
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA photoresponsive antibody–siRNA conjugate for activatable immunogene therapy of cancer†
Xingxing
Wang‡
,
Xiao
Xiao‡
,
Yi
Feng
,
Jinbo
Li
 * and
Yan
Zhang
*
* and
Yan
Zhang
*
State Key Laboratory of Analytical Chemistry for Life Sciences, Jiangsu Key Laboratory of Advanced Organic Materials, School of Chemistry and Chemical Engineering, Chemistry and Biomedicine Innovation Center (ChemBIC), Nanjing University, Nanjing 210023, China. E-mail: jinboli@nju.edu.cn; njuzy@nju.edu.cn
First published on 21st April 2022
Abstract
Tumor-targeted delivery of small-interfering RNAs (siRNAs) for cancer therapy still remains a challenging task. While antibody–siRNA conjugates (ARCs) provide an alternative way to address this challenge, the uncontrollable siRNA release potentially leads to undesirable off-tumor side effects, limiting their in vivo therapeutic efficacy. Here, we report a photoresponsive ARC (PARC) for tumor-specific and photoinducible siRNA delivery as well as photoactivable immunogene therapy. PARC is composed of an anti-programmed death-ligand 1 antibody (αPD-L1) conjugated with a siRNA against intracellular PD-L1 mRNA through a photocleavable linker. After targeting cancer cells through the interaction between αPD-L1 and membrane PD-L1, PARC is internalized and it liberates siPD-L1 upon light irradiation to break the photocleavable linker. The released siPD-L1 then escapes from the lysosome into the cytoplasm to degrade intracellular PD-L1 mRNA, which combines the blockade of membrane PD-L1 by αPD-L1 to boost immune cell activity. Owing to these features, PARC causes effective cancer suppression both in vitro and in vivo. This study thus provides a useful conditional delivery platform for siRNAs and a novel means for activatable cancer immunogene therapy.
Introduction
Small-interfering RNAs (siRNAs) are synthetic double-stranded RNAs capable of silencing gene expression via the RNA-interference (RNAi) pathway.1 By virtue of their potential for targeting any known genes, the use of siRNAs to knock down undruggable oncogenes represents a promising anti-cancer approach.2 When designed to target immune-related genes, siRNAs can also shape a tumor immune microenvironment.3 This immunogene therapy strategy has proved useful in orchestrating both the innate and adaptive immune systems to fight against cancers.4–6 However, due to their poor cell membrane permeability and undesired toxicity, safe and efficient delivery of siRNAs into target cells is a major barrier to advance their clinical applications.7 Currently, ligand conjugation8 and nanoparticle encapsulation9 are two prevailing ways to address siRNA delivery challenges. Although N-acetylgalactosamine conjugates and lipid nanoparticles have been approved for RNAi-based treatment of liver disorders,10 extrahepatic (e.g., tumor) delivery of siRNAs is still an unmet demand. Furthermore, to avoid side effects resulting from nonspecific siRNA accumulation, RNAi activity needs to be confined to tumor sites.3,11Antibody–siRNA conjugates (ARCs) are a novel class of bivalent macromolecules consisting of monoclonal antibodies as carriers and siRNAs as payloads, which can be noncovalently or covalently assembled.12–14 Based on the specific interaction between antibodies and cell-membrane receptors, ARCs efficiently deliver siRNAs into target cells upon internalization.15 Using antibodies against tumor-associated membrane antigens, various ARCs have been constructed to direct tumor-specific delivery of siRNAs to downregulate oncogenes.16–25 Notably, synergistic anti-cancer treatment is feasible using all-in-one ARCs combining therapeutic antibodies and siRNAs together.26–28 Despite these promising accomplishments, ARCs have been rarely developed to manipulate immunotargets for anti-cancer immunotherapy. Moreover, no smart activatable ARC has been developed to minimize on-target/off-tumor side effects. This is crucial because ARCs could also be unintendedly taken up by non-target cells29,30 due to the nonexclusive antigen expression, and siRNA release from ARCs generally lacks controllability.
Herein, we report a novel photoresponsive antibody–siRNA conjugate (PARC) for remote-controllable siRNA delivery and cancer immunogene therapy (Scheme 1). As a non-invasive external stimulus, light is practical for spatiotemporal regulation of biological processes31 and is thereby chosen as the trigger for photoactivation. PARC comprises an anti-programmed death-ligand 1 antibody (αPD-L1) conjugated with a PD-L1-targeted siRNA (siPD-L1) through a photocleavable linker. PD-L1 is a membrane protein overexpressed on cancer cells to hamper immune cell function.32 αPD-L1 that blocks membrane PD-L1 to reinvigorate immune cell function has been approved for clinical treatment of some solid cancers.33 Of note, αPD-L1 can get internalized into cancer cells upon interaction with membrane PD-L1,34 which has been exploited for targeted delivery of cytotoxic chemo-drugs.35,36 On the other hand, PD-L1 is persistently produced inside cancer cells for replenishment of membrane PD-L137,38 and secretion as circulating PD-L1 to exert pro-tumor effects.39,40 Therefore, in addition to membrane PD-L1, intracellular PD-L1 is also a pivotal target, which however is unreachable by αPD-L1. We thus integrated siPD-L1 with αPD-L1 in the PARC for not only targeted drug delivery but also synergistic inhibition of PD-L1 to amplify immunotherapeutic efficacy.
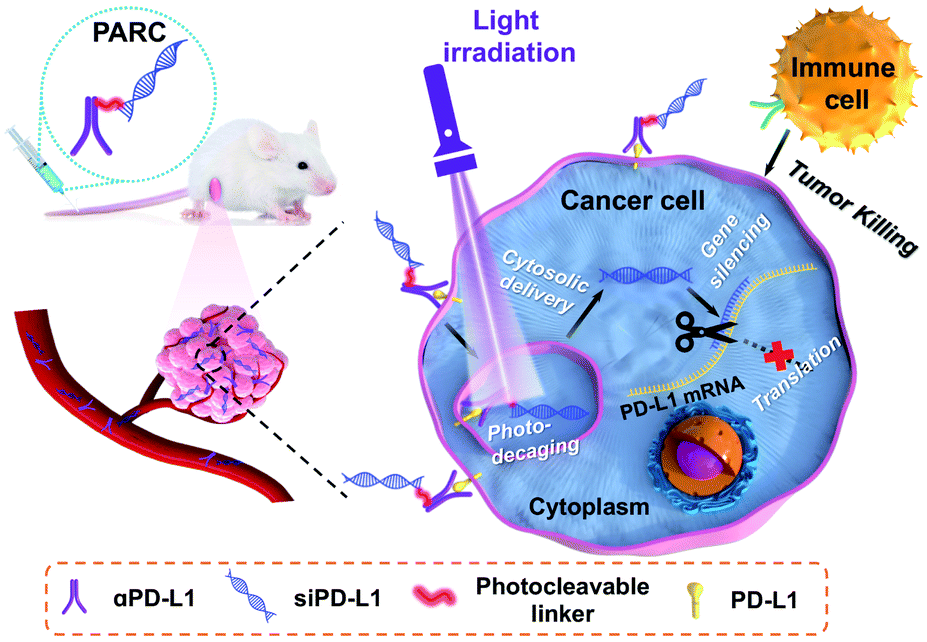 | ||
| Scheme 1 Schematic illustration of using a photoresponsive antibody–siRNA conjugate (PARC) for tumor-specific delivery of siPD-L1. Photoirradiation on tumors liberates siPD-L1 from PARC to silence intracellular PD-L1 expression in the cytoplasm, which combines with the αPD-L1-mediated blockade of membrane PD-L1 to boost anti-cancer immunotherapy. | ||
The mechanism of PARC-mediated activatable cancer immunogene therapy is proposed as follows. After systemic administration, PARC can accumulate in tumor tissues and specifically bind to PD-L1 on cancer cells, giving rise to the resurgence of immune cell activity. Following endocytosis, photoirradiation on tumors to break the cleavable linker in PARC leads to a tumor-specific release of siPD-L1. The free siPD-L1 then translocates into the cytoplasm to degrade PD-L1 mRNA, which prevents the continuous production of intracellular PD-L1 to further augment anti-cancer immunity. As a result, under light irradiation, PARC affords a synergetic blockade of PD-L1 to invoke efficacious cancer suppression.
