
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCell surface photoengineering enables modeling of glycocalyx shedding dynamics†
Sean C.
Purcell‡
a,
Michelle H.
Zhang‡§
a,
Daniel J.
Honigfort¶
a,
Hans Jefferson C.
Ng
a,
Austen L.
Michalak
a and
Kamil
Godula
 *ab
*ab
aDepartment of Chemistry and Biochemistry, University of California San Diego, 9500 Gilman Drive, La Jolla, CA 92093, USA. E-mail: kgodula@ucsd.edu
bGlycobiology Research and Training Center, University of California San Diego, 9500 Gilman Drive, La Jolla, CA 92093, USA
First published on 12th May 2022
Abstract
The cellular glycocalyx, composed of membrane associated glycoproteins and glycolipids, is a complex and dynamic interface that facilitates interactions between cells and their environment. The glycocalyx composition is continuously changing through biosynthesis of new glycoconjugates and membrane turnover. Various glycocalyx components, such as mucins, can also be rapidly shed from the cell surface in response to acute events, such as pathogenic threat. Mucins, which are large extended glycoproteins, deliver important protective functions against infection by creating a physical barrier at the cell surface and by capturing and clearing pathogens through shedding. Evaluating these mucin functions may provide better understanding of early stages of pathogenesis; however, tools to tailor the composition and dynamics of the glycocalyx with precision are still limited. Here, we report a chemical cell surface engineering strategy to model the shedding behavior of mucins with spatial and temporal control. We generated synthetic mucin mimetic glycopolymers terminated with a photolabile membrane anchor, which could be introduced into the membranes of living cells and, subsequently, released upon exposure to UV light. By tuning the molecular density of the artificial glycocalyx we evaluated lectin crosslinking and its effect on shedding, showing that lectins can stabilize the glycocalyx and limit release of the mucin mimetics from the cell surface. Our findings indicate that endogenous and pathogen-associated lectins, which are known to interact with the host-cell glycocalyx, may alter mucin shedding dynamics and influence the protective properties of the mucosal barrier. More broadly, we present a method which enables photoengineering of the glycocalyx and can be used to facilitate the study of glycocalyx dynamics in other biological contexts.
Introduction
The cellular glycocalyx, composed of membrane-associated glycoproteins and glycolipids, provides an important functional interface regulating the interactions between cells and various components of its external environment.1 The composition of the glycocalyx is defined by the availability of monosaccharide precursors and the activity of glycan biosynthesis enzymes and undergoes continuous reconfiguration through membrane recycling and glycoconjugate turnover. Various components of the glycocalyx, such as proteoglycans or mucins, can also be rapidly released from the cell surface through proteolytic ectodomain shedding to regulate cellular interactions and signaling.2 Acceleration of shedding is often associated with various pathophysiological events and pathogenic threats.3,4Mucins, a class of extended, highly glycosylated proteins, which are expressed at high levels on mucosal epithelial cells and project away from the membrane, form a physical shield that protects cells from pathogenic challenge (Fig. 1A). Mucins do so by either limiting the access of pathogens to their cell-surface receptors5 or by presenting decoy receptors to capture the pathogens and clear them from the cell surface via shedding.6 In response, pathogens have evolved mechanisms to overcome the barrier functions of the glycocalyx, such as by expressing enzymes that can break down mucins and expose the cell surface.7 Another possible mechanism through which pathogens may resist clearance through shedding is by exploiting glycan-binding proteins (GBPs) to crosslink to multiple cell surface glycoconjugates.8 Crosslinking of the glycocalyx by extracellular lectins, such as galectins, has been previously shown to contribute to its stabilization and to reduce endocytic turnover of native9 and synthetic10 glycoconjugates. However, the effects of glycocalyx crosslinking by oligomeric GBPs, including pathogen associated lectins, on mucin shedding are yet to be investigated; likely due to the lack of tools to induce mucin shedding from cells with spatial and temporal control.
 | ||
| Fig. 1 Shedding of the mucosal glycocalyx. (A) Cell-surface mucins provide a protective physical barrier against infection. Pathogens disrupt this barrier by inducing proteolytic mucin glycocalyx degradation or shedding. (B) Synthetic mucin mimetics with photocleavable membrane anchors enable modeling of mucin glycocalyx shedding behavior. | ||
Genetic tools to control mucin structure and expression to tailor the physical characteristics of the mucosal glycocalyx are rapidly emerging.11 While the recent isolation and characterization of a suite of mucinase enzymes12 enables selective removal of mucins from the glycocalyx, this leads to complete digestion of the mucin structure and does not fully recapitulate the process of shedding which leaves the glycosylated mucin ectodomains largely intact and capable of interacting with crosslinking lectins.
Synthetic glycopolymers, which approximate the structure of mucins and can be introduced into cell membranes, have provided a useful tool for the modeling the mucinous glycocalyx to study its biological functions.13 Here we describe cell surface engineering with mucin mimetics bearing photocleavable membrane anchors to model mucosal glycocalyx shedding with spatial and temporal control using light. The synthetic mucin mimetics showed membrane-density dependent crosslinking by the oligomeric lectin, Ricinus communis agglutinin, which resulted in increased protection against shedding from the cell surface induced by light. This strategy is poised to enable future investigations into the regulation of mucin shedding by host- and pathogen-associated lectins and provide new insights into the protective functions of the mucosal barrier.
Results and discussion
Generation of mucin mimetics with photo-cleavable membrane anchors
To model mucin glycoprotein shedding from cell surfaces, we designed mucin mimetic glycopolymers that can be presented on the plasma membrane of cells and subsequently released upon application of an external stimulus, such as light (Fig. 1B). The membrane targeting mucin-mimetic glycopolymers comprised a poly(ethylene oxide) (PEO) backbone glycosylated to produce the mucin-mimetic glycodomain and terminated with a hydrophobic anchor linked through a photocleavable nitrobenzyl group. Additionally, small percentage of the polymer sidechains (∼1%) were functionalized with a fluorescent reporter (Cy5) for visualization.The glycopolymer synthesis began by generating an azide-terminated poly(epichlorohydrin) scaffold P1 primed for copper-catalyzed azide–alkyne cycloaddition (CuAAC) with cholesterol alkyne 1 containing a previously reported photocleavable (PCL) nitrobenzyl linker group14 (Fig. 2A). Monomer-activated anionic ring opening polymerization of ECH8 in the presence of tetrabutylammonium azide (0.3 mol%) initiator and triisobutyl aluminum activator (0.7 mol%) furnished polymer precursor P1 near the target molecular weight (Mw = 29 kDa, DP ∼ 300) and narrow chain-length distribution (Đ = 1.23). Treatment of P1 with 1 (10 equiv.) in the presence of a copper(I) iodide catalyst (1 equiv. per end group) and diisopropylethylamine, afforded a photocleavable cholesterol end group-modified ECH polymer intermediate P2-PCL. Introduction of the cholesterol end-group was difficult to observe directly by 1H NMR spectroscopy; however, it could be confirmed by the disappearance of the IR characteristic, albeit weak, azide group absorption at ν = 2100 cm−1 (Fig. S9†). Following chain end functionalization, the chloromethyl side chains were primed for glycosylation by reaction with sodium azide to generate azidomethyl side chain modified polymer P3-PCL. Quantitative side-chain conversion was confirmed by 1H NMR and IR spectroscopy. The assembly of the desired mucin mimetic glycopolymer GP-PCL was accomplished through a sequential copper-click reaction with sub-stoichiometric (1 mol%) alkynyl-Cy5 to introduce the fluorescent label followed by excess propargyl lactoside (1.5 eq.) as a model glycan. We assessed fluorescent labeling efficiency of GPs after reconstitution in water by UV-Vis spectroscopy at λ = 633 nm to be ∼2–3 fluorophores per GP, as expected for a polymer DP = 300. IR spectroscopy confirmed full consumption of the azide side chains following glycan attachment (Fig. S10†), and the molecular weight of GP-PCL was calculated based on polymer length.
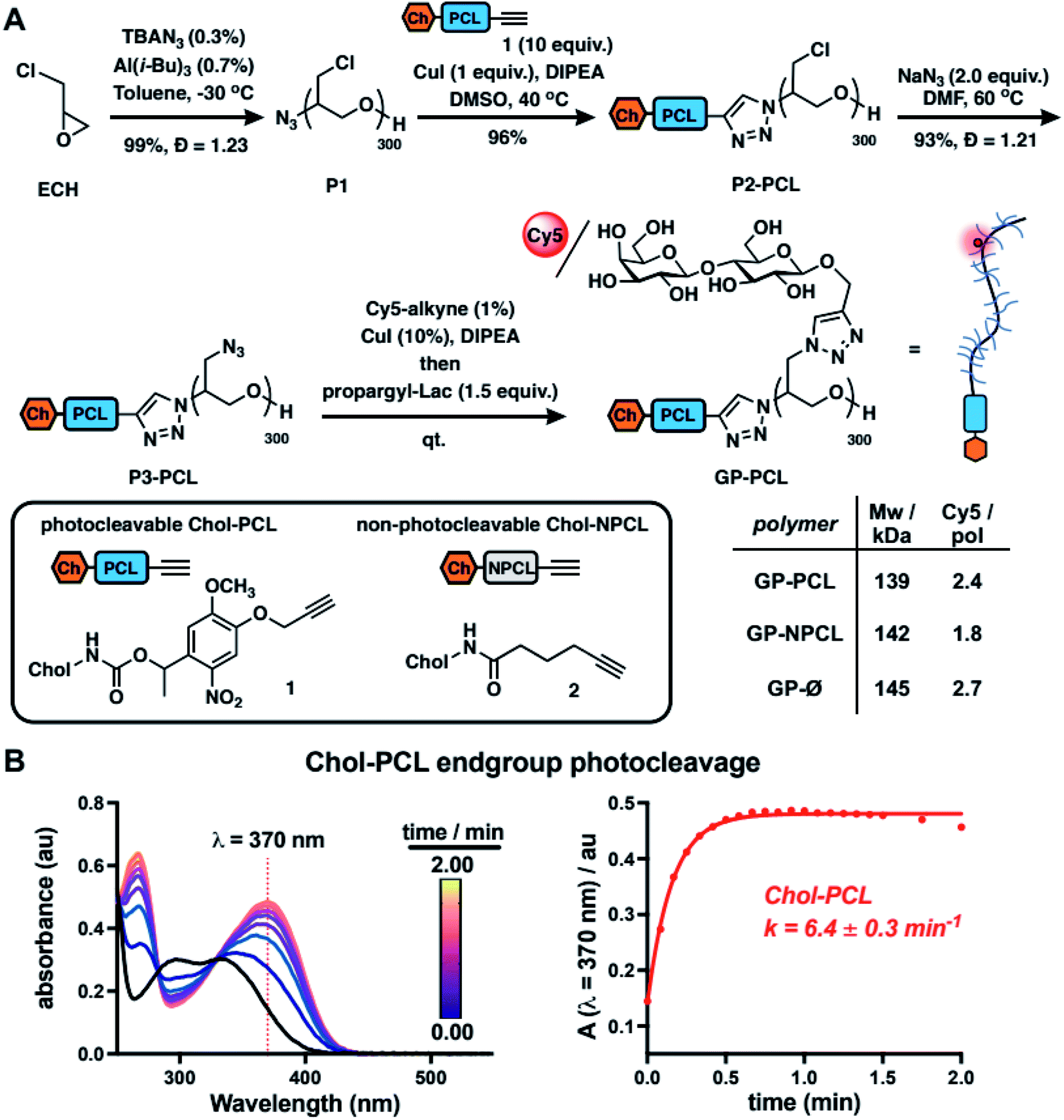 | ||
| Fig. 2 Synthesis and characterization of mucin mimetics with photocleavable membrane anchors. (A) Mucin mimetic glycopolymers (GPs) terminated with photocleavable (PCL) and non-photo cleavable (NPCL) cholesterol anchors were elaborated from a common poly(epichlorohydrin) precursor (P1). A sequential end- and side-chain modification via the CuAAC reaction was used to introduce cholesterol anchors 1 and 2 and to construct a mucin mimetic domain comprised of lactosylated side chains and a fluorescent probe for visualization (Cy5, ∼2–3 per GP). B) The photocleavage of cholesterol anchor Chol-PCL (1, 10 μg mL−1 in chloroform) with light at λ = 365 nm was analyzed by UV spectroscopy. The change in absorbance at λ = 370 nm over time was used to determine the rate of photocleavage (k = 6.4 ± 0.3 min−1, n = 3). | ||
We also generated two analogous mucin-mimetic glycopolymer controls (for details see ESI, Fig. S1–S11†). The first was glycopolymer GP-NPCL, in which the cholesterol anchor was connected to the backbone through a non-photocleavable alkyl chain linker (introduced via 5-hexynoyl cholesteroamide 2, Fig. 2A) to serve as a mucin mimetic control resistant to cleavage by UV light. The second was glycopolymer GP-Ø lacking the cholesterol end group used to confirm hydrophobic anchor-dependent membrane incorporation of the mucin mimetics. All three GPs originated from the common polymer precursor P1 and thus had the same length, with molecular weights estimated based on GPC analysis of polymers P3 and separated by less than 5% (Fig. 2A and S11†).
To characterize photocleavage of the Chol-PCL linker, the UV absorption spectrum was recorded following irradiation of 1 in chloroform (λ = 365 nm) at increasing time intervals (Fig. 2B). The formation of a new peak at λ = 370 nm is indicative of photocleavage and this absorbance was used to determine the rate of photolysis (k = 6.4 ± 0.3 min−1). Photocleavage of 1 neared completion within 0.75 min of UV exposure. We observed a similar photocleavage rate for the intermediate P2-PCL (k = 5.1 ± 2.3 min−1, Fig. S12†). These experiments confirmed the photolysis of the membrane anchor after UV irradiation and suggested that GP-PCL, in which the end group photolysis could not be detected directly in aqueous solution, should be suitable for cell-surface engineering.
Photoengineering of the mucin-mimetic glycocalyx
For the construction of mucin glycocalyx models using our glycopolymer mimetics, we chose mutant Chinese hamster ovary cells, CHO Lec8, depleted in Golgi uridine diphosphate galactose (UDP-Gal) pools due to impaired transport of the nucleotide sugar from the cytosol.15 As a result, these cells do not incorporate Gal into their cell surface glycans, thus providing a suitable cell system for membrane engineering with lactosylated mucin mimetics presenting β1,4-linked Gal residues. To establish optimal concentrations of the glycopolymers for cell-membrane remodeling, suspended CHO Lec8 cells were incubated with the Cy5-labeled GP-PCL and GP-NPCL at increasing polymer concentrations (0.63–20.00 μM, Fig. 3A) at 4 °C. After one hour, the cells were washed to remove unincorporated polymers and analyzed by flow cytometry based on glycopolymer fluorescence. Both polymers inserted into the cell membrane with similar efficiency, nearing signal saturation at ∼5 μM. The mucin mimetic GP-Ø lacking the cholesterol anchor exhibited no signal above untreated cell background, indicating the requirement for this group for membrane insertion and further confirming successful end-group functionalization of polymer precursors P2-PCL and P2-NPCL, which was difficult to detect using spectroscopic techniques (Fig. 2A, S9 and S10†). We observed slightly greater total fluorescence intensity for cells remodeled with GP-PCL compared to GP-NPCL, consistent with the ∼30% higher fluorophore labeling of the GPs (Fig. 2A). | ||
| Fig. 3 Photo-engineering of the mucin-mimetic glycocalyx in cells. (A) Mucin mimetics containing photo-cleavable (GP-PCL) and non-photocleavable (GP-NPCL) cholesterol anchors incorporate into the plasma membranes of CHO Lec8 cells in a concentration dependent manner. Glycopolymers lacking the cholesterol anchor (GP-Ø) showed no association with the cells surface. (B) Photo-shedding of the mucin mimetics upon irradiation with UV light (365 nm) was observed only for GP-PCL containing the photo-cleavable (PCL) anchor. Flow cytometry was used to determine the rate of photocleavage from the cell surface (k = 2.5 ± 0.6 min−1) and the half-life for mucin mimetic shedding (t1/2 = 0.28 ± 0.1 min, n = 3). Loss of fluorescence was not observed for the non-photocleavable mucin mimetic GP-NPCL. (C) Fluorescence micrographs of CHO cells remodeled with Cy5-labeled GP-PCL and GP-NPCL (cGP = 5 μM) before and after UV irradiation (λ = 365 nm, 3 min). Cell nuclei were stained with Hoechst 33342 dye. (D) Spatial photopatterning of CHO Lec8 cells remodeled with mucin mimetics GP-PCL was accomplished through application of a mask during UV irradiation (λ = 365 nm, 3 min). A plot of average fluorescence intensity per cell area with respect to the positioning of the photomask indicates mucin mimetic photo-shedding was specific to the subset of cells carrying the photocleavable GP-PCL and exposed to UV light (scale bars = 200 μm). | ||
Cell surface remodeling was performed at 4 °C to limit temperature-dependent endocytosis and polymer internalization, which has previously been observed with similar materials.16 The low temperature can decrease membrane fluidity and limit polymer insertion. Therefore, we assessed the cell-surface incorporation of GP-PCL (∼5 μM) in CHO Lec8 monolayers at 4, 21, and 37 °C. After a 1 hour incubation with the polymer, the cells were washed and analyzed via fluorescence microscopy (Fig. S13†). Remodeling was observed at all temperatures assayed and minimal endocytosis was observed at 4 °C.
We next evaluated light-induced shedding of the mucin mimetics from the plasma membrane by exposing cells remodeled with both GPs to ultraviolet light (λ = 365 nm, Fig. 3B). CHO Lec8 cells remodeled in suspension with GP-PCL and GP-NPCL (5 μM) on ice were exposed to UV light at increasing time intervals for up to 3 min and the loss of cell fluorescence was measured by flow cytometry (Fig. 3B). We only observed a light- and time-dependent reduction in Cy5 intensity for GP-PCL, indicating clearance of the mucin mimetics from the cell surface via photo-induced cleavage of the nitrobenzyl cholesterol anchor 1. The UV treatment of cells remodeled with the non-photocleavable polymer GP-NPCL resulted in no loss of Cy5 intensity, indicating resistance of the fluorophore to photobleaching under these conditions (Fig. 3B). Within 2 minutes of UV exposure, mucin mimetic density at the cell surface was reduced by more than 70%, with minimal further photocleavage observed after additional exposure. The small fraction of UV-cleavage resistant mucin mimetics may result from polymer internalization by the cells or, possibly, through crosslinking of the excited state radical intermediate to other membrane components. The rate of GP-PCL photocleavage from the cell surface (k = 2.5 ± 0.6 min−1) was similar to that measured for precursor P2-PCL in chloroform (k = 5.1 ± 2.3 min−1). The UV light treatment in the presence or absence of the GPs resulted in little apparent cytotoxicity, as determined by a live-dead staining assay (>93% cell viability, Fig. S14†).
Next, we assessed the light-dependent mucin mimetic shedding from CHO Lec8 cells in adherent culture via microscopy (Fig. 3C). The cells were incubated with all three GPs at sub-saturation conditions (2 μM) at 4 °C for 1 h. Unincorporated polymers were washed and a subset of the cells was exposed to UV light (λ = 365 nm), after which all cells were washed again and treated with a nuclear stain for imaging. Mucin mimetics GP-PCL and GP-NPCL, but not GP-Ø, showed robust cell surface labeling in the absence of UV light, confirming cholesterol-dependent membrane remodeling (Fig. 3C). While cells treated with the non-photocleavable mucin mimetic, GP-NPCL, retained their fluorescence after UV exposure, most of the GP-PCL signal was lost. The cytosolic punctate staining that remained visible was consistent with polymer internalization and the flow cytometry analysis (Fig. 3B).
The light responsiveness of the mucin mimetic GP-PCL enables patterning of the glycocalyx within a subpopulation of cells. To demonstrate this concept, CHO Lec8 cell monolayers remodeled with either GP-PCL or GP-NPCL (2 μM) were exposed to UV light (λ = 365 nm) on ice for 3 min in the presence of a photomask. After illumination, the cells were washed, treated with a nuclear stain and imaged (Fig. 3D). The fluorescence micrographs show a clear drop in Cy5 signal beyond the photomask for GP-PCL, which was quantified by plotting fluorescence intensity averaged over 100-pixel bins extending in both directions from the boundary. Cells remodeled with GP-NPCL, which is resistant to photocleavage, showed uniform fluorescence distributions. These experiments demonstrate the applicability of the light-responsive mucin mimetics for tailoring of the glycocalyx composition across a cell population with spatial resolution, which is difficult to achieve using existing glycan engineering techniques.
Effects of lectin interactions on mucin-mimetic glycocalyx shedding
The capture and shedding of pathogens by cell surface mucins are important defense mechanisms by which host cells can limit entry and infection.17 Pathogens, which often exploit lectin interactions to bind to glycoconjugates on host cells,18 may counteract the shedding process by stabilizing the glycocalyx. Better understanding how lectin crosslinking affects mucin shedding from cells may reveal new insights into this important aspect of mucosal barrier function.To evaluate whether lectin crosslinking can stabilize the mucinous glycocalyx and prevent its shedding, we investigated the interactions and photoshedding of cell-surface displays of GP-PCL in the presence or absence of Ricinus communis agglutinin (RCA). This lectin, with specificity for terminal β1,4-linked galactosides, is known for its ability to crosslink glycoconjugates and induce cell agglutination.19 In the absence of endogenous Gal on the surfaces of the mutant CHO Lec8 cells, RCA binding after remodeling and photocleavage could be attributed solely to the presence of the lactose-bearing GP-PCL mucin mimetics (Fig. S15 and S16†).
We first established an optimal concentration of RCA for use in binding assays (Fig. S17†). Accordingly, a suspension of wild type CHO Pro5 cells was incubated with biotinylated RCA (0–20 μg mL−1) on ice, stained with excess AlexaFluor488-streptavidin, and analyzed by flow cytometry. We observed concentration-dependent RCA staining with maximal signal intensity and no evidence of cell aggregation at lectin concentration of 5 μg mL−1 (Fig. S17a†). CHO Pro5 cells in monolayer culture were then stained with RCA at this concentration and analyzed by fluorescence microscopy to confirm robust staining for imaging (Fig. S17b†).
Using the optimized RCA staining conditions, we evaluated the binding of the lectin to CHO Lec8 cells remodeled with increasing concentrations of the photocleavable mucin mimetic (cGP-PCL = 0–2 μM). As expected, flow cytometry analysis revealed mucin mimetic concentration-dependent RCA binding (Fig. 4A). Anticipating that RCA crosslinking may be affected by the membrane-density of the mucin mimetic, we plotted the ratio of GP-PCL and RCA fluorescence intensities as a function of polymer concentration (Fig. 4B). We observed an increase in the polymer/RCA ratio, indicating more extensive crosslinking with increasing polymer density, until the saturation of glycan binding sites for the lectin.
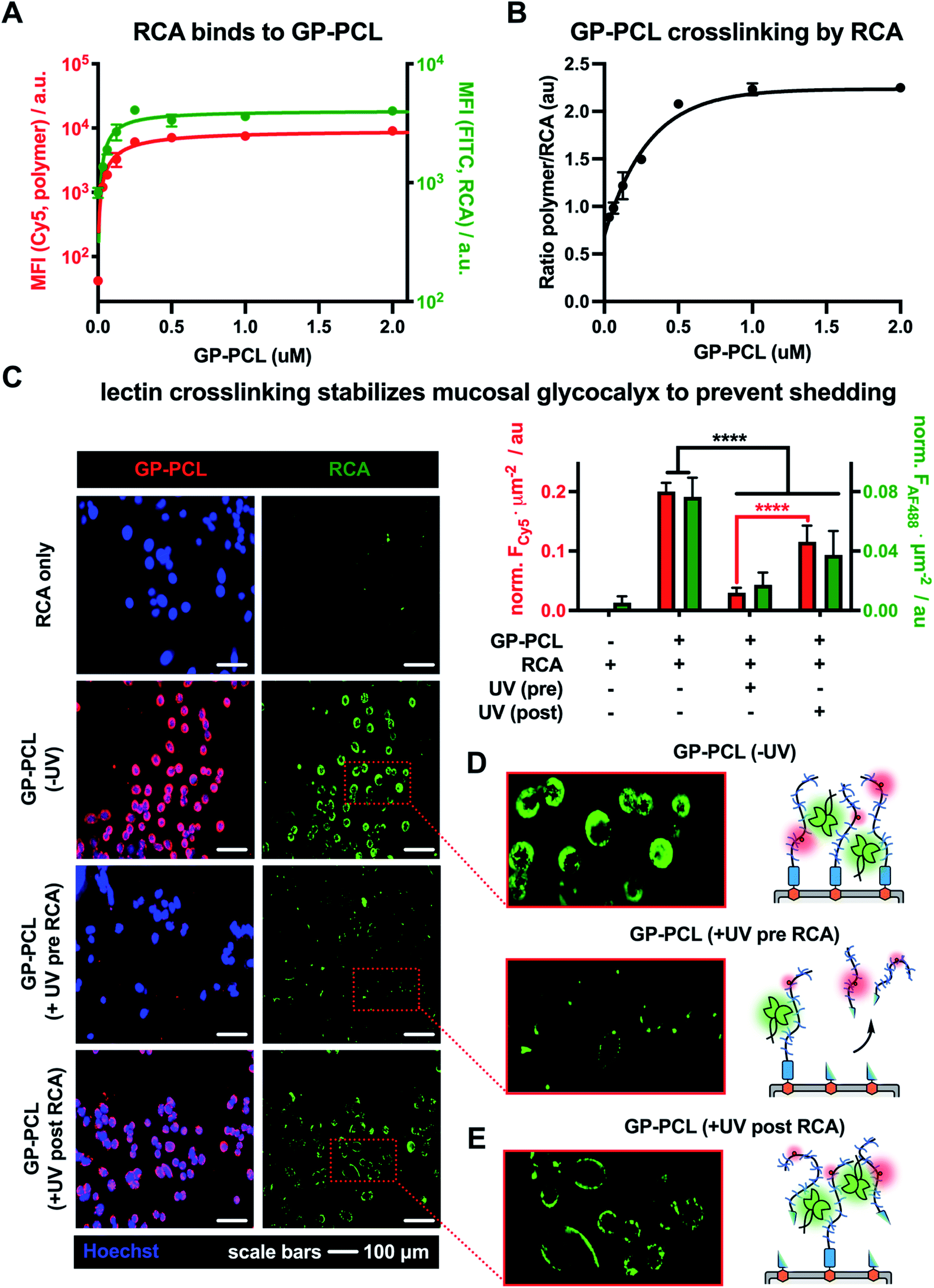 | ||
| Fig. 4 Lectin crosslinking limits photo-shedding of mucin mimetic glycocalyx. (A) Remodeling of CHO Lec8 cells with mucin mimetic GP-PCL (red) introduces galactose binding sites for RCA (green) on the cells surface in a concentration-dependent manner. (B) The plot of fluorescence intensity ratios for RCA and GP-PCL indicates enhanced lectin crosslinking with increasing polymer density in the membrane. (C) Fluorescence micrographs and bar graph representations of CHO Lec8 cells remodeled with GP-PCL and irradiated either before (UV pre) or after (UV post) RCA crosslinking. (D) Compared to a non-irradiated GP-PCL remodeled cells, photo-shedding of the mucin mimetics prior to RCA incubation reduces the number of available binding sites for the lectin. (E) RCA crosslinking of the mucin mimetic prior to irradiation stabilizes the glycocalyx and limits glycopolymer photo-shedding of the polymer from the cell surface. | ||
Next, we evaluated the effects of RCA crosslinking on mucin-mimetic shedding. We induced cleavage of GP-PCL from the cell surface either before or after crosslinked by the lectin (Fig. 4C–E).
CHO Lec8 cells in monolayer were remodeled with GP-PCL at a concentration sufficient to induce maximal RCA crosslinking (2 μM), stained with RCA (5 μg mL−1), and analyzed by fluorescence microscopy (Fig. 4C and Fig. S18†). We observed robust labeling of the remodeled cells by RCA prior to exposure to UV light. Pearson's correlation analysis showed strong colocalization of the RCA and GP-PCL signals, confirming association of the lectin with the mucin mimetic in the glycocalyx. When RCA was added to the remodeled cells following UV treatment (3 min), minimal binding was observed in agreement with the decrease in availability of lectin binding sites after photo-induced shedding of the mucin mimetic (Fig. 4D). When RCA was added before illumination, a significant portion of GP-PCL remained on the cell surface (Fig. 4E), which was quantified by measuring the mean Cy5 fluorescence intensity per cell area. This indicates that crosslinking by the lectin prior to shedding prevents clearance of the mucin mimetics from the cell surface, presumably through tethering to remaining polymers or entanglement with native glycocalyx structures. In the native environment of the mucosal glycocalyx, oligomeric lectins can bridge mucins with other endogenous glycoconjugates present at the cell surface and further decrease the efficiency glycocalyx shedding.
Conclusions
In this study, we have developed light-responsive glycomimetic materials to engineer the glycocalyx of live cells with spatial and temporal control and to model the process of glycoconjugate shedding. The introduction of a photocleavable cholesterol anchor into the glycomimetics enabled their installation into the plasma membranes to augment the glycocalyx of cells and allowed for their subsequent photo-release. By employing light-responsive mucin mimetic materials, we have demonstrated the utility of this approach in the photo-patterning and dynamic tuning of glycocalyx structures approximating those found on the mucosal epithelium. Utilizing interactions between the mucin mimetics and crosslinking lectins, we were able to establish that such lectins can enhance the retention of glycoconjugates on the cell surface and counteract the shedding process. This glycocalyx engineering approach may provide new insights into the roles of mucins in regulating host–pathogen interactions and the contributions from endogenous and pathogen-associated lectins to the protective functions of the mucosal barrier. More broadly, this strategy can be extended to other classes of membrane-associated glycoconjugates that undergo shedding from the cell surface as part of the cellular response during various pathophysiologies.Methods
General materials and methods
All chemicals, unless otherwise stated, were purchased from Sigma Aldrich and used as received. Cuprisorb resin was purchased from SeaChem Labs. Reaction progress was monitored by analytical thin-layer chromatography (TLC, Merck silica gel plates) with UV illumination or via CAM, ninhydrin, or KMnO4 staining. Column chromatography was performed on a Biotage Isolera One automated flash chromatography system. Nuclear magnetic resonance (1H and 13C NMR) spectra were recorded on Bruker 300 MHz and Jeol 500 MHz NMR spectrometers. Spectra were recorded in CDCl3 or D2O at 293 K and are reported in parts per million (ppm) on the δ scale relative to residual solvent as an internal standard (for 1H NMR: CDCl3 = 7.26 ppm, D2O = 4.79 ppm, for 13C NMR: CDCl3 = 77.0 ppm, CD3OD = 49.0 ppm). HRMS (high-resolution mass spectrometry) analysis was performed on an Agilent 6230 ESI-TOFMS in positive ion mode. UV-Vis spectra were collected in a quartz cuvette using a Thermo Scientific Nanodrop2000c spectrophotometer. IR spectroscopy was performed on a Nicolet 6700 FT-IR spectrophotometer. Size exclusion chromatography (SEC) was performed on a Hitachi Chromaster system equipped with an RI detector and two 5 μm, mixed bed, 7.8 mm I.D. × 30 cm TSK gel columns in series (Tosoh Bioscience) using an isocratic method with a flow rate of 0.7 mL min−1 in DMF (0.2% LiBr, 70 °C).Synthesis of azide-terminated poly(epichlorohydrin), P1
Epichlorohydrin was polymerized according to published procedures.20 Briefly, to a 10 mL flame-dried Schlenk flask equipped with a magnetic stirrer was added tetrabutylammonium azide (TBAN3, 30.0 mg, 60.0 μmol) under Ar atmosphere. Distilled epichlorohydrin (1.29 mL, 16.50 mmol) in anhydrous toluene (4.00 mL) and triisobutylaluminum in toluene (1.07 M, 0.10 μL, 0.11 mmol) were then added at −30 °C. The reaction was stirred for 4 hours before quenching with ethanol. The resulting polymer P1 was precipitated into hexanes and dried under vacuum to yield a clear viscous oil (1500 mg, 99%). The polymer was analyzed by SEC (0.2% LiBr in DMF): Mw = 29![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, Mn = 27700, Đ = 1.23 and 1H NMR (CDCl3, 500 MHz, Fig. S5†).
000, Mn = 27700, Đ = 1.23 and 1H NMR (CDCl3, 500 MHz, Fig. S5†).
Synthesis of poly(epichlorohydrin) polymers, P2
To separate 1-dram vials with a magnetic stirrer were added p(ECH) polymer P1 (7.50 mg, 0.25 μmol, 1 equiv.) and anhydrous DMSO (500 μL). Photocleavable cholesterol anchor 1 (1.70 mg, 2.50 μmol, 10 equiv.) or non-photocleavable cholesterol anchor 2 (1.20 mg, 2.50 μmol, 10 equiv.) was added, followed by CuI (0.05 mg, 0.30 μmol, 1.0 equiv.) and one drop diisopropylethyl amine (∼5 μL). The reactions were stirred at 40 °C for 12 hours before quenching with DCM and mixing with Cuprisorb beads (18 h) to sequester copper. The polymers were filtered through Celite, concentrated under vacuum, and triturated with chloroform in EtOH (30% v/v) to remove residual 1 or 2. The resultant polymers P2 were dried under vacuum to yield P2-PCL (7.2 mg, 96%) and P2-NPCL (6.7 mg, 89%). P2 were characterized by 1H NMR (CDCl3, 500 MHz, Fig. S6†), IR spectroscopy (Fig. S9†), and UV-Vis spectroscopy (Fig. S12†).Synthesis of cholesterol poly(glycidyl azide) polymers, P3
Three separate 1-dram vials were charged with cholesterol-terminated p(ECH) polymers P2 (6.7–7.2 mg, 0.22–0.24 μmol, 1 equiv.) in anhydrous DMF (200 μL) and a magnetic stirrer. To the solutions was then added NaN3 (2.0 mg, ∼2.0 equiv.) and the reactions was stirred at 60 °C for 72 h under Ar. The reaction solutions were filtered, dried, and concentrated from DCM to yield p(GA) polymers P3-PCL (7.0 mg, 93%), P3-NPCL (6.70 mg, 89%), and P3-Ø (7.18 mg, 88%). P3 were characterized by 1H NMR (CDCl3, 500 MHz, Fig. S7†), size exclusion chromatography (Fig. S11†), and IR spectroscopy (Fig. S10†).Synthesis of glycopolymers, GP
To three separate 1-dram vials were added p(GA) polymers P3 (7.50 mg, 0.075 mmol) dissolved in anhydrous DMSO (0.25 mL) and magnetic stirrers. Solutions of Cy5-alkyne (7.50 mg, 0.75 μmol) in DMSO (75 μL) were added, followed by CuI (1.67 mg, 7.50 μmol) and DIPEA (13.3 μL, 0.075 mmol). After 2 hours at 40 °C under Ar, propargyl lactose16b (50 μL, 0.113 mmol, 1.5 eq per azide side-chain) in anhydrous DMSO was added to the reactions and stirred at 40 °C overnight. The glycopolymers were diluted in water and treated with Cuprisorb beads for 18 hours to sequester copper before filtration over Celite and lyophilization. Methanol was used to remove excess glycoside and GPs were again lyophilized to yield the Cy5-labeled glycopolymers as pale blue solids GP-PCL (7.50 mg, quant.), GP-NPCL (7.50 mg, quant.), and P2-Ø (7.50 mg, quant.). GPs were characterized by 1H NMR (D2O, 500 MHz, Fig. S8†) and Cy5 labeling efficiency was quantified via UV-Vis spectroscopy (λmax = 633 nm, ∼2–3 fluorophores per polymer).General cell culture
All biological reagents were purchased from Gibco (ThermoFisher) unless otherwise stated. CHO Lec8 and CHO Pro5 cells used were obtained from ATCC (CRL-1737 and CRL-1781, respectively). Biotin-labeled Ricinus communis agglutinin I was purchased from Vector Labs (B-1085-5). Cells were cultured at 37 °C and 5% CO2 following standard tissue culture practices. CHO (Chinese Hamster Ovary cells, Pro5 and Lec8) were cultured in MEMα medium supplemented with 10% FBS, 100 U mL−1 penicillin, and 100 U mL−1 streptomycin. Cells were suspended utilizing 0.25% trypsin–EDTA and passaged every 2–4 days to achieve desired confluency on tissue-culture treated lab plastics. Live cell flow cytometry analysis was performed using a FACSCalibur or FACSCanto II system (BD Biosciences). Microscopy was performed on either a Keyence BZX800 epifluorescent microscope or a ThermoScientific EVOS imaging system and images were analyzed using ImageJ.GP membrane incorporation
Flow cytometry: CHO Lec8 cells were suspended, washed, and 106 cells were pelleted into Eppendorf tubes. GPs prepared in DPBS (0–20 μM, 100 μL) were added to the cell pellets, mixed, and incubated on ice for 1 hour. Following two washes with DPBS, cells were resuspended and analyzed by flow cytometry. Microscopy: CHO Lec8 cells grown in 12-well plates were rinsed with DPBS and GPs (2 μM) were added. Plates were incubated on ice for 1 hour before three DPBS washes. Nuclei were stained using Hoechst 33342 (10 μg mL−1, 10 min) followed by two additional DPBS washes. Fluorescent micrographs were captured on a Keyence epifluorescent microscope.Photo-induced shedding of GPs from cell surface
Flow cytometry: Remodeled CHO Lec8 cells in clear plastic tubes were subjected to ultraviolet light (λ = 365 nm, 0–3 min) using a handheld 15 W lamp. Following irradiation cells were washed twice with DPBS and resuspended for flow cytometry analysis. Microscopy: Remodeled CHO Lec8 cells were irradiated in well plates using a handheld 15 W lamp (λ = 365 nm, 3 min) directly below the plate. Cells were then washed three times with 1 mL of DPBS and nuclei were stained using Hoechst 33342 (10 μg mL−1, 10 min). Following two additional DPBS washes fluorescent micrographs were captured.Photopatterning of GPs in cell membrane
CHO Lec8 cells were grown until confluent in 6-well plates. Standard GP membrane incorporation protocols were utilized and fluorescent micrographs were captured within a selected region suitable for stitching. A portion of the well was masked before irradiation using a handheld 15 W lamp (λ = 365 nm, 0–3 min). Following mask removal, images were collected within the region, stitched using Keyence BZX Analyzer software, and quantified by ImageJ.RCA binding to GP-PCL remodeled cells
CHO Lec8 cells were suspended (0.25% trypsin–EDTA), washed, and 106 cells were pelleted in Eppendorf tubes. GP-PCL prepared in DPBS (0–5 μM, 100 μL) were added to the cell pellets and incubated on ice for 1 h. Following two DPBS washes cells were incubated in RCA-biotin (5 μg mL−1, 300 μL) for 40 min on ice. After two additional washes cells were incubated in excess AlexaFluor488 labeled streptavidin (300 μL, 1:750) for 20 min, washed twice with DPBS, and resuspended for flow cytometry analysis.
Lectin crosslinking during GP shedding
CHO Lec8 cells grown in 12-well plates were remodeled with GP-PCL (2 μM) on ice for 1 hour. Cells were then washed three times with DPBS and incubated with RCA-biotin (5 μg mL−1) either before (pre RCA) or after (post RCA) UV irradiation using a 15 W lamp (λ = 365 nm, 3 min). After an additional three DPBS washes, cells were incubated on ice for 30 min with an excess of AlexaFluor488 labeled streptavidin (300 μL, 1:750) and nuclei were stained with Hoechst 33342 (10 μg mL−1, 10 min) for visualization. Following two additional DPBS washes fluorescent micrographs were captured on a Keyence epifluorescent microscope and ImageJ was used to analyze micrographs.
Data availability
A complete description of reagents, expanded methods, characterization data for all chemical intermediates and GPs, and expanded biological data are provided in the ESI.†Author contributions
K. G. conceived of the research; S. C. P., M. H. Z., D. J. H. and K. G. designed the research; S. C. P., M. H. Z., H. J. C. N., D. J. H., and A. L. M. performed the research; S. C. P., M. H. Z., H. J. C. N., and K. G. analyzed data; S. C. P. and K. G. wrote the manuscript and ESI;† all authors approved the manuscript for publication.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was supported in part by the NIH Director's New Innovator Award (NICHD: 1DP2HD087954-01). We thank the UCSD Microscopy Core Facility for assistance with fluorescence microscopy (via NINDS P30 Grant: P30NS047101) and the UCSD Glycobiology Research and Training Center for providing access to critical analytical instrumentation. We also thank Garland Jackson for assistance with confocal microscopy and discussions. KG was supported by the Alfred P. Sloan Foundation (FG-2017-9094) and the Research Corporation for Science Advancement via the Cottrell Scholar Award (grant #24119); S. C. P. was supported by a GAANN fellowship (U.S. Dept. of Education, P200A150251).References
- (a) A. Varki and P. Gagneux, in Essentials of Glycobiology, ed. A. Varki, R. D. Cummings and J. D. Esko, Cold Spring Harbor Laboratory Press, New York USA, 3rd edn, 2017, ch. 7 Search PubMed; (b) S. K. Linden, P. Sutton, N. G. Karlsson, V. Korolik and M. A. McGuckin, Mucosal Immunol., 2008, 1, 183–197, DOI:10.1038/mi.2008.5.
- B. F. Becker, M. Jacob, S. Leipert, A. H. J. Salmon and D. Chappell, Br. J. Clin. Pharmacol., 2015, 80(3), 389–402, DOI:10.1111/bcp.12629.
- T. Manon-Jensen, Y. Itoh and J. R. Couchman, FEBS J., 2010, 277(19), 3876–3889, DOI:10.1111/j.1742-4658.2010.07798.x.
- C. Reily, T. J. Stewart, M. B. Renfrow and J. Novak, Nat. Rev. Nephrol., 2019, 15, 346–366, DOI:10.1038/s41581-019-0129-4.
- (a) S. C. Purcell and K. Godula, Interface Focus, 2018, 9, e20180080, DOI:10.1098/rsfs.2018.0080; (b) M. A. McGuckin, S. K. Linden, P. Sutton and T. H. Florin, Nat. Rev. Microbiol., 2011, 9, 265–278, DOI:10.1038/nrmicro2538.
- (a) D. J. Honigfort, M. O. Altman, P. Gagneux and K. Godula, Proc. Natl. Acad. Sci. U. S. A., 2021, 118(40) DOI:10.1073/pnas.2107896118; (b) S. C. Delaveris, R. E. Webster, S. M. Banik, G. Boxer and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2020, 17(23), 12643–12650, DOI:10.1073/pnas.1921962117.
- M. Cohen, X. Q. Zhang, H. P. Senaati, H. W. Chen, N. M. Varki, R. T. Schooley and P. Gagneux, Virol. J., 2013, 10(321) DOI:10.1186/1743-422X-10-321.
- W. Van Breedam, S. Pöhlmann, H. W. Favoreel, R. J. DeGroot and H. J. Nauwynck, FEMS Microbiol. Rev., 2014, 38(4), 598–632, DOI:10.1111/1574-6976.12052.
- P. Argueso, A. Guzman-Aranguez, F. Mantelli, Z. Cao, J. Ricciuto and N. Panjwani, J. Biol. Chem., 2009, 284(34), 23037–23045, DOI:10.1074/jbc.M109.033332.
- (a) B. Belardi, G. P. O'Donoghue, A. W. Smith, J. T. Groves and C. R. Bertozzi, J. Am. Chem. Soc., 2012, 134(23), 9549–9552, DOI:10.1021/ja301694s; (b) B. Belardi and C. R. Bertozzi, J. Chem. Biol., 2015, 22(8), 983–993, DOI:10.1016/j.chembiol.2015.07.009.
- C. R. Shurer, M. J. Colville, V. K. Gupta, S. E. Head, F. Kai, J. N. Lakins and M. J. Paszek, ACS Biomater. Sci. Eng., 2018, 4(2), 388–399, DOI:10.1021/acsbiomaterials.7b00037.
- (a) D. J. Shon, S. A. Malaker, K. Pedram, E. Yang, V. Krishnan, O. Dorigo and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2020, 117(35), 21299–21307, DOI:10.1073/pnas.2012196117; (b) S. A. Malaker, K. Pedram, M. J. Ferracane, B. A. Bensing, V. Krishnan, C. Pett, J. Yu, E. C. Woods, J. R. Kramer, U. Westerlind, O. Dorigo and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2019, 116(15), 7278–7287, DOI:10.1073/pnas.1813020116.
- (a) J. R. Kramer, B. Onoa, C. Bustamante and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2015, 112(41), 12574–12579, DOI:10.1073/pnas.1516127112; (b) H. Pan, M. J. Colville, N. T. Supekar, P. Azadi and M. J. Paszek, ACS Synth. Biol., 2019, 8(10), 2315–2326, DOI:10.1021/acssynbio.9b00127.
- (a) P. Gaur, O. A. Kucherak, Y. G. Ermakova, V. V. Shvadchak and D. A. Yushchenko, Chem. Commun., 2019, 55(82), 12288–12291, 10.1039/c9cc05602e; (b) S. Kaneko, H. Nakayama, Y. Yoshino, D. Fushimi, K. Yamaguchi, Y. Horiike and J. Nakanishi, Phys. Chem. Chem. Phys., 2011, 13(9), 4051–4059, 10.1039/c0cp02013c.
- S. Oelman, P. Stanley and R. Gerardy-Schahn, J. Biol. Chem., 2001, 276(28), 26291–26300, DOI:10.1074/jbc.M011124200.
- (a) E. C. Woods, N. A. Yee, J. Shen and C. R. Bertozzi, Angew. Chem., 2015, 52(54), 15782–15788, DOI:10.1002/anie.201508783; (b) D. Rabuka, M. B. Forstner, J. T. Groves and C. R. Bertozzi, J. Am. Chem. Soc., 2008, 130(18), 5947–5953, DOI:10.1021/ja710644g.
- (a) P. Dhar and J. McAuley, Front. Cell. Infect. Microbiol., 2019, 9(117) DOI:10.3389/fcimb.2019; (b) J. L. McAuley, L. Corcilius, H. X. Tan, R. J. Payne, M. A. McGuckin and L. E. Brown, Mucosal Immunol., 2017, 10, 1581–1593, DOI:10.1038/mi.2017.16; (c) J. Turner, Nat. Rev. Immunol., 2009, 9, 799–809, DOI:10.1038/nri2653.
- (a) A. P. Corfield, Biochim. Biophys. Acta, 2015, 1850(1), 236–252, DOI:10.1016/j.bbagen.2014.05; (b) C. E. Wagner, K. M. Wheeler and K. Ribbeck, Annu. Rev. Cell Dev. Biol., 2018, 6(34), 189–215, DOI:10.1146/annurev-cellbio-100617-062818M.
- (a) R. D. Cummings and R. L. Schnaar, in Essentials of Glycobiology, ed. A. Varki, R. D. Cummings and J. D. Esko, Cold Spring Harbor Laboratory Press, New York USA, 3rd edn, 2017, ch. 31 Search PubMed; (b) D. Gupta, H. Kaltner, X. Dong, H. J. Gabius and C. F. Brewer, Glycobiology, 1996, 6(8), 843–849, DOI:10.1093/glycob/6.8.843.
- (a) M. Gervais, A. Labbé, S. Carlotti and A. Deffieux, Macromolecules, 2009, 42, 2395–2400, DOI:10.1021/ma802063s; (b) D. J. Honigfort, M. H. Zhang, S. Verespy and K. Godula, Faraday Discuss., 2019, 219, 138–153, 10.1039/C9FD00024K.
Footnotes |
| † Electronic supplementary information (ESI) available: A complete description of reagents and expanded methods section: detailed protocols for GP membrane anchor and propargyl lactose synthesis, characterization data for all chemical intermediates and GPs. Detailed protocols for biological experiments, cell membrane incorporation and photocleavage optimization, lectin (RCA) binding and colocalization analysis, cytotoxicity assays, and expanded microscopy panels. See https://doi.org/10.1039/d2sc00524g |
| ‡ These authors contributed equally. |
| § Current address: Western Michigan University, 300 Portage St., Kalamazoo, MI, 49007, USA. |
| ¶ Current address: Element Biosciences, 9880 Campus Point Dr., San Diego, CA, 92121, USA. |
| This journal is © The Royal Society of Chemistry 2022 |