
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTowards the rational design of Pt-based alloy catalysts for the low-temperature water-gas shift reaction: from extended surfaces to single atom alloys†
Yuqi
Yang‡
 ,
Tonghao
Shen‡
and
Xin
Xu
*
,
Tonghao
Shen‡
and
Xin
Xu
*
Collaborative Innovation Center of Chemistry for Energy Materials, Shanghai Key Laboratory of Molecular Catalysis and Innovative Materials, MOE Key Laboratory of Computational Physical Sciences, Department of Chemistry, Fudan University, Shanghai 200433, People's Republic of China. E-mail: xxchem@fudan.edu.cn
First published on 5th May 2022
Abstract
The rational design of Pt-based catalysts for the low-temperature water-gas-shift (LT-WGS) reaction is an active research field because of its important role played in the fuel cell-based hydrogen economy, especially in mobile applications. Previous theoretical analyses have suggested that Pt alloys, leading to a weaker CO binding affinity than the Pt metal, could help alleviate CO poisoning and thus should be promising catalysts of the LT-WGS reaction. However, experimental research along this line was rather ineffective in the past decade. In the present work, we employed the state-of-the-art kinetic Monte Carlo (KMC) simulations to examine the influences of the electronic effect by introducing sub-surface alloys and/or core–shell structures, and the synergetic effect by introducing single atom alloys on the catalytic performance of Pt-alloy catalysts. Our KMC simulations have highlighted the importance of the OH binding affinity on the catalyst surfaces to reduce the barrier of water dissociation as the rate determining step, instead of the CO binding affinity as has been emphasized before in conventional mean-field kinetic models. Along this new direction of catalyst design, we found that Pt–Ru synergetic effects can significantly increase the activity of the Pt metal, leading to Ru1–3@Pt alloys with a tetrahedron site of one surface-three subsurface Ru atoms on the Pt host, showing a turnover frequency of about five orders of magnitude higher than the Pt metal.
1 Introduction
The water-gas-shift (WGS) reaction (CO + H2O ⇌ CO2 + H2) is one of the most important catalytic processes in the hydrogen economy, because of its indispensable role in hydrogen generation and purification.1–3 The WGS reaction is mildly exothermic (ΔH = −41 kJ mol−1), and, in practice, it is typically carried out in two steps: a high temperature WGS (HT-WGS) step, which is favorable for reaction kinetics (faster reaction rate), and a low temperature WGS (LT-WGS) step, which is favorable for thermodynamics (higher CO conversion ratio).1 In large scale industrial hydrogen production, the HT-WGS and LT-WGS processes often employ Fe2O3–Cr2O3 and Cu–ZnO catalysts, respectively, while in fuel cell applications, one pillar of the hydrogen economy, only LT-WGS is applicable, and the catalysts must be active over a wide range of temperature, air-insensitive, non-pyrophoric, and stable during frequent shutdown–restart operation cycles.2,3 Noble metal catalysts, in particular Pt-based catalysts, meet these criteria, and, therefore, they have attracted most of the interest in recent years.4–6It has been proved that the Pt species can be highly active under the reaction conditions of LT-WGS, if its oxidized state can be properly modified by introducing oxidized ligands,5,7 or reducible supports such as TiO2 or CeO2.8,9 Along this line, a significant breakthrough has been achieved in experiments.4–6,10,11 However, confirming the active sites, as well as understanding the reaction mechanisms on these composite catalysts, is quite difficult in both theoretical and experimental approaches, because of the complexity of metal–ligand/support interactions,4,5,9,12 which has become an obstacle for the rational design of new catalysts.
Alternatively, a rational way of catalyst design is to modify the metal composition of the catalyst.13–17 On one hand, chemical reactions usually follow the same mechanism on transition metal surfaces.18–20 On the other hand, the adsorption energies of key intermediates, as well as the energy barriers, on metal surfaces often linearly correlate with the electronic structure of surface metal atoms,21,22 which can be conveniently controlled by alloying different metals.23 Therefore, in both theoretical and experimental approaches, metallic Pt alloys provide not only a clean model to investigate the promoting effects, but also a practical way to systematically improve the catalytic performance of Pt-based catalysts.24
As an essential process in chemical industry, the reaction mechanism, active sites, and reaction kinetics of WGS have been widely investigated by various theoretical and experimental techniques.12,25–28 It is believed that, on transition metal surfaces, the WGS reaction generally follows the carboxyl mechanism, and the rate-determining step (RDS) is the decomposition of H2O into adsorbed H and OH species.25–27 In the catalyst design, reducing the energy barrier of the RDS should drastically increase the reaction rate in general. Therefore, the metals on which H2O decomposes easily are expected to be helpful in enhancing the LT-WGS reaction.9 Meanwhile, Pt-group metals would be poisoned by CO soon under the LT-WGS reaction conditions, which can completely block the H2O decomposition and thus shutdown the whole reaction network on metal surfaces.29,30 As a result, decreasing the CO adsorption energy, which mitigates the CO poisoning, has been considered as an effective way to improve the catalytic performance of Pt catalysts.29
The concern of CO poisoning has remarkably affected the direction of rational design, especially in the theoretical understanding, of alloy catalysts for the LT-WGS reaction in the past few years.3,29–31 Alloys such as Pt–Cu or Pt–Re, which significantly reduces the CO binding affinity without notably increasing the barrier of the RDS, were therefore predicted as promising candidates.29,31,32 However, despite that some promoting effects have been observed in LT-WGS experiments,31 the direct influence of decreasing CO adsorption energy on the catalytic performance is rather limited,6,10 and there is still a large discrepancy between the theoretical expectation and experimental achievement up to now.
After reviewing the theoretical approaches for investigations of the LT-WGS reaction in the last decade,25,26,29,30,32–36 we realized that these approaches perhaps have overemphasized the influence of CO poisoning on the Pt metal. Most of the existing theoretical approaches in kinetic studies rely on density functional theory (DFT) calculations and mean-field microkinetic modeling (MF-MKM).15,37 It has been found that the generalized gradient approximation (GGA) functionals such as Perdew–Burke–Ernzerhof (PBE)38 and PW91,39,40 which were frequently utilized in investigating the LT-WGS reaction,25,26,29,32–36 can significantly overestimate the adsorption energy of CO on the Pt metal surface.41–44 Moreover, the conventional MF-MKM can result in an unphysically high CO coverage and an incorrect description of CO poisoning on the Pt metal surface because of ignoring the adsorbate–adsorbate interactions.45 Although some corrections on CO adsorption energy were made by hand in some of the investigations,20,30 the overemphasis on the effect of CO poisoning would have shaded the impacts of other key properties, which might have led to an improper design of catalysts over the past decade.
Nowadays, more advanced DFT functionals and more sophisticated MKM methods, which are expected to capture the essence of CO adsorption on metal surfaces more correctly, have been developed and become more and more popular in the catalyst design.37,46 Therefore, a comprehensive investigation of accurate reaction kinetics, as well as the key properties that can improve the activity of Pt-based catalysts, is mandatory both from a fundamental and a practical point of views. In the present work, we employed the state-of-the-art scaling relation kinetic Monte Carlo (KMC) simulations,47 which offer an accurate description of all elementary reactions and adsorbate–adsorbate interactions on catalyst surfaces, to investigate the activities of Pt alloys. The energetic parameters needed in KMC simulations were calculated by using the more recently developed Bayesian error estimation functional with van der Waals corrections (BEEF-vdW)48 that can accurately reproduce experimental CO adsorption energy on Pt metal surfaces.42,43 Furthermore, the time-consuming KMC simulations were powered by the recently developed extended phenomenological kinetic (XPK) method if applicable,49 which extends KMC simulations to more complex reaction networks. On the basis of simulation results, we tried to understand the influence of the electronic structure of surface Pt atoms by introducing sub-surface alloys and/or core–shell structures, as well as synergetic effects between the host Pt and the promoter metals by introducing single atom alloys (SAAs), on the catalytic performance, and to screen the most active catalyst candidate.
2 Methods and models
In the present work, all DFT calculations and KMC simulations were performed on the Pt(111) surface, which is one of the most commonly used models of close-packed structures in catalyst design. In the investigations of Pt alloys, promoters could replace some of the Pt atoms in the model. In the electronic calculations for all gaseous species and surface intermediates including adsorbate states and transition states, the BEEF-vdW functional was used.48 For some key energetic parameters, three DFT functionals, including the PBE functional,38 the extended Perdew–Burke–Ernzerhof (xPBE) functional50 and the van der Waals Density Functional (vdW-DF),51 were employed to confirm the results of BEEF-vdW calculations. The zero-point energy (ZPE) corrections were considered through harmonic analysis for both gaseous species and surface intermediates. Besides, the entropy corrections for surface reactions and the strong adsorption of CO were considered in harmonic approximation, while for the adsorption of H2O, the entropy correction was calculated by using the Campbell–Sellers equation.52 All DFT calculations in the present work were carried out by using the Vienna ab initio simulation package (VASP).53,54 More computational details can be found in Section S1.1 in the ESI.†Following the carboxyl mechanism, there were six elementary chemical reactions in total, as shown in eqn (1)–(6). These elementary reactions were taken into account in the KMC simulations.
| CO(g) + * ⇌ CO* | (1) |
| H2O(g) + * + * ⇌ OH* + H* | (2) |
| CO* + OH* ⇌ COOH* + * | (3) |
 | (4) |
 | (5) |
| H* + H* → H2(g) + * + * | (6) |
| CO* + * ⇌ * + CO* | (7) |
| COOH* + * ⇌ * + COOH* | (8) |
| H* + * ⇌ * + H* | (9) |
Because of the very small adsorption energy of CO2, it is quite easy for CO2 to desorb from the surface, and thus its diffusion was not taken into account in the present work. More details for the reaction model are discussed in Section S1 in the ESI.†
We employed subsurface alloys PtMPt (M = Ag, Au, Cu, Ir, Os, Pd, Pt, Re, Rh and Ru), which modify the subsurface with M but leave the Pt atoms on the surface intact (see S1.2 and Fig. S1 in the ESI†), to investigate how the electronic structure of surface Pt atoms is influenced by the subsurface atoms M to affect the catalytic activity. To carry out the scaling relation KMC simulations, the linear scaling relations between the adsorption energies of different adsorbates,22 as well as that between energy barriers and reaction heats, i.e. the Brønsted–Evans–Polanyi (BEP) relations,21 were built on different subsurface alloys. In the construction of the activity map, we chose the adsorption energies of two adsorbates as the energetic descriptors; they are the adsorption energy of CO that represents the CO poisoning effect, and the adsorption energy of OH that is closely related to the energy barrier of the RDS. This choice is in line with that suggested in previous MF-MKM studies.30,56 Then, a two-dimensional (2D) volcano-shaped surface was built on the basis of reaction rates obtained in the KMC simulations, to understand the electronic effect on activity and to screen the potential catalysts following the idea of the Nørskov–Bligaard method.15,21
The synergetic effects between the host Pt and the promoter metal atoms were studied by introducing six types of metal atoms on the Pt(111) surface, in which one surface Pt atom is replaced by a promoter metal M to form SAAs, denoted as M1@Pt (M = Au, Ag, Cu, Ru, Rh, Pd). Because different elementary steps may prefer different active sites on SAAs, the net rate of the RDS associated with a surface atom was considered as its TOF in the analysis of the KMC results, as suggested in our previous work.28 For simplicity, we further assumed that the electronic and synergetic effects are additive, i.e. the activity of promoter M is determined by the energy difference between M and the host Pt on the surface. In this manner, the promoting effect of the promoter M was described by an additional 2D activity map.
On the basis of the analysis of SAAs, we picked up the most active candidate, and discussed the influence of the chemical environment on the activity. Finally, the predictions of the volcano-shaped surfaces, which were based on the linear scaling relations, were confirmed in the explicit DFT calculations and KMC simulations. In the present work, the XPK approach was employed to accelerate the time-consuming KMC simulations if applicable, and all the kinetic simulations were carried out by using the XPK code.57 We refer to Section S2.1 in the ESI† for more details of the simulations.
3 Results and discussion
3.1 Thermodynamics of the WGS reaction
The calculation of the thermodynamic parameters is the first step in applying KMC simulations. The calculated results with the BEEF-vdW functional with respect to the empty surface, gaseous CO and H2O are summarized in Fig. 1a. It can be seen that on the Pt(111) surface, the adsorption of CO (eqn (1)) is strongly exothermic with an adsorption energy of −1.46 eV. As compared to the PBE result (−1.76 eV), this value is significantly lower in magnitude and much closer to the experimentally measured value (−1.43 eV) reported in the literature.58 Meanwhile, the dissociative adsorption of H2O (eqn (2)) is mildly endothermic with a dissociative adsorption energy of 0.55 eV, while the energy barrier of this step is 0.83 eV. The formation of COOH* (eqn (3)) and its decomposition into H* and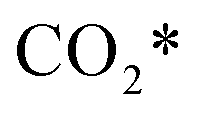 (eqn (4)) are both exothermic, with the reaction heats of −0.18 and −0.12 eV, respectively, and the energy barriers of 0.37 and 0.59 eV, respectively. The adsorption energies of the final products, CO2 and H2, are −0.18 and −0.41 eV, respectively. The consideration of entropic corrections would significantly decrease the free energies of the adsorption processes, but has little impact on the reactions that involve surface species only. The detailed values of thermodynamic parameters and their calculations are summarized in Section S3.1 in the ESI.† According to the landscape of the energy curve, we may conclude that CO with the highest adsorption energy is the major surface species, and the dissociative adsorption of H2O with the highest energy barrier is the RDS.
(eqn (4)) are both exothermic, with the reaction heats of −0.18 and −0.12 eV, respectively, and the energy barriers of 0.37 and 0.59 eV, respectively. The adsorption energies of the final products, CO2 and H2, are −0.18 and −0.41 eV, respectively. The consideration of entropic corrections would significantly decrease the free energies of the adsorption processes, but has little impact on the reactions that involve surface species only. The detailed values of thermodynamic parameters and their calculations are summarized in Section S3.1 in the ESI.† According to the landscape of the energy curve, we may conclude that CO with the highest adsorption energy is the major surface species, and the dissociative adsorption of H2O with the highest energy barrier is the RDS.