Results and discussion
We prepared PARC by connecting siPD-L1 to αPD-L1 through a photocleavable o-nitrophenyl linker (Fig. 1a). While covalent ARCs have been reported before, conditional release and activation of siRNA have been barely achieved. This is due to the preferred use of a siRNA passenger strand for bioconjugation, leaving the guide strand free to execute gene silencing function.29,41,42 In this study, we selected the guide strand of siRNA for covalent attachment, which would allow restoration of the gene silencing function only after siRNA release. Briefly, we synthesized the siPD-L1 guide strand with an o-nitrophenyl moiety at its 3′-end using a standard solid-phase synthesis protocol,43 followed by conversion of the terminal amine group to a maleimide group (Fig. S1 and S2†). Commercial αPD-L1 was functionalized with Traut's reagent to furnish a free sulfhydryl group (Fig. S1†). After annealing with the passenger strand, siPD-L1 was coupled to αPD-L1 via the thiol–maleimide reaction, finally providing PARC under optimal conditions (Fig. S3†).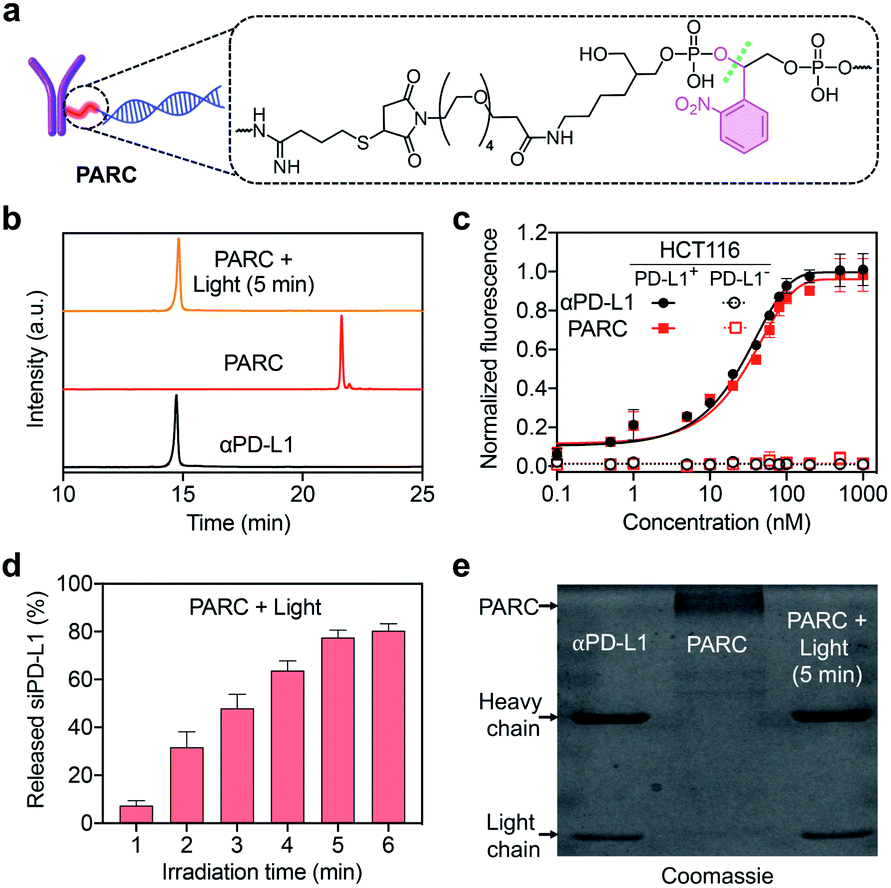 | ||
| Fig. 1 (a) Chemical structure of the photocleavable linker in the PARC. (b) IEC analysis of αPD-L1, PARC, or PARC irradiated with 365 nm light (10 mW cm−2) for 5 min. (c) Normalized fluorescence of PD-L1-positive or -negative HCT116 cells treated with αPD-L1 or PARC (0–1000 nM) and a fluorescent secondary antibody. (d) siPD-L1 release profiles of PARC irradiated with 365 nm light (10 mW cm−2) for 1–6 min. (e) SDS-PAGE analysis of αPD-L1 and PARC without or with irradiation with 365 nm light (10 mW cm−2) for 5 min. Data are shown as mean ± SD (n = 3). | ||
After ultrafiltration to remove free siPD-L1, PARC was obtained and characterized. Ion exchange chromatography (IEC) analysis revealed a significantly prolonged retention of PARC relative to αPD-L1 (Fig. 1b), indicating the successful addition of siPD-L1 to αPD-L1. We determined the average siPD-L1/αPD-L1 ratio in PARC to be 2.2 using a previously reported method (Fig. S4†).44 Noticeably, PARC and αPD-L1 displayed comparable binding profiles towards human colon cancer HCT116 cells that express a high level of membrane PD-L1,45 with half maximal effective concentration (EC50) values of ∼22 nM and ∼19 nM, respectively (Fig. 1c). By contrast, such binding was undetectable when siPD-L1 was pre-transfected into HCT116 cells to knock down PD-L1 (Fig. 1c and S5†). These results suggest a negligible impact of siPD-L1 bioconjugation on the PD-L1-binding activity of αPD-L1 in PARC. We then investigated the photo-responsiveness of PARC. Upon exposure to 365 nm light, PARC gradually unleashed siPD-L1, reaching a plateau (∼78%) at 5 min (Fig. 1d). Meanwhile, sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) analysis also revealed a gradual liberation of αPD-L1 from PARC under photoirradiation (Fig. S6†). A complete release of αPD-L1 was observed after irradiation for 5 min (Fig. 1e), which was also confirmed by the IEC analysis (Fig. 1b). These findings could be ascribed to photocleavage of the o-nitrophenyl moiety in PARC, as judged by mass spectrometry analysis of the released siPD-L1 (Fig. S7†) and long-term stability of PARC in the dark (Fig. S8†).
We next explored the targeted delivery of siPD-L1 by PARCin vitro. To this end, we prepared a fluorescently labeled PARC with siPD-L1 and αPD-L1 tagged by Cy3 and Cy5, respectively (Fig. 2a and S1†). Cellular uptake of Cy5/Cy3-labeled PARC was monitored using a confocal fluorescence microscope. As shown in Fig. S9†, Cy5 and Cy3 fluorescence signals were detected both on the membrane and inside of HCT116 cells, which reached a maximum at 1 h post incubation. Notably, the obvious overlap of Cy5 and Cy3 signals indicates that PARC does not disassemble during internalization, warranting conditional release of siPD-L1. In comparison, we found no significant uptake of Cy5/Cy3-labeled PARC in PD-L1-negative HCT116 cells even after 24 h incubation (Fig. S10†). These results suggest a specific and effective cellular uptake of PARC mediated by the αPD-L1-PD-L1 interaction. We then irradiated Cy5/Cy3-labeled PARC-treated HCT116 cells with 365 nm light for 5 min at 1 h after incubation. Cy5 and Cy3 fluorescence signals were found to gradually separate (Fig. S11†). At 12 h post incubation, Cy3 signals were uniformly distributed throughout the cytoplasm, whereas Cy5 signals were mainly located on the cell membrane (Fig. 2b). Meanwhile, when co-staining the lysosome, we found that the colocalization ratio between LysoTracker and Cy3 was only ∼18% (Fig. 2c). In contrast, under dark conditions, Cy5 and Cy3 signals remained overlapped, and the colocalization ratio between LysoTracker and Cy3 was ∼90% (Fig. 2b and c). These data thus demonstrate that, after cellular uptake and light irradiation, PARC could effectively release siPD-L1, which then successfully translocates from the lysosome into the cytoplasm. Additionally, since lysosomal trapping is a great limiting factor suffered by ARC-mediated siRNA delivery,42 our results also suggest that timely release of siRNAs may facilitate their lysosomal escape. In short, PARC allows tumor-targeted and photocontrollable cytosolic siRNA delivery, warranting further gene silencing applications.
 | ||
| Fig. 2 (a) Schematic illustration of a Cy5/Cy3-labeled PARC structure. (b) Confocal fluorescence images of HCT116 cells treated with Cy5/Cy3-labeled PARC (150 nM) for 12 h. Cells were irradiated with 365 nm light (10 mW cm−2) for 5 min at 1 h. The nucleus and lysosome were stained with DAPI and LysoTracker, respectively. (c) Colocalization ratios of fluorescence signals between Cy3 and LysoTracker. Data are shown as mean ± SD. | ||
Next, we assessed the biological activity of PARCin vitro. HCT116 cells were treated with PARC and irradiated with 365 nm light, followed by analysis of PD-L1 expression. As shown in Fig. S12†, PARC decreased PD-L1 expression in both photoirradiation time- and dose-dependent manners. At an irradiation time of 5 min and concentration of 150 nM, PD-L1 mRNA and protein levels dwindled to ∼28% (Fig. 3a) and 0 (Fig. 3b) by PARC, respectively. In contrast, PD-L1 expression was unaffected by αPD-L1 or PARC without light irradiation, suggesting that the downregulation of PD-L1 expression results from the photoinduced release of siPD-L1 from PARC. To further confirm this, a control siRNA (si-ctrl) with a random sequence was used to replace siPD-L1 in PARC, giving a control ARC termed CARC (Fig. S1 and S2†). Comparatively, CARC could not regulate PD-L1 expression even under photoirradiation (Fig. 3a and b), validating the specificity of PD-L1 knockdown by PARC. These results thus demonstrate that PARC allows photoactivatable gene silencing.
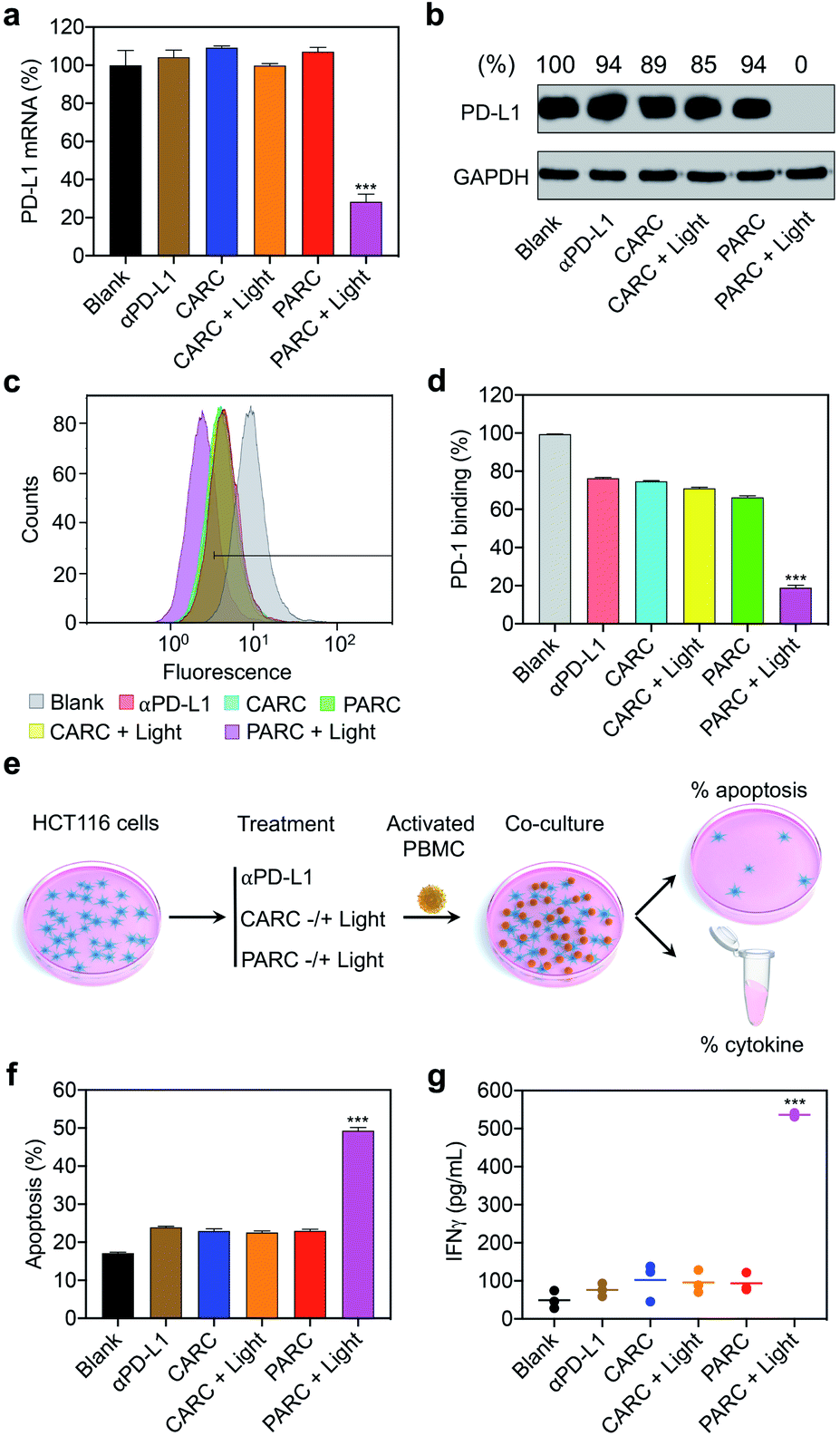 | ||
Fig. 3 (a) Reverse-transcription quantitative PCR (RT-qPCR) analysis of PD-L1 mRNA and (b) western blotting analysis of PD-L1 protein levels in HCT116 cells. GAPDH served as the internal control. Relative intensity ratios of PD-L1/GAPDH are shown at the top. (c and d) Flow cytometry analysis of immunostained PD-1 on HCT116 cells. (e) Schematic illustration of the experimental procedures for immune cell killing assay; PBMC, peripheral blood mononuclear cell. (f) Apoptosis rate of HCT116 cells determined by lactate dehydrogenase release assay. (g) IFNγ levels in the culture media. HCT116 cells were treated with αPD-L1 (150 nM), CARC (150 nM), or PARC (150 nM) for 48 h. Cells were irradiated with 365 nm light (10 mW cm−2) for 5 min at 1 h. For co-culture assay, PBMCs were added to HCT116 cells at an effector-to-target ratio of 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and incubated for another 24 h. Data are shown as mean ± SD (n = 3). ***P < 0.001, relative to other groups. :1 and incubated for another 24 h. Data are shown as mean ± SD (n = 3). ***P < 0.001, relative to other groups. | ||
It has been established that PD-L1 on cancer cells mediates immune cell tolerance through the interaction with PD-1, and its suppression can activate anti-cancer immunity.32 To test whether PARC-mediated PD-L1 suppression may impair PD-1 binding, a PD-1 protein was used to stain HCT116 cells treated with PARC. Flow cytometry analysis showed that PARC alone reduced binding of PD-1 to a cancer cell membrane by ∼30% (Fig. 3c and d), similar to αPD-L1 and CARC. This result indicates that PARC and CARC retain the PD-L1-blockade activity of αPD-L1. In comparison, under light irradiation, PARC but not CARC significantly inhibited PD-1 binding by ∼81%, revealing that silencing intracellular PD-L1 could dramatically prevent PD-1 binding. Consistently, PD-L1 suppression increased the sensitivity of HCT116 cells to immune cell killing,46 as determined by co-culture with activated human peripheral blood mononuclear cells (PBMCs, Fig. 3e). PBMCs are mixed immune cells with abundant lymphocytes such as T cells and nature killer (NK) cells that are able to mediate anti-cancer immunity upon PD-1/PD-L1 blockade.32,47 Specifically, αPD-L1, CARC, and PARC slightly upregulated HCT116 cell apoptosis (∼24%, Fig. 3f), which could be ascribed to the αPD-L1-mediated blockade of membrane PD-L1. However, upon light irradiation, PARC remarkably increased the apoptotic rate to ∼50%, which coincides well with its inhibition of PD-L1 expression and PD-1 binding (Fig. 3a–d). Of note, treatment with a cocktail mixture of αPD-L1 and siPD-L1 complexed with a commercial transfection agent only elicited ∼40% apoptosis (Fig. S13†). These results suggest a synergistic inhibition of PD-L1 by PARC with photoirradiation that favors anti-cancer immunity. Furthermore, we measured immunocytokines in the culture media (Fig. 3e). As shown in Fig. 3g and S14†, under photoirradiation, PARC increased interferon γ (IFNγ) and tumor necrosis factor α (TNFα) levels by ∼10.8 and ∼7.9 fold, respectively, implying expected immune cell activation for tumor killing. Overall, these data jointly validate that PARC permits photoactivatable immunogene therapy.
Encouraged by the in vitro results, we further evaluated PARCin vivo. To find an optimal time point for light irradiation, we first performed fluorescence imaging in mice bearing subcutaneous HCT116 tumors. Cy5/Cy3-labeled PARC was used for tracing, revealing a maximum tumor accumulation at 4 h post tail vein injection (Fig. S15†). Then, immunodeficient NCG mice bearing subcutaneous HCT116 tumors were constructed and transplanted with activated PBMCs. These mice were intravenously injected with saline, αPD-L1, CARC, or PARC at a same dose of antibody (5 mg kg−1) and every 3 days for a total of 4 injections. For photoactivation, tumors were irradiated with 365 nm light for 5 min at 4 h post each injection. After 14 days, tumor volumes of the PARC without photoirradiation group increased from ∼150 mm3 to ∼1500 mm3, similar to the saline and αPD-L1 groups (Fig. 4a). The insignificant cancer suppression could be attributed to the moderate dose of αPD-L1.48 However, with light irradiation, tumor growth of the PARC group but not the CARC group was significantly suppressed. Notably, during the treatment, body weights of all groups remained nearly constant (Fig. 4b), suggesting general biosafety. At the end of treatment, tumors of the PARC with photoirradiation group were inhibited by ∼62% (Fig. 4c and S16†), compared to the other groups. Our results thus demonstrate that, under light irradiation, PARC can evoke potent anti-cancer effects.
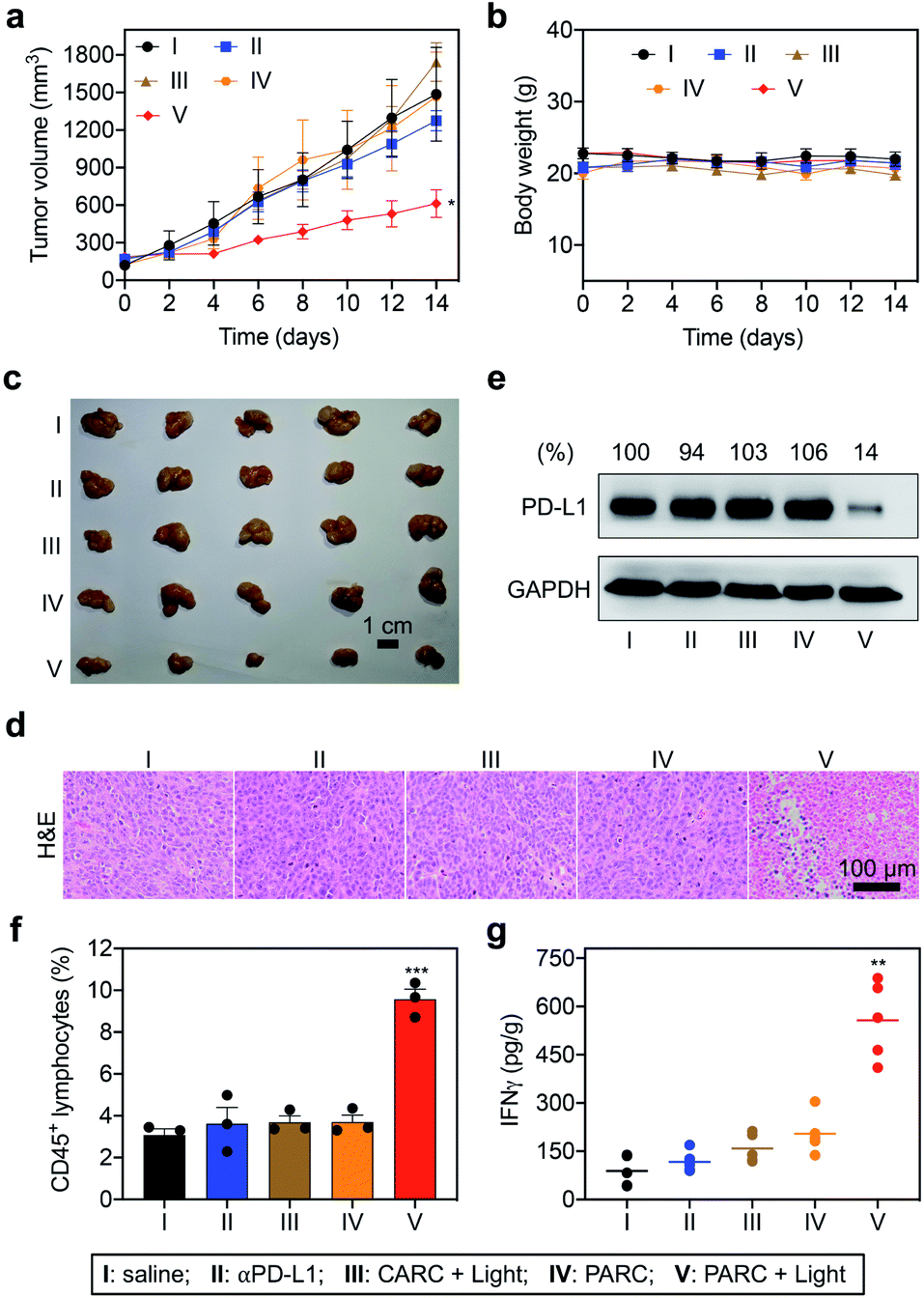 | ||
| Fig. 4 (a) Tumor growth curves and (b) body weights of HCT116 tumor-bearing NCG mice that were intravenously injected with saline, αPD-L1 (0.7 nmol), CARC (0.7 nmol), or PARC (0.7 nmol) every 3 days for a total of 4 injections. Tumors were irradiated with 365 nm light (50 mW cm−2) for 5 min at 4 h post each injection. (c) Optical images, (d) H&E staining, (e) western blotting analysis of PD-L1, (f) flow cytometry analysis of CD45+ lymphocytes, and (g) IFNγ levels. (e) Relative intensity ratios of PD-L1/GAPDH are shown at the top. Data are shown as mean ± SD (n = 3–5). *P < 0.05, **P < 0.01, and ***P < 0.001, relative to the other groups. | ||
To validate the immunotherapeutic efficacy of PARC, we further analyzed tumor tissues. The histological examination with haematoxylin and eosin (H&E) staining showed obvious cell death in tumor tissues of the PARC with photoirradiation group compared to the other groups (Fig. 4d). TdT-mediated dUTP nick end labelling (TUNEL) staining also demonstrated tumor cell apoptosis in the PARC with photoirradiation group (Fig. S17†). The apoptosis was mainly detected in the area with 5 mm depth where 365 nm light can penetrate,49 suggesting a local anti-cancer effect. However, we found no significant damage in normal tissues (heart, liver, spleen, lung, and kidney) of all groups as indicated by H&E staining (Fig. S18†), further confirming its biosafety. Western blotting analysis showed that PARC with photoirradiation significantly decreased PD-L1 protein levels by ∼86% compared to the other groups (Fig. 4e), verifying the photoactivatable gene silencing in vivo. To unveil whether lymphocytes in PBMCs mediate anti-cancer immunity, we evaluated their invasion in tumor tissues. Flow cytometry analysis showed that PARC with photoirradiation increased the tumor infiltration of CD45+ lymphocytes (Fig. 4f and S19†) and CD3+ T cells (Fig. S20†) ∼2.6 and ∼3.6 fold, respectively, relative to the other groups. Meanwhile, IFNγ (Fig. 4g) and TNFα (Fig. S21†) levels in tumor tissues of the PARC with photoirradiation group were upregulated ∼4.1 and ∼3.1 fold, respectively. Taken together, these ex vivo data prove photoactivatable immunogene therapy by PARC, which also match the in vivo anti-cancer results.
Conclusion
In summary, we developed a photoresponsive conjugate of αPD-L1 with siPD-L1 (PARC), which were connected through a photocleavable linker. PARC could target cancer cells through αPD-L1/PD-L1 interaction, allowing not only resurgence of immune cell activity but also intracellular transportation of siPD-L1. Under light irradiation to break the photocleavable linker, siPD-L1 could get released from PARC and escape from the lysosome into the cytoplasm, where intracellular PD-L1 expression was subsequently silenced. Eventually, PARC afforded a synergetic blockade of PD-L1 to potently activate immune cells to kill cancer cells. We demonstrated the potential of PARC both in vitro and in vivo. Our work thus presents a novel approach to achieve tumor-targeted and photoinduced siRNA delivery, which also promises photoactivatable immunogene therapy. To further the in vivo applications of PARC, optimized or alternative photocleavable linkers50 with long-wavelength excitation that ensure deep penetration of light as well as safe and fast release of siRNA deserve future exploration. Overall, given its high biocompatibility and versatility to assemble different antibody/siRNA combos, we believe this photoresponsive ARC will open new avenues for cancer treatment.Experimental section
Preparation of PARC and CARC
αPD-L1 (2 mg mL−1) was mixed with Traut's reagent (0–10 eq.) in the EDTA-PB (100 nM phosphate buffer containing 2 mM EDTA, pH = 6.8) buffer, and the mixture was incubated at 37 °C for 1 h. The small molecules were removed by ultrafiltration (molecular weight cut-off, 3 kDa) with EDTA-PB 5 times. The supernatant was collected and mixed with maleimide-labeled siPD-L1 or si-ctrl (10 eq.) in EDTA-PB and incubated at 37 °C for 2 h. Excess siRNAs were removed by ultrafiltration (molecular weight cut-off, 50 kDa) with EDTA-PB 5 times. To prepare Cy5/Cy3-labeled PARC, αPD-L1 was first labeled with Cy5 and further labeled with Cy3-modified siPD-L1. Briefly, αPD-L1 (2 mg mL−1) was mixed with Cy5-NHS (0.7 mM) in PBS buffer and the mixture was incubated at 37 °C for 1 h. Small molecules were removed by ultrafiltration (molecular weight cut-off, 3 kDa) with EDTA-PB buffer 5 times. Then, Cy5-labeled αPD-L1 was further modified with maleimide/Cy3-labeled siPD-L1 using the same method as described above.Detection of siPD-L1 release
PARC (100 nM) in PBS was irradiated by using an 8 W hand-held 365 nm UV lamp (10 mW cm−2) for 0–6 minutes. The released siPD-L1 was collected by ultrafiltration (molecular weight cut-off, 50 kDa), and the concentration was determined using a NanoDrop 2000 (Thermo Scientific). The siPD-L1 release percentages were calculated by dividing the measured amount by the initial amount. Then, the collected siRNA was desalted, lyophilized, and characterized by mass spectrometry.Stability test
PARC (150 nM) was incubated in PBS at 37 °C. IEC analysis was performed at 0, 6, 12, and 24 h time points to indicate the stability of PARC.Cell culture
HCT116 cells were cultured in high glucose DMEM containing 10% FBS and 1% penicillin/streptomycin, and maintained in 5% CO2 at 37 °C.Western blotting
After reaching ∼80% confluency, HCT116 cells were transfected with siPD-L1 (200 nM) or si-ctrl (200 nM) using Lipofectamine 2000 according to the manufacturer's protocol. Or, HCT116 cells were treated with αPD-L1 (150 nM), CARC (150 nM), or PARC (150 nM). For the photoirradiation group, after incubation for 1 h, HCT116 cells were further irradiated by using an 8 W hand-held 365 nm UV lamp (10 mW cm−2) for 5 min. At 48 h, cell lysates were extracted using RIPA lysis buffer (150 mM NaCl, 10 mM Tris, pH 7.5, 1% NP40, 1% deoxycholate, 0.1% SDS, and 1× protease inhibitor cocktail). To measure the expression levels of PD-L1 in tumors, total proteins were extracted from 100 mg tumor tissues. The protein concentration was determined using a BCA protein assay kit. A total of 40 μg of protein was denatured in 1× SDS loading buffer and resolved by 10% SDS-PAGE, followed by transferring to a PVDF membrane and block in 5% nonfat milk in TBST. The PVDF membrane was then stained with primary antibodies for PD-L1 and GAPDH. After washing, the membrane was further incubated with the appropriate secondary antibody and finally visualized using ECL reagents.RT-qPCR
After reaching ∼80% confluency, HCT116 cells were transfected with siPD-L1 (200 nM) or si-ctrl (200 nM) using Lipofectamine 2000 according to the manufacturer's protocol. Or, HCT116 cells were treated with αPD-L1 (150 nM), CARC (150 nM), or PARC (150 nM). For the photoirradiation group, after incubation for 1 h, HCT116 cells were further irradiated by using an 8 W hand-held 365 nm UV lamp (10 mW cm−2) for 5 min. After incubation for 48 h, total RNA was isolated from HCT116 cells with TRIZOL reagent (Invitrogen). cDNA was synthesized using a SuperScript III kit (Invitrogen), and qPCR was performed with a SYBR Green PCR Master Mix (Vazyme Biotech). Primers: PD-L1: forward: 5′-TTTGCTGAACGCCCCATA-3′; reverse: 5′-TGC-TTGTCCAGATGACTTCG-3′; GAPDH: forward: 5′-GGACCTGACCT-GCCGTCTAG-3′; reverse: 5′-GTAGCCCAGGATGCCCTTGA-3′. GAPDH served as the internal control.Confocal fluorescence imaging
1 × 105 HCT116 cells were seeded on 35 mm glass-bottom culture dishes. When reaching ∼80% confluency, cells were incubated with Cy5/Cy3-labeled PARC (150 nM). To explore the effect of light irradiation, at 1 h time point, cells were irradiated by using an 8 W hand-held 365 nm UV lamp (10 mW cm−2) for 5 min. After incubation for different time points, cells were washed with PBS three times. Then, cells were stained with DAPI and Lysotracker according to the manufacturer's instructions. The imaging acquisitions were carried out immediately using a Leica confocal microscope.PD-1 binding analysis
