
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRh(I)-catalyzed dimerization of ene-vinylidenecyclopropanes for the construction of spiro[4,5]decanes and mechanistic studies†
Chao
Ning
a,
Kang-Hua
Rui
a,
Yin
Wei
 *b and
Min
Shi
*ab
*b and
Min
Shi
*ab
aKey Laboratory for Advanced Materials and Institute of Fine Chemicals, Key Laboratory for Advanced Materials, Feringa Nobel Prize Scientist Joint Research Center, School of Chemistry & Molecular Engineering, East China University of Science and Technology, Meilong Road No. 130, Shanghai, 200237, China. E-mail: mshi@mail.sioc.ac.cn
bState Key Laboratory of Organometallic Chemistry, Center for Excellence in Molecular Synthesis, University of Chinese Academy of Sciences, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China. E-mail: weiyin@sioc.ac.cn
First published on 1st June 2022
Abstract
Rh(I) complex catalyzed dimerization of ene-vinylidenecyclopropanes took place smoothly to construct a series of products containing spiro[4,5]decane skeletons featuring a simple operation procedure, mild reaction conditions, and good functional group tolerance. In this paper, the combination of experimental and computational studies reveals a counterion-assisted Rh(I)–Rh(III)–Rh(V)–Rh(III)–Rh(I) catalytic cycle involving tandem oxidative cyclometallation/reductive elimination/selective oxidative addition/selective reductive elimination/reductive elimination steps; in addition, a pentavalent spiro-rhodium intermediate is identified as the key intermediate in this dimerization reaction upon DFT calculation.
Introduction
Spiro[4,5]decanes and polycyclic natural products containing the spiro[4,5]decane moiety possessing useful biological properties are important resources in medicinal chemistry and the pharmaceutical industry.1,2 One of the representative bioactive molecules containing the spiro[4,5]decane moiety is β-vetivone (Fig. 1), which was first isolated from Vetiveria zizanioides in 1939 by Pfau and Plattner.2a Since then, more and more natural products containing the spiro[4,5]decane skeleton with biological activity have been isolated and synthesized by organic chemists.2d Several representative compounds are shown in Fig. 1. For example, hinesol may represent a novel medicinal drug having indications in the treatment of leukemia. Sequosempervirin A isolated from Sequoia sempervirens is also expected to exhibit useful biological activity.2f In addition, the guaianolide-type sesquiterpenoid dimers are known as a unique array of sesquiterpenoid natural products and show biological antitumor and anti-HIV activity. In view of their excellent biological activity, it is of significant interest to introduce the spiro[4,5]decane skeleton into pharmaceutically active molecules for further research.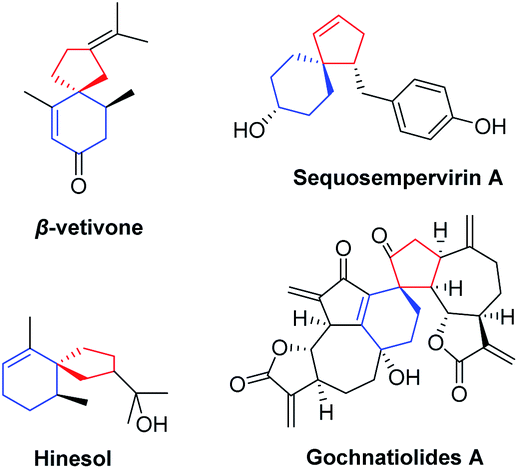 | ||
| Fig. 1 Representative bioactive compounds containing the spiro[4,5]decane moiety. | ||
It has always been challenging to construct the spiro quaternary carbon stereocenter, because the formation of the spiro quaternary carbon stereocenter needs to overcome strong steric hindrance and ring strain.3 Therefore, in the past few decades, significant efforts have been devoted to the development of novel strategies to build all-carbon spirocyclic ring systems.4 The transition metal-catalyzed domino cyclization of 1,n-enynes and 1,n-dienes serves as a useful strategy for the synthesis of cyclic products.5 In the metal-catalyzed domino cyclization reaction, the formation of multiple C–C bonds can be controlled by choosing different metals (e.g., Rh,6 Co,7 Ni,8etc.9) and ligands to control the chemo-, regio- and stereo-selectivity, involving interesting mechanisms. In the metal-catalyzed domino cyclization process, the cyclometallation of the [1,n]-π-system provides the key intermediate, which subsequently undergoes migration and insertion of the unsaturated bonds of alkenes, alkynes, or allenes via an intramolecular or intermolecular mode (Scheme 1). In most cases, the products containing fused rings are usually accessed through metal-catalyzed domino cyclization reactions. There are only a few reports on the construction of spirocyclic compounds through rational design of substrates and novel mechanisms.10
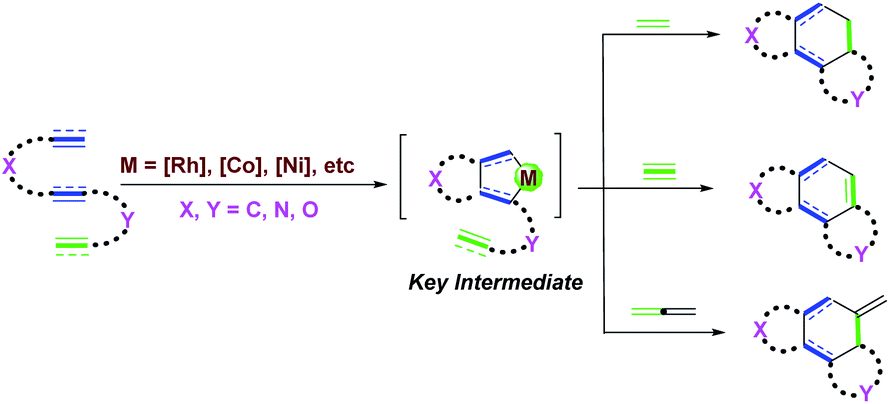 | ||
| Scheme 1 General model of the transition metal-catalyzed domino cyclization of 1,n-enynes and 1,n-dienes. | ||
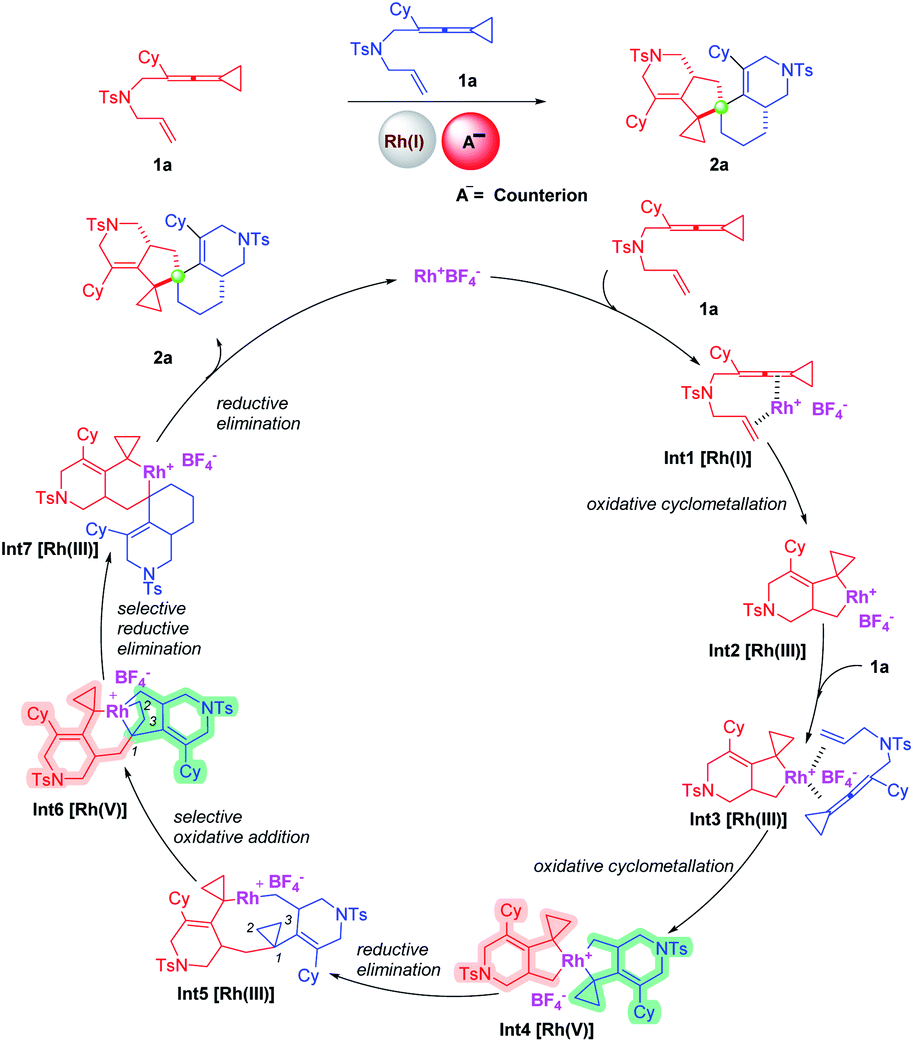
In this work, we report a dimerization reaction of ene-vinylidenecyclopropanes (ene-VDCPs) as an effective and facile method to provide a series of compounds containing spiro[4,5]decane skeletons (Scheme 2, this work). A plausible mechanism is proposed as outlined in Scheme 2. First, the coordination of Rh(I) with the allene moiety and olefin moiety in an ene-VDCP leads to the formation of intermediate Int1.
 | ||
| Scheme 2 This work and proposed mechanism. | ||
Subsequent cyclometallation then gives the rhodacyclic intermediate Int2.11 The intermediate Int2 is coordinated with another molecular ene-VDCP to produce intermediate Int3, which undergoes a cyclometallation to afford the spirocyclic rhodium(V) intermediate Int4.12,13 Reductive elimination affords medium-membered rhodacyclic intermediate Int5. The rhodium(III) inserts into the C1–C2 bond or C1–C3 of the distal cyclopropane to generate the rhodium(V) intermediate Int6. Subsequently, the rhodium(V) intermediate Int6 undergoes reductive elimination to afford intermediate Int7, which then produces product 2a through reductive elimination and regenerates the catalyst. Herein, the exploration of the substrate scope of this novel domino dimerization process and the detailed mechanistic investigations will be performed to study the suggested reaction mechanism in this context.
Results and discussion
Experimental investigations
At the start of our studies, ene-VDCP 1a was used as the model substrate to test the reaction outcome. Interestingly, the reaction successfully took place in the presence of 5 mol% Rh(cod)2BF4 in anhydrous toluene at 80 °C after 8 hours and the dimerization product 2a was obtained in 40% isolated yield (Table 1, entry 1). Upon lowering the reaction temperature to 60 °C, the target product was obtained in 60% yield (entry 2). Gratifyingly, taking the combination of [Rh(cod)Cl]2 and AgBF4 could afford the desired product 2a in 85% yield at 60 °C (entry 3). Accordingly, a series of additives were screened, such as Ag salts, Lewis acids and NaBArF4, and none of them showed a better yield to obtain the desired product than the combination of [Rh(cod)Cl]2 and AgBF4 (entries 4–10). Screening different solvents implied that toluene was the most optimal solvent for this reaction (entry 3 vs. entries 11–15). Increasing the amount of additive to 20 mol% did not have a great effect on the yield (entry 16). Next, we tried to explore the asymmetric variants of this dimerization reaction by using chiral binaphthyl bisdiarylphosphine ligands. When the ligand was added, the yield of the desired product slightly decreased; the corresponding stereoselective product was not obtained, suggesting that the ligands did not play roles in this reaction, probably due to the key intermediate Int2 preferring to coordinate with another substrate molecule (for more details, see Table S1 on page S5 in the ESI†).|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Additive | T (°C) | Solvent | Yieldb (%) |
| a Reaction conditions: 1a (0.20 mmol), catalyst (5 mol%), additive (10 mol%), solvent (2 mL), 8 h, Ar. All the reactions were carried out on a 0.20 mmol scale in solvent (2 mL) at 60 °C for 8 h unless otherwise specified. b Isolated yield. c 20 mol% additive was added. | |||||
| 1 | Rh(cod)2BF4 | — | 80 | Toluene | 40 |
| 2 | Rh(cod)2BF4 | — | 60 | Toluene | 60 |
| 3 | [Rh(cod)Cl] 2 | AgBF 4 | 60 | Toluene | 85 |
| 4 | [Rh(cod)Cl]2 | AgOTf | 60 | Toluene | 62 |
| 5 | [Rh(cod)Cl]2 | AgNTf2 | 60 | Toluene | — |
| 6 | [Rh(cod)Cl]2 | AgSbF6 | 60 | Toluene | — |
| 7 | [Rh(cod)Cl]2 | Sc(OTf)3 | 60 | Toluene | 34 |
| 8 | [Rh(cod)Cl]2 | Yb(OTf)3 | 60 | Toluene | 32 |
| 9 | [Rh(cod)Cl]2 | In(OTf)3 | 60 | Toluene | 28 |
| 10 | [Rh(cod)Cl]2 | NaBArF4 | 60 | Toluene | — |
| 11 | [Rh(cod)Cl]2 | AgBF4 | 60 | PhCI | 60 |
| 12 | [Rh(cod)Cl]2 | AgBF4 | 60 | DCE | 78 |
| 13 | [Rh(cod)Cl]2 | AgBF4 | 60 | THE | 70 |
| 14 | [Rh(cod)Cl]2 | AgBF4 | 60 | Dioxane | 72 |
| 15 | [Rh(cod)Cl]2 | AgBF4 | 60 | MeCN | Trace |
| 16c | [Rh(cod)Cl]2 | AgBF4 | 60 | Toluene | 86 |
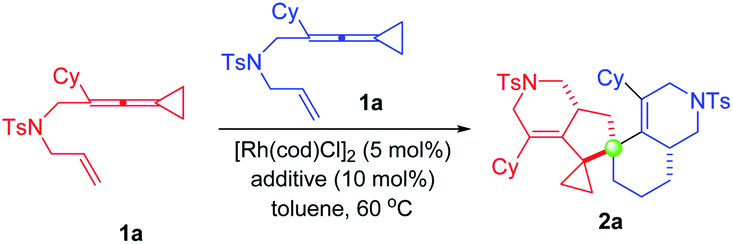
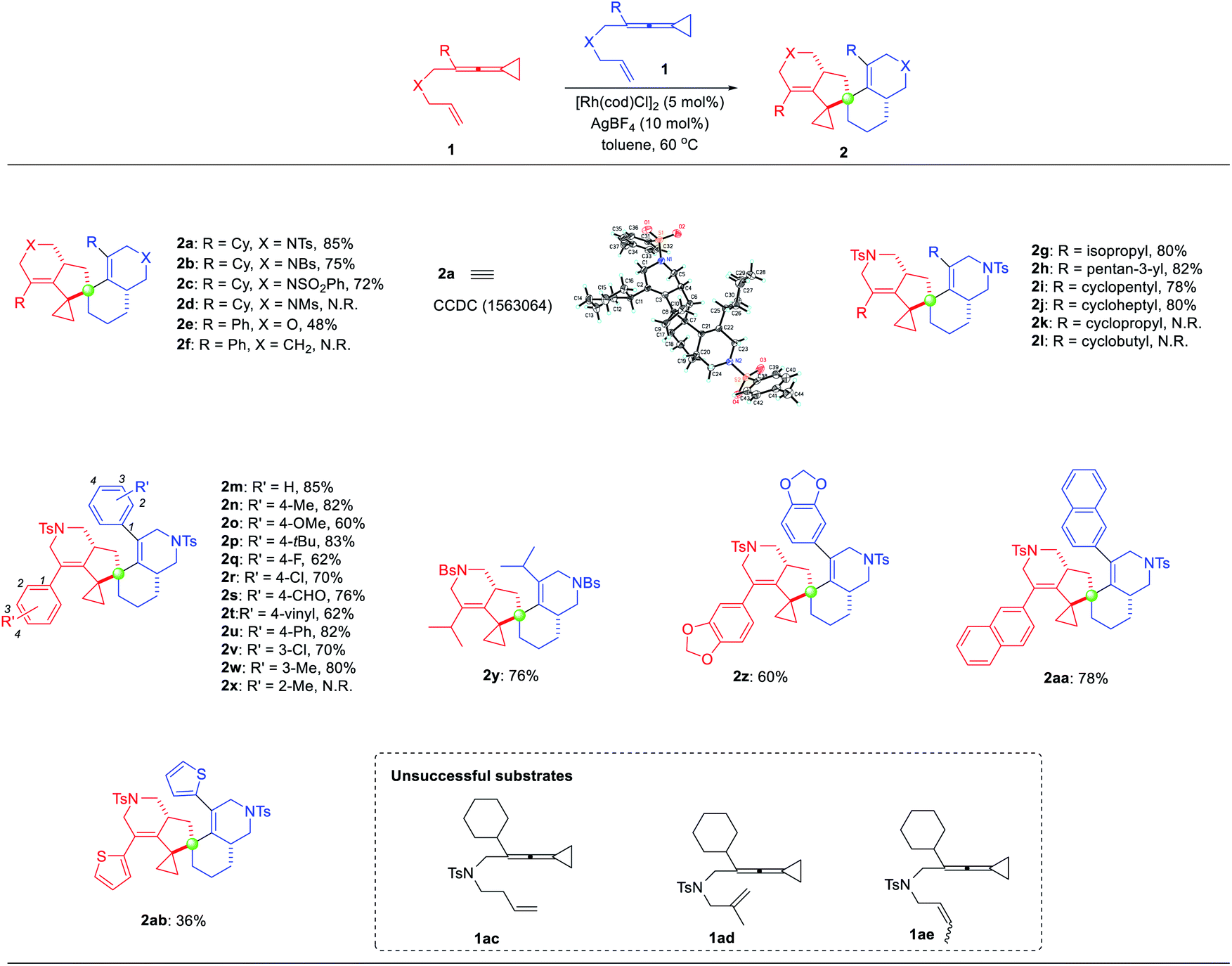
With the optimized reaction conditions identified, the substrate scope for the dimerization of ene-VDCPs 1 was then explored and the results are shown in Table 2. The X-ray diffraction pattern of 2a is shown in Table 2, whose structure had been unambiguously determined. The related CIF data of 2a are shown in the ESI.† Firstly, changing the N-linked ene-VDCPs to N-p-bromobenzenesulfonyl (NBs) and NSO2Ph yielded similar results, producing the dimerization products 2b and 2c in 75% yield and 72% yield, respectively. However, upon changing the linker as N-methylsulfonyl (NMs), the expected product 2d was not detected. To our delight, upon changing the linker as an oxygen atom, the desired product 2e was obtained in 48% yield. Unfortunately, no desired reaction occurred using the CH2-linked substrate 1f presumably due to the skeleton structure of the full carbon chain not being conducive to the generation of cyclometalated species. Next, the R moiety of substrates 1 was investigated. For substrates containing alkyl substituents including cycloalkane substituents, the reactions took place smoothly, giving the target products 2g–2j in 78–82% yields. However, employing substrates 1k and 1l with a cyclopropyl and a cyclobutyl group as a substituent, the desired products 2k and 2l were not obtained under the standard conditions. Then, various substituents on the benzene ring were tested. Neither electron-rich nor electron-poor aromatic rings have an obvious impact on the reaction activity, and the desired products 2m–2w were obtained in good yields ranging from 60% to 85%. Substrate 1x with an ortho-substituted methyl group did not proceed under the standard reaction conditions, probably due to the steric hindrance of the ortho-substituent. As for substrate 1y, the reaction proceeded smoothly to give the desired product 2y in 76% yield. Product 2z, bearing a benzo[d][1,3]dioxole moiety, could also be obtained in 60% yield. We further conducted an investigation on other aromatic rings such as naphthalene or thiophene substituted substrates and obtained the corresponding products 2aa and 2ab in 78% yield and 36% yield, respectively. Upon extending the carbon linker as for substrate 1ac or introducing a substituent at the olefin moiety as for substrates 1ad and 1ae, none of the desired spirocyclic dimers were formed, presumably due to the steric issue.
| a Reaction conditions: 1 (0.20 mmol), catalyst (5 mol%), additive (10 mol%), solvent (2 mL), 60 °C, 8 h, Ar. Yields were determined from isolated products. |
|---|
|
In order to investigate the regio- and chemoselectivity of the dimerization of ene-vinylidenecyclopropanes, we attempted the cross-over experiments. Under the standard conditions, we performed cross-over experiments on substrates 1a and 1m. We obtained a mixture of products 2a, 2m and cross-over products 2am and 2ma (Scheme 3a). The products 2a, 2m and cross-over products 2am and 2ma were identified by high-resolution mass spectrometry [HRMS (ESI) m/z: (M + H)+ calcd for C44H53N2O4S2: 737.3447, found: 737.3448] (Scheme 3a). Based on the new signals of cyclopropanes in the 1H NMR spectra of the mixture, the cross-over products were also confirmed (for more details, see page S7 in the ESI†). Unfortunately, because the polarities of these compounds are too close, it is difficult for us to separate these compounds. We further used substrates 1c without the Ts functional group and 1p to conduct the cross-over experiment. Similarly, we obtained a mixture of products 2c, 2p and cross-over products 2cp and 2pc (Scheme 3b). The products 2c, 2p and cross-over products 2cp and 2pc were also identified by high-resolution mass spectrometry [HRMS (ESI) m/z: (M + H)+ calcd for C47H59N2O4S2: 779.3916, found: 779.3914] (Scheme 3b). Based on the 1H NMR spectra of the mixture, we found the signals of a new Ts group and tert-butyl group, which confirmed the formation of cross-over products (see page S9 in the ESI†). However, the products were not isolated due to the close polarities of these compounds.
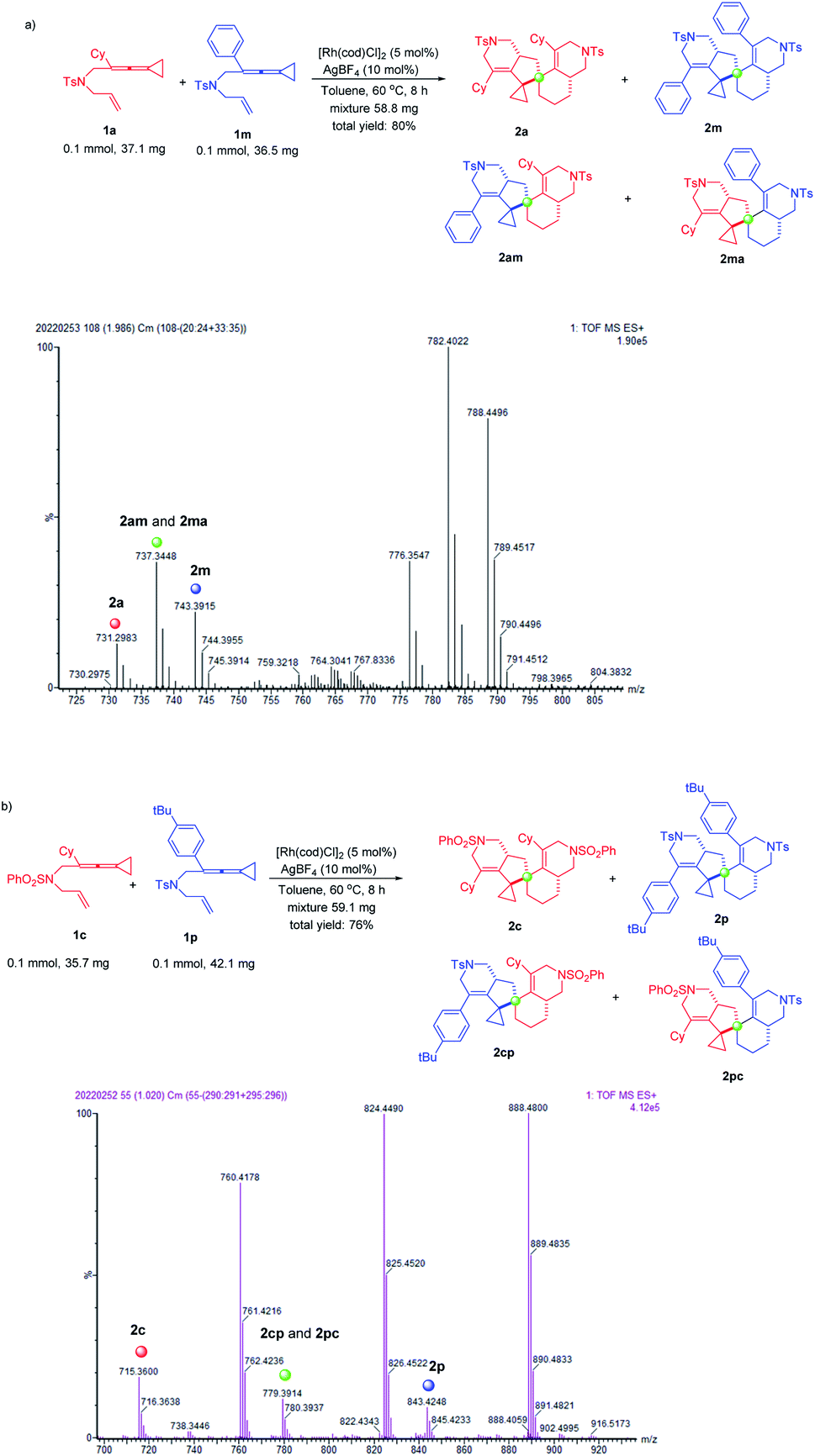 | ||
| Scheme 3 Cross-over experiments of (a) 1a and 1m, and (b) 1c and 1p. | ||
Synthetic applications
In order to explore the synthetic applicability of this protocol, scale-up (half-gram) reactions were carried out. As shown in Scheme 4, these reactions were conducted employing 2.00 mmol of 1a or 1m, producing 0.563 g of 2a and 0.584 g of 2m in 76% yield and 80% yield under the standard conditions, respectively (Scheme 4a). Furthermore, we demonstrated the synthetic utility of the spiro[4,5]decane products, showing that the N-tosyl group of 2m was removed upon treating with sodium naphthalenide in THF at −78 °C, affording product 3 in only 36% yield, probably due to its instability (Scheme 4b). Several further synthetic transformations were attempted; however, we did not obtain the desired products (for details, see the ESI from page S11 to page S13†).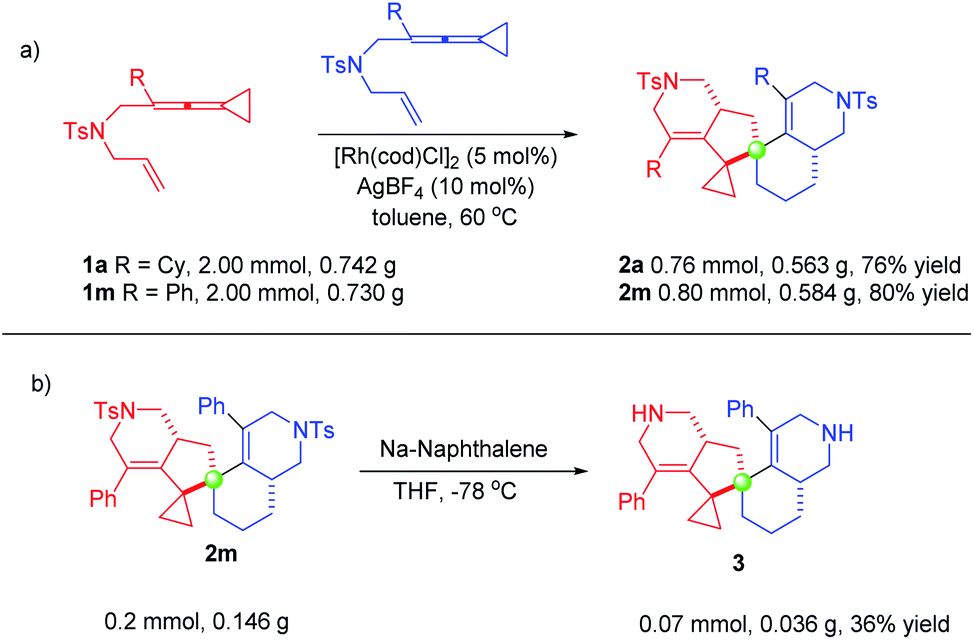 | ||
| Scheme 4 (a) Scale-up (half-gram) experiments; (b) synthetic transformations. | ||
Mechanistic studies
To further understand the mechanism, we carried out a series of control experiments and density functional theory (DFT) calculations. We first proposed a possible mechanism involving rhodium carbenoid and silver carbenoid intermediates as key intermediates (see Scheme S1 in the ESI†). However, the suggested key silver carbene intermediate is not stable and cannot be located by DFT calculations (for details, see the ESI†), so we excluded this mechanism. Then, we proposed another mechanism involving a rhodium carbenoid intermediate accompanied by a [2 + 2 + 1] cycloaddition reaction with another molecular substrate14 (for more details, see Scheme S1 in the ESI†) and a mechanism involving the spiro rhodium(V) intermediate shown in Scheme 2. We attempted to capture the possible rhodium carbenoid intermediates by trapping experiments with styrene or pyridine N-oxides; however, we did not succeed in these trapping experiments (Scheme 5a). We subsequently embarked on DFT calculations to gain insight into the reaction mechanism. All calculations have been performed at the SMD/B3LYP/6-311+G(d,p)/Lanl2dz//B3LYP/6-31G(d)/Lanl2dz level with the Gaussian 16 program. We investigated the reaction pathway starting from a stable rhodium complex Int1 (shown in Scheme 5b), in which both the alkene and the allene units of 1a are coordinated to the rhodium catalyst. Int1 can undergo an oxidative cyclometallation to give a rhodacyclic intermediate Int2 through TS1 with an energy barrier of 23.7 kcal mol−1. It should be mentioned here that without the assistance of a counterion (BF4−) the intermediate Int2 is unstable; this result may account for why an amount of additive (AgBF4) is necessary in experiments. Alternatively, Int1 can be transformed into intermediate Int2′viaTS1′, with an energy barrier of 29.9 kcal mol−1 in a highly endothermic process (ΔG = 22.1 kcal mol−1). The intermediate Int2′ subsequently undergoes cyclometallation to generate an intermediate Int3′via transition state TS2′. Passing through transition state TS3′ with an extremely high energy barrier of 52.3 kcal mol−1, the intermediate Int3′ can transform into a very stable rhodium carbene intermediate Int4′. Although the rhodium carbene intermediate Int4′ is thermodynamically stable, an extremely high energy barrier needs to be overcome, which cannot be achieved under our experimental conditions. Thus, the suggested mechanism involving the rhodium carbene intermediate (see page S15 in the ESI†) is excluded. We also investigated whether the intermediate Int2 can undergo a β-C elimination. This step is not a canonical β-C elimination, but instead consists of a distal opening of the cyclopropyl ring (C⋯C bond distance of 2.16 Å in TS2′′), a process reminiscent to that previously proposed for cyclopropyl Rh(III) complexes and other transition metal derivatives. Passing through transition state TS2′′ with an energy barrier of 22.2 kcal mol−1, a TMM metal complex Int3′′ is generated.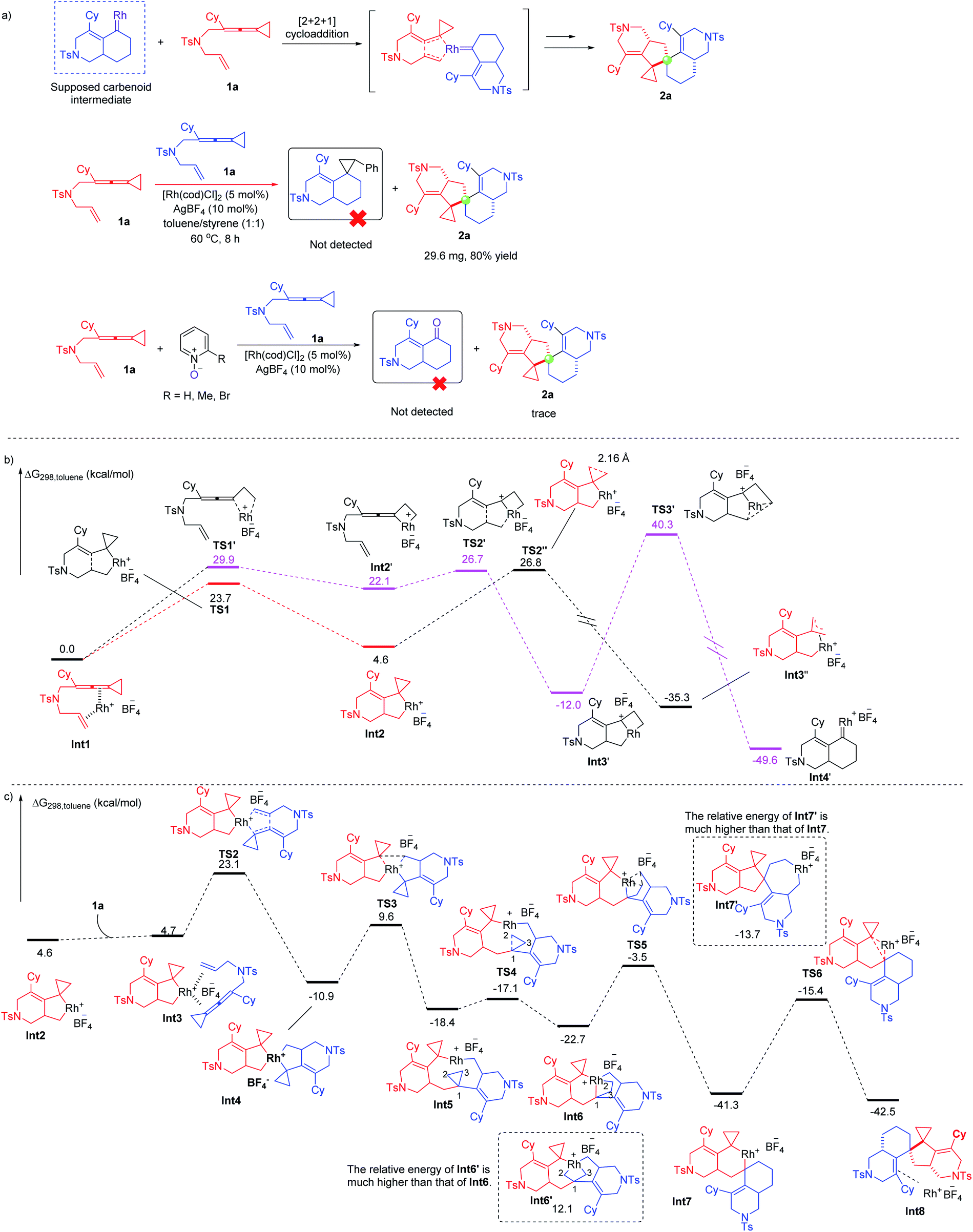 | ||
| Scheme 5 (a) Control experiments; (b) DFT calculations on the possible pathways for generation of key intermediates; (c) DFT calculations on the possible pathways for formation of product 2a. | ||
Next, we continued to investigate the following reaction steps through computational studies (Scheme 5c). The rhodacyclic intermediate Int2 can associate with another molecule of substrate 1a to generate a rhodium complex Int3 since there is no ligand in the presence of the reaction system. Interestingly, Int3 can undergo another cyclometallation to generate an uncommonly stable spiro rhodium(V) intermediate Int4via transition state TS2 in an exothermic process (ΔG = −15.6 kcal mol−1). The energy barrier for cyclometallation is 18.4 kcal mol−1, which is lower than that of the competitive pathway involving the so-called β-C elimination step (22.2 kcal mol−1viaTS2′′). Thus, we exclude the β-C elimination pathway, which is similar to the Co-catalyzed cycloaddition reaction of ACPs.7b The subsequent reductive elimination step takes place through transition state TS3 with an energy barrier of 20.5 kcal mol−1 to afford intermediate Int5 having a nine-membered ring. Similarly, the competitive pathway involving the so-called β-C elimination of Int4 is ruled out due to the higher energy barrier (for details, see Scheme S4 on page S123 in the ESI†). Instead of insertion into the neighboring cyclopropane, the rhodium(III) prefers to insert into the C–C bond of the distal cyclopropane to generate another rhodium(V) intermediate Int6viaTS4 with a small energy barrier (1.3 kcal mol−1), probably due to the flexibility of the geometry of the medium-sized ring.15 An alternative intermediate Int6′ was also investigated; however, its energy is much higher than that of Int6 by 34.8 kcal mol−1. Subsequently, the rhodium(V) intermediate Int6 undergoes a reductive elimination step to produce intermediate Int7via transition state TS5; another possible intermediate Int7′ is also investigated. However, intermediate Int7′ is discarded since its energy is much higher than that of Int7 by 27.6 kcal mol−1. Finally, intermediate Int7 undergoes another reductive elimination step to produce the product complex Int8 through transition state TS6 with an energy barrier of 25.9 kcal mol−1, which is the highest barrier in the whole reaction pathway. The calculation results provide theoretical evidence for the proposed mechanism shown in Scheme 2. We also investigated another possible reaction pathway where int2 inserts into the allene moiety of another 1a, and β-C elimination takes place to open the cyclopropane ring; then the resulting intermediate inserts back into the allene and then the alkene, followed by reductive elimination to afford the product (for details, see Schemes S2 and S3 on pages S16 and S17 in the ESI†). However, the whole process involving Rh(I)–Rh(III)–Rh(I) involves intermediates and transition states that have the higher energies than the key intermediates and transition states in the redox-neutral Rh(I)–Rh(III)–Rh(V)–Rh(III)–Rh(I) catalytic cycle. Thus, a redox-neutral Rh(I)–Rh(III)–Rh(V)–Rh(III)–Rh(I) catalytic cycle assisted by a counterion, involving tandem oxidative cyclometallation/reductive elimination/selective oxidative addition/selective reductive elimination/reductive elimination steps for this dimerization reaction, was deduced based on a series of mechanistic investigations.
Conclusions
In conclusion, we have developed an efficient protocol for the Rh(I)-catalyzed dimerization reaction of ene-vinylidenecyclopropanes, delivering products containing spiro[4,5]decane skeletons in moderate to good yields with a wide substrate scope and good functional group tolerance. In addition, this reaction was achieved on a half-gram scale, and the corresponding products could be further transformed into synthetically interesting compounds. Systematic mechanistic studies, supported by DFT calculations, allowed us to propose a plausible counterion-assisted Rh(I)–Rh(III)–Rh(V)–Rh(III)–Rh(I) reaction mechanism. Further investigations with regard to the construction of related spirocyclic compounds, which may have useful applications in the synthesis of drug-like substances, and other functional materials under Rh(I) catalysis are underway in our laboratory.Data availability
Experimental and computational data have been made available as ESI.†Author contributions
C. N. and K.-H. R. contributed to the experimental work. Y. W. contributed to the computational work. C. N., Y. W. and M. S. contributed to ideation and writing of the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are thankful for the financial support from the National Natural Science Foundation of China (21121062, 21302203, 21372250, 21772037, 21772226, 21861132014, 91956115 and 22171078), the project supported by Shanghai Municipal Science and Technology Major Project (Grant No. 2018SHZDZX03) and the Fundamental Research Funds for the Central Universities (222201717003).References
- (a) M. Vandewalle and P. De Clercq, Tetrahedron, 1985, 41, 1765–1831 CrossRef; (b) W. G. Dauben and D. J. Hart, J. Am. Chem. Soc., 1977, 99, 7307–7314 CrossRef CAS; (c) A. R. Díaz-Marrero, G. Porras, Z. Aragón, J. M. de la Rosa, E. Dorta, M. Cueto, L. D'Croz, J. Maté and J. Darias, J. Nat. Prod., 2011, 74, 292–295 CrossRef PubMed; (d) S. Kováčová, S. K. Adla, L. Maier, M. Babiak, Y. Mizushina and K. Paruch, Tetrahedron, 2015, 71, 7575–7582 CrossRef; (e) J.-R. Huang, M. Sohail, T. Taniguchi, K. Monde and F. Tanaka, Angew. Chem., Int. Ed., 2017, 56, 5853–5857 CrossRef CAS PubMed.
- (a) A. S. Pfau and P. A. Plattner, Helv. Chim. Acta, 1939, 22, 640–654 CrossRef; (b) J. A. Marshall and P. C. Johnson, J. Am. Chem. Soc., 1967, 89, 2750–2751 CrossRef CAS; (c) E. Wenkert, B. L. Buckwalter, A. A. Craveiro, E. L. Sanchez and S. S. Sathe, J. Am. Chem. Soc., 1978, 100, 1267–1273 CrossRef CAS; (d) Y. Ito, K. Takahashi, H. Nagase and T. Honda, Org. Lett., 2011, 13, 4640–4643 CrossRef CAS PubMed; (e) C. Li, L. Dian, W. Zhang and X. Lei, J. Am. Chem. Soc., 2012, 134, 12414–12417 CrossRef CAS PubMed; (f) H. B. Park, Y.-J. Kim, J. K. Lee, K. R. Lee and H. C. Kwon, Org. Lett., 2012, 14, 5002–5005 CrossRef CAS PubMed; (g) L. K. Smith and I. R. Baxendale, Org. Biomol. Chem., 2015, 13, 9907–9933 RSC; (h) P. Luo, Y. Cheng, Z. Yin, C. Li, J. Xu and Q. Gu, J. Nat. Prod., 2019, 82, 349–357 CrossRef CAS PubMed.
- (a) V. A. D'yakonov, O. A. Trapeznikova, A. de Meijere and U. M. Dzhemilev, Chem. Rev., 2014, 114, 5775–5814 CrossRef PubMed; (b) Y. Li and S. Xu, Chem.–Eur. J., 2018, 24, 16218–16245 CrossRef CAS PubMed; (c) Z. Wang, Z.-P. Yang and G. C. Fu, Nat. Chem., 2021, 13, 236–242 CrossRef CAS PubMed.
- (a) R. Rios, Chem. Soc. Rev., 2012, 41, 1060–1074 RSC; (b) A. K. Franz, N. V. Hanhan and N. R. Ball-Jones, ACS Catal., 2013, 3, 540–553 CrossRef CAS; (c) L. K. Smith and I. R. Baxendale, Org. Biomol. Chem., 2015, 13, 9907–9933 RSC; (d) M.-Y. Wu, W.-W. He, X.-Y. Liu and B. Tan, Angew. Chem., Int. Ed., 2015, 54, 9409–9413 CrossRef CAS PubMed; (e) D.-Z. Chen, W.-J. Xiao and J.-R. Chen, Org. Chem. Front., 2017, 4, 1289–1293 RSC; (f) J.-H. Ye, L. Zhu, S.-S. Yan, M. Miao, X.-C. Zhang, W.-J. Zhou, J. Li, Y. Lan and D.-G. Yu, ACS Catal., 2017, 7, 8324–8330 CrossRef CAS; (g) A. Ding, M. Meazza, H. Guo, J. W. Yang and R. Rios, Chem. Soc. Rev., 2018, 47, 5946–5996 RSC; (h) P. Bunse, C. Schlepphorst, F. Glorius, M. Kitamura and B. Wünsch, J. Org. Chem., 2019, 84, 13744–13754 CrossRef CAS PubMed; (i) J. Ma, F. Schäfers, C. Daniliuc, K. Bergander, C. A. Strassert and F. Glorius, Angew. Chem., Int. Ed., 2020, 59, 9639–9645 CrossRef CAS PubMed.
- (a) Y. Yamamoto, Chem. Rev., 2012, 112, 4736–4769 CrossRef CAS PubMed; (b) Y. Shibata and K. Tanaka, Synthesis, 2012, 44, 323–350 CrossRef CAS; (c) A. Lledó, A. Pla-Quintana and A. Roglans, Chem. Soc. Rev., 2016, 45, 2010–2023 RSC; (d) G. Domínguez and J. Pérez-Castells, Chem.–Eur. J., 2016, 22, 6720–6739 CrossRef PubMed; (e) A. Roglans, A. Pla-Quintana and M. Solà, Chem. Rev., 2021, 121, 1894–1979 CrossRef CAS PubMed.
- (a) T. Sato, Nippon Kagaku Zassi, 1971, 92, 277–296 CrossRef CAS; (b) Y. Wang and Z.-X. Yu, Acc. Chem. Res., 2015, 48, 2288–2296 CrossRef CAS PubMed; (c) Y. Ohta, S. Yasuda, Y. Yokogawa, K. Kurokawa and C. Mukai, Angew. Chem., Int. Ed., 2015, 54, 1240–1244 CrossRef CAS PubMed; (d) K. Masutomi, H. Sugiyama, H. Uekusa, Y. Shibata and K. Tanaka, Angew. Chem., Int. Ed., 2016, 55, 15373–15376 CrossRef CAS PubMed; (e) H. Ueda, K. Masutomi, Y. Shibata and K. Tanaka, Org. Lett., 2017, 19, 2913–2916 CrossRef CAS PubMed; (f) S. Yoshizaki, Y. Shibata and K. Tanaka, Angew. Chem., Int. Ed., 2017, 56, 3590–3593 CrossRef CAS PubMed; (g) Z. Tian, Q. Cui, C. Liu and Z. Yu, Angew. Chem., Int. Ed., 2018, 57, 15544–15548 CrossRef CAS PubMed; (h) L. Deng, Y. Fu, S. Y. Lee, C. Wang, P. Liu and G. Dong, J. Am. Chem. Soc., 2019, 141, 16260–16265 CrossRef CAS PubMed; (i) S.-H. Hou, X. Yu, R. Zhang, L. Deng, M. Zhang, A. Y. Prichina and G. Dong, J. Am. Chem. Soc., 2020, 142, 13180–13189 CrossRef CAS PubMed; (j) Y. Wang, W. Liao, Y. Wang, L. Jiao and Z.-X. Yu, J. Am. Chem. Soc., 2022, 144, 2624–2636 CrossRef CAS PubMed.
- (a) A. Geny, S. Gaudrel, F. Slowinski, M. Amatore, G. Chouraqui, M. Malacria, C. Aubert and V. Gandon, Adv. Synth. Catal., 2009, 351, 271–275 CrossRef CAS; (b) E. Da Concepción, I. Fernández, J. L. Mascareñas and F. López, Angew. Chem., Int. Ed., 2021, 60, 8182–8188 CrossRef PubMed; (c) T. Yasui, R. Tatsumi and Y. Yamamoto, ACS Catal., 2021, 11, 9479–9484 CrossRef CAS; (d) J. H. Herbort, R. F. Lalisse, C. M. Hadad and T. V. RajanBabu, ACS Catal., 2021, 11, 9605–9617 CrossRef CAS PubMed; (e) X. Xiao and Z. Yu, Chem.–Eur. J., 2021, 27, 7176–7182 CrossRef CAS PubMed.
- (a) M. Shanmugasundaram, M.-S. Wu and C.-H. Cheng, Org. Lett., 2001, 3, 4233–4236 CrossRef CAS PubMed; (b) T. Kawasaki, S. Saito and Y. Yamamoto, J. Org. Chem., 2002, 67, 4911–4915 CrossRef CAS PubMed; (c) M. Shanmugasundaram, M.-S. Wu, M. Jeganmohan, C.-W. Huang and C.-H. Cheng, J. Org. Chem., 2002, 67, 7724–7729 CrossRef CAS PubMed; (d) T. N. Tekavec and J. Louie, J. Org. Chem., 2008, 73, 2641–2648 CrossRef CAS PubMed; (e) L. Saya, I. Fernández, F. López and J. L. Mascareñas, Org. Lett., 2014, 16, 5008–5011 CrossRef CAS PubMed.
- (a) E. S. Johnson, G. J. Balaich and I. P. Rothwell, J. Am. Chem. Soc., 1997, 119, 7685–7693 CrossRef CAS; (b) J. A. Varela, S. G. Rubín, C. González-Rodríguez, L. Castedo and C. Saá, J. Am. Chem. Soc., 2006, 128, 9262–9263 CrossRef CAS PubMed; (c) M. Gulías, A. Collado, B. Trillo, F. López, E. Oñate, M. A. Esteruelas and J. L. Mascareñas, J. Am. Chem. Soc., 2011, 133, 7660–7663 CrossRef PubMed.
- (a) M. Hatano and K. Mikami, J. Am. Chem. Soc., 2003, 125, 4704–4705 CrossRef CAS PubMed; (b) L. Bai, Y. Yuan, J. Liu, J. Wu, L. Han, H. Wang, Y. Wang and X. Luan, Angew. Chem., Int. Ed., 2016, 55, 6946–6950 CrossRef CAS PubMed; (c) T. Shu, L. Zhao, S. Li, X.-Y. Chen, C. von Essen, K. Rissanen and D. Enders, Angew. Chem., Int. Ed., 2018, 57, 10985–10988 CrossRef CAS PubMed; (d) Z. Ding, Y. Wang, W. Liu, Y. Chen and W. Kong, J. Am. Chem. Soc., 2021, 143, 53–59 CrossRef CAS PubMed.
- K.-H. Rui, S. Yang, Y. Wei and M. Shi, Org. Chem. Front., 2019, 6, 2506–2513 RSC.
- (a) L. Xu, Q. Zhu, G. Huang, B. Cheng and Y. Xia, J. Org. Chem., 2012, 77, 3017–3024 CrossRef CAS PubMed; (b) Y. Park, J. Heo, M.-H. Baik and S. Chang, J. Am. Chem. Soc., 2016, 138, 14020–14029 CrossRef CAS PubMed; (c) Y.-F. Yang, K. N. Houk and Y.-D. Wu, J. Am. Chem. Soc., 2016, 138, 6861–6868 CrossRef CAS PubMed; (d) S. Liu, X. Qi, L.-B. Qu, R. Bai and Y. Lan, Catal. Sci. Technol., 2018, 8, 1645–1651 RSC; (e) S. Vásquez-Céspedes, X. Wang and F. Glorius, ACS Catal., 2018, 8, 242–257 CrossRef; (f) Y. Li, H. Chen, L.-B. Qu, K. N. Houk and Y. Lan, ACS Catal., 2019, 9, 7154–7165 CrossRef CAS; (g) H. Xu, M. Bian, Z. Zhou, H. Gao and W. Yi, ACS Omega, 2021, 6, 17642–17650 CrossRef CAS PubMed.
- (a) C.-Q. Wang, Y. Zhang and C. Feng, Angew. Chem., Int. Ed., 2017, 56, 14918–14922 CrossRef CAS PubMed; (b) H. Xu, W. Chen, M. Bian, H. Xu, H. Gao, T. Wang, Z. Zhou and W. Yi, ACS Catal., 2021, 11, 14694–14701 CrossRef CAS.
- B. Wang, Y. Wang, Z. Wang and J. Wang, Org. Chem. Front., 2019, 6, 2329–2333 RSC.
- (a) D. Cassú, T. Parella, M. Solà, A. Pla-Quintana and A. Roglans, Chem.–Eur. J., 2017, 23, 14889–14899 CrossRef PubMed; (b) Y. Kawaguchi, A. Nagata, K. Kurokawa, H. Yokosawa and C. Mukai, Chem.–Eur. J., 2018, 24, 6538–6542 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data of new compounds. CCDC 1563064. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d1sc06986a |
| This journal is © The Royal Society of Chemistry 2022 |