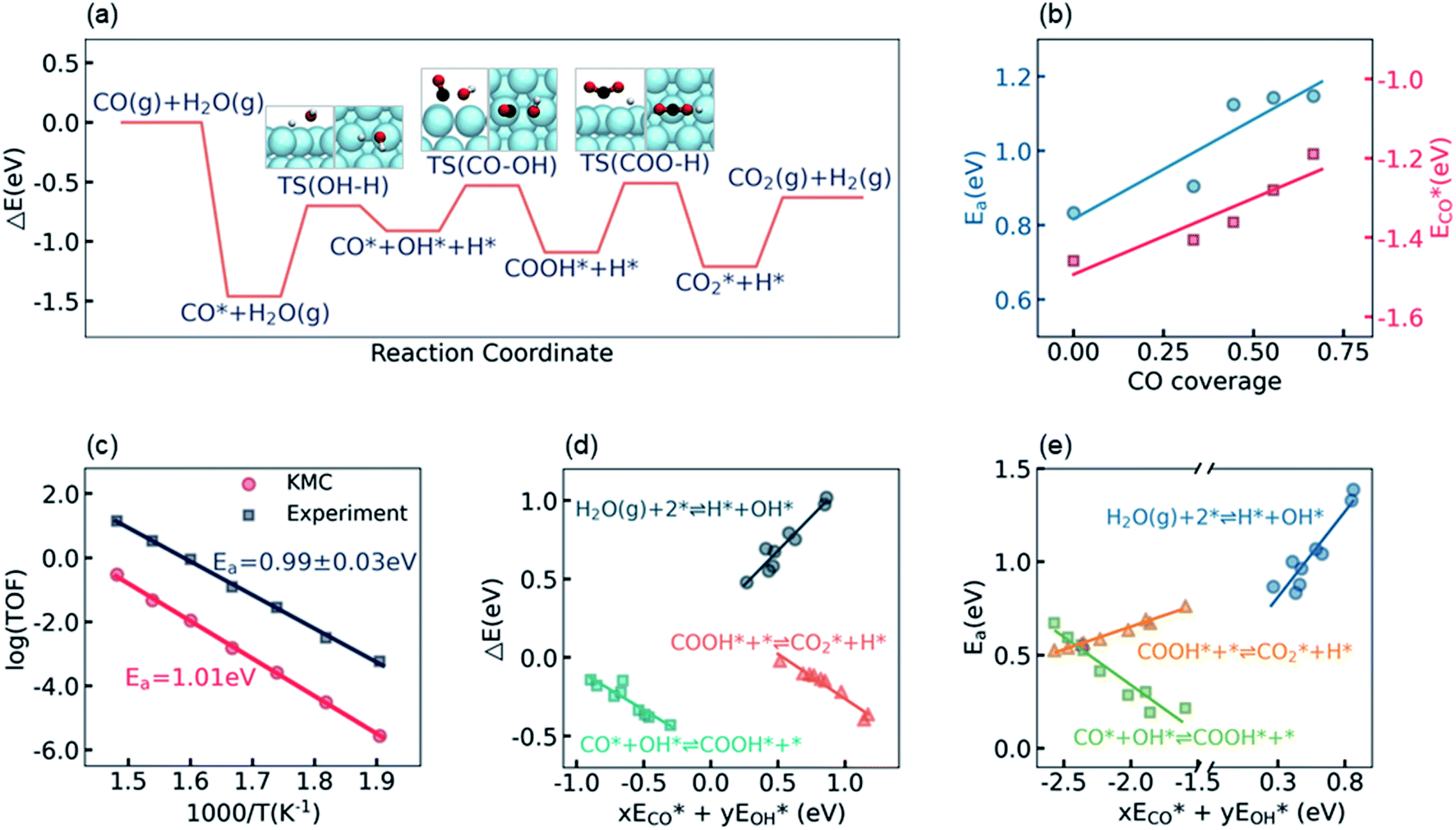 | ||
| Fig. 1 (a) Electronic potential energy curve for the WGS reaction on the Pt(111) surface. (b) The influence of CO coverage on the energy barrier of the RDS (blue circle) and the CO adsorption energy (red square). (c) Comparison of the apparent activation energies on the Pt(111) surface obtained from the experimental data (black line) and from the theoretical simulations (red line) in the present work. (d) The linear scaling relations between reaction heats and the combined energetic descriptor (see the main text for the definition). (e) The linear scaling relations between the energy barriers and the combined energetic descriptor. | ||
Under the LT-WGS reaction conditions, the strong CO binding affinity of Pt would result in a high CO coverage on the catalyst surface, which could poison the Pt catalyst. Yet, the high CO coverage on the surface would lead to a strong adsorbate–adsorbate interaction, which, in turn, influences the thermodynamic parameters. On one hand, as shown by ECO* in Fig. 1b, the CO–CO interaction decreases its adsorption energy in magnitude as the CO coverage increases, i.e. the adsorption energy of CO on the Pt surface with a high CO coverage is lower in magnitude than the one at the zero-coverage limit in a common DFT calculation, which keeps the CO coverage at about 2/3 monolayer (ML), instead of blocking all active sites on the Pt(111) surface, under the LT-WGS reaction conditions, and thus partly provides a way of self-cure for the CO poisoning. On the other hand, the interactions between CO and the other surface intermediates change the reaction heats and energy barriers. As shown by Ea in Fig. 1b, for instance, the energy barrier for the dissociative adsorption of H2O increases as the CO coverage increases, which inhibits this reaction on the Pt surface with a high CO coverage. These observations on top of BEEF-vdW calculations are consistent with DFT calculations reported in the previous literature,44,59 which are also confirmed by calculations using other functionals as PBE, xPBE and vdW-DF. More details and discussion on the adsorbate–adsorbate interactions can be found in Section S3.2 in the ESI.†
To validate/test the settings and calculations in our model, we performed KMC simulations on Pt(111) surfaces at different temperatures in the range from 525 K to 675 K, and compared the calculated kinetic properties with the experimental data. It is usually difficult to directly compare the calculated turnover frequency (TOF) with the experimental one,60,61 as the values of the TOF are very sensitive to the errors associated with both DFT calculations and experimental measurements. Therefore, instead of the TOF, herein we focused on the systematic variation of the TOF, i.e. the apparent activation energy, which has been concluded to be a more robust criterion for benchmarking theoretical modeling.28,60 The apparent activation energy was obtained from an Arrhenius plot, as shown in Fig. 1c. The experimental data reported by Williams et al.55 in the same temperature range are plotted for comparison. It can be seen that, despite that the current KMC simulations systematically underestimated the TOF by about two orders of magnitude, the theoretically predicted apparent activation energy, 1.01 eV, is very close to the experimentally measured value, 0.99 ± 0.03 eV. The good agreement between the calculated and the experimental apparent activation energy verifies the reliability of the present kinetic model.
The linear scaling relations between the thermodynamic parameters and the energy descriptors were then built by a series of DFT calculations on the subsurface alloys. We assumed a bilinear dependence of the parameters on the adsorption/binding energies of CO* and OH*,
| ΔEi = a(xECO* + yEOH*) + b, | (10) |
3.2 Influence of the electronic structure on activity
Using the knowledge of all the thermodynamic parameters and their dependences on the energy descriptors, ECO* and EOH*, we carried out KMC simulations to construct a 2D volcano-shaped activity map as shown in Fig. 2a. In these simulations, only the electronic energies varied according to their dependence on ECO* and EOH*, while the other energetic and kinetic settings such as thermodynamic corrections, temperature, partial pressures, etc., were kept the same as that on the Pt(111) surface. Therefore, the 2D activity map, which represents the activity of different subsurface/core–shell alloys, reflects the electronic effects of surface Pt atoms on activity influenced by introducing the subsurface atoms. On the activity map, the location of the Pt(111) surface is labeled with a black cross, while its contour line of activity is colored in gray. The core–shell structured PtRe alloy and the subsurface PtCuPt alloy, which have been suggested as promising candidates of LT-WGS catalysts due to their lower CO adsorption energies in magnitude,29,31 are also marked for comparison. It is surprising that the activities of PtRe (red square) and PtCuPt (yellow triangle) are distinctly different as compared to the Pt metal, even though, for both of the former, the CO binding affinities are much weaker than that for the Pt metal based on DFT calculations. It can be seen that, in Fig. 2a, the core–shell structured PtRe is about 65.4 fold more active than the Pt metal, which is consistent with the experimental observation in the literature.31 In contrast, the PtCuPt subsurface alloy is inactive as compared to the Pt metal, which is quite different from the conclusion in a previous theoretical study.29 It is worth noting that in the BEEF-vdW calculations, the CO adsorption energy on PtCuPt is 0.25 eV lower than that on Pt, which is close to the corresponding result from PW91 reported in the literature (0.33 eV),29 as well as the values from PBE (0.20 eV), xPBE (0.24 eV), and vdW-DF (0.31 eV) calculations in the present work (see Table S10† for more details). Therefore, the discrepancy between the present calculation and the previous prediction in the literature indicates that the CO adsorption energy alone does not determine the activity in this region of the volcano-shaped surface. | ||
| Fig. 2 (a) 2D activity map of Pt alloys for the LT-WGS reaction obtained in KMC simulations at 400 K. The triangles refer to the subsurface alloys PtMPt, while the squares and circles refer to the core–shell structured alloys of Pt–M and Pt–MPt3, respectively. The contour line of Pt is shown in gray. (b) Dependences of the logarithmic TOF on the OH* binding energy (black line) and the CO* adsorption energy (red line) along the corresponding arrows on the activity map of (a). (c) Dependence of the energy barrier of the RDS on the CO* adsorption energy. The dark cyan line and the yellow line stand for the contributions from linear scaling relations and repulsive interactions with CO*, respectively, while the red line shows the total effect. The total effect is evaluated by accumulating the effect of linear scaling relations, which is calculated with respect to the energy barrier on the Pt(111) surface, to the repulsion contribution. (d) 2D activity map constructed by MF-MKM. (e) Dependences of the logarithmic TOF on the OH* binding energy and the CO* adsorption energy corresponding to (d). (f) Linear correlations between the d-band center of the surface Pt atoms and the OH* binding energy (in red) and the CO* adsorption energy (in blue). | ||
To investigate the influences of energy descriptors on activity, we plotted the dependence of the logarithmic TOF on ECO* and EOH* in Fig. 2b, where the activity of the Pt metal is labeled with a vertical dashed line, which is taken as the reference. The red/black line in Fig. 2b represents the change of activity when ECO*/EOH* varies along the direction of the red/black dashed arrow in Fig. 2a. Hence, the CO*/OH* binding affinity is decreased/increased from left to right in Fig. 2b. As indicated by the red line, in the region where the CO adsorption is stronger than that on Pt (i.e., on the left side of the vertical dashed line), decreasing the CO* binding affinity would monotonically increase the TOF, while a further decrease of ECO* in magnitude beyond that on Pt has a limited impact on the TOF. In comparison, the increase of the OH* binding affinity would dramatically increase the TOF on the activity map as indicated by the black line, and its slope is much larger than that of the red one. Considering that EOH* is closely related to the energy barrier of the RDS, it is concluded that decreasing the energy barrier of the RDS, i.e. the decomposition of H2O, is the key to improving the performance of Pt-based catalysts by tuning their electronic structure. Any trade-off between the energy barrier of the RDS and the CO adsorption energy, such as what occurs on the PtCuPt alloy, is unfavorable for the LT-WGS reaction. In fact, we found that the impact of the CO binding affinity can be roughly interpreted by its energetic effect on the barrier of the RDS, as shown in Fig. 2c. On one hand, the yellow line shows that the energy barrier of the RDS is linearly dependent on the CO adsorption energy; on the other hand, a stronger CO adsorption results in a higher CO coverage and thus higher repulsive interactions, which changes the energy barrier of the RDS, as represented by the dark cyan line. The overall effect of these two contributions on the energy barrier of the RDS was shown by the red line, in which the net effect of linear scaling relation with respect to the barrier on the Pt(111) surface was accumulated to the barrier with repulsive interaction. It can be seen that the trends of these two contributions are in the opposite directions, leading to a minimum of the total impact on the energy barrier of the RDS at about −1.3 eV. This minimum is close to the location of the top for the volcano-shaped surface, −1.30 eV, at which it can be seen that the landscape of the energy barrier curve is in good agreement with the logarithmic TOF shown in Fig. 2b. A detailed analysis showed that the influence of the spatial correlation of empty sites for H2O dissociation is responsible for the deviation of the activity from the energy barrier curve in the high CO adsorption energy region. Other details in constructing the activity map are summarized in Section S4.1 in the ESI.† Nevertheless, for the purpose of improving the activity of Pt catalysts, we focus on the region that is more active than Pt, i.e. the region with lower ECO* in magnitude than that on Pt (−1.46 eV in BEEF-vdW calculation), and therefore the energy analysis is sufficiently effective in the present discussion.
These observations are distinctly different from that obtained in the MF-MKM without considering the adsorbate–adsorbate interactions. To illustrate the difference between MF-MKM solutions and KMC results, the 2D activity map given by MF-MKM based on the same thermodynamic parameters is shown in Fig. 2d. It is clear that decreasing CO binding affinity does increase the activity drastically, and the PtCuPt alloy is shown to be indeed more active than Pt from Fig. 2d in the MF picture. As shown in Fig. 2e, the slope of the red line is larger than that of the black line, which means that the influence of ECO* on activity is more significant than that of EOH*, such that ECO* dominates the activity on this activity map. As a result, in the case of MF-MKM, it seems reasonable to decrease the OH* binding affinity a bit in exchange for a lower CO* binding affinity in the catalyst design. This improper conclusion from MF-MKM can be understood by comparing the MF-MKM and the KMC results shown in Fig. 2b and e. It is clear that the MF-MKM solution provides a linear dependence of activity on EOH* similar to the KMC simulations, i.e. the logarithmic TOFs in KMC and MF-MKM increase by 10.7 and 11.6, respectively, as EOH* changes from 1.10 to 0.10 eV. In contrast, the MF-MKM significantly overestimates the influence of ECO* on activity, i.e. the MF-MKM predicts that the logarithmic TOF increases by 22.3 as ECO* changes from −1.80 to −0.80 eV, while the corresponding increase from KMC simulations is only 2.43. According to the discussions above and those in Section S4 in the ESI,† it is concluded here that the neglect of the adsorbate–adsorbate interactions, which leads to an inappropriate description of the spatial correlation of the empty sites at a high CO coverage, is responsible for the failure of MF-MKM.
With the messages delivered by KMC simulations, we are now able to understand the observations in experiments and to screen more active Pt alloys. It is easy to find that the higher activity of the core–shell structured PtRe alloy than Pt should be primarily attributed to its higher OH binding affinity, while its lower CO binding affinity has only a minor contribution, whereas the PtCuPt alloy is less active than Pt due to its lower OH binding affinity, in spite of its lower CO binding affinity. The latter, to the best of our knowledge, coincides with the fact that a highly active PtCuPt alloy has not been reported for LT-WGS yet after years of effort. The screening of more active alloys, however, is much more difficult. We calculated the CO adsorption energies and the OH binding energies for a number of subsurface and core–shell structured bimetallic alloys, and projected them onto the activity map shown in Fig. 2a. It is found that moving towards the top of activity map located at ECO* = −1.30 eV is highly difficult, if not impossible, by tuning the electronic structure of the surface Pt atoms alone. Most of the subsurface and core–shell structured alloys are located in the rectangular region enclosed by the dashed lines in black, indicating that modifying the electronic structure of surface Pt atoms results in a movement along the diagonal line of the activity map from right bottom to left top. The observation can be understood via d-band center theory. As shown in Fig. 2f, both CO adsorption energy and OH binding energy show a good linear correlation with the position of the d-band center, while they vary in the opposite direction versus the change of the d-band center, which is undesired for moving towards the top of the activity map located at ECO* = −1.30 eV from the Pt(111) surface. As suggested by Xin and co-workers,62,63 the unusual dependence of the OH binding energy on the d-band center is attributed to the repulsion between the adsorbate states and the d-states of the metal such as Pt.
From Fig. 2a, the activity of the PtRePt alloy looks promising, which has the highest OH* binding affinity, and whose activity is 3.95 orders of magnitude higher than that of the Pt metal. To confirm this prediction from scaling relation KMC simulations, we then carried out explicit DFT and KMC simulations. Unfortunately, this validation simulation refuted the prediction. It was found that, as compared to DFT calculations, the linear relationship underestimated the barrier of the RDS by 0.26 eV on PtRePt, while it overestimated the barrier of the RDS by 0.10 eV on Pt. The accumulation in the error of the RDS, 0.36 eV, is large enough to reverse the relative activity of PtRePt and Pt, which was proved in the following KMC simulations by showing that the DFT corrected TOFs of PtRePt and Pt are 4.6 × 10−6 and 1.4 × 10−5 site−1 per s, respectively. Hence, it is important to carry out this prediction–validation procedure in the rational design of catalysts.
3.3 Improving catalytic performance by the synergetic effect
For the purpose of further improving the activity of Pt alloys, we considered the synergetic effect between the surface Pt atoms and the promoter metal M by using the SAA model, denoted as M1@Pt. The additional 2D activity map relative to the Pt metal is shown in Fig. 3a. It is seen that the top activity is located at the right bottom of the map, which coincides with discussion in Section 3.2 that the RDS prefers to occur on surface sites with a lower CO* binding affinity and a higher OH* binding affinity as compared to Pt. As marked with the crosses in Fig. 3a, most transition metal atoms on the surface show stronger binding with both CO* and OH*. The increase of the OH* binding affinity reduces the energy barrier of the RDS, which promotes the reaction from one side of the influencing factors, whereas a higher CO* binding affinity than that on Pt leads to specific CO poisoning on the promoter atom M, which seriously decreases the appearance of the empty site from the other side of the influencing factors. As a result, the SAAs are distributed near the white region, in which the activity is close to that of the Pt host. | ||
| Fig. 3 2D activity map of the promoter atoms in the SAA model relative to the host of (a) the Pt metal, (b) the PtCuPt alloy, (c) the PtRePt alloy, and (d) the PtRuPt alloy. Although the top activity is located at the right bottom of each map, the SAAs are distributed near the white region, in which the activity is close to the respective host. (e) The activities of these four hosts. (f) Linear dependences of the OH* binding energy (in red) and the CO* adsorption energy (in blue) on the d-band center of the promoter atoms. | ||
Similar phenomena can be observed in the calculations of promoters on the subsurface alloys M1@PtM’Pt, as shown in Fig. 3b, c and 3d. Here the promoter atom M replaces one surface Pt atom on the subsurface alloys PtM′Pt. The corresponding subsurface alloys PtCuPt, PtRePt, and PtRuPt, as well as the Pt metal, are projected onto the activity map shown in Fig. 3e. As can be seen from Fig. 3 a–e, the change of subsurface metal M′ simultaneously influences the electronic structures of both the promoter atom M and the Pt host. Thus, the activities of surface promoter M atoms relative to the respective host are similar on different subsurface alloys. Again, the distribution of the activity of different promoters can be explained by d-band center theory. As shown in Fig. 3f, the CO adsorption energies on different promoters follow a unique linear relation with respect to the d-band center of the promoter, while different linear correlations between the OH binding energies and the d-band center of the promoter are observed on these four hosts. Both the CO* and OH* binding affinities decrease as the d-band center upshifts toward the Fermi level. Therefore, they vary in the same direction when the d-band center moves, which leads to specific CO poisoning as the side effect of lowering the energy barrier of the RDS, and the promoters are distributed along the diagonal from left bottom to right top.
With the knowledge of electronic and synergetic effects, it is time to screen active Pt alloys. Considering the difficulties in the theoretical modeling and the experimental synthesis of a multi-component alloy, here we focused on bimetallic alloys, including transition metal promoters on Pt, i.e. M1@Pt, and the subsurface alloys with the same promoter metal, i.e., M1@PtMPt. It is interesting to note that the exposure of the Cu atom on the surface would increase CO* and OH* binding affinities and thus enhance the catalytic performance of the host Pt, especially on the PtCuPt subsurface alloy (see Discussion in S5.3 in the ESI† for more details). This enhancement, however, cannot compensate for the loss of activity for the host PtCuPt as compared to the Pt metal, such that the PtCu alloy is excluded. The PtRePt alloy has been considered as the best subsurface alloy before due to its high OH* binding affinity, which is a good starting point of design. However, the Re atom on the surface would be oxidized soon owing to its high oxygen affinity.32 Such an oxidation tendency would complicate the study, and the discussion of metal oxide formation is out of the scope of this work. Finally, we noticed that the surface Ru atom can significantly increase the OH* binding affinity on both Pt and PtRuPt hosts, which indicates that PtRu is a promising catalyst candidate for LT-WGS, if the specific poisoning problem on the Ru surface atoms can be reduced by adjusting its ratio and its environment.
3.4 Rational design of the PtRu alloy catalyst
The final step of the present theoretical catalyst design is to explore the influence of the environment on the activity of the Ru surface atoms, and look for the most active sites that can be reached in experiment. Recalling the analysis in Section 3.3 and S5.4 in the ESI,† our task is to diminish the difference of ECO* between the promoter Ru and the host Pt, while keeping a high OH* binding affinity on Ru to promote the RDS. It is known that a subsurface Ru atom can decrease the CO* binding affinities for both surface Ru and Pt simultaneously. Therefore, a non-uniform subsurface layer mixed with Ru and Pt is necessary to achieve this target. Starting from the Pt metal host, it is expected that the existence of a neighboring subsurface Ru atoms can decrease the CO* binding affinity of the surface Ru, as shown in Fig. 3a, d, and e. To illustrate the influence of the neighboring atom on the ECO* of a surface Ru atom, we calculated ECO* in different surface chemical environments and the results are summarized in Fig. 4a. It can be seen that the introduction of the neighboring Ru in the subsurface significantly decreases the CO* binding affinity, while the effect of the neighboring Ru on the surface is considerably small. Furthermore, a linear correlation was observed between ECO* and the number of neighboring Ru atoms, which could be understood by realizing the linear dependence of ECO* on the d-band center as shown in Fig. 3f. According to the discussion above, the most active site for LT-WGS is the surface Ru single atom located at the hollow site constituted by three subsurface Ru atoms. This Ru tetrahedron site on the Pt host is labeled Ru1–3@Pt, and its location on the activity map is shown in Fig. 4b. As a comparison, the activities of other types of PtRu alloys are also marked in Fig. 4b. It can be seen that using a surface Ru atom as the promoter, the CO* binding affinity significantly decreases as the number of subsurface Ru atoms increases, while the OH* binding affinity only increases slightly. As a result, the locations of these alloys move towards the top on the activity map, and Ru1–3@Pt is expected to be the most active site of the PtRu alloys.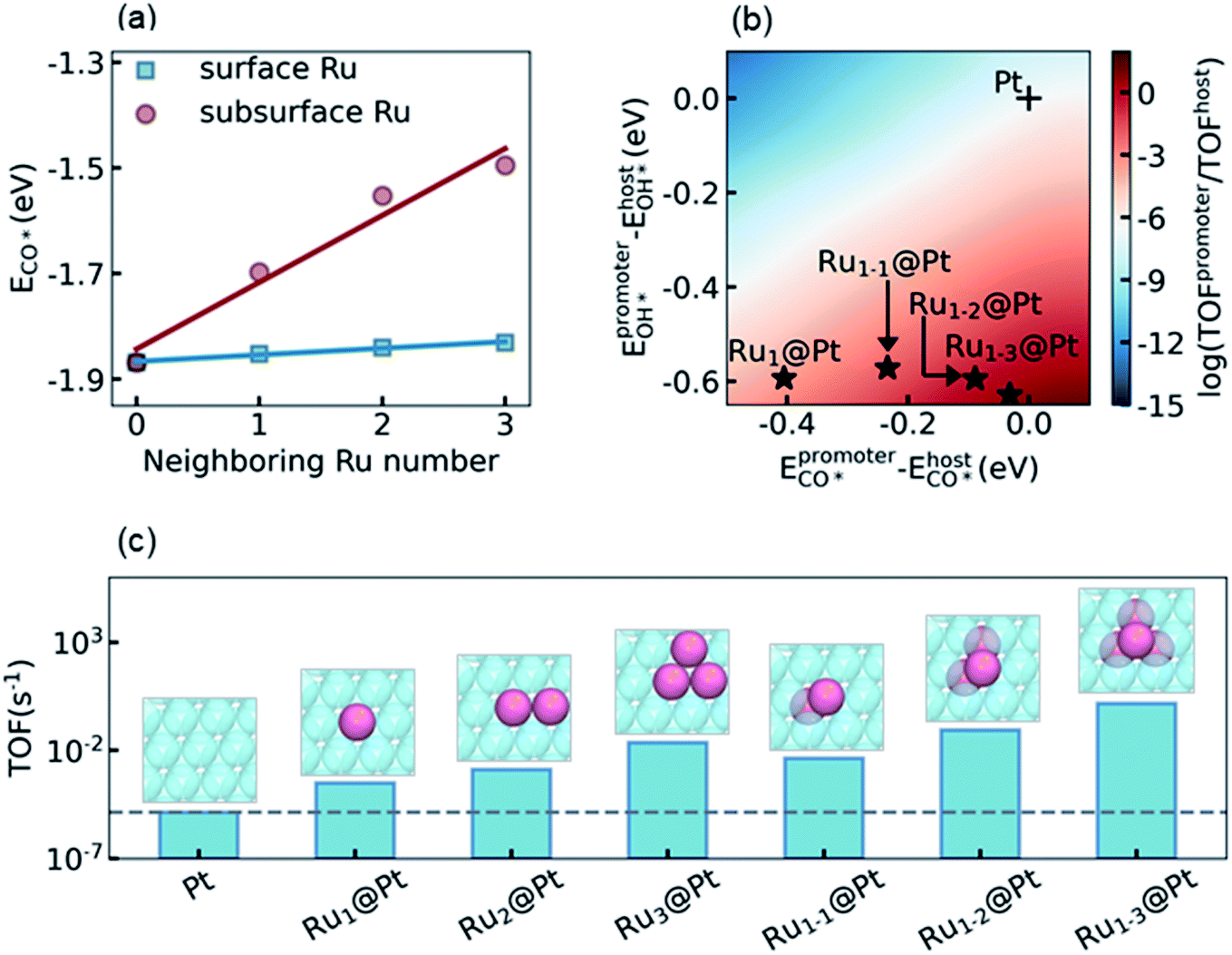 | ||
| Fig. 4 (a) CO* adsorption energy on a surface Ru promoter in different chemical environments. The influence of the neighboring surface Ru atoms is labeled with blue squares, while the influence of the neighboring Ru in the subsurface is marked with red circles. (b) Locations of different types of PtRu alloys on the 2D activity map relative to the Pt metal. (c) Activities of the PtRu alloys calculated by explicit DFT and KMC simulations. The Ru1–3@Pt alloy with a tetrahedron site of one surface-three subsurface Ru atoms on the Pt host is predicted here as the most active Pt alloys, showing a turnover frequency of about five orders of magnitude higher than the Pt metal for LT-WGS. | ||
To confirm this prediction from scaling relation KMC simulations, we now carried out explicit DFT and KMC simulations to evaluate the activities of different types of PtRu alloys. The results are summarized in Fig. 4c. As expected, the activity increases with the increase of the number of neighboring Ru atoms in the subsurface, and Ru1–3@Pt is the most active site, whose activity is about five orders of magnitude higher than that of the Pt metal. It is interesting to note that the activities of the surface PtRu alloys, Ru2@Pt and Ru3@Pt, in which two and three neighboring surface Pt atoms are substituted by Ru atoms, showed higher activity than Ru1@Pt (4.27 and 74.4 fold higher, respectively), though their values of ECO* and EOH* are almost the same. The enhancement of Ru2@Pt and Ru3@Pt is, therefore, attributed to the spatial correlation effect of the neighboring surface Ru atoms, which reduces the poisoning by CO–CO interactions (more detailed description is given in S5.4 in the ESI†). Nevertheless, the direct decrease of the CO* binding affinity by modifying the electronic structure of Pt is more efficient, e.g. the activity of the Ru1–2@Pt alloy is 4-fold higher than that of Ru3@Pt, although both of them contain three Ru atoms in the active site. We also examined the stability and availability of these candidates in experiment. The DFT calculations showed that a Ru atom that appeared in the subsurface is 0.61 eV more stable than that exposed on the surface, which is consistent with the observation reported in the literature.64 Moreover, the Ru–Ru bond is more stable than the Ru–Pt bond, and thus the agglomeration of Ru atoms in the Pt host is energetically favorable (see discussion in Section S5.5 in the ESI†). Considering that the mix of Ru and Pt atoms in the PtRu alloy also increases the entropy contribution, the PtRu alloy with small Ru clusters embedded on the Pt surface is practically feasible in experiment if the concentration of Ru is well controlled.
4 Concluding remarks
In the present work, we have performed the state-of-the-art scaling relation KMC simulations on top of thermodynamic parameters calculated by using the BEEF-vdW functional, to investigate the reaction kinetics of the LT-WGS reaction on Pt-alloy catalysts. A 2D map of activity was constructed to evaluate the electronic effects of the surface Pt atoms on the catalytic performance influenced by the subsurface atoms. It was found that the shape of the activity map is significantly influenced by the adsorbate–adsorbate interactions and the spatial correlations between adsorbates, while microkinetic modeling that does not contain all these messages, such as traditional MF-MKM, would lead to qualitatively incorrect predictions of the reaction kinetics. The 2D activity map showed that decreasing the energy barrier of the RDS is the most effective way to enhance the activity of the surface Pt atoms, while the influence of decreasing the CO adsorption energy, a key conclusion from MF-MKM, is much smaller. As a result, the PtCuPt alloy, which has been suggested as a promising catalyst candidate of LT-WGS due to its low CO adsorption energy,29 is actually much less active than the Pt metal. On the activity map, the PtRePt alloy is predicted to be the most active subsurface alloy owing to the highest OH binding affinity, which indicates the lowest energy barrier of the RDS by linear scaling relations. Our results resolve the long-standing disparity between the theoretical prediction and the experimental observation that whether reducing CO binding affinity alone holds the key to significantly improving the catalytic performance for LT-WGS on Pt-based alloy catalysts.The synergetic effect between the Pt host and the transition metal promoter atoms exposed on the surface has been investigated by using the SAA model. A good promoter atom was found to have a lower energy barrier of the RDS, as well as a lower CO adsorption energy to avoid specific poisoning. Among the studied alloys modeled by using SAAs, the PtRu alloy was suggested to be the best catalyst candidate for LT-WGS, which was confirmed in explicit DFT and KMC simulations. Moreover, we found that the Ru atoms in the PtRu alloy tend to agglomerate into small clusters, which further increases the activity if a small portion of Ru atoms are exposed on the surface. It is worth noting that the synergetic effect between the Ru and the Pt atoms, which can promote the catalytic performance of Pt, has already attracted some attention in recent experiments.65,66 In particular, the promoting effect of Ru on the performance of the Pt catalyst supported on reducible CeO2 has been observed in the WGS reaction,67 while the CO methanation reaction, a side reaction that often occurs on the Ru metal under the WGS reaction conditions, is negligible. Therefore, we predict here that the PtRu alloy would be a promising candidate for the LT-WGS reaction.
Finally, a prediction–validation framework has been employed in the current investigation, showing its applicability in general and is expected to be a powerful tool in the understanding of reaction mechanisms and in rational catalyst design.
Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
Yuqi Yang: investigation, methodology, formal analysis, visualization, and writing. Tonghao Shen: project administration, methodology, software, data curation, validation, and writing. Xin Xu: conceptualization, funding acquisition, resources, writing, and supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We appreciate the support from the National Natural Science Foundation of China (grant 21688102), the Science Challenge Project (grant TZ2018004), and the National Key Research and Development Program of China (grant 2018YFA0208600).References
- D. S. Newsome, The Water-Gas Shift Reaction, Catal. Rev., 2006, 21, 275–318 CrossRef.
- R. M. Navarro, M. A. Pena and J. L. Fierro, Hydrogen production reactions from carbon feedstocks: fossil fuels and biomass, Chem. Rev., 2007, 107, 3952–3991 CrossRef CAS PubMed.
- C. Ratnasamy and J. P. Wagner, Water Gas Shift Catalysis, Catal. Rev., 2009, 51, 325–440 CrossRef CAS.
- Q. Fu, H. Saltsburg and M. Flytzani-Stephanopoulos, Active nonmetallic Au and Pt species on ceria-based water-gas shift catalysts, Science, 2003, 301, 935–938 CrossRef CAS PubMed.
- Y. Zhai, D. Pierre, R. Si, W. Deng, P. Ferrin, A. U. Nilekar, G. Peng, J. A. Herron, D. C. Bell, H. Saltsburg, M. Mavrikakis and M. Flytzani-Stephanopoulos, Alkali-stabilized Pt–OHx species catalyze low-temperature water-gas shift reactions, Science, 2010, 329, 1633–1636 CrossRef CAS PubMed.
- X. Zhang, M. Zhang, Y. Deng, M. Xu, L. Artiglia, W. Wen, R. Gao, B. Chen, S. Yao, X. Zhang, M. Peng, J. Yan, A. Li, Z. Jiang, X. Gao, S. Cao, C. Yang, A. J. Kropf, J. Shi, J. Xie, M. Bi, J. A. van Bokhoven, Y. W. Li, X. Wen, M. Flytzani-Stephanopoulos, C. Shi, W. Zhou and D. Ma, A stable low-temperature H2-production catalyst by crowding Pt on alpha-MoC, Nature, 2021, 589, 396–401 CrossRef CAS.
- B. Zugic, S. Zhang, D. C. Bell, F. F. Tao and M. Flytzani-Stephanopoulos, Probing the low-temperature water-gas shift activity of alkali-promoted platinum catalysts stabilized on carbon supports, J. Am. Chem. Soc., 2014, 136, 3238–3245 CrossRef CAS PubMed.
- I. D. Gonzalez, R. M. Navarro, W. Wen, N. Marinkovic, J. A. Rodriguez, F. Rosa and J. L. G. Fierro, A comparative study of the water gas shift reaction over platinum catalysts supported on CeO2, TiO2 and Ce-modified TiO2, Catal. Today, 2010, 149, 372–379 CrossRef CAS.
- A. Bruix, J. A. Rodriguez, P. J. Ramirez, S. D. Senanayake, J. Evans, J. B. Park, D. Stacchiola, P. Liu, J. Hrbek and F. Illas, A new type of strong metal-support interaction and the production of H2 through the transformation of water on Pt/CeO2(111) and Pt/CeO(x)/TiO2(110) catalysts, J. Am. Chem. Soc., 2012, 134, 8968–8974 CrossRef CAS.
- Z. Li, Y. Cui, Z. Wu, C. Milligan, L. Zhou, G. Mitchell, B. Xu, E. Shi, J. T. Miller, F. H. Ribeiro and Y. Wu, Reactive metal-support interactions at moderate temperature in two-dimensional niobium-carbide-supported platinum catalysts, Nat. Catal., 2018, 1, 349–355 CrossRef CAS.
- N. M. Schweitzer, J. A. Schaidle, O. K. Ezekoye, X. Pan, S. Linic and L. T. Thompson, High activity carbide supported catalysts for water gas shift, J. Am. Chem. Soc., 2011, 133, 2378–2381 CrossRef CAS PubMed.
- K. Ding, A. Gulec, A. M. Johnson, N. M. Schweitzer, G. D. Stucky, L. D. Marks and P. C. Stair, Identification of active sites in CO oxidation and water-gas shift over supported Pt catalysts, Science, 2015, 350, 189–192 CrossRef CAS PubMed.
- R. T. Hannagan, G. Giannakakis, R. Réocreux, J. Schumann, J. Finzel, Y. Wang, A. Michaelides, P. Deshlahra, P. Christopher, M. Flytzani-Stephanopoulos, M. Stamatakis and E. C. H. Sykes, First-principles design of a single-atom-alloy propane dehydrogenation catalyst, Science, 2021, 372, 1444–1447 CrossRef CAS.
- J. Greeley and M. Mavrikakis, Alloy catalysts designed from first principles, Nat. Mater., 2004, 3, 810–815 CrossRef CAS.
- J. K. Nørskov, T. Bligaard, J. Rossmeisl and C. H. Christensen, Towards the computational design of solid catalysts, Nat. Chem., 2009, 1, 37–46 CrossRef.
- R. T. Hannagan, G. Giannakakis, M. Flytzani-Stephanopoulos and E. C. H. Sykes, Single-Atom Alloy Catalysis, Chem. Rev., 2020, 120, 12044–12088 CrossRef CAS PubMed.
- Z. J. Zhao, S. H. Liu, S. J. Zha, D. F. Cheng, F. Studt, G. Henkelman and J. L. Gong, Theory-guided design of catalytic materials using scaling relationships and reactivity descriptors, Nat. Rev. Mater., 2019, 4, 792–804 CrossRef.
- H. Falsig, B. Hvolbaek, I. S. Kristensen, T. Jiang, T. Bligaard, C. H. Christensen and J. K. Nørskov, Trends in the catalytic CO oxidation activity of nanoparticles, Angew. Chem., Int. Ed., 2008, 47, 4835–4839 CrossRef CAS PubMed.
- C. Ding, T. H. Shen, Y. Q. Yang and X. Xu, Involvement of the Unoccupied Site Changes the Kinetic Trend Significantly: A Case Study on Formic Acid Decomposition, ACS Catal., 2020, 10, 5153–5162 CrossRef CAS.
- J. S. Yoo, F. Abild-Pedersen, J. K. Nørskov and F. Studt, Theoretical Analysis of Transition-Metal Catalysts for Formic Acid Decomposition, ACS Catal., 2014, 4, 1226–1233 CrossRef CAS.
- J. K. Nørskov, T. Bligaard, A. Logadottir, S. Bahn, L. B. Hansen, M. Bollinger, H. Bengaard, B. Hammer, Z. Sljivancanin, M. Mavrikakis, Y. Xu, S. Dahl and C. J. H. Jacobsen, Universality in Heterogeneous Catalysis, J. Catal., 2002, 209, 275–278 CrossRef.
- A. Michaelides, Z.-P. Liu, C. J. Zhang, A. Alavi, D. A. King and P. Hu, Identification of general linear relationships between activation energies and enthalpy changes for dissociation reactions at surfaces, J. Am. Chem. Soc., 2002, 125, 3704–3705 CrossRef PubMed.
- J. R. Kitchin, J. K. Nørskov, M. A. Barteau and J. G. Chen, Modification of the surface electronic and chemical properties of Pt(111) by subsurface 3d transition metals, J. Chem. Phys., 2004, 120, 10240 CrossRef CAS PubMed.
- W. Yu, M. D. Porosoff and J. G. Chen, Review of Pt-based bimetallic catalysis: from model surfaces to supported catalysts, Chem. Rev., 2012, 112, 5780–5817 CrossRef CAS PubMed.
- A. A. Gokhale, J. A. Dumesic and M. Mavrikakis, On the mechanism of low-temperature water gas shift reaction on copper, J. Am. Chem. Soc., 2007, 130, 1402–1414 CrossRef PubMed.
- L. C. Grabow, A. A. Gokhale, S. T. Evans, J. A. Dumesic and M. Mavrikakis, Mechanism of the Water Gas Shift Reaction on Pt First Principles Experiments and Microkinetic Modeling, J. Phys. Chem. C, 2008, 112, 4608–4617 CrossRef CAS.
- N. C. Nelson, M. T. Nguyen, V. A. Glezakou, R. Rousseau and J. Szanyi, Carboxyl intermediate formation via an in situ-generated metastable active site during water-gas shift catalysis, Nat. Catal., 2019, 2, 916–924 CrossRef CAS.
- T. Shen, Y. Yang and X. Xu, Structure-Reactivity Relationship for Nano-Catalysts in the Hydrogenation/Dehydrogenation Controlled Reaction Systems, Angew. Chem., Int. Ed., 2021, 60, 26342–26345 CrossRef CAS PubMed.
- J. Knudsen, A. U. Nilekar, R. T. Vang, J. Schnadt, E. L. Kunkes, J. A. Dumesic, M. Mavrikakis and F. Besenbacher, A Cu/Pt near-surface alloy for water-gas shift catalysis, J. Am. Chem. Soc., 2007, 129, 6485–6490 CrossRef CAS PubMed.
- W. D. Williams, J. P. Greeley, W. N. Delgass and F. H. Ribeiro, Water activation and carbon monoxide coverage effects on maximum rates for low temperature water-gas shift catalysis, J. Catal., 2017, 347, 197–204 CrossRef CAS.
- A. S. Duke, K. Xie, A. J. Brandt, T. D. Maddumapatabandi, S. C. Ammal, A. Heyden, J. R. Monnier and D. A. Chen, Understanding Active Sites in the Water-Gas Shift Reaction for Pt–Re Catalysts on Titania, ACS Catal., 2017, 7, 2597–2606 CrossRef CAS.
- R. Carrasquillo-Flores, J. M. R. Gallo, K. Hahn, J. A. Dumesic and M. Mavrikakis, Density Functional Theory and Reaction Kinetics Studies of the Water-Gas Shift Reaction on Pt–Re Catalysts, ChemCatChem, 2013, 5, 3690–3699 CrossRef CAS.
- C.-H. Lin, C.-L. Chen and J.-H. Wang, Mechanistic Studies of Water-Gas-Shift Reaction on Transition Metals, J. Phys. Chem. C, 2011, 115, 18582–18588 CrossRef CAS.
- S. C. Ammal and A. Heyden, Water-Gas Shift Catalysis at Corner Atoms of Pt Clusters in Contact with a TiO2(110) Support Surface, ACS Catal., 2014, 4, 3654–3662 CrossRef CAS.
- J. P. Clay, J. P. Greeley, F. H. Ribeiro, W. N. Delgass and W. F. Schneider, DFT comparison of intrinsic WGS kinetics over Pd and Pt, J. Catal., 2014, 320, 106–117 CrossRef CAS.
- S. C. Ammal and A. Heyden, Water-Gas Shift Activity of Atomically Dispersed Cationic Platinum versus Metallic Platinum Clusters on Titania Supports, ACS Catal., 2016, 7, 301–309 CrossRef.
- B. W. J. Chen, L. Xu and M. Mavrikakis, Computational Methods in Heterogeneous Catalysis, Chem. Rev., 2021, 121, 1007–1048 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 6671–6687 CrossRef CAS PubMed.
- J. A. White and D. M. Bird, Implementation of gradient-corrected exchange-correlation potentials in Car-Parrinello total-energy calculations, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 4954–4957 CrossRef CAS PubMed.
- L. Schimka, J. Harl, A. Stroppa, A. Gruneis, M. Marsman, F. Mittendorfer and G. Kresse, Accurate surface and adsorption energies from many-body perturbation theory, Nat. Mater., 2010, 9, 741–744 CrossRef CAS PubMed.
- S. Gautier, S. N. Steinmann, C. Michel, P. Fleurat-Lessard and P. Sautet, Molecular adsorption at Pt(111). How accurate are DFT functionals?, Phys. Chem. Chem. Phys., 2015, 17, 28921–28930 RSC.
- G. T. K. K. Gunasooriya and M. Saeys, CO Adsorption Site Preference on Platinum: Charge Is the Essence, ACS Catal., 2018, 8, 3770–3774 CrossRef.
- G. T. K. K. Gunasooriya and M. Saeys, CO Adsorption on Pt(111): From Isolated Molecules to Ordered High-Coverage Structures, ACS Catal., 2018, 8, 10225–10233 CrossRef CAS.
- H. Wang, T. H. Shen, S. Duan, Z. Chen and X. Xu, Bistability for CO Oxidation: An Understanding from Extended Phenomenological Kinetics Simulations, ACS Catal., 2019, 9, 11116–11124 CrossRef CAS.
- A. H. Motagamwala and J. A. Dumesic, Microkinetic Modeling: A Tool for Rational Catalyst Design, Chem. Rev., 2021, 121, 1049–1076 CrossRef CAS.
- M. Jorgensen and H. Gronbeck, Scaling Relations and Kinetic Monte Carlo Simulations To Bridge the Materials Gap in Heterogeneous Catalysis, ACS Catal., 2017, 7, 5054–5061 CrossRef.
- J. Wellendorff, K. T. Lundgaard, A. Mogelhoj, V. Petzold, D. D. Landis, J. K. Nørskov, T. Bligaard and K. W. Jacobsen, Density functionals for surface science: Exchange-correlation model development with Bayesian error estimation, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 235149 CrossRef.
- Z. Chen, H. Wang, N. Q. Su, S. Duan, T. Shen and X. Xu, Beyond Mean-Field Microkinetics: Toward Accurate and Efficient Theoretical Modeling in Heterogeneous Catalysis, ACS Catal., 2018, 8, 5816–5826 CrossRef CAS.
- X. Xu and W. A. Goddard, The extended Perdew-Burke-Ernzerhof functional with improved accuracy for thermodynamic and electronic properties of molecular systems, J. Chem. Phys., 2004, 121, 4068–4082 CrossRef CAS.
- K. Lee, E. D. Murray, L. Z. Kong, B. I. Lundqvist and D. C. Langreth, Higher-accuracy van der Waals density functional, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 081101 CrossRef.
- C. T. Campbell and J. R. Sellers, The entropies of adsorbed molecules, J. Am. Chem. Soc., 2012, 134, 18109–18115 CrossRef CAS PubMed.
- G. Kresse and J. Furthmuller, Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmuller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- D. W. Flaherty, W.-Y. Yu, Z. D. Pozun, G. Henkelman and C. B. Mullins, Mechanism for the water–gas shift reaction on monofunctional platinum and cause of catalyst deactivation, J. Catal., 2011, 282, 278–288 CrossRef CAS.
- N. Schumacher, A. Boisen, S. Dahl, A. Gokhale, S. Kandoi, L. Grabow, J. Dumesic, M. Mavrikakis and I. Chorkendorff, Trends in low-temperature water gas shift reactivity on transition metals, J. Catal., 2005, 229, 265–275 CrossRef CAS.
- T. H. Shen and X. Xu, The XPK Package: A Comparison between the Extended Phenomenological Kinetic (XPK) Method and the Conventional Kinetic Monte Carlo (KMC) Method, Chinese J. Chem. Phys., 2019, 32, 143–150 CrossRef CAS.
- G. Ertl, M. Neumann and K. M. Streit, Chemisorption of Co on Pt(111) Surface, Surf. Sci., 1977, 64, 393–410 CrossRef CAS.
- Z. Chen, H. Wang, Z. Y. Liu and X. Xu, Dynamic and Intermediate-Specific Local Coverage Controls the Syngas Conversion on Rh(111) Surfaces: An Operando Theoretical Analysis, ACS Catal., 2021, 11, 3830–3841 CrossRef CAS.
- F. Hess, A. Farkas, A. P. Seitsonen and H. Over, ‘‘First-Principles’’ Kinetic Monte Carlo Simulations Revisited: CO Oxidation over RuO2(110), J. Comput. Chem., 2012, 33, 757–766 CrossRef CAS PubMed.
- H. Wang, T. Shen and X. Xu, A Good Prediction of the Overall Reaction Rate May Not Mean a Correct Description of the Reaction Kinetics: A Case Study for CO Oxidation on RuO2(110) Surfaces, J. Phys. Chem. C, 2021, 125, 9169–9177 CrossRef CAS.
- H. L. Xin, A. Vojvodic, J. Voss, J. K. Nørskov and F. Abild-Pedersen, Effects of d-band shape on the surface reactivity of transition-metal alloys, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 115114 CrossRef.
- H. L. Xin and S. Linic, Communications: Exceptions to the d-band model of chemisorption on metal surfaces: The dominant role of repulsion between adsorbate states and metal d-states, J. Chem. Phys., 2010, 132, 221101 CrossRef PubMed.
- K. K. Rao, Q. K. Do, K. Pham, D. Maiti and L. C. Grabow, Extendable Machine Learning Model for the Stability of Single Atom Alloys, Top. Catal., 2020, 63, 728–741 CrossRef CAS.
- T. Takeguchi, T. Yamanaka, K. Asakura, E. N. Muhamad, K. Uosaki and W. Ueda, Evidence of nonelectrochemical shift reaction on a CO-tolerant high-entropy state Pt–Ru anode catalyst for reliable and efficient residential fuel cell systems, J. Am. Chem. Soc., 2012, 134, 14508–14512 CrossRef CAS PubMed.
- P. Zhou, X. G. Hou, Y. G. Chao, W. X. Yang, W. Y. Zhang, Z. J. Mu, J. P. Lai, F. Lv, K. Yang, Y. X. Liu, J. Li, J. Y. Ma, J. Luo and S. J. Guo, Synergetic interaction between neighboring platinum and ruthenium monomers boosts CO oxidation, Chem. Sci., 2019, 10, 5898–5905 RSC.
- W. Xu, R. Si, S. D. Senanayake, J. Llorca, H. Idriss, D. Stacchiola, J. C. Hanson and J. A. Rodriguez, In situ studies of CeO2-supported Pt, Ru, and Pt–Ru alloy catalysts for the water-gas shift reaction: Active phases and reaction intermediates, J. Catal., 2012, 291, 117–126 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of energetic calculations; details of kinetic simulations; thermodynamics of the WGS reaction; details of the construction of activity maps; details of catalyst screening. See https://doi.org/10.1039/d2sc01729f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |