolawole, W. M. Lau and V. V. Khutoryanskiy, J. Pharm. Sci., 2019, 108, 3046–3053 CrossRef CAS PubMed
- G. Prosperi-Porta, S. Kedzior, B. Muirhead and H. Sheardown, Biomacromolecules, 2016, 17, 1449–1457 CrossRef CAS PubMed
- S. Hu, X. Pei, L. Duan, Z. Zhu, Y. Liu, J. Chen, T. Chen, P. Ji, Q. Wan and J. Wang, Nat. Commun., 2021, 12, 1689 CrossRef CAS PubMed
- K. Kim, K. Kim, J. H. Ryu and H. Lee, Biomaterials, 2015, 52, 161–170 CrossRef CAS PubMed
- C. Leichner, P. Wulz, R. A. Baus, C. Menzel, S. K. Götzfried, R. Gust and A. Bernkop-Schnürch, Mol. Pharm., 2019, 16, 1211–1219 CrossRef CAS PubMed
- C. Menzel, M. Hauser, A. Frey, M. Jelkmann, F. Laffleur, S. K. Götzfried, R. Gust and A. Bernkop-Schnürch, Eur. J. Pharm. Biopharm., 2019, 139, 161–167 CrossRef CAS PubMed
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T Le, R. T. A Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS
- G. Moad, A. E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58, 379–410 CrossRef CAS
- G. Moad, Polym. Chem., 2017, 8, 177–219 RSC
- E. E. Brotherton, C. P. Jesson, N. J. Warren, M. J. Smallridge and S. P. Armes, Angew. Chem., Int. Ed., 2021, 60, 12032–12037 CrossRef CAS PubMed
- E. E. Brotherton, M. J. Smallridge and S. P. Armes, Biomacromolecules, 2021, 22, 5382–5389 CrossRef CAS PubMed
- Y. Li and S. P. Armes, Angew. Chemie, 2010, 122, 4136–4140 CrossRef
- I. Bannister, N. C. Billingham, S. P. Armes, S. P. Rannard and P. Findlay, Macromolecules, 2006, 39, 7483–7492 CrossRef CAS
- M. K. Kocik, O. O. Mykhaylyk and S. P. Armes, Soft Matter, 2014, 10, 3984–3992 RSC
-
M. B. Smith and J. March, March's advanced organic chemistry: reactions, mechanisms, and structure, John Wiley & Sons, Inc., Hoboken, NJ, 2007 Search PubMed
- E. A. Mun, A. C. Williams and V. V. Khutoryanskiy, Int. J. Pharm., 2016, 512, 32–38 CrossRef CAS PubMed
- C. A. Withers, M. T. Cook, L. Methven, M. A. Gosney and V. V. Khutoryanskiy, Food Funct., 2013, 4, 1668–1674 RSC
- S. L. Canning, T. J. Neal and S. P. Armes, Macromolecules, 2017, 50, 6108–6116 CrossRef CAS PubMed
- D. B. Kaldybekov, P. Tonglairoum, P. Opanasopit and V. V. Khutoryanskiy, Eur. J. Pharm. Sci., 2018, 111, 83–90 CrossRef CAS PubMed
- V. V. Khutoryanskiy, Macromol. Biosci., 2011, 11, 748–764 CrossRef CAS PubMed
- J. Ilavsky and P. R. Jemian, J. Appl. Crystallogr., 2009, 42, 347–353 CrossRef CAS
- D. B. Kaldybekov, S. K. Filippov, A. Radulescu and V. V. Khutoryanskiy, Eur. J. Pharm. Biopharm., 2019, 143, 24–34 CrossRef CAS PubMed
- M. T. Cook, G. Tzortzis, D. Charalampopoulos and V. V Khutoryanskiy, Biomacromolecules, 2011, 12, 2834–2840 CrossRef CAS PubMed
- B. L. Symonds, C. I. Lindsay, N. R. Thomson and V. V Khutoryanskiy, RSC Adv., 2016, 6, 102206–102213 RSC
Footnotes |
| † Electronic supplementary information (ESI) available: Reaction schemes for the synthesis of PGEO5MAx and PGEO5MA13-P(HPxMA155-st-FMA0.15), TEM images and digital photographs recorded for PGEO5MA13-PHPMAy, PGEO5MA16-PHPMAy and periodate-treated PGEO5MA13-P(HPMA155-st-FMA0.15) diblock copolymer worms. Oscillatory rheology strain curves and variable temperature measurements for the original and periodate-treated PGEO5MA13-P(HPMA155-st-FMA0.15) diblock copolymer worms. See https://doi.org/10.1039/d2sc02074b |
| ‡ These authors contributed equally to this manuscript. |
| This journal is © The Royal Society of Chemistry 2022 |