
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnantioselective iodolactonization to prepare ε-lactone rings using hypervalent iodine†
Jenna L.
Payne‡
 ,
Zihang
Deng‡
,
Andrew L.
Flach‡
and
Jeffrey N.
Johnston
*
,
Zihang
Deng‡
,
Andrew L.
Flach‡
and
Jeffrey N.
Johnston
*
Department of Chemistry, Vanderbilt Institute of Chemical Biology, Vanderbilt University, Nashville, Tennessee 37235-1822, USA. E-mail: jeffrey.n.johnston@vanderbilt.edu
First published on 8th June 2022
Abstract
Despite the rapid growth of enantioselective halolactonization reactions in recent years, most are effective only when forming smaller (6,5,4-membered) rings. Seven-membered ε-lactones, are rarely formed with high selectivity, and never without conformational bias. We describe the first highly enantioselective 7-exo-trig iodolactonizations of conformationally unbiased ε-unsaturated carboxylic acids, effected by an unusual combination of a bifunctional BAM catalyst, I2, and I(III) reagent (PhI(OAc)2:PIDA).
Introduction
Conversions of alkenes to vicinal diols,1 aminoalcohols,2 and diamines3 are prized transformations for their utility in chemical synthesis, with applications ranging from small molecules to complex natural products.4,5 By analogy, alkene haloesterification reactions are pathways to these functional groups in two steps, and offer divergent pathways that leverage the carbon–halogen bond's intermediacy to sulfur-, phosphorous-, nitrogen-, and oxygen–carbon bond formation, in addition to reduction. Broad interest in alkene haloesterification reactions was reignited with the advent of enantioselective variants,6 but the intramolecular cyclizations are restricted to small ring-sizes that form β-, γ-, and δ-lactones.7,8 The synthetic impact of these functionalizations is evident when considering ring-opening reactions of the enantioenriched lactone products that furnish, for example, enantioenriched epoxides or dihydroxy acids from unsaturated acids (Fig. 1); enantioselective carbon–oxygen bond formation with an unactivated alkene is achieved, formally, in two steps (i.e. halolactonization/saponification) using organocatalysis.9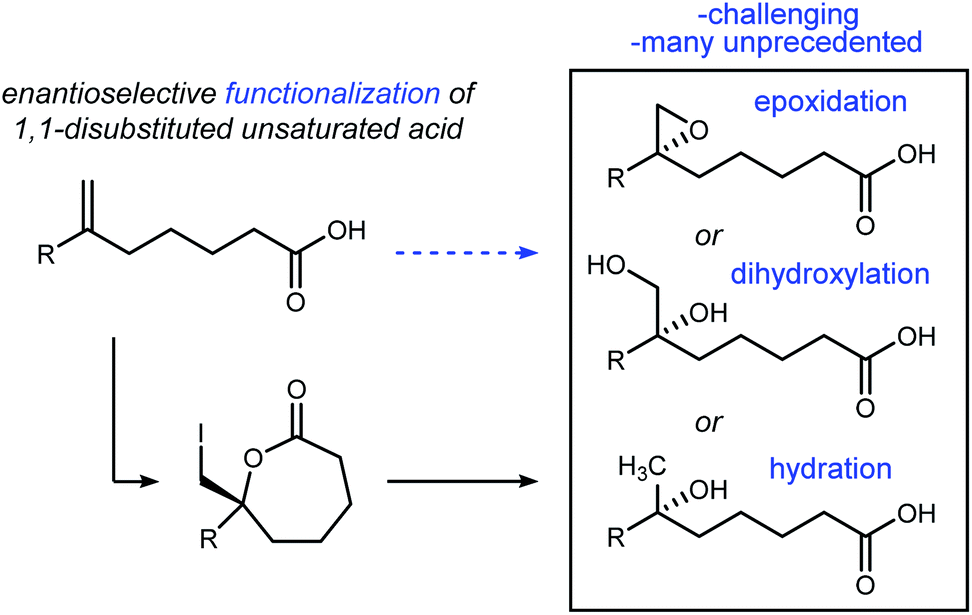 | ||
| Fig. 1 Formal enantioselective functionalization of remote alkenes using iodolactonization. | ||
The halolactonization reaction of unsaturated acids is currently limited to the formation of small (β, γ, and δ) lactones. One exception is Tang's use of enynes (Scheme 1) that deliver an allene exocyclic to a benzo-fused oxacycloheptanyl ester with excellent selectivity (94–98% ee).6d Efforts by others provide context for the challenge, including halolactonization to benzo-fused 7-membered rings in up to 10% ee for iodolactonization, and 31% ee for bromolactonization.10 Yeung recently reported enantioselective bromo- and chlorocyclizations that further generalize the use of an arylamine backbone,11 prompting us to disclose our findings. Importantly, all of these examples are limited to conformationally restricted unsaturated acids.12,13 Herein we report the use of a bis(amidine) [BAM] organocatalyst for the highly selective synthesis of enantioenriched ε-lactones. Notably, these substrates are conformationally unbiased, further extending the scope of halolactonization in enantioselective synthesis (Scheme 1).14,15
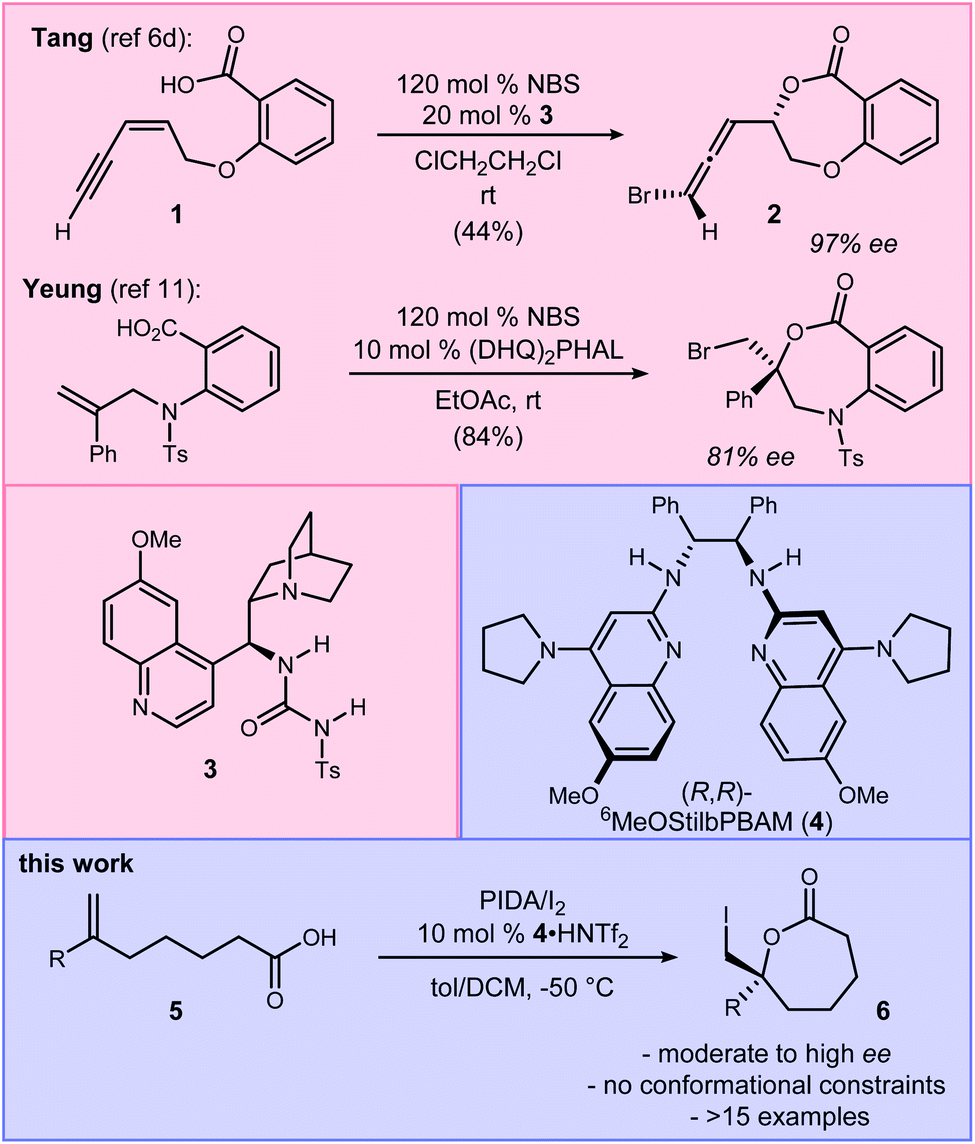 | ||
| Scheme 1 Tang's intramolecular alkene iodoesterification to a 7-membered oxylactone, and the development of an enantioselective ε-lactone synthesis (this work). | ||
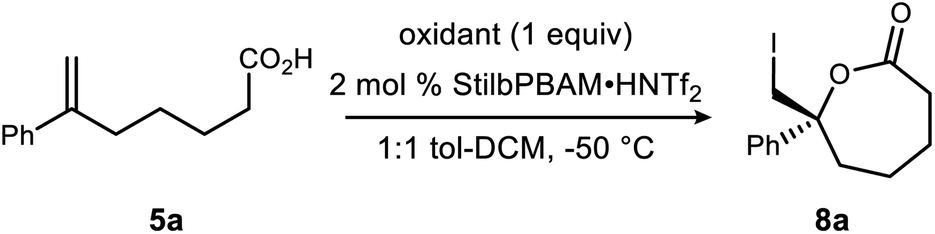
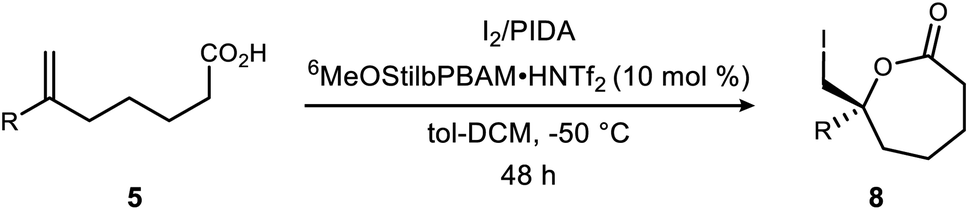
Initial attempts to extend our own protocols16 to ε-lactone synthesis were met with failure. Specifically, iodocyclization of unsaturated acid 5a led, at best, to a 6% yield of 8 with 20% ee (Scheme 2). By comparison, the corresponding δ-lactone synthesis (9 → 10) gives a 95% yield of the lactone in 98% ee. The low reactivity notwithstanding, we applied a very different iodonium-generating protocol that has been highly successful in unrelated work in our program.17–19 This protocol uses potassium iodide in combination with hypervalent iodine oxidant (PIDA),20 a combination initially investigated by Suárez.21–24 Our working hypothesis that BAM catalysis engages a carbonyl of the iodononium donor via a hydrogen bond in cyclizations of 9 was loosely extended to PIDA acetate oxygen, or acetyl hypoiodite (Scheme 2). A wide variety of activation modes for I(III) catalysis have accumulated, ranging from Brønsted acid to Lewis base activation, typically within alkene diacetoxylation studies.25–27 This analogy provided a promising result for δ-lactone formation (Scheme 2: 90% ee, 71% yield). The homologue 8 was formed in only 14% yield, but a promising 70% ee.
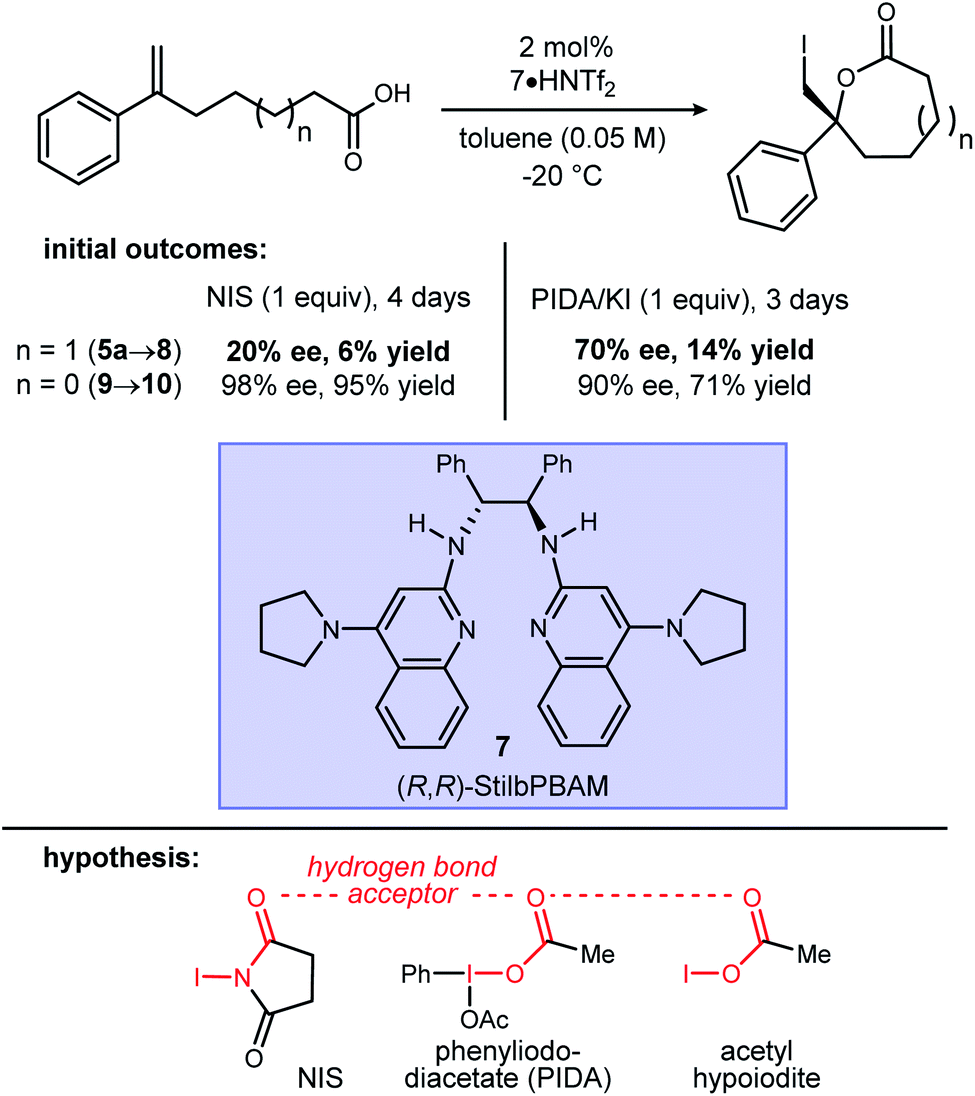 | ||
| Scheme 2 An intramolecular alkene iodoesterification to a 7-membered ring lactone. | ||
Exploration of alternative solvents led to the finding that a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 solution of toluene–dichloromethane impacted conversion substantially, delivering the lactone in 70% ee and 67% yield (Table 1, entry 2). The first point of investigation focused on the oxidant system as a means to improve lactone yield (Table 1). The low yields in many experiments summarized here were largely due to low conversion, but some evidence was collected for the formation of intermolecular iodoesterification products, highlighting the competitive rate of this process when iodolactonization is slow (not shown). Based on evidence that PIDA and iodine form acyl hypoiodite (IOAc) when combined,28–31 this reagent was generated directly from iodine and silver acetate.32 The yield of and ee of lactone 8a were both very low (Table 1, entry 3). The insolubility of the reagent in this experiment should be considered during interpretation of the outcome. Under conditions where NIS failed to form the desired lactone (Table 1, entry 4), replacement of NIS by the combination of PIDA/KI33,34 furnished an improved, but still low yield (14%) of lactone in 70% ee (Table 1, entry 1), confirming the trend identified in Scheme 2. Substitution of molecular iodine for KI further improved the yield to 40% despite a lower temperature (Table 1, entry 5) that increased selectivity to 80% ee.35 A brief investigation of alternative hypervalent iodine reagents,36 looking particularly for more homogeneous conditions, failed to improve yield, much less selectivity (Table 1, entries 6–11). Reagent Z4 delivered the lactone with similar selectivity and attenuated yield (Table 1, entry 11). Use of NIS/I2 to promote iodonium formation failed to form significant amounts of lactone product (Table 1, entry 12).
:1 solution of toluene–dichloromethane impacted conversion substantially, delivering the lactone in 70% ee and 67% yield (Table 1, entry 2). The first point of investigation focused on the oxidant system as a means to improve lactone yield (Table 1). The low yields in many experiments summarized here were largely due to low conversion, but some evidence was collected for the formation of intermolecular iodoesterification products, highlighting the competitive rate of this process when iodolactonization is slow (not shown). Based on evidence that PIDA and iodine form acyl hypoiodite (IOAc) when combined,28–31 this reagent was generated directly from iodine and silver acetate.32 The yield of and ee of lactone 8a were both very low (Table 1, entry 3). The insolubility of the reagent in this experiment should be considered during interpretation of the outcome. Under conditions where NIS failed to form the desired lactone (Table 1, entry 4), replacement of NIS by the combination of PIDA/KI33,34 furnished an improved, but still low yield (14%) of lactone in 70% ee (Table 1, entry 1), confirming the trend identified in Scheme 2. Substitution of molecular iodine for KI further improved the yield to 40% despite a lower temperature (Table 1, entry 5) that increased selectivity to 80% ee.35 A brief investigation of alternative hypervalent iodine reagents,36 looking particularly for more homogeneous conditions, failed to improve yield, much less selectivity (Table 1, entries 6–11). Reagent Z4 delivered the lactone with similar selectivity and attenuated yield (Table 1, entry 11). Use of NIS/I2 to promote iodonium formation failed to form significant amounts of lactone product (Table 1, entry 12).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Deviation | Iodine source | Oxidantb | eec (%) | Yieldd (%) | t (d) |
a General experimental details: the oxidant (100 mol%), KI (100 mol%), catalyst, and carboxylic acid were combined in solvent and stirred for the designated time, prior to workup and analysis; see ESI for complete details. Note that the uncatalyzed reaction, using PIDA/I2, −20 °C, 3 d, leads to 8 in 29% yield.
b Solubility observations under these reaction conditions: NIS, PIDA/KI minimally soluble; I2, PIFA/KI very soluble; PhIO/KI = unknown, but very high reactivity.
c Measured by HPLC using a chiral stationary phase.
d Measured by 1H NMR using an internal standard added to the crude reaction mixture after workup.
e Catalyst loading increased to 10 mol%.
|
||||||
| 1 | Tol. only, −20 °C | KI | PIDA | 70 | 14 | 3 |
| 2 | −20 °C | KI | PIDA | 70 | 67 | 3 |
| 3 | — | I2 | AgOAc | 12 | 2 | 4 |
| 4 | — | NIS | — | — | 0 | 2 |
| 5 | — | I2 | PIDA | 80 | 40 | 4 |
| 6 | — | I2 | PhI(O2CCCl3) | 0 | 13 | 2 |
| 7 | −20 °C | KI | Togni reagent (Z1) | — | 0 | 4 |
| 8 | −20 °C | I2 | Z1 | — | 5 | 2 |
| 9 | — | I2 | Koser's reagent (Z2) | 0 | 11 | 5 |
| 10 | — | I2 | Dess–Martin reagent (Z3) | 37 | 11 | 2 |
| 11 | — | I2 | Z4 | 80 | 24 | 4 |
| 12 | — | I2 | NIS | — | 8 | 2 |
| 13e | wu: SiO2 quench | I2 | PIDA | 78 | 85 | 3 |
| 14e | wu: aq. Na2S2O3 | I2 | PIDA | 85 | 74 | 3 |
| 15e | StilbPBAM | I2 | PIDA | 80 | 33 | 2 |
| 16e | StilbPBAM, NEt3 (100 mol%) | I2 | PIDA | — | 12 | 2 |
| 17e | 6MeOStilbPBAM·HNTf2 | I2 | PIDA | 87 | 81 | 2 |
| 18e | 6MeOStilbPBAM·HNTf2, (0.1 M) | I2 | PIDA | 87 | 66 | 2 |
| 19e | 6MeOStilbPBAM·HNTf2, (0.075 M) | I2 | PIDA | 91 | 70 | 2 |
| 20e | 6MeOStilbPBAM·HNTf2, (0.075 M), −40 °C | I2 | PIDA | 83 | 80 | 2 |
A substantial benefit to yield was realized by a careful evaluation of quench and workup procedures, as it was noted that the lactone was prone to decomposition in some experiments, a challenge that was faced as well during the development of scope (vide infra). A higher catalyst loading was used to encourage higher conversion for subsequent experiments. To simulate an incomplete reductive quench, a reaction was filtered through silica gel prior to full work-up. In this case, unreacted starting material may still react with the iodine source to form product. Since this reaction is still catalyzed, but at higher temperature, less enantioenriched material is produced. In the event (Table 1, entry 13), the lactone is retrieved in 78% ee and 85% yield. A thorough reductive quench with aqueous sodium thiosulfate is effective, across reaction scales, delivering the lactone in 85% ee and 74% yield (Table 1, entry 14).
The nature of the catalyst was scrutinized by evaluation of its free base form, which delivered the product in low yield (33%), but good selectivity (80% ee) (Table 1, entry 15). It is important to note in this case that the carboxylic acid substrate may play a role in forming a catalytically active, but less reactive (and/or selective) ligand-5a species. The finding that addition of a stoichiometric amount of amine base provided very little product is consistent with this hypothesis, as the substrate-NEt3 salt is likely formed (Table 1, entry 16). It was also noted that use of the doubly protonated ligand (L·2HNTf2) arrests the reaction with NIS, but merely lowers the yield when using PIDA/KI (93% ee, 28% yield). Finally, a broader investigation of common BAM catalysts identified 6MeOStilbPBAM as a ligand that provides slightly higher yield while maintaining selectivity (4·HNTf2, Table 1, entry 17). A lower concentration had a slightly positive effect on selectivity (87 → 91% ee), while maintaining yield (Table 1, entries 18–20). Raising the temperature slightly to −40 °C lowered selectivity, but more than anticipated (Table 1, entry 20) for reasons not yet clear.
This process of optimization established what appeared to be conditions most appropriate for a broader range of substrates: iodine and PIDA (1 equiv. each), 10 mol% catalyst, at 0.075 M in toluene–CH2Cl2 (1:1) at −50 °C for 48 hours. Application of these conditions to unsaturated acids is outlined in Table 2. The α-substituted styrene 5a provided lactone 8a in 71% isolated yield and 88% ee. The more hindered 2-naphthyl substrate 5b gave lactone 8b in 64% isolated yield and 87% ee. Aromatic ring substitution had little effect (Table 2, entries 3–4), except that an increase in temperature to −20 °C was helpful to the meta-substituted substrate 5d. ortho-Substituted 5e was quite different, with nearly no conversion observed. A series of halogenated derivatives exhibited high levels of enantioselection (91–96% ee) (Table 2, entries 6–9). In only one case was reactivity depressed (incomplete conversion), resulting in a 21% yield for 8h (Table 2, entry 8) that improved slightly at larger scale (vide infra, entry 24). Chlorinated substrate 5i produced a lactone (8i) exhibiting sufficient stability for purification, crystallization, and X-ray analysis. This led to the determination that the (R)-lactone is favored when using the (R,R)-catalyst (Fig. 2). However, decomposition of the crystals (brown/black discoloration) was noted after 24 hours despite refrigeration. Although a single trifluoromethyl substituent depressed reactivity and selectivity slightly, a pair of trifluoromethyl groups led to low reactivity and selectivity (c.f.Table 2, entries 10–11). An aldehyde-bearing aromatic ring in the substrate produced a lactone with excellent enantioselectivity and modest yield (Table 2, entry 12).
|
|
||||
|---|---|---|---|---|
| Entry | 5/8 | R | eeb (%) | Yieldc (%, isolated) |
| a General experimental details: the oxidant (100 mol%), iodine (100 mol%), and carboxylic acid were combined in solvent and stirred for 48 hours prior to workup and analysis; see ESI for complete details. b Measured by HPLC using a chiral stationary phase. c Isolated yield after chromatography. Although column chromatography was successfully used at room temperature, many of the lactone products were noted to turn yellow, red, and then brown during final concentration in pure form. As a result, care should be taken to avoid heating during workup, and storage of pure materials at low temperature (<0 °C), or as a frozen solution in benzene, is highly recommended as standard practice. Yields measured from the crude reaction mixture (internal standard, 1H NMR) were routinely higher. d Reaction temperature = −20 °C. e Reaction time = 4 days. f Reaction temperature = 25 °C. g Measured by 1H NMR analysis of the crude reaction mixture due to product instability. h Results at 1 mmol scale for substrate. | ||||
| 1 | a | C6H5 | 88 | 71 |
| 2 | b | 2Np | 87 | 64 |
| 3 | c | p MeC6H4 | 77 | 87 |
| 4d | d | m MeC6H4 | 84 | 76 |
| 5 | e | o MeC6H4 | — | Trace |
| 6 | f | p FC6H4 | 93 | 65 |
| 7 | g | m FC6H4 | 93 | 53 |
| 8e | h | m ClC6H4 | 96 | 21 |
| 9 | i | p ClC6H4 | 91 | 70 |
| 10 | j | p CF3C6H4 | 81 | 48 |
| 11f | k | 3,5(CF3)2C6H3 | 10 | 10 |
| 12 | l | m CHOC6H4 | 94 | 54 |
| 13 | m | p PhC6H4 | 84 | 72 |
| 14 | n | m MeOC6H4 | 90 | 63 |
| 15 | o | p MeOC6H4 | 49 | 53 |
| 16 | p | p (tBu)C6H4 | 83 | 66 |
| 17 | q | 5-(2-Methoxypyridine) | 75 | 73 |
| 18 | r | Me | 63 | 42 |
| 19 | s | Et | 72 | 58 |
| 20 | t | H | 44 | 24g |
|
||||
| Substrate | Product | |||
| 21 |

|
10 | 87 | 70 |
| 22 |
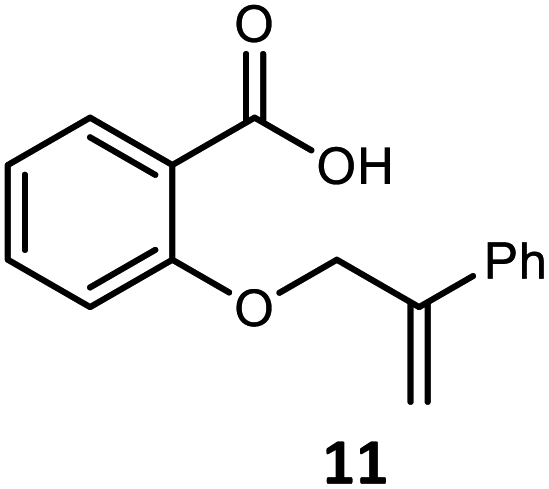
|
12 | 65 | 90 |
| 23d | 92 | 19 | ||
| 24h | h | m ClC6H4 | 96 | 26 |
| 25h | n | m MeOC6H4 | 87 | 61 |
 | ||
| Fig. 2 Absolute configuration assignment by X-ray analysis of 8i, and an interaction map of bifunctional activation hypothesized for enantioselective cyclization. | ||
Biphenyl substrate 5m performed well, forming lactone 8m in 72% yield and 84% ee (Table 2, entry 13). Electron-rich aromatic rings exhibited varying degrees of success, with methoxy substitution at the meta-position delivering lactone 8n in 63% yield and 90% ee, compared to 53% yield and 49% ee for para-substitution in 5o (Table 2, entries 14–15). Lactone 8o was noted to be prone to decomposition. An unsaturated acid with para–tert-butyl substitution (5p) gave the lactone in 66% yield and 83% ee (Table 2, entry 16). An initial attempt using a heterocyclic substituent (5q) led to a 73% yield of 8q in 75% ee (Table 2, entry 17). A substantial increase in reactivity was realized with simple alkyl (Me, 5r) substitution when compared to the styrene derivatives, providing lactone 8r in 42% yield, but with lower (63% ee) selectivity (Table 2, entry 18). Although an increase in size showed an increase in selectivity (5s, 72% ee), a terminal vinyl group produced lactone 8t in only 44% ee (c.f.Table 2, entries 19–20).
Heteroatom substitution in the backbone provided the oxazepinone (10) in 70% yield and 87% ee (Table 2, entry 21). A more conformationally restricted example was also successful, delivering the lactone in 90% yield and 65% ee (Table 2, entry 22). Interestingly, this case was found to be unusually sensitive to temperature, with product formed at −50 °C in only 19% yield and dramatically improved selectivity (92% ee, Table 2, entry 23). Two cases were examined at 1 mmol scale, with little change in selectivity or yield (Table 2, entries 24–25). A final look was given to these optimized conditions when applied to a selection of alternative halogenating agents, but DIDMH, NBS, or DBDMH resulted in recovery of only starting materials.
Lactones have long been valued for their synthetic versatility.37 Although these ε-lactones are unprecedented as enantioenriched materials in the literature, conversion of 8a to diol 13 proceeded in 75% yield (Scheme 3) using LiAlH4 (c.f. enantioselective alkene hydration in Fig. 1).38,39 Transesterification of 8a with methanol provides a pathway to epoxide formation (14), fulfilling the vision for enantioselective epoxidation, highlighted in Fig. 1 as an otherwise unmet need. Similarly, stannane reduction of 8i to 15 furnishes the product of enantioselective alkene hydroesterification (from 5i).
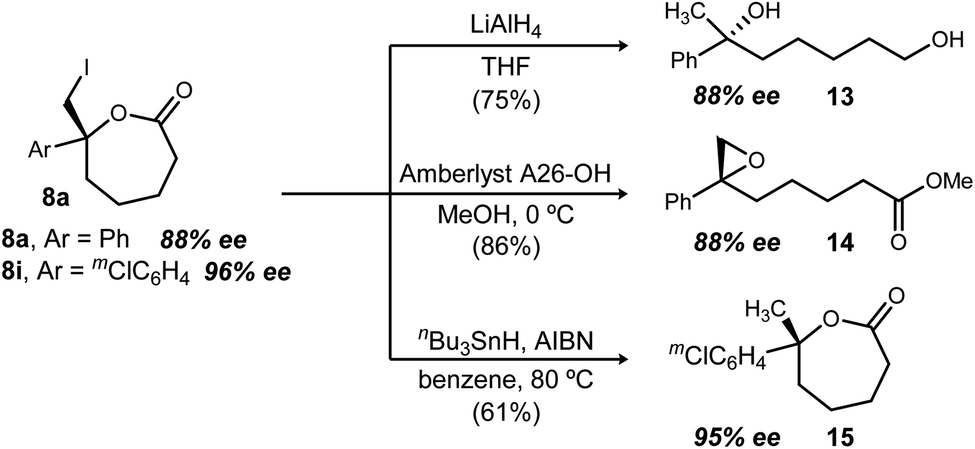 | ||
| Scheme 3 Conversions of ε-lactones 8. | ||
Discussion of mechanism
Our rationale for enantioselection using BAM catalysis is predicated on a bifunctional activation model in all cases except one40 to-date. In the study described here, the correlation between highest reactivity/selectivity and a 1:1 ligand–acid complex suggests that the resulting complex's amidinium hydrogen bond donor, and amidine hydrogen bond acceptor functionality, work in concert. We have found this behavior to be clear and consistent, albeit less well-defined when more Brønsted basic ligands and Brønsted acidic substrates are used, presumably since the substrate is a partial surrogate for the strong acid – here, triflimidic acid. The NIS/StilbPBAM·HNTf2 protocol works exceptionally well to form 6-membered lactones (Scheme 2), but for reasons still unclear, NIS is not effective in the formation of ε-lactones. This difference notwithstanding, the consistent π-face selectivity when using the (R,R)-catalyst suggests a generally conserved catalyst–substrate complex. PIDA/I2 overcomes the reactivity problem while maintaining the high selectivity, perhaps through a hydrogen bond donor–acceptor arrangement between catalyst and PIDA, respectively. The structural similarities between NIS and PIDA acceptors are highlighted in Scheme 2. In this proposition, PIDA may not merely function by oxidizing KI to I2, or I2 to IOAc, but instead participate in the catalyst–substrate complex. Consistent with this, there is no conversion if PIDA/KI is replaced with iodine alone,41 compared to lactone 8a formation in 42% yield and 41% ee if PIDA/I2 is used. The experiment to form AcOI directly41–43 suggested poor performance of AcOI (Table 1, entry 3).44 These outcomes are provided for consideration, but they do not yet converge on a single definition for X–I in Fig. 2. Given the similar outcomes for KI/PIDA and I2/PIDA, and our hypothesis that the acetate is a conserved feature for high enantioselection, we speculate that PhI(OAc)(I) may be a highly reactive and selective electrophilic iodine source that is formed slowly by this reagent combination, thereby matching the slower rate of ε-lactonization.
The critical bifunctional character of the catalyst is underscored by the finding that DMAP, when used alone as a catalyst replacement, provided ε-lactone in only 2% yield, and the product of intermolecular iodoesterification (the tertiary acetate) in 39% yield. Together, these experiments suggest that the catalyst is critical to acceleration of both intramolecular cyclization and selective activation of one alkene π-face. Although simple pyridine–iodonium complexes are established reagents,12,13 we have no reason to exclude additional counterions and the critical role they may play (i.e., X of X–I in Fig. 2).
Conclusion
ε-Lactones are prepared in moderate yields and high enantioselectivity using a bifunctional BAM·HNTf2 catalyst. The conformationally unbiased nature of these substrates is remarkable, filling a meaningful gap in the otherwise limited structure space pioneered by Tang6d and Yeung.11 The technology also enables several 1,1-disubstituted alkene functionalization reactions, including epoxidation, that do not yet have more direct solutions despite the potential directing ability of the remote carboxylic acid. Essential to this success is the discovery that PIDA/I2 is uniquely effective at generating the reactive iodonium species, one that engages the catalyst for productive and selective odolactonization.Data availability
All experimental and characterization data in this article are available in the ESI.† Crystallographic data for compound 8i has been deposited in the Cambridge Crystallographic Data Centre (CCDC) under accession number CCDC 2160104.Author contributions
A. F. and J. J. conceived the project, A. F., J. P., and Z. D. completed the experimental work. All authors wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the National Institute of General Medical Sciences (NIH GM 084333) for financial support. The Indiana University Mass Spectrometry Facility acknowledges support from the NSF (CHE1726633). Nathan Schley (VU) is gratefully acknowledged for X-ray diffraction support, and we thank Jared Mendiola (Colorado College, NSF-REU (CHE 2051011)) and Corri Calandra for preparation of several starting materials.References
- (a) C. J. R. Bataille and T. J. Donohoe, Chem. Soc. Rev., 2011, 40, 114–128 RSC; (b) M. C. Noe, M. A. Letavic and S. L. Snow, Org. React., 2005, 66, 109–625 CAS; (c) M. R. Monaco, S. Prévost and B. List, Angew. Chem., Int. Ed., 2014, 53, 8142–8145 CrossRef CAS PubMed.
- (a) A. Parra, Chem. Rev., 2019, 119, 12033–12088 CrossRef CAS PubMed; (b) A. Yoshimura and V. V. Zhdankin, Chem. Rev., 2016, 116, 3328–3435 CrossRef CAS PubMed.
- S. De Jong, D. G. Nosal and D. J. Wardrop, Tetrahedron, 2012, 68, 4067–4105 CrossRef CAS PubMed.
- (a) J. H. Lee, S. Choi and K. B. Hong, Molecules, 2019, 24, 2634 CrossRef PubMed; (b) J.-C. Kizirian, Chem. Rev., 2008, 108, 140–205 CrossRef CAS PubMed; (c) D. Lucet, T. L. Gall and C. Mioskowski, Angew. Chem., Int. Ed., 1998, 37, 2580–2627 CrossRef CAS.
- L. A. Segura-Quezada, K. R. Torres-Carbajal, Y. Satkar, K. A. J. Ornelas, N. Mali, D. B. Patil, R. Gámez-Montaño, J. R. Zapata-Morales, S. Lagunas-Rivera, R. Ortiz-Alvarado and C. R. Solorio-Alvarado, Mini-Rev. Org. Chem., 2021, 18, 159 CrossRef CAS.
- (a) D. C. Whitehead, R. Yousefi, A. Jaganathan and B. Borhan, J. Am. Chem. Soc., 2010, 132, 3298–3300 CrossRef CAS PubMed; (b) K. Murai, T. Matsushita, A. Nakamura, S. Fukushima, M. Shimura and H. Fujioka, Angew. Chem., Int. Ed., 2010, 49, 9174 CrossRef CAS PubMed; (c) G. E. Veitch and E. N. Jacobsen, Angew. Chem., Int. Ed., 2010, 49, 7332–7335 CrossRef CAS PubMed; (d) W. Zhang, S. Zheng, N. Liu, J. B. Werness, I. A. Guzei and W. Tang, J. Am. Chem. Soc., 2010, 132, 3664–3665 CrossRef CAS PubMed; (e) L. Zhou, J. Chen, C. K. Tan and Y.-Y. Yeung, J. Am. Chem. Soc., 2011, 133, 9164–9167 CrossRef CAS PubMed.
- (a) K. D. Ashtekar, A. Jaganathan, B. Borhan and D. C. Whitehead, Org. React., 2021, 1–266 Search PubMed; (b) R. Kristianslund, J. E. Tungen and T. V. Hansen, Org. Biomol. Chem., 2019, 17, 3079–3092 RSC; (c) J. M. J. Nolsøe and T. V. Hansen, Eur. J. Org. Chem., 2014, 2014, 3051–3065 CrossRef.
- (a) M. Fujita, M. Wakita and T. Sugimura, Chem. Commun., 2011, 47, 3983–3985 RSC; (b) T. H. Wöste and K. Muñiz, Synthesis, 2016, 48, 816–827 CrossRef.
- For examples of enantioselective epoxidation and hydration of homoallylic alcohols, using NIS, CO2, and an organocatalyst, see: (a) B. A. Vara, T. J. Struble, W. Wang, M. C. Dobish and J. N. Johnston, J. Am. Chem. Soc., 2015, 137, 7302–7305 CrossRef CAS PubMed. Similarly, for an example of formal enantioselective hydration of a homoallylic amine, see: (b) R. Yousefi, T. J. Struble, J. L. Payne, M. Vishe, N. D. Schley and J. N. Johnston, J. Am. Chem. Soc., 2019, 141, 618–625 CrossRef CAS PubMed.
- S. Jana, A. Verma, V. Rathore and S. Kumar, Synlett, 2019, 30, 1667–1672 CrossRef CAS.
- X. Jiang, X. Xu, W. Xu, P. Yu and Y.-Y. Yeung, Org. Lett., 2021, 23, 6316–6320 CrossRef CAS PubMed.
- (a) B. Simonot and G. Rousseau, J. Org. Chem., 1993, 58, 4–5 CrossRef CAS; (b) B. Simonot and G. Rousseau, J. Org. Chem., 1994, 59, 5912–5919 CrossRef CAS.
- (a) Y. A. Cheng, T. Chen, C. K. Tan, J. J. Heng and Y.-Y. Yeung, J. Am. Chem. Soc., 2012, 134, 16492–16495 CrossRef CAS PubMed; (b) P.-T. Tang, Y.-X. Shao, L.-N. Wang, Y. Wei, M. Li, N.-J. Zhang, X.-P. Luo, Z. Ke, Y.-J. Liu and M.-H. Zeng, Chem. Commun., 2020, 56, 6680–6683 RSC.
- G. Rousseau, Tetrahedron, 1995, 51, 2777–2849 CrossRef CAS.
- A. Parenty, X. Moreau and J. M. Campagne, Chem. Rev., 2006, 106, 911–939 CrossRef CAS PubMed.
- (a) M. C. Dobish and J. N. Johnston, J. Am. Chem. Soc., 2012, 134, 6068–6071 CrossRef CAS PubMed; (b) Y. Toda, M. Pink and J. N. Johnston, J. Am. Chem. Soc., 2014, 136, 14734–14737 CrossRef CAS PubMed; (c) T. J. Struble, H. M. Lankswert, M. Pink and J. N. Johnston, ACS Catal., 2018, 8, 11926–11931 CrossRef CAS PubMed.
- M. W. Danneman, K. B. Hong and J. N. Johnston, Org. Lett., 2015, 17, 3806–3809 CrossRef CAS PubMed.
- M. W. Danneman, K. B. Hong and J. N. Johnston, Org. Lett., 2015, 17, 2558–2561 CrossRef CAS PubMed.
- (a) K. Tokumaru, K. Bera and J. N. Johnston, Synthesis, 2017, 49, 4670–4675 CrossRef CAS PubMed; (b) K. Tokumaru and J. N. Johnston, Chem. Sci., 2017, 8, 3187–3191 RSC; (c) M. S. Crocker, H. Foy, K. Tokumaru, T. Dudding, M. Pink and J. N. Johnston, Chem, 2019, 5, 1248–1264 CrossRef CAS PubMed.
- (a) T. Wirth and U. H. Hirt, Synthesis, 1999, 1999, 1271–1287 CrossRef; (b) U. H. Hirt, M. F. H. Schuster, A. N. French, O. G. Wiest and T. Wirth, Eur. J. Org. Chem., 2001, 2001, 1569–1579 CrossRef; (c) T. Wirth, Angew. Chem., Int. Ed., 2005, 44, 3656–3665 CrossRef CAS PubMed.
- R. Freire, J. J. Marrero, M. S. Rodríguez and E. Suárez, Tetrahedron Lett., 1986, 27, 383–386 CrossRef CAS.
- A. Keizo and O. Yoshiro, Bull. Chem. Soc. Jpn., 1968, 41, 1476–1477 CrossRef.
- For studies focused on the development of chiral hypervalent iodine reagents, see: (a) S. Ghosh, S. Pradhan and I. Chatterjee, Beilstein J. Org. Chem., 2018, 14, 1244–1262 CrossRef CAS PubMed; (b) M. Fujita, Heterocycles, 2018, 96, 563–594 CrossRef CAS; (c) C. Röben, J. A. Souto, Y. González, A. Lishchynskyi and K. Muñiz, Angew. Chem., Int. Ed., 2011, 50, 9478–9482 CrossRef PubMed; (d) K. Muñiz, L. Barreiro, R. M. Romero and C. Martínez, J. Am. Chem. Soc., 2017, 139, 4354–4357 CrossRef PubMed.
- H. Jeon, D. Kim, J. H. Lee, J. Song, W. S. Lee, D. W. Kang, S. Kang, S. B. Lee, S. Choi and K. B. Hong, Adv. Synth. Catal., 2018, 360, 779–783 CrossRef CAS.
- Y.-B. Kang and L. H. Gade, J. Am. Chem. Soc., 2011, 133, 3658–3667 CrossRef CAS PubMed.
- S. Haubenreisser, T. H. Wöste, C. Martínez, K. Ishihara and K. Muñiz, Angew. Chem., Int. Ed., 2016, 55, 413–417 CrossRef CAS PubMed.
- Leading references: (a) R. Weiss and J. Seubert, Angew. Chem., Int. Ed., 1994, 33, 891–893 CrossRef; (b) K. Aertker, R. J. Rama, J. Opalach and K. Muñiz, Adv. Synth. Catal., 2017, 359, 1290–1294 CrossRef CAS; (c) A. F. Tierno, J. C. Walters, A. Vazquez-Lopez, X. Xiao and S. E. Wengryniuk, Chem. Sci., 2021, 12, 6385–6392 RSC.
- H. Togo and M. Katohgi, Synlett, 2001, 0565–0581 CrossRef CAS.
- T. Hokamp, A. T. Storm, M. Yusubov and T. Wirth, Synlett, 2018, 29, 415–418 CrossRef CAS.
- T. K. Achar, S. Maiti and P. Mal, Org. Biomol. Chem., 2016, 14, 4654–4663 RSC.
- H. Gottam and T. K. Vinod, J. Org. Chem., 2011, 76, 974–977 CrossRef CAS PubMed.
- R. Giri and J.-Q. Yu, in Encyclopedia of Reagents for Organic Synthesis, 2008 Search PubMed.
- H. Wang, M. Frings and C. Bolm, Org. Lett., 2016, 18, 2431–2434 CrossRef CAS PubMed.
- M. Daniel, F. Blanchard, S. Nocquet-Thibault, K. Cariou and R. H. Dodd, J. Org. Chem., 2015, 80, 10624–10633 CrossRef CAS PubMed.
- For an example of I(III)/I2 for C–H amination, see: C. Martínez and K. Muñiz, Angew. Chem., Int. Ed., 2015, 54, 8287–8291 CrossRef PubMed.
- X. Li, P. Chen and G. Liu, Beilstein J. Org. Chem., 2018, 14, 1813–1825 CrossRef CAS PubMed.
- (a) S.-K. Khim, M. Dai, X. Zhang, L. Chen, L. Pettus, K. Thakkar and A. G. Schultz, J. Org. Chem., 2004, 69, 7728–7733 CrossRef CAS PubMed; (b) A. G. Schultz, Chem. Commun., 1999, 1263–1271 RSC.
- M. Date, Y. Tamai, T. Hattori, H. Takayama, Y. Kamikubo and S. Miyano, J. Chem. Soc., Perkin Trans. 1, 2001, 645–653 RSC.
- Assignment of absolute configuration was made initially by chemical correlation with 13 reported in ref. 38 (see ESI† for details). However, direct assignment by X-ray analysis of 8i is opposite, as depicted throughout this manuscript. The reason for the discrepancy is unclear, but note that the material used in ref. 38 was 43% ee, with a rotation relatively small in magnitude.
- M. T. Knowe, M. W. Danneman, S. Sun, M. Pink and J. N. Johnston, J. Am. Chem. Soc., 2018, 140, 1998–2001 CrossRef CAS PubMed.
- R. Freire, J. J. Marrero, M. S. Rodríguez and E. Suárez, Tetrahedron Lett., 1986, 27, 383–386 CrossRef CAS.
- G. Doleschall and G. Tóth, Tetrahedron, 1980, 36, 1649–1665 CrossRef CAS.
- H. J. Kim, S. H. Cho and S. Chang, Org. Lett., 2012, 14, 1424–1427 CrossRef CAS PubMed.
- (a) P. A. Bartlett and J. Myerson, J. Am. Chem. Soc., 1978, 100, 3950–3952 CrossRef CAS; (b) P. A. Bartlett, D. P. Richardson and J. Myerson, Tetrahedron, 1984, 40, 2317–2327 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Complete experimental details (PDF); NMR and HPLC trace data (PDF); data for 8i (CIF). CCDC 2160104. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc01587k |
| ‡ These authors (JLP, ZD, ALF) contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |