
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePrecisely synthesized segmented polyurethanes toward block sequence-controlled drug delivery†
Yuanqing
Song
a,
Chuandong
Sun
a,
Chenxu
Tian
a,
Hao
Ming
a,
Yanjun
Wang
a,
Wenkai
Liu
a,
Nan
He
a,
Xueling
He
b,
Mingming
Ding
 *a,
Jiehua
Li
a,
Feng
Luo
a,
Hong
Tan
*a and
Qiang
Fu
a
*a,
Jiehua
Li
a,
Feng
Luo
a,
Hong
Tan
*a and
Qiang
Fu
a
aCollege of Polymer Science and Engineering, State Key Laboratory of Polymer Materials Engineering, Sichuan University, Chengdu 610065, China. E-mail: hongtan@scu.edu.cn; dmmshx@scu.edu.cn
bLaboratory Animal Center of Sichuan University, Sichuan University, Chengdu 610065, China
First published on 5th April 2022
Abstract
The construction of polyurethanes (PUs) with sequence-controlled block structures remains a serious challenge. Here, we report the precise synthesis of PUs with desirable molecular weight, narrow molecular weight distribution, and controlled block sequences from commercially available monomers. The synthetic procedure is derived from a liquid-phase synthetic methodology, which involves diisocyanate-based iterative protocols in combination with a convergent strategy. Furthermore, a pair of multifunctional PUs with different sequence orders of cationic and anion segments were prepared. We show that the sequence order of functional segments presents an impact on the self-assembly behavior and results in unexpected surface charges of assembled micelles, thereby affecting the protein absorption, cell internalization, biodistribution and antitumor effect of the nanocarriers in vitro and in vivo. This work provides a versatile platform for the development of precise multiblock PUs with structural complexity and functional diversity, and will greatly facilitate the clinical translation of PUs in biomedicine.
Introduction
Polyurethanes (PUs) have been extensively applied in many aspects of biomedical fields, such as drug delivery and tissue engineering, by virtue of their adjustable mechanical properties, good biocompatibility and excellent molecular tailorability.1,2 They are compatible with diverse structural blocks and facilitate the construction of multifunctional material systems by incorporating various functional moieties into the polymeric chains.3–6 Moreover, the multiple blocks with thermodynamic incompatibility could form phase-separated structures at the molecular level and further self-organize into three-dimensional supramolecular assemblies, which have the ability to arrange functional domains at the nanoscale and provide advanced features for potential drug delivery applications.7–9 However, as new segments are added, the structures of multifunctional PUs will become more complex and heterogeneous because of the inherent attribute of PU synthesis.10 Traditionally, multiblock PUs are prepared by a one-pot step-growth polyaddition process, where the reactions between highly reactive diisocyanate and active hydrogen compounds (e.g. polyols and polyfunctional chain extenders) lack controllability and selectivity.11 The multi-step competitive bond-forming reactions inevitably lead to the formation of random polymers with statistical nature of uncertain molecular weights, broad molecular weight distributions, random sequence structures, and non-uniform functionalities among the batches.12 This issue not only results in a basic obstacle to the understanding of structure–property relationships of multiblock PUs, but also limits their industrial and commercial translation.13–15On the other hand, biomacromolecules including proteins have demonstrated charming and precise primary structures guaranteed by the exquisite biological polymerization process in highly evolved biosystems. Only by controlling the sequence structure of limited monomers, a variety of high-level organized architectures and sophisticated biological functions can be regulated.16,17 This has provided considerable motivation for the exploration of sequence-controlled polymerization.18–22 For instance, several promising approaches have been developed for the precise synthesis of sequence-controlled PUs using multistep iterative synthesis (e.g. solid-phase synthesis).23–28 Nonetheless, most of these protocols deviate from classical diisocyanate methods and depend on the use of special monomers involving monoisocyanate23,24 and non-isocyanate molecules.25–28 Moreover, despite their well-defined structures, the oligocarbamate products generally show low molecular weights (Mn < 2000), and have been frequently used as peptidomimetics in biochemistry29,30 or as intermediates for the further preparation of macromolecules.24,27 To date, the manipulation of block structures of multiblock PUs at the macromolecular level, thus to further develop precise PUs with controlled functional domains, still remains a longstanding challenge in chemical research.
Here we report a model precisely synthesized multiblock PU with relatively high molecular weight (Mn > 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), narrow molecular weight distribution (Đ ∼ 1.10) and controlled block sequences. Enlightened by the liquid-phase synthetic methodology (LPS) that integrates the advantages of the high conversion efficiency of homogeneous solution chemistry and the simplified workup procedures of solid-phase synthesis,31–33 we used the iterative reactions of classical diisocyanate chemistry, combined with convergent strategies, to build a series of PU oligomers and polymers with precise structures. The structural modules were commercially available monomers without modification or protection, and polyethylene glycol (PEG) as the soluble polymer support remained in the final chains without cleaving off. Based on the established platform, we further designed and prepared two complex multifunctional PUs with different sequence orders of cationic and anion segments. We found that the segmented copolymers with the same composition ratio but different segment sequence arrangements exhibited distinct and counterintuitive performances in the surface charge of self-assembled micelles, cell internalization and drug delivery in vitro and in vivo.
000), narrow molecular weight distribution (Đ ∼ 1.10) and controlled block sequences. Enlightened by the liquid-phase synthetic methodology (LPS) that integrates the advantages of the high conversion efficiency of homogeneous solution chemistry and the simplified workup procedures of solid-phase synthesis,31–33 we used the iterative reactions of classical diisocyanate chemistry, combined with convergent strategies, to build a series of PU oligomers and polymers with precise structures. The structural modules were commercially available monomers without modification or protection, and polyethylene glycol (PEG) as the soluble polymer support remained in the final chains without cleaving off. Based on the established platform, we further designed and prepared two complex multifunctional PUs with different sequence orders of cationic and anion segments. We found that the segmented copolymers with the same composition ratio but different segment sequence arrangements exhibited distinct and counterintuitive performances in the surface charge of self-assembled micelles, cell internalization and drug delivery in vitro and in vivo.
Experimental methods
Synthesis of three-segment PUs (1, 2, 8, 9, 10, and 11)
The synthetic diagram of compound 1 is shown in ESI Scheme S1.† Briefly, hydroxyl-PEG monomethyl ether (mPEG-OH, T, Mn 2000, 1 mmol) and L-lysine ethyl ester diisocyanate (LDI, B, 3 mmol) with an organic bismuth catalyst (0.1 wt%) were added into a three-necked flask for 2 h of reaction with stirring at 60 °C under a nitrogen environment. The reaction process was monitored by thin layer chromatography (TLC) analysis. After mPEG-OH was almost converted, anhydrous tetrahydrofuran (THF, 10 mL) was added into the reaction mixture, and the solution with diethyl ether (20 mL) added was refrigerated at −24 °C for 1 h. The purified intermediate product (TB) was quickly collected by suction filtration and washed twice with diethyl ether. Then the reaction of dried TB and poly(ε-caprolactone) (PCL, A, Mn 2000, 3 mmol) with organic bismuth was carried out at 60 °C for 2 h, and at 80 °C for another 3 h in a nitrogen atmosphere. After the reaction, the above THF/diethyl ether (1:1, v/v) purification process was repeated to remove parts of the impurities, and the product was obtained through column chromatography (silica gel, 200–300 mesh) with the gradient eluent system of chloroform/methanol. The target collecting ratio of compound 1 (TBA) was about 35:1 (chloroform/methanol, v/v) to give white powder (∼32% yield). 1H NMR (600 MHz, CDCl3, ppm): δ = 4.27 (t, 4H, A), 4.20 (m, 4H, B + T), 4.06 (t, 33H, A), 3.64 (t, 190H, T), 3.38 (s, 3H, T), 3.15 (d, 2H, B), 2.32 (t, 35H, A), 1.65 (m, 69H, A), 1.38 (m, 37H, A), 1.27 (m, 5H, B).
The synthetic diagram of compound 2 is shown in ESI Scheme S2.† In brief, amino-PEG monomethyl ether (mPEG-NH2, T′, Mn 2000, 1 mmol) dissolved in anhydrous THF (10%, wt/v) was added dropwise into the LDI (B, 3 mmol)/THF solution (30%, wt/v) with stirring, and the reaction was performed at 30 °C for 3 h under a nitrogen environment. After mPEG-NH2 was almost converted, the reaction solution was added into diethyl ether, and the solution was refrigerated at −24 °C for 1 h. The purified intermediate product (T′B) was quickly collected by suction filtration and washed twice with diethyl ether. Then dried T′B dissolved in anhydrous THF was added into the desalted cystamine (Cysa, C, 3 mmol)/THF solution (30%, wt/v) with an ice bath, and this reaction proceeded at 30 °C for 3 h. Finally, the similar purification process was conducted for compound 2 (T′BC), and the target collecting ratio was about 10:1 (v/v) to give the product as slightly yellow powder (∼21% yield). 1H NMR (600 MHz, CDCl3, ppm): δ = 4.20 (q, 2H, B), 3.64 (t, 156H, T′), 3.38 (s, 3H, T′), 3.20–2.90 (m, 4H, B + C), 2.81 (m, 4H, C), 1.27 (m, 5H, B).
The other three-segment PUs (8 (TBK), 9 (TBQ), 10 (A′BQ) and 11 (A′BK)) were synthesized according to the diisocyanate-based protocols with synthetic segments: anionic segment (K), cationic segment (Q) and mono-functional PCL (A′). See ESI Section S2.2.2† for detailed procedures and characterization results.
Synthesis of five-segment PUs (3, 4, and 5)
Five-segment PUs were prepared based on the synthetic three-segment compounds via iterative reactions of diisocyanate chemistry. Take compound 3 for example, the synthetic diagram is shown in Fig. 1B(a). Briefly, 1 (TBA, 1 mmol) and LDI (B, 5 mmol) with organic bismuth were reacted at 60 °C for 3 h under a nitrogen environment. After TBA was almost converted, anhydrous THF/diethyl ether (1:1, v/v) was used for refrigeration at −24 °C for 2 h. The purified intermediate product (TBAB) was quickly collected by suction filtration and washed twice with diethyl ether. Then the reaction of dried TBAB and desalted Cysa (C, 5 mmol) dissolved in anhydrous THF (30%, wt/v) proceeded for 3 h at 30 °C. Finally, a similar purification process was conducted for compound 3 (TBABC), and the target collecting ratio was about 20:1 (v/v) to give the product as slightly yellow solid (∼25% yield). 1H NMR (600 MHz, CDCl3, ppm): δ = 4.27 (t, 4H, A), 4.20 (m, 6H, B + T), 4.06 (t, 43H, A), 3.64 (t, 174H, T), 3.38 (s, 3H, T), 3.15 (d, 4H, B), 2.81 (m, 4H, C), 2.32 (t, 42H, A), 1.65 (m, 87H, A), 1.38 (m, 46H, A), 1.27 (m, 9H, B).
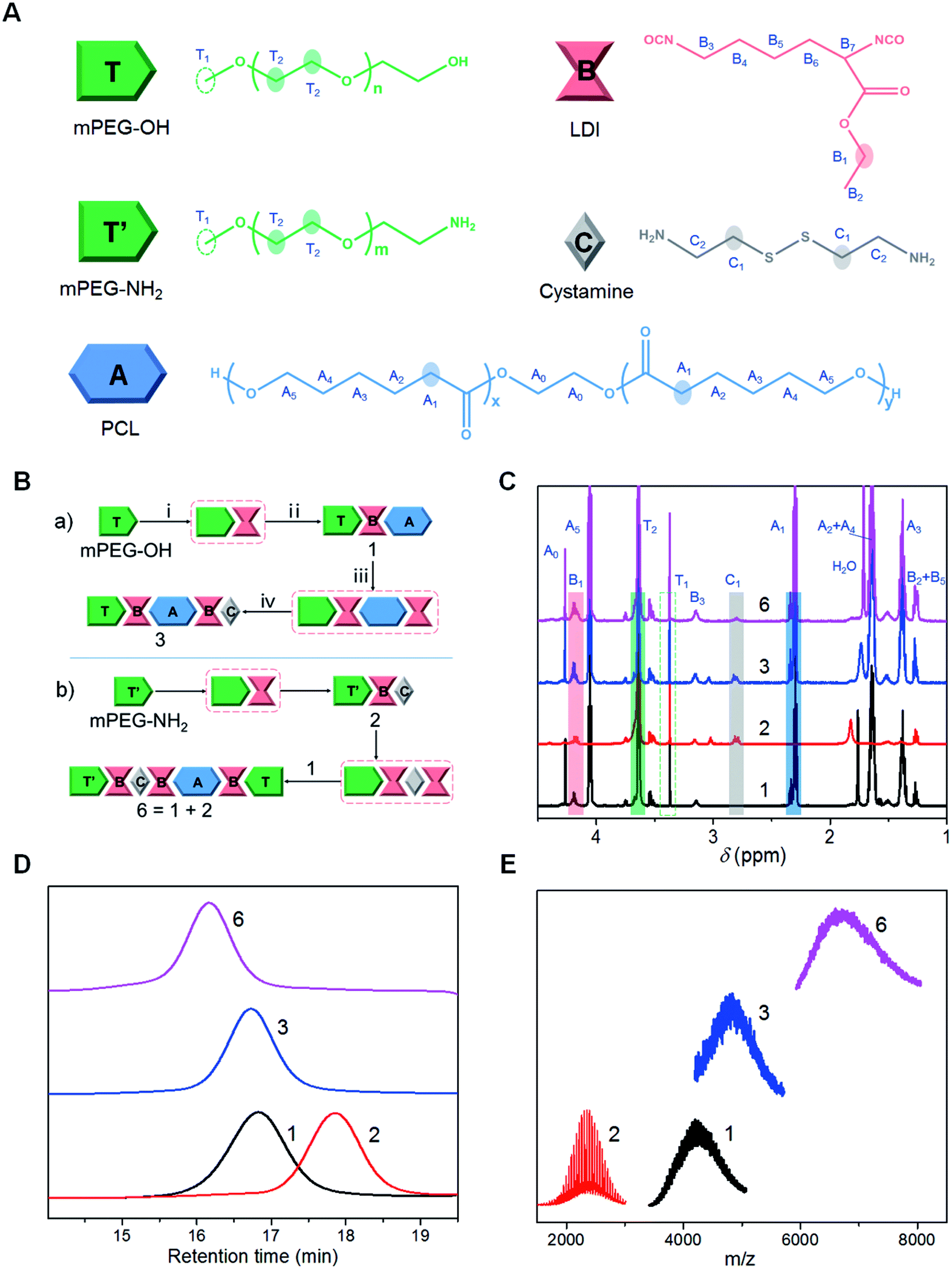 | ||
| Fig. 1 Precise synthesis of PUs. (A) Structural representations of different modules involved in the synthetic platform. (B) Synthetic schematic diagrams of 3 (TBABC) and 6 (T′BCBABT) via the diisocyanate protocols. The intermediates are denoted with dashed boxes. Experimental reagents and conditions for compound 3: (i) anhydrous T, B (3 eq.), bismuth catalysts, N2, 60 °C, 2 h, THF frozen precipitation; (ii) anhydrous TB, A (3 eq.), N2, 60 °C, 2 h, then 80 °C, 3 h, column chromatography for 1 (TBA); (iii) anhydrous TBA, B (5 eq.), bismuth catalysts, N2, 60 °C, 3 h, THF frozen precipitation; (iv) anhydrous TBAB, C (5 eq.) in THF, N2, 30 °C, 3 h, column chromatography for 3 (TBABC). (C) Stacked 1H NMR spectra (CDCl3, 600 MHz, δ 4.50–1.00 ppm) of synthetic PUs with the characteristic protons highlighted. (D and E) GPC traces (D) and MALDI-TOF MS spectra (E) of different synthetic PUs. | ||
Compounds 4 (T′BCBA) and 5 (TBABA) were synthesized by similar approaches. See ESI Section S2.1.2† for detailed procedures and characterization results.
Synthesis of seven-segment (6) and fourteen-segment PUs (7)
The convergent strategies were used for the rapid construction of multiblock PUs with higher molecular weights. The synthetic diagram for compound 6 based on diisocyanate chemistry is shown in Fig. 1B(b). In brief, 2 (T′BC, 1 mmol) and LDI (B, 3 mmol) dissolved in dehydrated THF were reacted at 30 °C for 3 h under a nitrogen environment. After T′BC was almost converted, the reaction solution was added into anhydrous diethyl ether, and the solution was refrigerated at −24 °C for 1 h. Then, the purified intermediate product (T′BCB) was quickly collected by suction filtration and washed twice with diethyl ether. Then the reaction of dried T′BCB and 1 (TBA, 1 mmol) with organic bismuth was performed at 60 °C for 2 h, and at 80 °C for another 3 h in a nitrogen atmosphere. Finally, a similar purification process was conducted for compound 6 (T′BCBABT), and the target collecting ratio was about 20:1 (v/v) to give the product as white solid (∼28% yield). 1H NMR (600 MHz, CDCl3, ppm): δ = 4.27 (t, 4H, A), 4.20 (m, 8H, B + T), 4.06 (t, 30H, A), 3.64 (t, 364H, T + T′), 3.38 (s, 6H, T + T′), 3.15 (d, 6H, B), 2.81 (m, 4H, C), 2.32 (t, 30H, A), 1.65 (m, 65H, A), 1.38 (m, 38H, A), 1.27 (m, 14H, B).
The synthesis of multiblock PU 7 with fourteen segments (TBABABC′-C′′BABABT) depended on the convergent approach via the classical click chemistry: copper-catalyzed azido-alkyne cycloaddition reaction, where C′ and C′′ refer to the clickable monomers of propargylamine and 2-azidoethylamine, respectively (see ESI Section S2.1.3† for detailed procedures and characterization results).
Synthesis of multifunctional seven-segment PUs (12 and 13)
As shown in Fig. 2B, the convergent synthetic process of compound 12 was similar to that of compound 6 based on diisocyanate chemistry. Briefly, the reaction of dried 8 (TBK, 1 mmol) and LDI (B, 5 mmol) with organic bismuth dissolved in anhydrous dimethyl sulfone (DMSO, 10 mL) was performed at 60 °C for 2 h under a nitrogen environment. After TBK was almost converted, the reaction solution was added into anhydrous diethyl ether with stirring, and the solution was refrigerated at −24 °C for 2 h. The intermediate product (TBKB) was quickly collected by suction filtration and washed twice with diethyl ether. Then dried TBKB and 10 (A′BQ, 1 mmol) with organic bismuth dissolved in DMSO was reacted at 60 °C for 5 h in a nitrogen atmosphere. Finally, a similar purification procedure was conducted for compound 12 (TBKBQBA′, KQ), and the target collecting ratio was about 7:1 (v/v) to give white solid (∼19% yield). 1H NMR (600 MHz, CDCl3, ppm): δ = 4.75 (m, 2H, K + A′), 4.20 (m, 8H, B + T), 4.06 (t, 47H, A′), 3.64 (t, 185H, T), 3.38 (s, 3H, T), 3.22 (s, 6H, Q), 2.32 (t, 47H, A′), 1.65 (m, 102H, A′), 1.38 (m, 58H, A′), 0.88 (t, 4H, Q).
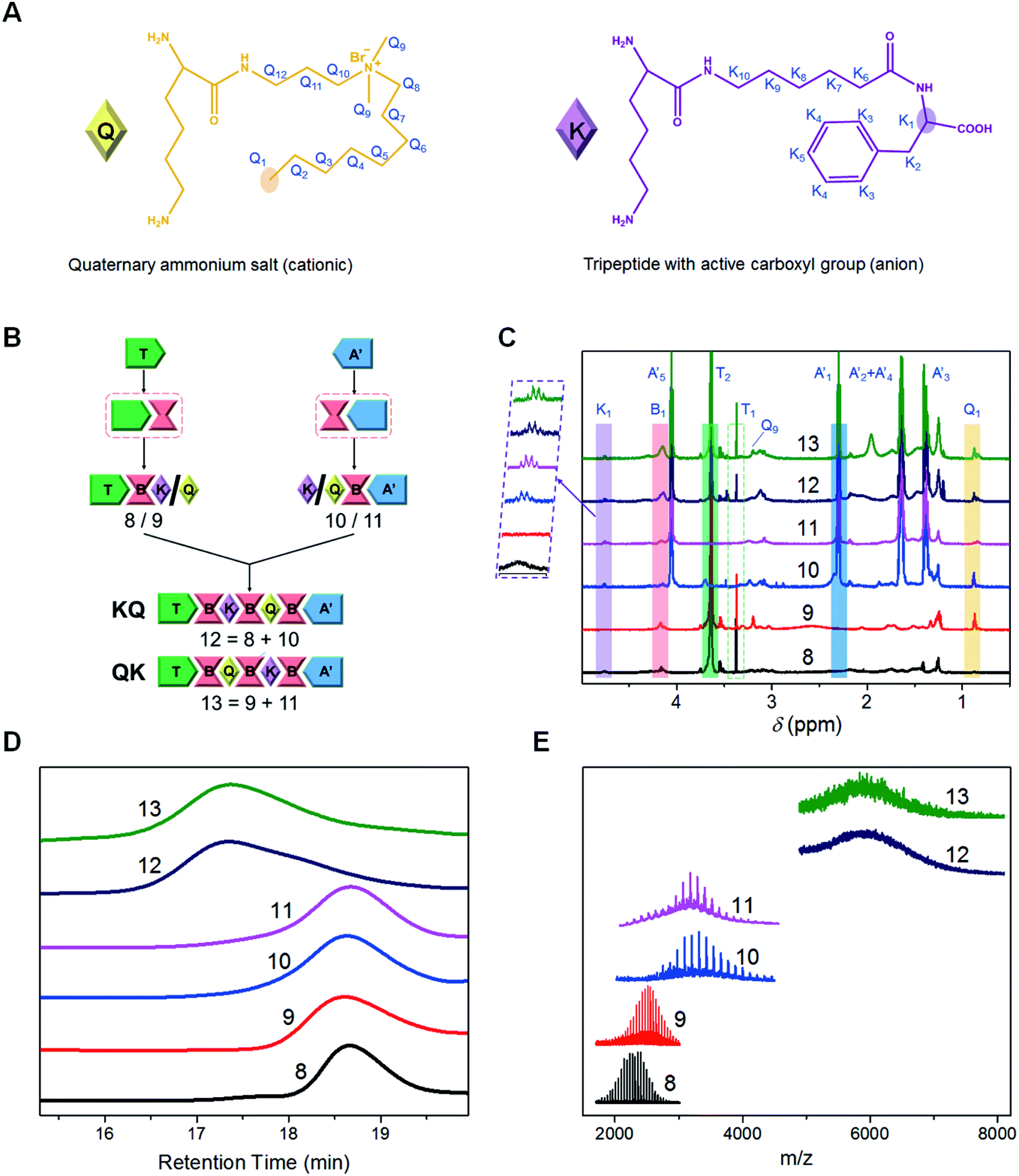 | ||
| Fig. 2 Precise synthesis of multifunctional PUs with defined block sequence orders. (A) Structural diagrams of cationic (Q) and anion (K) functional segments. (B) Brief schematic illustrations depicting the sequence-controlled synthesis of 12 (TBKBQBA′, KQ) and 13 (TBQBKBA′, QK). (C) Stacked 1H NMR spectra (CDCl3, 600 MHz, δ 5.00–0.50 ppm) of synthetic PUs with the characteristic protons highlighted. (D and E) GPC traces (D) and MALDI-TOF MS spectra (E) of different synthetic PUs. | ||
With the same procedure, compound 13 (TBQBKBA′, QK) was synthesized via the short chains: 9 (TBQ) and 11 (A′BK). See ESI Section S2.2.3† for detailed procedures and characterization results.
Results and discussion
Synthetic platform of precise PUs
To construct a versatile platform for the precise synthesis of PUs, mPEG-OH (T) or mPEG-NH2 (T′) was chosen as soluble polymer supports for the ordered incorporation of PCL (A, polyester diol) and Cysa (C, bifunctional diamine) using LDI (B) as a linkage segment (Fig. 1A and S19†). The procedures were derived from conventional LPS with unprotected monomers, and the segment sequence was manipulated by the reaction of a monofunctional support polymer with excessive bifunctional segments in every step.34,35 Relying on the solubility difference of PEG in different solvents, the excess feedstocks were removed conveniently through selective precipitation. Moreover, the target products were further purified by conventional column chromatography. All the synthesis and purification processes could be well monitored by TLC (Fig. S20†). A proof-of-principle synthetic protocol of the iterative diisocyanate chemistry is illustrated in Fig. 1B. Each two-step cyclic reaction increased the segment number by two, where one was the linker (B) and the other was the target module. The representative PUs with accurate structures involved in the synthetic platform are three-segment (1 and 2), five-segment (3, 4, and 5), seven-segment (6) and fourteen-segment polymers (7). The synthesis routes of [T-1-3] and [T′-2-4] demonstrate that starting from a monofunctional macromer (PEG) containing a hydroxyl group or an amino group, an arbitrary diol or diamine segment can be introduced into the molecular chains continuously and orderly based on the iterative diisocyanate procedures (see ESI Section S2.1.2†). The compounds 3 and 4 could be regarded as a pair sharing the same molecular composition but different segment sequence structures. Besides, the synthesis of compound 6 based on the diisocyanate protocol and compound 7via a click chemistry reveals that different convergent strategies were compatible for rapid growth of multiblock PU chains.The resultant PUs were first characterized by 1H NMR spectroscopy. The sequential appearance of the characteristic chemical shifts of mPEG (C![[H with combining low line]](https://www.rsc.org/images/entities/i_char_0048_0332.gif) 3–O–, 3.38 ppm; –O–C2C2–O–, 3.64 ppm), LDI (CH3C2–O–, 4.20 ppm; C3CH2–O–, 1.26 ppm), PCL (–O–C2C2–O–, 4.27 ppm; –O–C2CH2CH2–, 4.06 ppm; –CH2C2CO–, 2.32 ppm; –O–CH2C2CH2C2CH2–CO–, 1.64 ppm; –O–CH2CH2C2CH2CH2–CO–, 1.38 ppm) and Cysa (–CH2C2–S–S–C2CH2–, 2.82 ppm) with increasing segment numbers demonstrates the stepwise successive introduction of different structural modules into the polymeric chains (Fig. 1C, S19 and S21A†). According to the integration areas of mPEG (C3–O–, 3.38 ppm; –O–C2C2–O–, 3.64 ppm), LDI (CH3C2–O–, 4.20 ppm), PCL (–O–C2C2–O–, 4.27 ppm; –CH2C2–CO–, 2.32 ppm) and Cysa (–CH2C2–S–S–C2CH2–, 2.82 ppm), the accurate compositions were determined and the number-average molecular weights were calculated (see ESI Section S2.1 and Table S1†). The results are in good agreement with the theoretical predictions (Tables 1 and S5†), manifesting the successful construction of the synthetic platform for the preparation of precise multiblock PUs with a regulated block sequence.
3–O–, 3.38 ppm; –O–C2C2–O–, 3.64 ppm), LDI (CH3C2–O–, 4.20 ppm; C3CH2–O–, 1.26 ppm), PCL (–O–C2C2–O–, 4.27 ppm; –O–C2CH2CH2–, 4.06 ppm; –CH2C2CO–, 2.32 ppm; –O–CH2C2CH2C2CH2–CO–, 1.64 ppm; –O–CH2CH2C2CH2CH2–CO–, 1.38 ppm) and Cysa (–CH2C2–S–S–C2CH2–, 2.82 ppm) with increasing segment numbers demonstrates the stepwise successive introduction of different structural modules into the polymeric chains (Fig. 1C, S19 and S21A†). According to the integration areas of mPEG (C3–O–, 3.38 ppm; –O–C2C2–O–, 3.64 ppm), LDI (CH3C2–O–, 4.20 ppm), PCL (–O–C2C2–O–, 4.27 ppm; –CH2C2–CO–, 2.32 ppm) and Cysa (–CH2C2–S–S–C2CH2–, 2.82 ppm), the accurate compositions were determined and the number-average molecular weights were calculated (see ESI Section S2.1 and Table S1†). The results are in good agreement with the theoretical predictions (Tables 1 and S5†), manifesting the successful construction of the synthetic platform for the preparation of precise multiblock PUs with a regulated block sequence.
| Samples | M n1 (g mol−1) | M n2 (g mol−1) | M n3 (g mol−1) | M n4 (g mol−1) | Đ (=Mw/Mn)d |
|---|---|---|---|---|---|
| a Theoretical molecular weights from additive molecular weights of all raw segments. b Molecular weights calculated by integration of NMR peaks. c Molecular weights detected by MALDI-TOF MS. d Molecular weights and molecular weight distributions measured by GPC. | |||||
| 1 | 4500 | 4400 | 4300 | 9700 | 1.09 |
| 2 | 2200 | 2200 | 2300 | 6000 | 1.08 |
| 3 | 4900 | 5000 | 4900 | 12000 |
1.07 |
| 6 | 6900 | 6700 | 6800 | 17000 |
1.06 |
Further gel permeation chromatography (GPC) traces evidenced single elution peaks and narrow molecular weight distributions (Đ 1.06–1.13) of the synthetic PUs (Fig. 1D, S21C and Table S2†), which are indicative of a high purity. As expected, the peaks gradually shifted to higher molecular weight ranges with the iterative reactions proceeding. The increase of molecular weights was also confirmed by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS). All the target peaks of model PU structures were observed clearly in the corresponding regions (Fig. 1E and S21D†). The lower-molecular weight ranges were eliminated because the substantial matrix and impurity noises resulted from the laser power required for ionization of high molecular weight compounds. Notably, detailed spectra analysis for block copolymers containing both PEG and PCL was not possible, because the non-equivalent masses of the repeat units lead to a complex distribution in which individual characteristics were no longer resolved (see Fig. S22† for more details).36,37 The accurate molecular weight values agree well with those determined by NMR (Tables 1 and S5†), further verifying the well-defined structures of PUs. In combination with an efficient click reaction, the calculated absolute molecular weight of multiblock PUs further increased to Mn = 13700 g mol−1 (Table S1†) for compound 7, which is favorable for traditional usage of PUs.
Synthesis of PUs with controlled sequences of functional segments
Based on the pre-established synthetic platform, we further explored the capacity of synthesizing precise PUs with controlled sequences of functional moieties. The incorporation of functional moieties into the polymeric chains with a tailored sequence is helpful to elucidate the sequence effect on the bulk and self-assembly behaviors and provide a fundamental understanding on the structure–property relationship of sequence-controlled multifunctional copolymers.38–40 To this end, we constructed a pair of seven-segment PUs with different sequence orders of cationic and anion segments: compounds 12 (TBKBQBA′, KQ) and 13 (TBQBKBA′, QK), taking a cell penetrating quaternary ammonium salt with a single-carbon chain (Q) and a tripeptide with an active carboxyl anion group (K) as model functional segments (Fig. 2A), and a synthetic mono-hydroxyl PCL (A′) as another soluble support (Fig. S10†).41,42 The two well-defined PUs were precisely synthesized through the convergent pathway similar to that of compound 6 (Fig. 2B), where various precise short chains were matched to give the products with different segment arrangements. As shown in the 1H NMR spectra (Fig. 2C, see ESI Section S2.2.3† for details), the characteristic peaks of different segments appeared specifically with sequential proceeding of iterative and convergent reactions, confirming that the sequence of functional monomers was controlled successfully. The integration of the peak areas indicates that the two polymers have identical composition (Table S3†). The structures of the final products were also confirmed by FT-IR and 13C NMR measurements (Fig. S23 and S24†). GPC (Fig. 2D and Table S4†) and MALDI-TOF MS (Fig. 2E and Table S5†) tests showed similar molecular weights for compounds 12 and 13. However, the molecular weight distributions differed slightly, possibly due to the different interactions between the functional segments and GPC chromatographic columns.43Sequence-controlled self-assembly and drug delivery properties
We further studied the influence of functional segment sequences on the self-assembly behaviors of amphiphilic seven-segment PUs in aqueous solution. In principle, the hydrophobic PCL and hydrophilic PEG segments form the core and corona, respectively, while the mixed-charged middle block (BKBQB or BQBKB) is expected to form an interlayer between the core and corona (Fig. 3A).4,44 The core–shell micelle-like structure was first confirmed with 1H NMR spectra (Fig. S25†), where only the characteristic peaks of PEG were apparent and the signals of other segments were significantly weakened in D2O compared with those in CDCl3.45 To further verify the micelle formation, fluorescence analysis was carried out using pyrene as a probe. We found a red-shift of the (0, 0) absorption band in the excitation spectra of pyrene with an increase of PU concentration (Fig. S26A†). The result indicates the transfer of pyrene into a hydrophobic microenvironment of the micellar core.45 Accordingly, the critical aggregation concentrations were calculated to be ∼4 μg mL−1 (Fig. S26B and Table S6†). Further dynamic light scattering (DLS) and static light scattering (SLS) measurements support the formation of spherical micellar-like structures, since their characteristic parameters (ρ) were close to 0.775 (Fig. S27 and Table S6†).46 The size of KQ was relatively larger than that of QK, as evidenced by DLS and transmission electron microscopy (TEM) (Fig. 3B–D and Table S6†). Note that the sizes derived from TEM are smaller than those determined by DLS because of the shrinkage and dehydration of particles in a dry state.45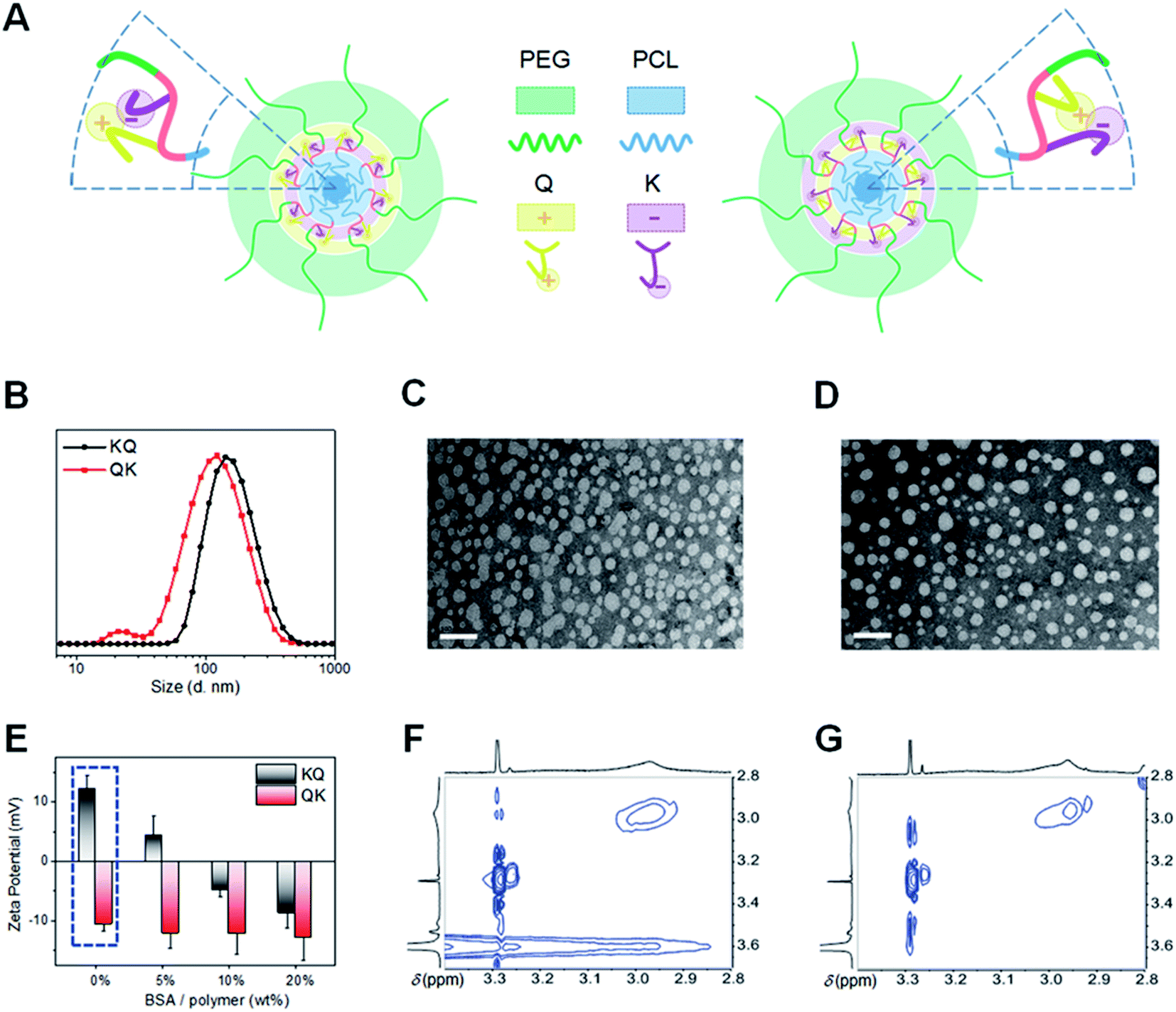 | ||
| Fig. 3 Self-assembly of multifunctional PUs with controlled sequences of functional segments (KQ: 12 and QK: 13). (A) Schematic illustrations of KQ micelles (left) and QK micelles (right). (B) Particle size distributions of PU assemblies at the concentration of 1 mg mL−1 in ultrapure water. (C and D) TEM images of KQ micelles (C) and QK micelles (D). The scale bars are 100 nm. (E) Zeta potentials of multifunctional PU micelles before and after incubation with different amounts of BSA protein. (F and G) Enlarged 1H–1H NOESY NMR spectra of KQ micelles (F) and QK micelles (G) in water with 10% D2O (600 MHz, horizontal 1H: δ 3.40–2.80 ppm, vertical 1H: δ 3.70–2.80 ppm). | ||
Of interest, the surface charges of self-assembled micelles showed large differences. We originally postulated that QK micelles would exhibit more positive charges than KQ because the quaternary ammonium group (Q) in QK is closer to the hydrophilic segment (T), which is more likely to be exposed to the micellar surface. However, to our surprise, the zeta potential of QK was negative (−10.2 ± 3.6 mV) and that of KQ was positive (13.2 ± 6.4 mV) (Fig. 3E and Table S6†). The unexpected surface charges of PU micelles were also confirmed by a protein absorption experiment. It was found that QK micelles remained a negatively charged surface whereas KQ particles underwent a positive-to-negative charge transition with an increasing content of mixed bovine serum albumin (BSA) (Fig. 3E). Furthermore, KQ absorbed more proteins than QK assemblies (Fig. S28†), suggesting a higher amount of positive charges on the surface of KQ micelles. According to the surface properties, the probable schematic illustrations of the assembled core–shell micelles of PUs are presented in Fig. 3A.44,45 As the ionized parts of K (–COO−) and Q (–CH3N+CH3–) possess an intrinsic tendency of migrating to the hydrophilic surface, it is hypothesized that the functional moieties closer to the hydrophobic PCL were more likely to appear on the surface during phase separation because of their high incompatibility with core segments, while the other part would be invaginated and shielded to some degree due to the competitive chain arrangement. To validate this hypothesis, the 1H–1H nuclear Overhauser enhancement spectroscopy (NOESY) of the micellar solutions was performed (Fig. 3F and G).17,46 Evidently, a distinct NOE signal between Q (–C3N+C3–, 2.96 ppm) and mPEG (–O–C2C2–O–, 3.61 ppm) was observed for KQ micelles, while this correlation was negligible in the spectrum of QK micelles. The result implies that the Q group, despite its chemical nearness with the PCL block, showed a spatial proximity to the hydrophilic PEG segment in KQ micelles. The detailed mechanism for this interesting chain folding behavior is still largely unclear. In addition, the length, rigidity, and hydrophilicity of the spacers between the functional groups should also be taken into account. More evidence for elaborating the self-assembly structures of sequence-regulated multifunctional segmented PUs is imperative in follow-up work.
To investigate whether the sequence of functional moieties impacted the drug delivery properties of PU micelles, DOX as a hydrophobic antitumor drug was loaded into the nanocarriers (DOX@KQ and DOX@QK). It was found that DOX@QK displayed slightly higher loading efficiency (Fig. S29A†) and a similar release profile in phosphate buffer solution (PBS, pH 7.4) compared with DOX@KQ (Fig. S29B†). Moreover, as indicated by confocal laser scanning microscopy (CLSM) (Fig. 4A and S30A†) and flow cytometry tests (Fig. S30B†), DOX@KQ micelles showed faster cellular uptake by HeLa cells in the first 3 h, which may be due to their positive surface charge. As a result, the half-maximal inhibitory concentration (IC50) of DOX@KQ (0.33 μg mL−1) for 72 h of incubation was lower than that of DOX@QK (0.53 μg mL−1) (Fig. S31†). In addition, the drug-free assemblies did not show any noticeable inhibition effect against both L929 mouse fibroblasts and HeLa cells (Fig. S32†), suggesting a good cytocompatibility of PU micelles.
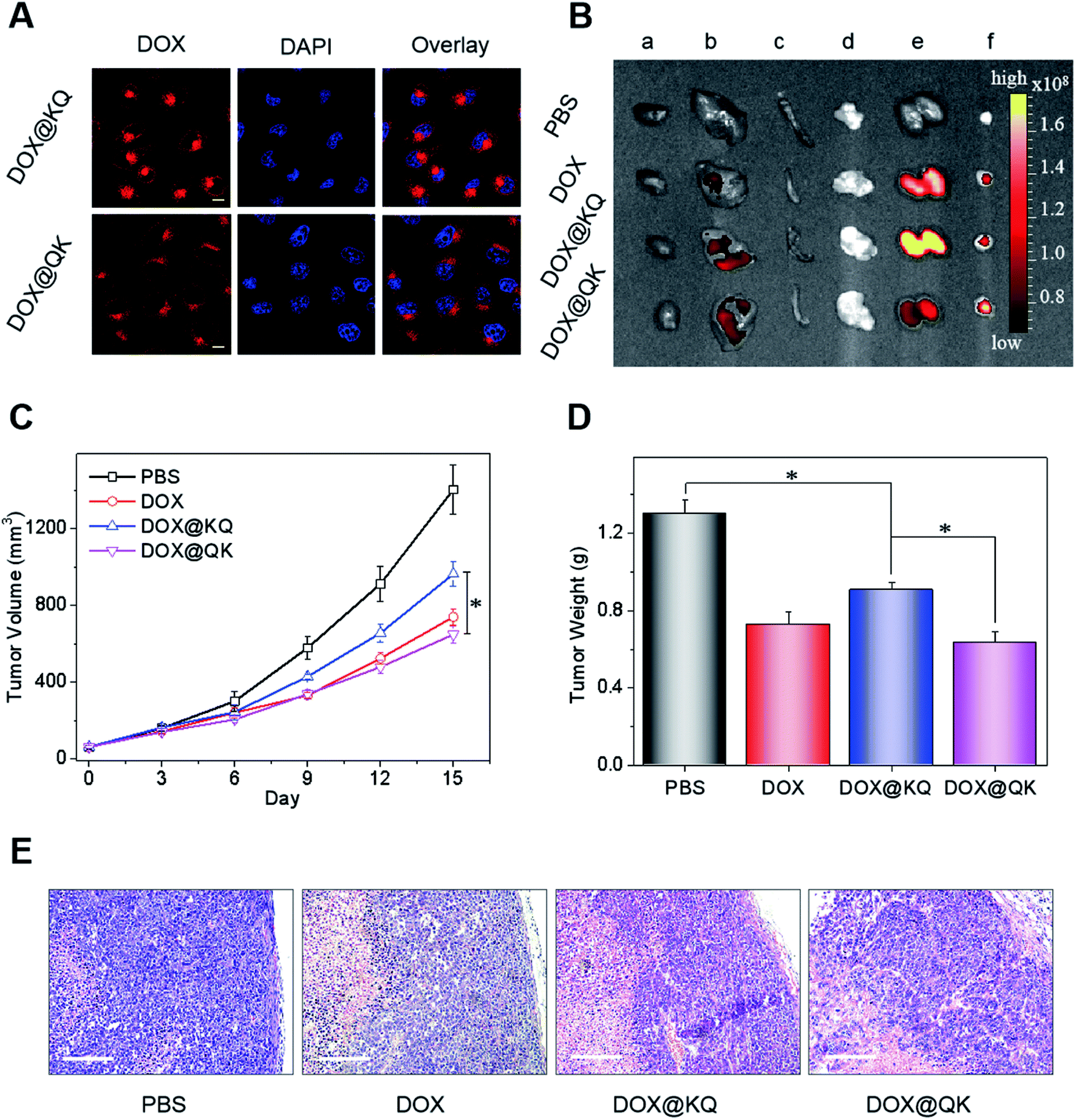 | ||
| Fig. 4 Biological evaluations of sequence-controlled multifunctional PUs as drug carriers. (A) Confocal images of HeLa cells incubated with DOX-loaded micelles for 3 h. The scale bars are 10 μm. (B) Ex vivo fluorescence images of excised organs of HeLa tumor-bearing nude mice after intravenous injection with DOX-labeled micelles for 3 h, where a, b, c, d, e, and f represent heart, liver, spleen, lung, kidney, and tumor, respectively. Mice receiving PBS were set as control. (C) Changes of tumor volumes of nude mice receiving intravenous administration of different drug formulations at a DOX dose of 2 mg kg−1. (D) Mean weights of tumors separated from mice treated with various DOX formulations for 2 weeks. (E) Ex vivo histological analyses of tumor sections stained with hematoxylin and eosin (H&E). The scale bars are 100 μm. Statistical significance: (*) P < 0.05. | ||
To assess the fate and antitumor performance of sequence-controlled multifunctional PU formulations in vivo, DOX@KQ and DOX@QK were intravenously administered into the tail vein of mice bearing HeLa tumors, taking free DOX as a control. During the treatment period, drug-free nanoparticles showed no obvious effects on tumor growth (Fig. S33†), and the administration of drug formulations presented negligible influences on the normal life of mice (Fig. S34†). The fluorescence images (Fig. 4B) and semi-quantitative analysis of excised organs (Fig. S35†) evidenced an improved tissue distribution of DOX@QK with remarkable DOX accumulation in the tumor tissues compared to free DOX, because of the enhanced permeability and retention (EPR) effect.47 DOX@KQ displayed worse tissue distribution, which may be due to that the higher plasma protein absorption of positively charged KQ micelles led to rapid clearance by the reticuloendothelial system (RES).48–50 Consequently, DOX@QK exhibited a more potent inhibitory effect on the growth of tumors (Fig. 4C), with a tumor weight inhibition (TWI, 51.3%) much higher than that of DOX@KQ (30.3%) (Fig. 4D). Moreover, DOX@QK displayed higher efficiency concerning a greater extent of apoptosis and tissue necrosis as revealed by the histological analysis of tumor sections (Fig. 4E). Despite the relatively lower antitumor effect compared with other micellar DOX formulations due to the low dosage and lack of functionality,5,7 the significant difference in the therapeutic efficacy between QK and KQ micelles demonstrates that the sequences of cationic and anion segments play an indispensable role in determining the performances of polymeric materials for in vivo applications. Hence, the insight arising from this work offers potential guidelines for the development of multifunctional polymeric nanomedicines. Future work will incorporate other functional moieties, such as targeting ligands and imaging groups into the sequence-controlled PUs and carry out systematical structure–property relationship studies. In addition, the further optimization of the synthetic platform is needed to produce multiblock PUs with higher yield, better-defined structures and sequence-tailored functionalities that are easier to be approved by FDA and translated into the clinic.
Conclusions
In summary, we have developed a synthetic platform, derived from a liquid-phase synthesis via the iterative diisocyanate chemistry, for the preparation of precise PUs with regularly-growing high molecular weight, narrow polydispersity and orderly stacking of different commercial segments along molecular chains. The established platform, although not elegant for the moment, allows access to the sequence-controlled synthesis of multifunctional PUs containing cationic and anion segments. The subsequent fundamental structure–property relationship study elucidates the great impacts of segment sequences on the surface charges of self-assembled nanoparticles and antitumor therapy of PUs in vivo. Overall, the versatile platform facilitates efficient preparation of complex multifunctional PUs with the desired block sequence, which will open new perspectives for the structural and functional designs of next-generation materials.Data availability
Data are available within the article or in the ESI file.†Author contributions
Conceptualization, M. D. and H. T.; investigation, Y. S. and C. S.; resources, C. T. and X. H.; writing – original draft, Y. S. and M. D.; writing – review & editing, H. M., Y. W., W. L. and N. H.; funding acquisition, M. D. and H. T.; supervision, M. D., J. L., F. L., H. T. and Q. F.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge Prof. Pengchi Deng (Analytical and Testing Center, Sichuan University) for NMR measurement, Jiao Lu (College of Biomedical Engineering, Sichuan University) for MALDI-TOF MS detection, Tao Fu (West China School of Basic Medical Sciences and Forensic Medicine, Sichuan University) for TEM observation and Guolong Meng (College of Biomedical Engineering, Sichuan University) for the CLSM test. The research was supported by the National Natural Science Foundation of China (52022062, 51873122 and 51733005).Notes and references
- J.-F. Lutz, J.-M. Lehn, E. W. Meijer and K. Matyjaszewski, Nat. Rev. Mater., 2016, 1, 1–14 CrossRef
.
- M. Ding, J. Li, H. Tan and Q. Fu, Soft Matter, 2012, 8, 5414–5428 RSC
- Y. Matsushita, Polym. J., 2008, 40, 177–183 CrossRef CAS
- M. Ding, N. Song, X. He, J. Li, L. Zhou, H. Tan, Q. Fu and Q. Gu, ACS Nano, 2013, 7, 1918–1928 CrossRef CAS PubMed
- J. Wei, X. Shuai, R. Wang, X. He, Y. Li, M. Ding, J. Li, H. Tan and Q. Fu, Biomaterials, 2017, 145, 138–153 CrossRef CAS PubMed
- C. Weng, H. Chen, T. Xu, Z. Li, X. Liu, M. Ding, Q. Zhang, H. Tan and Q. Fu, ACS Mater. Lett., 2020, 2, 602–609 CrossRef CAS
- R. Yang, Y. Zheng, X. Shuai, F. Fan, X. He, M. Ding, J. Li, H. Tan and Q. Fu, Adv. Sci., 2020, 7, 1902701 CrossRef CAS PubMed
- Y. Zheng, C. Weng, C. Cheng, J. Zhao, R. Yang, Q. Zhang, M. Ding, H. Tan and Q. Fu, Macromolecules, 2020, 53, 5992–6001 CrossRef CAS
- T. H. Epps III and R. K. O'Reilly, Chem. Sci., 2016, 7, 1674–1689 RSC
- Y. Matsuo, R. Konno, T. Ishizone, R. Goseki and A. Hirao, Polymers, 2013, 5, 1012–1040 CrossRef
- P. Krol, Prog. Mater. Sci., 2007, 52, 915–1015 CrossRef CAS
- M. Ali and S. Brocchini, Adv. Drug Delivery Rev., 2006, 58, 1671–1687 CrossRef CAS PubMed
- K. Matyjaszewski, Prog. Polym. Sci., 2005, 30, 858–875 CrossRef CAS
- P. Mi, K. Miyata, K. Kataoka and H. Cabral, Adv. Ther., 2020, 4, 2000159 CrossRef
- B. Voit, Angew. Chem., Int. Ed., 2017, 56, 2810–2811 CrossRef CAS PubMed
- M. F. Perutz, Nature, 1962, 194, 914–917 CrossRef CAS PubMed
- Y. Zheng, Z. Wang, Z. Li, H. Liu, J. Wei, C. Peng, Y. Zhou, J. Li, Q. Fu, H. Tan and M. Ding, Angew. Chem., Int. Ed., 2021, 60, 22529–22536 CrossRef CAS PubMed
- J. F. Lutz, M. Ouchi, D. R. Liu and M. Sawamoto, Science, 2013, 341, 1238149 CrossRef PubMed
- J. De Neve, J. J. Haven, L. Maes and T. Junkers, Polym. Chem., 2018, 9, 4692–4705 RSC
- N. Badi and J.-F. Lutz, Chem. Soc. Rev., 2009, 38, 3383–3390 RSC
- L. Hartmann and H. G. Borner, Adv. Mater., 2009, 21, 3425–3431 CrossRef CAS PubMed
- N. Zaquen, M. Rubens, N. Corrigan, J. Xu, P. B. Zetterlund, C. Boyer and T. Junkers, Prog. Polym. Sci., 2020, 107, 101256 CrossRef CAS
- A. A. Denholm, M. H. George, H. C. Hailes, P. J. Tiffin and D. A. Widdowson, J. Chem. Soc., Perkin Trans. 1, 1995, 5, 541–547 RSC
- M.-C. Kuo, R.-J. Jeng, W.-C. Su and S. A. Dai, Macromolecules, 2008, 41, 682–690 CrossRef CAS
- C. Cho, E. Moran, S. Cherry, J. Stephans, S. Fodor, C. Adams, A. Sundaram, J. Jacobs and P. Schultz, Science, 1993, 261, 1303–1305 CrossRef CAS PubMed
- U. S. Gunay, B. E. Petit, D. Karamessini, A. Al Ouahabi, J.-A. Amalian, C. Chendo, M. Bouquey, D. Gigmes, L. Charles and J.-F. Lutz, Chem, 2016, 1, 114–126 CAS
- E. A. Hoff, G. X. De Hoe, C. M. Mulvaney, M. A. Hillmyer and C. A. Alabi, J. Am. Chem. Soc., 2020, 142, 6729–6736 CrossRef CAS PubMed
- S. D. Dahlhauser, S. R. Moor, M. S. Vera, J. T. York, P. Ngo, A. J. Boley, J. N. Coronado, Z. B. Simpson and E. V. Anslyn, Cell Rep. Phys. Sci., 2021, 2, 100393 CrossRef CAS PubMed
- C. Y. Cho, R. S. Youngquist, S. J. Paikoff, M. H. Beresini, A. R. Hebert, L. T. Berleau, C. W. Liu, D. E. Wemmer, T. Keough and P. G. Schultz, J. Am. Chem. Soc., 1998, 120, 7706–7718 CrossRef CAS
- P. A. Wender, J. B. Rothbard, T. C. Jessop, E. L. Kreider and B. L. Wylie, J. Am. Chem. Soc., 2002, 124, 13382–13383 CrossRef CAS PubMed
- E. Bayer and M. Mutter, Nature, 1972, 237, 512–513 CrossRef CAS PubMed
- P. H. Toy and K. D. Janda, Acc. Chem. Res., 2000, 33, 546–554 CrossRef CAS PubMed
- L. Hartmann, E. Krause, M. Antonietti and H. G. Börner, Biomacromolecules, 2006, 7, 1239–1244 CrossRef CAS PubMed
- R. N. Zuckermann, J. M. Kerr, S. B. H. Kent and W. H. Moos, J. Am. Chem. Soc., 1992, 114, 10646–10647 CrossRef CAS
- T. T. Trinh, C. Laure and J.-F. Lutz, Macromol. Chem. Phys., 2015, 216, 1498–1506 CrossRef CAS
- Y. Li, B. Zhang, J. N. Hoskins and S. M. Grayson, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1086–1101 CrossRef CAS
- B. Zhang, H. Zhang, Y. Li, J. N. Hoskins and S. M. Grayson, ACS Macro Lett., 2013, 2, 845–848 CrossRef CAS
- W. Zhang, X. Lu, J. Mao, C. H. Hsu, G. Mu, M. Huang, Q. Guo, H. Liu, C. Wesdemiotis, T. Li, W. B. Zhang, Y. Li and S. Z. D. Cheng, Angew. Chem., Int. Ed., 2017, 56, 15014–15019 CrossRef CAS PubMed
- L. Gabrielli, D. Núñez-Villanueva and C. A. Hunter, Chem. Sci., 2020, 11, 561–566 RSC
- T. M. C. Warnock, S. Rajkumar, M. Fitzpatrick, C. J. Serpell, P. Dingwall and P. C. Knipe, Chem. Sci., 2021, 12, 15632–15636 RSC
- Z. C. Pan, D. X. Fang, N. J. Song, Y. Q. Song, M. M. Ding, J. H. Li, F. Luo, H. Tan and Q. Fu, ACS Appl. Mater. Interfaces, 2017, 9, 2138–2149 CrossRef CAS PubMed
- M. Ding, J. Li, X. He, N. Song, H. Tan, Y. Zhang, L. Zhou, Q. Gu, H. Deng and Q. Fu, Adv. Mater., 2012, 24, 3639–3645 CrossRef CAS PubMed
- M. Ding, Z. Qian, J. Wang, J. Li, H. Tan, Q. Gu and Q. Fu, Polym. Chem., 2011, 2, 885–891 RSC
- M. Ding, X. He, Z. Wang, J. Li, H. Tan, H. Deng, Q. Fu and Q. Gu, Biomaterials, 2011, 32, 9515–9524 CrossRef CAS PubMed
- M. Ding, L. Zhou, X. Fu, H. Tan, J. Li and Q. Fu, Soft Matter, 2010, 6, 2087 RSC
- H. Liu, R. Wang, J. Wei, C. Cheng, Y. Zheng, Y. Pan, X. He, M. Ding, H. Tan and Q. Fu, J. Am. Chem. Soc., 2018, 140, 6604–6610 CrossRef CAS PubMed
- V. Torchilin, Adv. Drug Delivery Rev., 2011, 63, 131–135 CrossRef CAS PubMed
- Z. Pan, X. He, N. Song, D. Fang, Z. Li, J. Li, F. Luo, J. Li, H. Tan and Q. Fu, ACS Appl. Mater. Interfaces, 2019, 11, 16421–16429 CrossRef CAS PubMed
- D. Owensiii and N. Peppas, Int. J. Pharm., 2006, 307, 93–102 CrossRef PubMed
- M. Lundqvist, J. Stigler, G. Elia, I. Lynch, T. Cedervall and K. A. Dawson, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 14265–14270 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Additional materials and characterization, experimental protocols, synthetic procedures with NMR spectra (Fig. S1–S18), figures (Fig. S19–S35) and tables (Tables S1–S6). See https://doi.org/10.1039/d1sc06457f |
| This journal is © The Royal Society of Chemistry 2022 |