After reaching ∼80% confluency, HCT116 cells were treated with αPD-L1 (150 nM), CARC (150 nM), or PARC (150 nM). For the photoirradiation group, after incubation for 1 h, HCT116 cells were further irradiated by using an 8 W hand-held 365 nm UV lamp (10 mW cm−2) for 5 min. At 48 h, cells were incubated with 5 μg mL−1 recombinant human PD-1 FC chimera protein at room temperature for 30 min. After washing with PBS 3 times, cells were further incubated with an anti-human IgG Alexa Fluor 488 dye conjugated antibody (ThermoFisher) (1:200) for another 30 min. Following washing with PBS 3 times, Alexa Fluor 488-positive cells were analyzed by flow cytometry.
Immune cell killing assay
After reaching ∼80% confluency, HCT116 cells were treated with αPD-L1 (150 nM), CARC (150 nM), or PARC (150 nM). For the photoirradiation group, after incubation for 1 h, HCT116 cells were further irradiated by using an 8 W hand-held 365 nm UV lamp (10 mW cm−2) for 5 min. At 24 h, PBMCs (MT-BIO) were resuscitated and activated with an anti-CD3 antibody (100 ng mL−1), an anti-CD28 antibody (100 ng mL−1), and IL2 (10 ng mL−1) for 24 h. Then, activated PBMCs were incubated with HCT116 cells at an effector-to-target ratio of 5:1 at 37 °C for another 24 h. Then, lactate dehydrogenase analysis was carried out according to the manufacturer's protocol.
In vivo and ex vivo imaging
Animal care and euthanasia were carried out with the approval of the Institutional Animal Care and Use Committee (IACUC) of Nanjing University. A total of 2 × 106 HCT116 cells were subcutaneously injected into the right back flank of 4–5 weeks old female nude mice (Nanjing Biomedical Research Institute of Nanjing University). When tumor diameters reached ∼1 cm, these tumor-bearing mice were injected via the tail vein with Cy5/Cy3-labeled PARC (0.7 nmol). Fluorescence imaging was then performed with an IVIS Lumina XR III small animal imaging system. To acquire fluorescence signals from siPD-L1, a Cy3 filter was used. To acquire fluorescence signals from αPD-L1, a Cy5 filter was used. Identical illumination settings (lamp voltage, filters, f/stop, field of views, and binning) were used for acquiring all images. Fluorescence images were taken at 0, 1, 2, 4 and 8 h post injection. The images were then analyzed using Living Image 3.2 software (Caliper, Hopkinton, MA). The fluorescence emission was normalized to photons per second per centimeter squared per steradian (p per s per cm2 per sr). For ex vivo imaging, tumor and other major tissues were dissected and put on black paper. The ex vivo images were acquired immediately using an IVIS Lumina XR III system with the same illumination setting as in vivo imaging. The images were also processed with the same method as described above.In vivo cancer therapy
Animal care and euthanasia were carried out with the approval of the Institutional Animal Care and Use Committee (IACUC) of Nanjing University. A total of 2 × 106 HCT116 cells were subcutaneously injected into the right back flank of 4–5 weeks old female immune-deficient NOD/Prkdcnull/IL2RGnull (NCG) mice (Nanjing Biomedical Research Institute of Nanjing University). When tumor volumes reached ∼100–200 mm3, each mouse received 1 × 107 activated human PBMCs via tail vein injection to generate a PBMC-humanized mouse. IL-2 (2.75 μg per mouse) was intraperitoneally injected for 3 consecutive days. These mice were then randomly divided into five groups (n = 5/group) and respectively treated with saline, αPD-L1 (0.7 nmol), CARC (0.7 nmol), or PARC (0.7 nmol) through tail vein injection every 3 days for a total of 4 injections. Mice treated with CARC or PARC were subject to irradiation with a LED 365 nm UV lamp (50 mW cm−2) for 5 min at 4 h after each injection. Tumor volumes and body weights were recorded every two days. Tumor volumes were calculated using the following formula: V = (width2 × length)/2. After 14 days, these mice were sacrificed, and tissues were collected for further analysis.Immune cell infiltration
Tumors were cut into small pieces and subjected to mechanical disruption and separation, followed by passing through 70 μm strainers and treatment with the erythrocyte lysis solution (BioLegend) according to the manufacturer's instructions. The single-cell suspensions were then stained with an FITC-labeled anti-CD3 antibody and PE-labeled anti-CD45 antibody, respectively. Cells were further analyzed by flow cytometry.Immunocytokine detection
After reaching ∼80% confluency, HCT116 cells were treated with αPD-L1 (150 nM), CARC (150 nM), or PARC (150 nM). For the photoirradiation group, these treated cells were further subjected to 5 min’ 365 nm light irradiation (10 mW cm−2) after incubation for 1 h. At 24 h, PBMCs (MT-BIO, Shanghai) were resuscitated and activated with an anti-CD3 antibody (100 ng mL−1), an anti-CD28 antibody (100 ng mL−1), and IL2 (10 ng mL−1) for 24 h. Activated PBMCs were incubated with HCT116 cells at an effector-to-target ratio of 5:1 at 37 °C for another 24 h. Then, culture media were collected for analysis. To detect the expression of TNFα and IFNγ in tumors, tumor tissues were digested. The supernatants were collected and diluted. TNFα and IFNγ levels were analyzed using ELISA kits according to the manufacturer's protocols.
Statistical analysis
Statistical comparisons between groups were evaluated by Student's t-test. Data are shown as mean ± SD. *P < 0.05, **P < 0.01, and ***P < 0.001 were considered to be statistically significant.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Author contributions
X. Wang and X. Xiao performed the experiments. Y. Feng drew the schematic figures. J. Li and Y. Zhang designed the project and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to acknowledge the financial support from the Natural Science Foundation of Jiangsu Province on project BK20202004, the National Natural Science Foundation of China (21877058, 22077063, 21977043, and 91753116), and the Fundamental Research Funds for the Central Universities (020514380226 and 020514380251). The animal study was carried out in strict accordance with the guidelines for care and use of laboratory animals of Nanjing University and was approved by the Institutional Animal Care and Use Committee (IACUC) of Nanjing University (Nanjing, China).References
- T. C. Roberts, R. Langer and M. J. A. Wood, Nat. Rev. Drug Discovery, 2020, 19, 673–694 CrossRef CAS PubMed
.
- B. Kim, J. H. Park and M. J. Sailor, Adv. Mater., 2019, 31, e1903637 CrossRef PubMed
- C. Zhang and K. Pu, Chem. Soc. Rev., 2020, 49, 4234–4253 RSC
- D. W. Zheng, F. Gao, Q. Cheng, P. Bao, X. Dong, J. X. Fan, W. Song, X. Zeng, S. X. Cheng and X. Z. Zhang, Nat. Commun., 2020, 11, 1985 CrossRef CAS PubMed
- H. Xiao, Y. Guo, B. Li, X. Li, Y. Wang, S. Han, D. Cheng and X. Shuai, ACS Cent. Sci., 2020, 6, 1208–1222 CrossRef CAS PubMed
- K. W. Huang, F. F. Hsu, J. T. Qiu, G. J. Chern, Y. A. Lee, C. C. Chang, Y. T. Huang, Y. C. Sung, C. C. Chiang, R. L. Huang, C. C. Lin, T. K. Dinh, H. C. Huang, Y. C. Shih, D. Alson, C. Y. Lin, Y. C. Lin, P. C. Chang, S. Y. Lin and Y. Chen, Sci. Adv., 2020, 6, eaax5032 CrossRef CAS PubMed
- R. L. Setten, J. J. Rossi and S. P. Han, Nat. Rev. Drug Discovery, 2019, 18, 421–446 CrossRef CAS PubMed
- S. Benizri, A. Gissot, A. Martin, B. Vialet, M. W. Grinstaff and P. Barthelemy, Bioconjugate Chem., 2019, 30, 366–383 CrossRef CAS PubMed
- M. J. Mitchell, M. M. Billingsley, R. M. Haley, M. E. Wechsler, N. A. Peppas and R. Langer, Nat. Rev. Drug Discovery, 2021, 20, 101–124 CrossRef CAS PubMed
- J. A. Kulkarni, D. Witzigmann, S. B. Thomson, S. Chen, B. R. Leavitt, P. R. Cullis and R. van der Meel, Nat. Nanotechnol., 2021, 16, 630–643 CrossRef CAS PubMed
- C. Yu, L. Li, P. Hu, Y. Yang, W. Wei, X. Deng, L. Wang, F. R. Tay and J. Ma, Adv. Sci., 2021, 8, 2100540 CrossRef CAS PubMed
- J. Dugal-Tessier, S. Thirumalairajan and N. Jain, J. Clin. Med., 2021, 10, 838 CrossRef CAS PubMed
- I. Dovgan, O. Koniev, S. Kolodych and A. Wagner, Bioconjugate Chem., 2019, 30, 2483–2501 CrossRef CAS PubMed
- I. V. Chernikov, V. V. Vlassov and E. L. Chernolovskaya, Front. Pharmacol., 2019, 10, 444 CrossRef CAS PubMed
- N. Baumer, W. E. Berdel and S. Baumer, Mol. Pharm., 2017, 14, 1339–1351 CrossRef PubMed
- D. Klein, S. Goldberg, C. S. Theile, R. Dambra, K. Haskell, E. Kuhar, T. Lin, R. Parmar, M. Manoharan, M. Richter, M. Wu, J. Mendrola Zarazowski, V. Jadhav, M. A. Maier, L. Sepp-Lorenzino, K. O'Neil and V. Dudkin, Mol. Ther., 2021, 29, 2053–2066 CrossRef CAS PubMed
- A. R. Nanna, A. V. Kel'in, C. Theile, J. M. Pierson, Z. X. Voo, A. Garg, J. K. Nair, M. A. Maier, K. Fitzgerald and C. Rader, Nucleic Acids Res., 2020, 48, 5281–5293 CrossRef CAS PubMed
- S. J. Shi, L. J. Wang, D. H. Han, J. H. Wu, D. Jiao, K. L. Zhang, J. W. Chen, Y. Li, F. Yang, J. L. Zhang, G. X. Zheng, A. G. Yang, A. Z. Zhao, W. J. Qin and W. H. Wen, Theranostics, 2019, 9, 1247–1263 CrossRef CAS PubMed
- N. Baeumer, N. Appel, L. Terheyden, F. Buchholz, C. Rossig, C. Mueller-Tidow, W. E. Berdel and S. Baeumer, Nat. Protoc., 2016, 11, 22–36 CrossRef CAS PubMed
- K. Jiang, J. Li, J. Yin, Q. Ma, B. Yan, X. Zhang, L. Wang, L. Wang, T. Liu, Y. Zhang, Q. Fan, A. Yang, X. Qiu and B. Ma, Biomaterials, 2015, 59, 77–87 CrossRef CAS PubMed
- Y. Su, L. Yu, N. Liu, Z. Guo, G. Wang, J. Zheng, M. Wei, H. Wang, A. G. Yang, W. Qin and W. Wen, Cancer Lett., 2013, 338, 282–291 CrossRef CAS PubMed
- Y. D. Yao, T. M. Sun, S. Y. Huang, S. Dou, L. Lin, J. N. Chen, J. B. Ruan, C. Q. Mao, F. Y. Yu, M. S. Zeng, J. Y. Zang, Q. Liu, F. X. Su, P. Zhang, J. Lieberman, J. Wang and E. Song, Sci. Transl. Med., 2012, 4, 130ra148 Search PubMed
- S. Dou, Y. D. Yao, X. Z. Yang, T. M. Sun, C. Q. Mao, E. W. Song and J. Wang, J. Controlled Release, 2012, 161, 875–883 CrossRef CAS PubMed
- Y. Ma, C. M. Kowolik, P. M. Swiderski, M. Kortylewski, H. Yu, D. A. Horne, R. Jove, O. L. Caballero, A. J. Simpson, F. T. Lee, V. Pillay and A. M. Scott, ACS Chem. Biol., 2011, 6, 962–970 CrossRef CAS PubMed
- E. Song, P. Zhu, S. K. Lee, D. Chowdhury, S. Kussman, D. M. Dykxhoorn, Y. Feng, D. Palliser, D. B. Weiner, P. Shankar, W. A. Marasco and J. Lieberman, Nat. Biotechnol., 2005, 23, 709–717 CrossRef CAS PubMed
- W. Fu, H. Sun, Y. Zhao, M. Chen, L. Yang, X. Yang and W. Jin, Mol. Immunol., 2018, 99, 124–133 CrossRef CAS PubMed
- Y. Lu, L. Liu, Y. Wang, F. Li, J. Zhang, M. Ye, H. Zhao, X. Zhang, M. Zhang, J. Zhao, B. Yan, A. Yang, H. Feng, R. Zhang and X. Ren, Biomaterials, 2016, 76, 196–207 CrossRef CAS PubMed
- S. Baumer, N. Baumer, N. Appel, L. Terheyden, J. Fremerey, S. Schelhaas, E. Wardelmann, F. Buchholz, W. E. Berdel and C. Muller-Tidow, Clin. Cancer Res., 2015, 21, 1383–1394 CrossRef PubMed
- O. Zavoiura, B. Brunner, P. Casteels, L. Zimmermann, M. Ozog, C. Boutton, M. W. Helms, T. Wagenaar, V. Adam, J. Peterka, C. Metz-Weidmann, P. Deschaght, S. Scheidler and K. Jahn-Hofmann, Mol. Pharm., 2021, 18, 1048–1060 CrossRef CAS PubMed
- D. Peer, P. Zhu, C. V. Carman, J. Lieberman and M. Shimaoka, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 4095–4100 CrossRef CAS PubMed
- J. Li, H. Kong, C. Zhu and Y. Zhang, Chem. Sci., 2020, 11, 3390–3396 RSC
- S. L. Topalian, J. M. Taube and D. M. Pardoll, Science, 2020, 367, eaax0182 CrossRef CAS PubMed
- J. Xin Yu, J. P. Hodge, C. Oliva, S. T. Neftelinov, V. M. Hubbard-Lucey and J. Tang, Nat. Rev. Drug Discovery, 2020, 19, 163–164 CrossRef PubMed
- M. L. Burr, C. E. Sparbier, Y. C. Chan, J. C. Williamson, K. Woods, P. A. Beavis, E. Y. N. Lam, M. A. Henderson, C. C. Bell, S. Stolzenburg, O. Gilan, S. Bloor, T. Noori, D. W. Morgens, M. C. Bassik, P. J. Neeson, A. Behren, P. K. Darcy, S. J. Dawson, I. Voskoboinik, J. A. Trapani, J. Cebon, P. J. Lehner and M. A. Dawson, Nature, 2017, 549, 101–105 CrossRef CAS PubMed
- C. W. Li, S. O. Lim, E. M. Chung, Y. S. Kim, A. H. Park, J. Yao, J. H. Cha, W. Xia, L. C. Chan, T. Kim, S. S. Chang, H. H. Lee, C. K. Chou, Y. L. Liu, H. C. Yeh, E. P. Perillo, A. K. Dunn, C. W. Kuo, K. H. Khoo, J. L. Hsu, Y. Wu, J. M. Hsu, H. Yamaguchi, T. H. Huang, A. A. Sahin, G. N. Hortobagyi, S. S. Yoo and M. C. Hung, Cancer Cell, 2018, 33, 187–201 CrossRef CAS PubMed
- S. Sau, A. Petrovici, H. O. Alsaab, K. Bhise and A. K. Iyer, Cancers, 2019, 11, 232 CrossRef CAS PubMed
- Y. Wu, W. Chen, Z. P. Xu and W. Gu, Front. Immunol., 2019, 10, 2022 CrossRef CAS PubMed
- Y. Wang, H. Wang, H. Yao, C. Li, J. Y. Fang and J. Xu, Front. Pharmacol., 2018, 9, 536 CrossRef PubMed
- G. Chen, A. C. Huang, W. Zhang, G. Zhang, M. Wu, W. Xu, Z. Yu, J. Yang, B. Wang, H. Sun, H. Xia, Q. Man, W. Zhong, L. F. Antelo, B. Wu, X. Xiong, X. Liu, L. Guan, T. Li, S. Liu, R. Yang, Y. Lu, L. Dong, S. McGettigan, R. Somasundaram, R. Radhakrishnan, G. Mills, Y. Lu, J. Kim, Y. H. Chen, H. Dong, Y. Zhao, G. C. Karakousis, T. C. Mitchell, L. M. Schuchter, M. Herlyn, E. J. Wherry, X. Xu and W. Guo, Nature, 2018, 560, 382–386 CrossRef CAS PubMed
- M. Poggio, T. Hu, C. C. Pai, B. Chu, C. D. Belair, A. Chang, E. Montabana, U. E. Lang, Q. Fu, L. Fong and R. Blelloch, Cell, 2019, 177, 414–427 CrossRef CAS PubMed
- T. Sugo, M. Terada, T. Oikawa, K. Miyata, S. Nishimura, E. Kenjo, M. Ogasawara-Shimizu, Y. Makita, S. Imaichi, S. Murata, K. Otake, K. Kikuchi, M. Teratani, Y. Masuda, T. Kamei, S. Takagahara, S. Ikeda, T. Ohtaki and H. Matsumoto, J. Controlled Release, 2016, 237, 1–13 CrossRef CAS PubMed
- T. L. Cuellar, D. Barnes, C. Nelson, J. Tanguay, S.-F. Yu, X. Wen, S. J. Scales, J. Gesch, D. Davis, A. van Brabant Smith, D. Leake, R. Vandlen and C. W. Siebel, Nucleic Acids Res., 2015, 43, 1189–1203 CrossRef CAS PubMed
- L. Chen, Y. Sun, J. Li and Y. Zhang, Chem. Commun., 2020, 56, 627–630 RSC
- X. X. Wang, Y. Wang, J. Li, T. Tian, J. B. Li, Z. J. Guo and Y. Zhang, Adv. Ther., 2022, 5, 2100161 CrossRef CAS
- Q. Liu, L. Jiang, K. Li, H. Li, G. Lv, J. Lin and L. Qiu, Cancer Immunol. Immunother., 2021, 70, 1721–1733 CrossRef CAS PubMed
- H. Wang, H. Yao, C. Li, H. Shi, J. Lan, Z. Li, Y. Zhang, L. Liang, J. Y. Fang and J. Xu, Nat. Chem. Biol., 2019, 15, 42–50 CrossRef CAS PubMed
- J. Hsu, J. J. Hodgins, M. Marathe, C. J. Nicolai, M. C. Bourgeois-Daigneault, T. N. Trevino, C. S. Azimi, A. K. Scheer, H. E. Randolph, T. W. Thompson, L. Zhang, A. Iannello, N. Mathur, K. E. Jardine, G. A. Kirn, J. C. Bell, M. W. McBurney, D. H. Raulet and M. Ardolino, J. Clin. Invest., 2018, 128, 4654–4668 CrossRef PubMed
- Y. Zhang, C. Fang, R. E. Wang, Y. Wang, H. Guo, C. Guo, L. Zhao, S. Li, X. Li, P. G. Schultz, Y. J. Cao and F. Wang, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 15889–15894 CrossRef CAS PubMed
- J. Wang, Y. Liu, Y. Liu, S. Zheng, X. Wang, J. Zhao, F. Yang, G. Zhang, C. Wang and P. R. Chen, Nature, 2019, 569, 509–513 CrossRef CAS PubMed
- R. Weinstain, T. Slanina, D. Kand and P. Klan, Chem. Rev., 2020, 120, 13135–13272 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc01672a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |