
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModular synthesis of α-arylated carboxylic acids, esters and amides via photocatalyzed triple C–F bond cleavage of methyltrifluorides†
Sifan
Li
ab,
Paul W.
Davies
 *b and
Wei
Shu
*a
*b and
Wei
Shu
*a
aShenzhen Grubbs Institute, Department of Chemistry, and Guangdong Provincial Key Laboratory of Catalysis, Southern University of Science and Technology, Shenzhen 518055, Guangdong, China. E-mail: shuw@sustech.edu.cn
bSchool of Chemistry, University of Birmingham, Edgbaston, Birmingham, B15 2TT, UK
First published on 12th May 2022
Abstract
α-Arylated carboxylic acids, esters and amides are widespread motifs in bioactive molecules and important building blocks in chemical synthesis. Thus, straightforward and rapid access to such structures is highly desirable. Here we report an organophotocatalytic multicomponent synthesis of α-arylated carboxylic acids, esters and amides from exhaustive defluorination of α-trifluoromethyl alkenes in the presence of alkyltrifluoroborates, water and nitrogen/oxygen nucleophiles. This operationally simple strategy features a unified access to functionally diverse α-arylated carboxylic acids, esters, and primary, secondary, and tertiary amides through backbone assembly from simple starting materials enabled by consecutive C–F bond functionalization at room temperature. Preliminary mechanistic investigations reveal that the reaction operates through a radical-triggered three-step cascade process, which involves distinct mechanisms for each defluorinative functionalization of the C–F bond.
Introduction
α-Arylated carboxylic acids and amides are ubiquitous substructures and indispensable building blocks in chemical synthesis.1 The wide existence of such structural motifs in biologically active molecules2 (Scheme 1A) has inspired much effort to develop efficient methods towards the synthesis of α-arylated carboxylate derivatives.3 Pioneered by Buchwald and Hartwig, methods relying on the use of transition-metal catalyzed α-arylation of enolates with aryl halides have been extensively developed.4 Later, metal-catalyzed cross-coupling of α-halo carbonyl electrophiles with aryl nucleophiles has also been described.5 However, these methods are typically not applicable to direct arylation of aliphatic carboxylic acids and amides with acidic N–H bonds.6 To address this issue, Hartwig a reported an elegant Pd-catalyzed α-arylation of carboxylic acids and secondary amides through the in situ formation of the corresponding disilyl intermediates in the presence of a base and TMSCl (Scheme 1B).7 In 2020, Engle's group described the synthesis of α-arylated amides enabled by Pd-catalyzed α-selective hydroarylation of acrylamides (Scheme 1B).8 These methods suffer from the use of transition metals, multi-step synthesis of advanced starting materials, and/or harsh conditions. Different approaches for the non-transition metal catalyzed synthesis of α-arylated amides have also been reported. Nevado, Greaney, Clayden and Studer have independently reported aryl migration strategies,9 while Miyata and Maulide have each shown the umpolung α-arylation of amides.10 Despite these significant advances, a straightforward and unified method providing rapid access to α-arylated carboxylic acids, and primary, secondary, and tertiary amides from readily available precursors is highly desirable.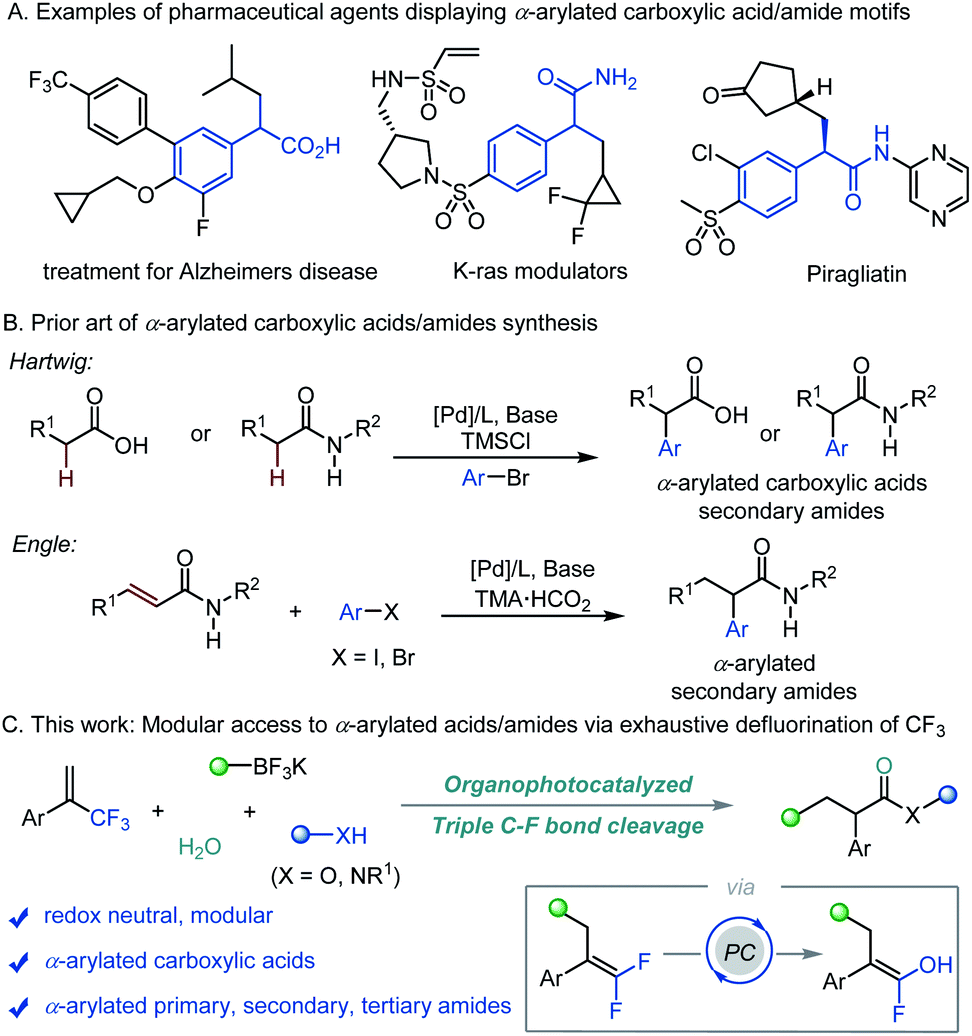 | ||
| Scheme 1 Significance and synthetic design for α-arylated carboxylic acids and amides. | ||
Over the past decade, photoredox catalysis has served as a powerful platform for numerous challenging organic transformations. Notably, manipulation of strong C–F bonds in multifluorinated compounds under photoredox conditions has attracted considerable attention due to the potential applications in accessing novel bond forming strategies and for the degradation of fluorinated pollutants.11 Great efforts have been devoted to the cleavage of one or two of the C–F bonds in α-trifluoromethyl alkenes for the synthesis of di- or monofluoroalkenes, respectively.12,13 To date, no example of the consecutive cleavage of three C–F bonds in α-trifluoromethyl alkenes to access versatile products has been reported. Here we report an organophotoredox-catalyzed triple defluorination of α-trifluoromethyl alkenes to furnish a variety of functionally diverse α-arylated carboxylic acids, and primary, secondary, and tertiary amides in the presence of alkyltrifluoroborates, water and nitrogen/oxygen nucleophiles (Scheme 1C). This metal-free protocol consists of a defluorinative alkylation, defluorinative hydroxylation, and defluorinative amination/hydroxylation cascade, providing access to α-arylated carboxylate derivatives via assembly of the carbon skeleton from simple starting materials under redox-neutral conditions.
Results and discussion
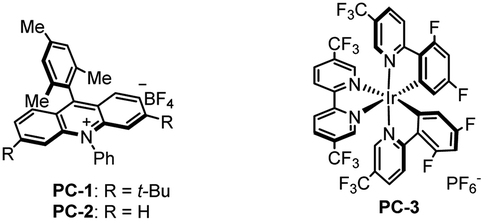
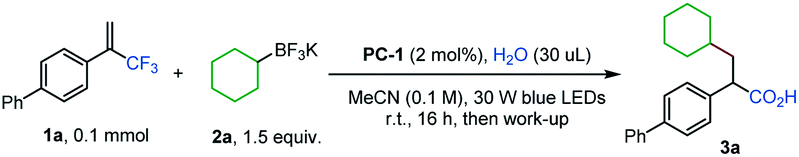
Our initial study focused on the reaction of α-trifluoromethyl alkene 1a with cyclohexyltrifluoroborate 2a to test the feasibility of this proposal (Table 1).14 After evaluation of various parameters, the desired α-arylated carboxylic acid 3a was obtained in 80% yield using acridinium PC-1 as a photocatalyst with H2O in acetonitrile. Other photocatalysts could mediate the transformation, albeit in lower efficiencies (Table 1, entries 2 and 3). Replacing acetonitrile with CH2Cl2 or Et2O gave 3a in diminished yields, and 3a was not detected when 1,4-dioxane was used as the solvent (Table 1, entries 4–6). Varying the amount of H2O led to lower yields of 3a (Table 1, entries 7 and 8). Control experiments revealed that photocatalyst PC-1 and H2O were both necessary for this reaction while a reduced yield was obtained when the base/acid work-up was not performed (Table 1, entries 9–11).15|
|
||
|---|---|---|
| Entry | Deviation from standard conditions | Yield of 3ab (%) |
| a Reaction conditions: 1a (0.1 mmol), 2a (0.15 mmol), PC-1 (0.002 mmol), and H2O (30 μL) in MeCN (0.1 M) under 30 W blue LED irradiation at room temperature for 16 h, then NaOH/HCl work-up. b Yields were determined by 1H NMR of the crude reaction mixture with mesitylene as the internal standard, and isolated yield is shown in parentheses. | ||
| 1 | None | 88 (80) |
| 2 | PC-2 instead of PC-1 | 44 |
| 3 | PC-3 instead of PC-1 | 16 |
| 4 | DCM instead of MeCN | 43 |
| 5 | Et2O instead of MeCN | 37 |
| 6 | 1,4-Dioxane instead of MeCN | 0 |
| 7 | 10 μL of H2O | 56 |
| 8 | 100 μL of H2O | 76 |
| 9 | W/O PC-1 | 0 |
| 10 | W/O H2O | 0 |
| 11 | W/O work-up | 53 |
|
||
With the optimized reaction conditions in hand, the scope of α-trifluoromethyl alkenes 1 and alkyltrifluoroborates 2 was investigated (Scheme 2). para-Substituted styrenyl methyltrifluorides were well-tolerated in the exhaustive defluorinative reaction, forming the desired α-arylated carboxylic acid in 49–84% yields (3a–3g). Electron-donating groups (3b and 3c) and a fluoride substituent (3d) are compatible under the reaction conditions. Terminal ethynyl-substituted α-arylated carboxylic acid 3g was obtained in 68% yield when an α-trifluoromethyl alkene with a 4-(trimethylsilyl)ethynylphenyl group was used. Cleavage of the silyl group is likely due to the release of fluoride anions during the reaction. 3-Methoxy, 3-allyloxy and 3,5-dimethyl groups were well-tolerated (3h–3j). The use of fused aryl and heteroaryl vinylmethyltrifluorides allowed the incorporation of naphthyl (3k), benzothiophenyl (3l) and dibenzofuranyl (3m) substituted carboxylic acids in good yields (61–90%). Notably, α-pyridinyl carboxylic acid (3n) was effectively obtained in this reaction. A cyclic alkene was also transformed into α,β-cyclic-α-arylated carboxylic acid 3o in 57% yield. However, α-trifluoromethyl alkenes with electron-withdrawing arenes failed to deliver the corresponding α-arylated carboxylic acids, with the reaction stalling at the corresponding gem-difluoroalkenes.16 A wide range of alkyltrifluoroborates (2) were found to be suitable for this reaction. Five-, six- and seven-membered cyclic secondary alkyl groups (3p, 3q, and 3r) with diverse substitution patterns can be readily incorporated by this route. Piperidine (3s) and tetrahydropyran (3t) motifs can also be introduced in this exhaustive defluorination reaction (43% and 83% yield, respectively). Acyclic secondary (3u) and tertiary alkyl radical precursors (3v–3x) participated in this reaction with good efficiencies (68–82%). The formation of a 1,6-keto acid 3y showcased a new approach to this desirable functional motif. Primary α-alkoxymethyltrifluoroborates can also be used, providing access to γ-alkoxy acids (3z–3ab).
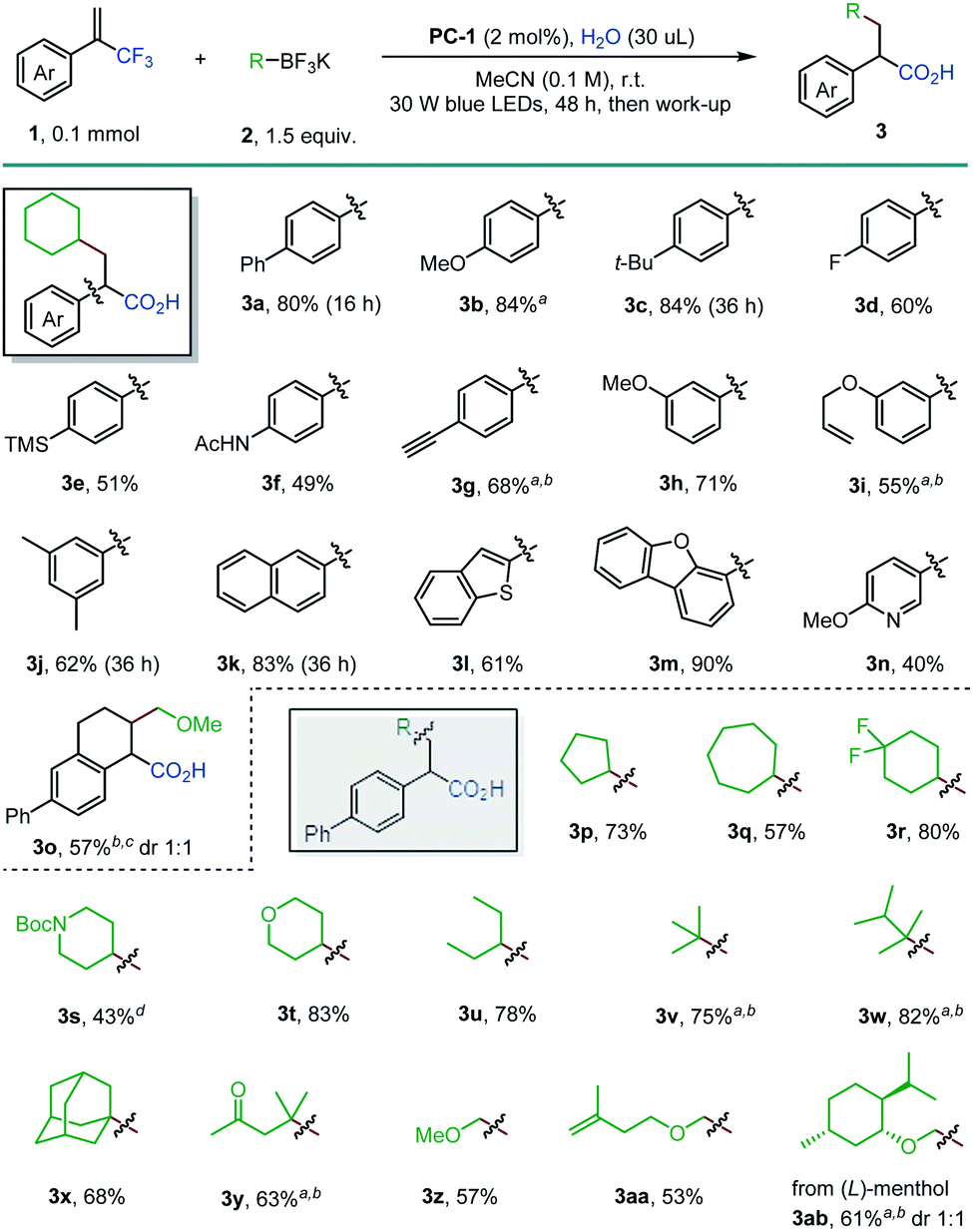 | ||
| Scheme 2 Reaction scope for the triple defluorinative formation of α-arylated carboxylic acids. Reaction conditions: 1 (0.1 mmol), 2 (0.15 mmol), PC-1 (0.002 mmol), and H2O (30 μL, 17 equiv.) in MeCN (0.1 M) under 30 W blue LED irradiation at room temperature, then NaOH/HCl work-up. Isolated yields after flash chromatography are reported. a 50 μL of H2O was used. b 5 mol% of PC-1 was used. c Reaction was run on 0.2 mmol scale. d 100 μL of H2O was used. | ||
We then questioned whether amines could be compatible in the reaction, thus allowing the direct formation of amides from vinylmethyltrifluorides via triple C–F bond functionalizations. Pleasingly, the desired α-arylated amides were obtained efficiently via exhaustive defluorinative C–N coupling (Scheme 3), despite the competing potential for single-electron oxidation of amines by the excited-state photocatalyst.17 Primary amines with a wide range of functional groups and structural elements worked well (5a–5j). Alongside linear and branched substituents (5a–5e), allylic (5f) and propargyl (5g) amines were good substrates. Moreover, trifluoromethyl and nitrile containing primary amines delivered the corresponding α-arylated amides 5h and 5i in 72% and 67% yields, respectively. Notably, free amino alcohol was compatible in the reaction, furnishing the corresponding hydroxyl amide 5j in 72% yield. Interestingly, a diamide product 5k was formed in 75% yield when 1,2-diaminoethane was used. Acyclic and cyclic secondary amines were also shown to be suitable coupling partners in this reaction (5l–5s, 55–90% yields), including piperidines bearing ketal (5o), ester (5p), free carboxylic acid (5q), morpholine (5n) and a piperazine derivative (5s). Electron-rich and electron-deficient anilines participated smoothly in the reaction, affording 5t–5v in 51–75% yields. Various alkyltrifluoroborates including primary, secondary, and tertiary alkyl groups, and silyl and ketone groups were also good precursors for the triple defluorinative amide formation (5w–5aa, 48–87% yields). The desilylated amide 5ab was obtained in 43% yield when α-trifluoromethyl alkene with a 4-(trimethylsilyl)ethynylphenyl group was used. In addition, structurally complex substrates derived from natural products, such as estrone and (+)-dehydroabietylamine, were readily incorporated in this reaction, affording 5ac and 5ad in 67% and 62% yields, respectively.
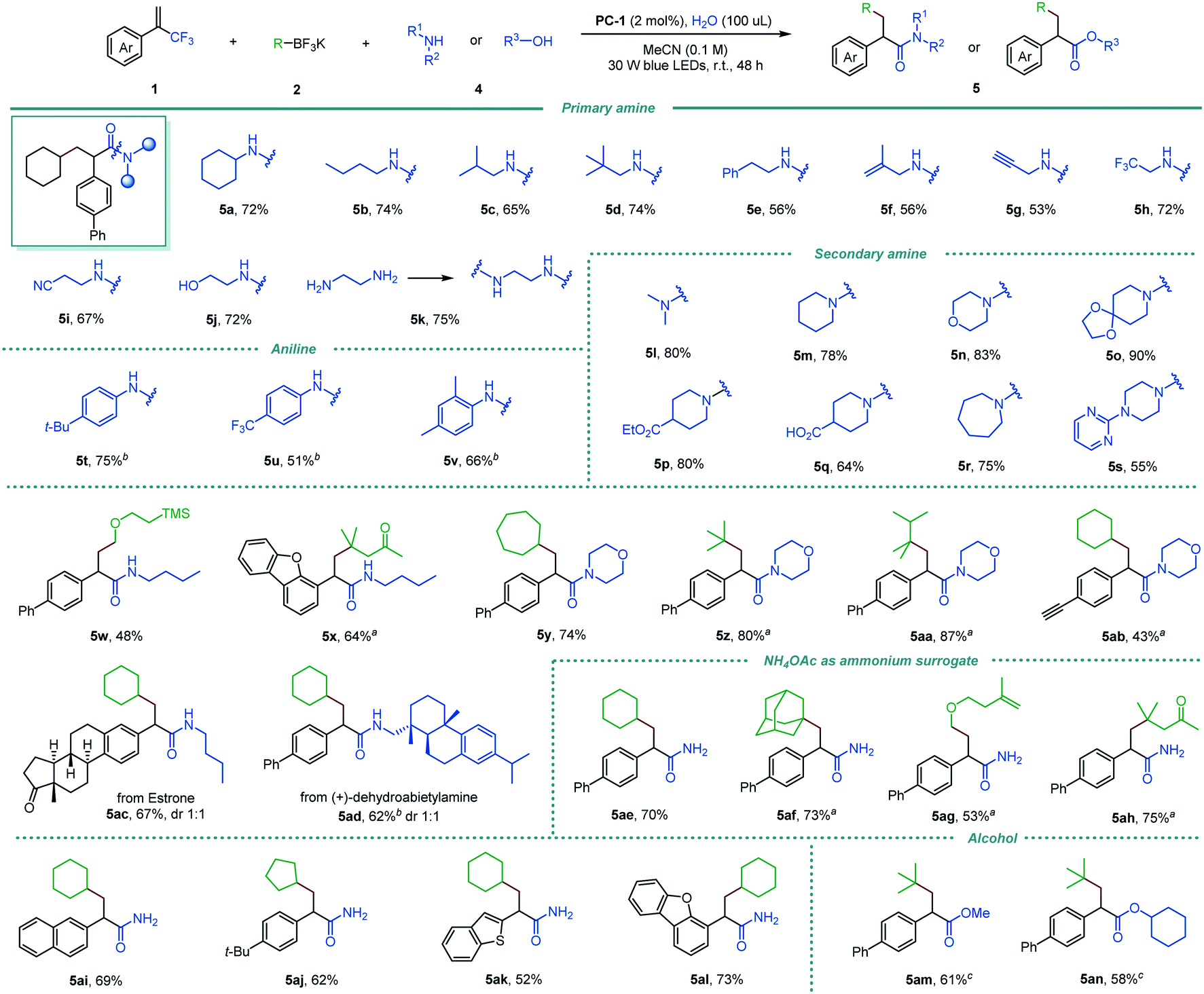 | ||
Scheme 3 Reaction scope for the triple defluorinative formation of α-arylated amides and esters. Reaction conditions: 1 (0.1 mmol), 2 (0.15 mmol), 4 (0.15 mmol, 0.20 mmol for NH4OAc, and 0.50 mmol for alcohol), PC-1 (0.002 mmol), and H2O (100 μL) in MeCN (0.1 M) under 30 W blue LEDs irradiation at room temperature. Isolated yields are reported. a 5 mol% of PC-1 was used. b MeCN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CH2Cl2 = 3:1. c 10 μL of H2O was used. :CH2Cl2 = 3:1. c 10 μL of H2O was used. | ||
Next, the feasibility of transforming α-trifluoromethyl alkenes into primary amides by cleaving three C–F bonds was also tested. Ammonium acetate was identified as a readily available and cost-effective ammonia surrogate, allowing efficient access to a series of primary amides (5ae–5al, 52–75% yields). This protocol is also suitable for the synthesis of α-arylated esters by using alcohols as nucleophiles, affording 5am and 5an in good yields (61% and 58% yield, respectively).
To further demonstrate the utility of this protocol, product 3a was prepared on a 2.0 mmol scale in 82% yield (Scheme 4a). This new transformation allows for ready access into other useful motifs, such as β-arylated alcohol 6a in quantitative yield (Scheme 4a), α-arylated nitrile 6b in 71% yield (Scheme 4b) and quinazoline 6c in 55% yield (Scheme 4c). In addition, one-step synthesis of glucokinase activator 6f from simple precursors was achieved, illustrating the great potential of this protocol to facilitate rapid analogue evaluation in medicinal chemistry (Scheme 4d).18
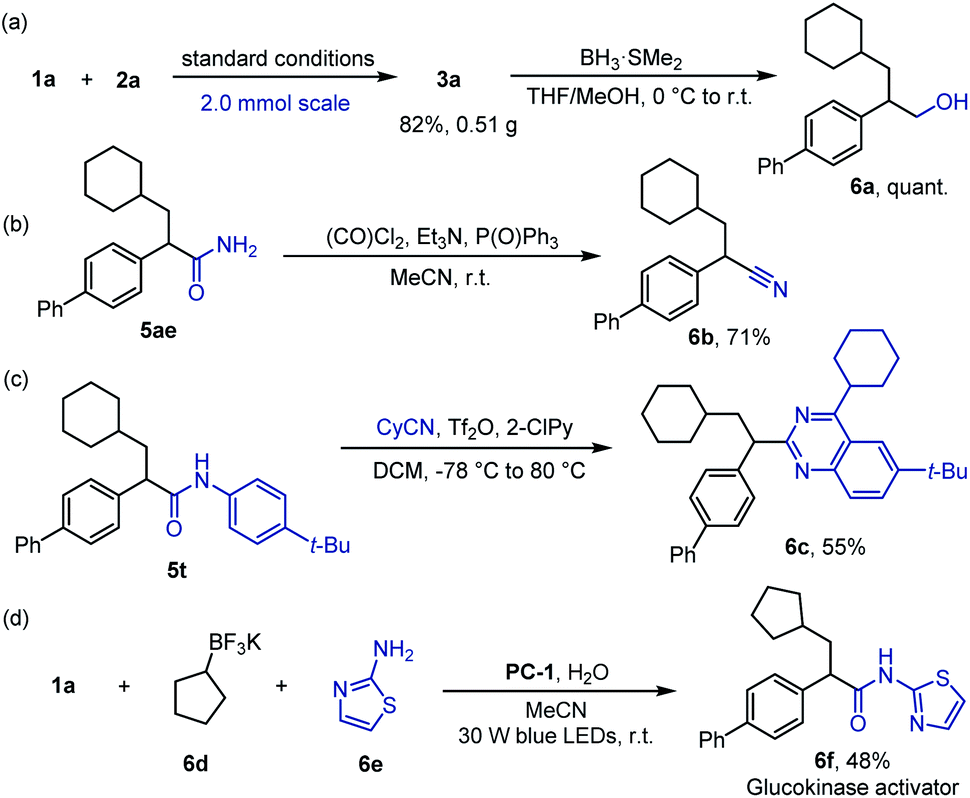 | ||
| Scheme 4 Synthetic applications. | ||
Control experiments were conducted to probe the reaction mechanism (Scheme 5). Addition of (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) into the reaction completely inhibited the formation of 3a, whilst the use of the radical precursor 7a led to the ring-opening product 7b in 42% yield (Scheme 5a and b). Conducting the reaction in the absence of H2O did not yield the product 3a, and gem-difluoroalkene 7c was obtained in 90% yield instead. Moreover, 7c was productive under the standard reaction conditions to afford 3a in good yield (Scheme 5c), suggesting that gem-difluoroalkene might be the intermediate of this reaction. When 1a and 2a were subjected to the standard conditions without a NaOH/HCl work-up procedure, acid 3a was formed in 53% yield alongside acyl fluoride 7d in 39% yield as detected by NMR spectroscopy and HRMS (Scheme 5d). Further evidence for the formation of an electrophilic acyl fluoride intermediate was obtained by replacing NaOH in the reaction work-up protocol with n-butylamine which led to the formation of 3a in 61% yield and amide 5b in 27% yield (Scheme 5e). gem-Difluoroalkene 7c was formed rather than amide 5t when H2O was excluded from the standard reaction conditions for the amide formation, indicating that water was required for the second defluorination step (Scheme 5f). These results support the involvement of alkyl radical species during the reaction, indicating that gem-difluoroalkene is an intermediate in this transformation that is converted into acyl fluoride species and not subject to a direct SNV process.19
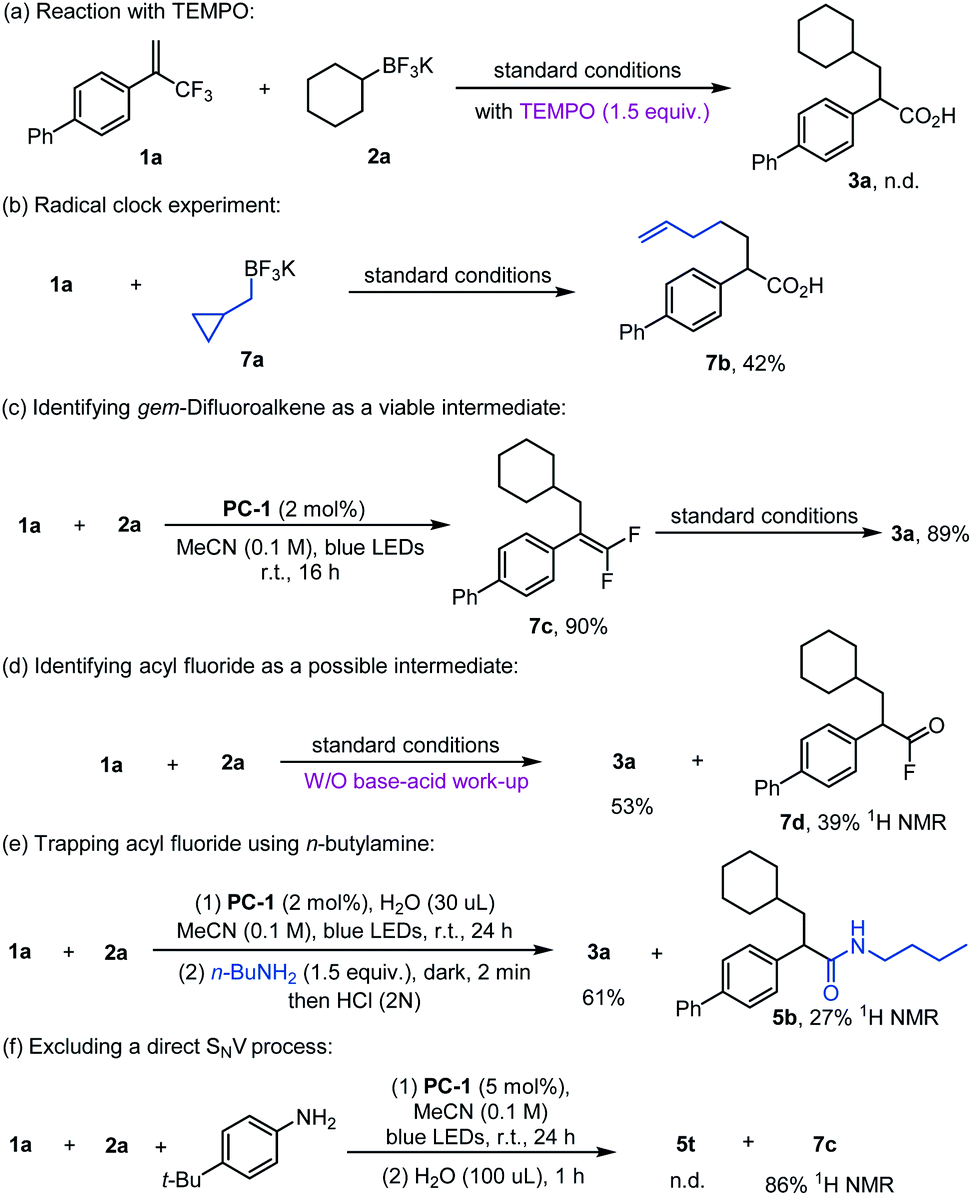 | ||
| Scheme 5 Control experiments. | ||
Stern–Volmer luminescence quenching studies were also performed using representative substrates 1a and 2a, cyclohexylamine (CyNH2), ammonium acetate and the intermediate gem-difluoroalkene 7c (Scheme 6). The results showed that gem-difluoroalkene 7c quenched the excited photocatalyst significantly, and that 2a and 1a were also suitable quenchers for the excited photocatalyst.
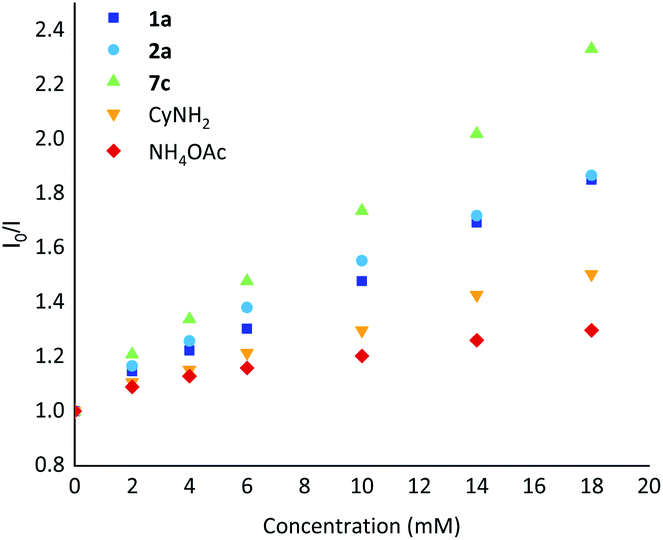 | ||
| Scheme 6 Stern–Volmer luminescence quenching studies with PC-1. | ||
Based on these results, a possible mechanism is depicted (Scheme 7). Upon blue LED irradiation, the excited acridinium photocatalyst *PCn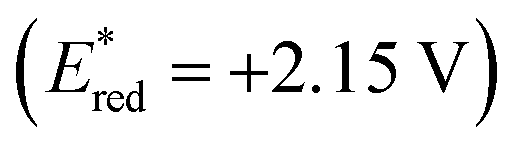 would undergo single electron transfer (SET) with 2 to generate alkyl radical species I and reduced photocatalyst PCn−1.14,20 Radical species I underwent radical addition with 1 to generate a benzylic radical II. Single-electron reduction of II by PCn−1 followed by β-fluoride elimination yielded gem-difluoroalkenes III and the photocatalyst PCn to complete the first catalytic cycle. After this stage, single-electron oxidation of intermediate III by the excited photocatalyst *PCn afforded a radical cationic species IV and reduced photocatalyst PCn−1.21IV would be trapped by H2O to generate intermediate V.22,23 The presence of strongly electron-withdrawing aryl substituents renders this step inefficient. The SET process between PCn−1 and V followed by second β-fluoride elimination affords the enol intermediate VI or its tautomer acyl fluoride species VII, and the photocatalyst PCn. The desired products were then obtained through acyl substitution with oxygen or nitrogen nucleophiles to cleave the third C–F bond.
would undergo single electron transfer (SET) with 2 to generate alkyl radical species I and reduced photocatalyst PCn−1.14,20 Radical species I underwent radical addition with 1 to generate a benzylic radical II. Single-electron reduction of II by PCn−1 followed by β-fluoride elimination yielded gem-difluoroalkenes III and the photocatalyst PCn to complete the first catalytic cycle. After this stage, single-electron oxidation of intermediate III by the excited photocatalyst *PCn afforded a radical cationic species IV and reduced photocatalyst PCn−1.21IV would be trapped by H2O to generate intermediate V.22,23 The presence of strongly electron-withdrawing aryl substituents renders this step inefficient. The SET process between PCn−1 and V followed by second β-fluoride elimination affords the enol intermediate VI or its tautomer acyl fluoride species VII, and the photocatalyst PCn. The desired products were then obtained through acyl substitution with oxygen or nitrogen nucleophiles to cleave the third C–F bond.
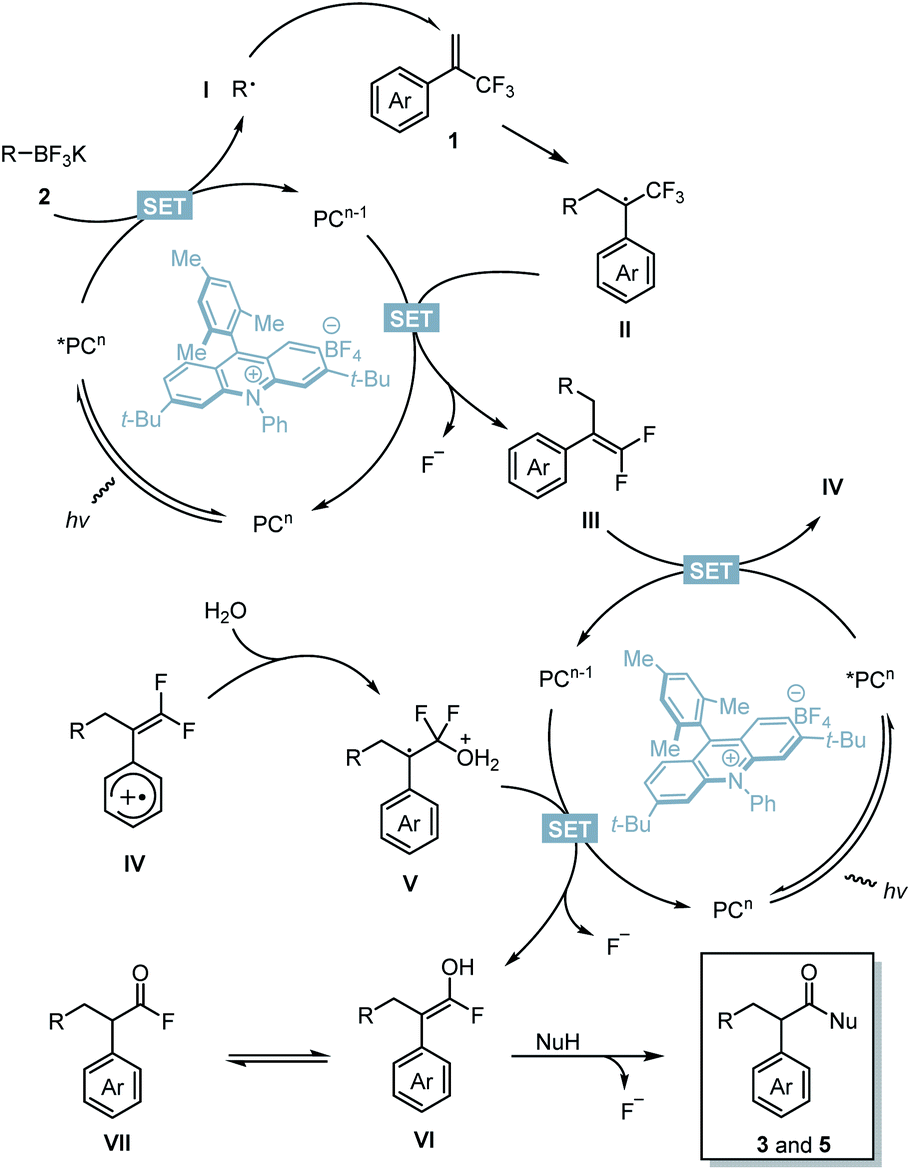 | ||
| Scheme 7 Proposed mechanism. | ||
Conclusions
In summary, a photocatalyzed multicomponent triple defluorinative functionalization of α-trifluoromethyl alkenes was achieved to afford a variety of α-arylated carboxylic acids, esters, and primary, secondary, and tertiary amides at room temperature. The reaction operates through three consecutive and distinct C–F bond cleavages opening an avenue for converting methyltrifluorides into synthetically valuable functional groups. α-Trifluoromethyl alkenes, themselves readily prepared in one step from boronic acids,24 can be combined with alkyltrifluoroborates, water, and alcohols/amines to prepare α-arylated carboxylic acids, esters and amides by the simple alternation of nucleophiles. As a result, these important motifs can now be assembled bearing significant structural and functional group variations in a modular way from simple and readily accessible molecular building blocks.Data availability
Experimental data has been provided as ESI.†Author contributions
S. L. discovered the reaction. W. S. and P. W. D. conceived and directed the project. S. L. performed the experiments and analysed the data. S. L., W. S. and P. W. D. prepared the manuscript. All authors discussed the experimental results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This paper is dedicated to Prof. Guochen Jia on his 60th birthday. Financial support from NSFC (21971101, 22171127, and 21801126), Guangdong Basic and Applied Basic Research Foundation (2022A1515011806 and 2019A1515011976), Department of Education of Guangdong Province (2021KTSCX106), the Stable Support Plan Program of Shenzhen Natural Science Fund (No. 20200925152608001), Thousand Talents Program for Young Scholars, the Pearl River Talent Recruitment Program (2019QN01Y261), Guangdong Provincial Key Laboratory of Catalysis (No. 2020B121201002) is sincerely acknowledged. We acknowledge the assistance of SUSTech Core Research Facilities. We thank Dr Yu-Feng Ren (SUSTech) for reproducing the results of 3j, 3v, 5j and 5w. P. W. D. gratefully acknowledges the Royal Society and the Leverhulme Trust for the award of a Senior Research Fellowship (SRF\R1\191033).Notes and references
- (a) T. Y. Shen, Angew. Chem., Int. Ed., 1972, 11, 460–472 CrossRef CAS PubMed; (b) W. B. Wright Jr, J. B. Press, P. S. Chan, J. W. Marsico, M. F. Haug, J. Lucas, J. Tauber and A. S. Tomcufcik, J. Med. Chem., 1986, 29, 523–530 CrossRef CAS PubMed.
- (a) G. Koenig and J.-F. Blain, US Pat., 2013106328A1, 2013 Search PubMed; (b) A. Pandey, S. K. Samanta, A. J. Duraiswamy, A. E. Maciag, D. Turner, M. A. J. Duncton, V. Kumari, A. R. Renslo, E. Low, C. Brassard, H. V. Adcock, D. Hamza and S. T. Onions, US Pat., 2019204505A2, 2019 Search PubMed; (c) R. Sarabu, F. T. Bizzaro, W. L. Corbett, M. T. Dvorozniak, W. Geng, J. F. Grippo, N.-E. Haynes, S. Hutchings, L. Garofalo, K. R. Guertin, D. W. Hilliard, M. Kabat, R. F. Kester, W. Ka, Z. Liang, P. E. Mahaney, L. Marcus, F. M. Matschinsky, D. Moore, J. Racha, R. Radinov, Y. Ren, L. Qi, M. Pignatello, C. L. Spence, T. Steele, J. Tengi and J. Grimsby, J. Med. Chem., 2012, 55, 7021–7036 CrossRef CAS PubMed.
- For selected reviews, see: (a) G. C. Lloyd-Jones, Angew. Chem., Int. Ed., 2002, 41, 953–956 CrossRef CAS; (b) D. A. Culkin and J. F. Hartwig, Acc. Chem. Res., 2003, 36, 234–245 CrossRef CAS PubMed; (c) C. C. C. Johansson and T. J. Colacot, Angew. Chem., Int. Ed., 2010, 49, 676–707 CrossRef CAS PubMed; (d) F. Bellina and R. Rossi, Chem. Rev., 2010, 110, 1082–1146 CrossRef CAS PubMed; (e) Y.-J. Hao, X.-S. Hu, Y. Zhou, J. Zhou and J.-S. Yu, ACS Catal., 2020, 10, 955–993 CrossRef CAS.
- For selected examples, see: (a) K. H. Shaughnessy, B. C. Hamann and J. F. Hartwig, J. Org. Chem., 1998, 63, 6546–6553 CrossRef CAS; (b) M. Kawatsura and J. F. Hartwig, J. Am. Chem. Soc., 1999, 121, 1473–1478 CrossRef CAS; (c) J. M. Fox, X. Huang, A. Chieffi and S. L. Buchwald, J. Am. Chem. Soc., 2000, 122, 1360–1370 CrossRef CAS; (d) S. Lee, N. A. Beare and J. F. Hartwig, J. Am. Chem. Soc., 2001, 123, 8410–8411 CrossRef CAS PubMed; (e) W. A. Moradi and S. L. Buchwald, J. Am. Chem. Soc., 2001, 123, 7996–8002 CrossRef CAS PubMed; (f) J. Cossy, A. de Filippis and D. G. Pardo, Org. Lett., 2003, 5, 3037–3039 CrossRef CAS PubMed; (g) T. Hama, X. Liu, D. A. Culkin and J. F. Hartwig, J. Am. Chem. Soc., 2003, 125, 11176–11177 CrossRef CAS PubMed; (h) X. Liu and J. F. Hartwig, J. Am. Chem. Soc., 2004, 126, 5182–5191 CrossRef CAS PubMed.
- E. Barde, A. Guérinot and J. Cossy, Synthesis, 2019, 51, 178–184 CrossRef CAS.
- (a) H. A. Chiong, Q.-N. Pham and O. Daugulis, J. Am. Chem. Soc., 2007, 129, 9879–9884 CrossRef CAS PubMed; (b) S.-C. Sha, J. Zhang and P. J. Walsh, Org. Lett., 2015, 17, 410–413 CrossRef CAS PubMed; (c) R. A. Altman, A. M. Hyde, X. Huang and S. L. Buchwald, J. Am. Chem. Soc., 2008, 130, 9613–9620 CrossRef CAS PubMed; (d) Z.-K. Xiao, H.-Y. Yin and L.-X. Shao, Org. Lett., 2013, 15, 1254–1257 CrossRef CAS PubMed.
- Z.-T. He and J. F. Hartwig, J. Am. Chem. Soc., 2019, 141, 11749–11753 CrossRef CAS PubMed.
- A. M. Vasquez, J. A. Gurak Jr, C. L. Joe, E. C. Cherney and K. M. Engle, J. Am. Chem. Soc., 2020, 142, 10477–10484 CrossRef CAS PubMed.
- (a) C. Hervieu, M. S. Kirillova, T. Suárez, M. Müller, E. Merino and C. Nevado, Nat. Chem., 2021, 13, 327–334 CrossRef CAS PubMed; (b) H. L. Barlow, P. T. G. Rabet, A. Durie, T. Evans and M. F. Greaney, Org. Lett., 2019, 21, 9033–9035 CrossRef CAS PubMed; (c) D. J. Leonard, J. W. Ward and J. Clayden, Nature, 2018, 562, 105–109 CrossRef CAS PubMed; (d) N. Radhoff and A. Studer, Angew. Chem., Int. Ed., 2021, 60, 3561–3565 CrossRef CAS PubMed.
- (a) N. Takeda, E. Futaki, Y. Kobori, M. Ueda and O. Miyata, Angew. Chem., Int. Ed., 2017, 56, 16342–16346 CrossRef CAS PubMed; (b) J. Li, M. Berger, W. Zawodny, M. Simaan and N. Maulide, Chem, 2019, 5, 1883–1891 CrossRef CAS.
- For selected examples, see: (a) C. Saboureau, M. Troupel, S. Sibille and J. Périchon, J. Chem. Soc., Chem. Commun., 1989, 1138–1139 RSC; (b) S. Yoshida, K. Shimomori, Y. Kim and T. Hosoya, Angew. Chem., Int. Ed., 2016, 55, 10406–10409 CrossRef CAS PubMed; (c) K. Chen, N. Berg, R. Gschwind and B. König, J. Am. Chem. Soc., 2017, 139, 18444–18447 CrossRef CAS PubMed; (d) H.-B. Wang and N. T. Jui, J. Am. Chem. Soc., 2018, 140, 163–166 CrossRef CAS PubMed; (e) C. Luo and J. S. Bandar, J. Am. Chem. Soc., 2019, 141, 14120–14125 CrossRef CAS PubMed; (f) Y.-J. Yu, F.-L. Zhang, T.-Y. Peng, C.-L. Wang, J. Cheng, C. Chen, K. N. Houk and Y.-F. Wang, Science, 2021, 371, 1232–1240 CrossRef CAS PubMed; (g) J. B. I. Sap, N. J. W. Straathof, T. Knauber, C. F. Meyer, M. Médebielle, L. Buglioni, C. Genicot, A. A. Trabanco, T. Noël, C. W. am Ende and V. Gouverneur, J. Am. Chem. Soc., 2020, 142, 9181–9187 CrossRef CAS PubMed; (h) S.-S. Yan, S.-H. Liu, L. Chen, Z.-Y. Bo, K. Jing, T.-Y. Gao, B. Yu, Y. Lan, S.-P. Luo and D.-G. Yu, Chem, 2021, 7, 3099–3113 CrossRef CAS; (i) M. W. Campbell, V. C. Polites, S. Patel, J. E. Lipson, J. Majhi and G. A. Molander, J. Am. Chem. Soc., 2021, 143, 19648–19654 CrossRef CAS PubMed; (j) J.-H. Ye, P. Bellotti, C. Heusel and F. Glorius, Angew. Chem., Int. Ed., 2022, 61, e202115456 CrossRef CAS PubMed; (k) S. Li and W. Shu, Chem. Commun., 2022, 58, 1066–1077 RSC; (l) G. Yan, K. Qiu and M. Guo, Org. Chem. Front., 2021, 8, 3915–3942 RSC.
- For selected examples, see: (a) T. Xiao, L. Li and L. Zhou, J. Org. Chem., 2016, 81, 7908–7916 CrossRef CAS PubMed; (b) S. B. Lang, R. J. Wiles, C. B. Kelly and G. A. Molander, Angew. Chem., Int. Ed., 2017, 56, 15073–15077 CrossRef CAS PubMed; (c) J. Qi, F.-L. Zhang, J.-K. Jin, Q. Zhao, B. Li, L.-X. Liu and Y.-F. Wang, Angew. Chem., Int. Ed., 2020, 59, 12876–12884 CrossRef CAS PubMed; (d) W.-J. Yue, C. S. Day and R. Martin, J. Am. Chem. Soc., 2021, 143, 6395–6400 CrossRef CAS PubMed.
- (a) L. Li, T. Xiao, H. Chen and L. Zhou, Chem.–Eur. J., 2017, 23, 2249–2254 CrossRef CAS PubMed; (b) H. Chen, Y. He and L. Zhou, Org. Chem. Front., 2018, 5, 3240–3244 RSC.
- J. K. Matsui, D. N. Primer and G. A. Molander, Chem. Sci., 2017, 8, 3512–3522 RSC.
- For more details on the condition evaluation, see ESI.†.
- See ESI† for unsuccessful substrates..
- (a) A. Adenier, M. M. Chehimi, I. Gallardo, J. Pinson and N. Vilà, Langmuir, 2004, 20, 8243–8253 CrossRef CAS PubMed; (b) H. G. Roth, N. A. Romero and D. A. Nicewicz, Synlett, 2016, 27, 714–723 CAS.
- W. L. Corbett, N.-E. Haynes and R. Sarabu, US Pat., 2001085706A1, 2001 Search PubMed.
- B. Wang, X. Zhao, Q. Liu and S. Cao, Org. Biomol. Chem., 2018, 16, 8546–8552 RSC.
- N. A. Romero, K. A. Margrey, N. E. Tay and D. A. Nicewicz, Science, 2015, 349, 1326–1330 CrossRef CAS PubMed.
- H. Liu, L. Ge, D.-X. Wang, N. Chen and C. Feng, Angew. Chem., Int. Ed., 2019, 58, 3918–3922 CrossRef CAS PubMed.
- G. Zhang, X. Hu, C.-W. Chiang, H. Yi, P. Pei, A. K. Singh and A. Lei, J. Am. Chem. Soc., 2016, 138, 12037–12040 CrossRef CAS PubMed.
- J. B. McManus, J. D. Griffn, A. R. White and D. A. Nicewicz, J. Am. Chem. Soc., 2020, 142, 10325–10330 CrossRef CAS PubMed.
- J. Walkowiak, T. M. del Campo, B. Ameduri and V. Gouverneur, Synthesis, 2010, 11, 1883–1890 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc01905a |
| This journal is © The Royal Society of Chemistry 2022 |