
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRepurposing of intestinal defensins as multi-target, dual-function amyloid inhibitors via cross-seeding†
Yijing
Tang
a,
Dong
Zhang
 a,
Xiong
Gong
b and
Jie
Zheng
*a
a,
Xiong
Gong
b and
Jie
Zheng
*a
aDepartment of Chemical, Biomolecular, and Corrosion Engineering, The University of Akron, Ohio, USA. E-mail: zhengj@uakron.edu
bSchool of Polymer Science and Polymer Engineering, The University of Akron, Ohio, USA
First published on 20th May 2022
Abstract
Amyloid formation and microbial infection are the two common pathological causes of neurogenerative diseases, including Alzheimer's disease (AD), type II diabetes (T2D), and medullary thyroid carcinoma (MTC). While significant efforts have been made to develop different prevention strategies and preclinical hits for these diseases, conventional design strategies of amyloid inhibitors are mostly limited to either a single prevention mechanism (amyloid cascade vs. microbial infection) or a single amyloid protein (Aβ, hIAPP, or hCT), which has prevented the launch of any successful drug on the market. Here, we propose and demonstrate a new “anti-amyloid and anti-bacteria” strategy to repurpose two intestinal defensins, human α-defensin 6 (HD-6) and human β-defensin 1 (HBD-1), as multiple-target, dual-function, amyloid inhibitors. Both HD-6 and HBD-1 can cross-seed with three amyloid peptides, Aβ (associated with AD), hIAPP (associated with T2D), and hCT (associated with MTC), to prevent their aggregation towards amyloid fibrils from monomers and oligomers, rescue SH-SY5Y and RIN-m5F cells from amyloid-induced cytotoxicity, and retain their original antimicrobial activity against four common bacterial strains at sub-stoichiometric concentrations. Such sequence-independent anti-amyloid and anti-bacterial functions of intestinal defensins mainly stem from their cross-interactions with amyloid proteins through amyloid-like mimicry of β-sheet associations. In a broader view, this work provides a new out-of-the-box thinking to search and repurpose a huge source of antimicrobial peptides as amyloid inhibitors, allowing the blocking of the two interlinked pathological pathways and bidirectional communication between the central nervous system and intestines via the gut–brain axis associated with neurodegenerative diseases.
1. Introduction
Amyloid formation and neuroinflammation are the two interlinked, pathological hallmarks of many neurodegenerative diseases, including Alzheimer's disease (AD), type II diabetes (T2D), medullary thyroid carcinoma (MTC), and Parkinson's disease (PD).1–3 The amyloid cascade4 is believed to contribute to the pathology of these diseases via (i) the formation of toxic amyloid aggregates of different sizes and morphologies with characteristic β-rich structures and (ii) subsequent accumulation and deposition of these β-structure-rich amyloid aggregates formed by amyloid-β (Aβ) in the brain, human islet amyloid polypeptide (hIAPP) in the pancreas, human calcitonin (hCT) in the thyroid, and α-synuclein (α-syn) in the brain, all of which cause cell degeneration/death, tissue dysfunction, and final behavioral disability. More importantly, apart from amyloid formation by the same amyloid proteins, different disease-related amyloid proteins, including Aβ and α-synuclein,5 Aβ and tau,6 Aβ and transthyretin,7 hIAPP and insulin,8 and hIAPP-α-synuclein,9 can co-aggregate and misfold into conformationally and pathologically similar amyloid fibrils (namely, amyloid cross-seeding).10,11 Amyloid cross-seeding is also considered as a pathological risk factor to mutually cause and promote each disease, i.e., one disease can be identified as a risk factor to increase the possibility of another disease, further complicating the amyloid cascade scenario.12 The neuroinflammation cascade for neurodegenerative diseases13 is closely linked to microbial infection by virus (e.g., HSV-1,14 HIV,15 and HHV-6A16), bacteria (e.g., gut bacteria,17 liver bacteria Helicobacter pylori,18 and Chlamydia pneumoniae19), and fungi (e.g., Candida species, Cladosporium, and Cryptococcus20), which will impair the blood–brain barrier (BBB), trigger a long-lasting immune response, and ultimately promote neurodegeneration.21,22 Similar to microbial threats, amyloid aggregates can also be recognized as undesirable targets by the immune system, which triggers the long-lasting immune response by stimulating the release of inflammatory cytokines23,24 and the activation of immune cells (e.g., microglia),25 ultimately leading to neurodegeneration. Particularly, bacterial amyloid proteins, as a subgroup of gut microbiota, share similar structural and aggregation characteristics to native amyloid proteins in the central nervous system (CNS). The exposure to bacterial amyloid proteins in the gut could cause the priming and enhancing of the immune response to the endogenous production of neuronal amyloids, which will translocate across the leaky gut-blood–brain barrier to interfere with the signaling of the brain in aged or gut-infected people.Apart from the independent contributions of amyloid formation and neuroinflammation to the pathogenesis of neurodegenerative diseases, a growing body of experimental and clinical data shows a pathogenic connection, even a loop, between amyloid formation and neuroinflammation via the bidirectional, constant communication between amyloid proteins and gut microbiota,13,26 suggesting a disease mechanism via the brain–gut–microbiota axis.27 While this spreading mechanism still remains elusive, several lines of evidence have shown this bidirectional communication between amyloid proteins and gut microbiota (e.g., bacterial amyloids), probably because they both exist in the blood circulation and cerebrospinal fluid, which raises the possibilities for their co-aggregation with each other or cross-seeding with other amyloidogenic proteins.28–30 Evidently, curli produced by Escherichia coli accelerated α-syn aggregation and motor impairment in a mouse model.31 CsgC as curli-specific operons inhibited the amyloid formation of both CsgA and α-syn by preventing the β-sheet transition, but had no effect on Aβ42 amyloid formation.32 FapC amyloid fragments (FapCS) from Pseudomonas aeruginosa demonstrated their cross-seeding capacity for interacting with Aβ and promoting Aβ aggregation at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 molar ratio in vitro and in a zebrafish model.26 Additionally, amyloid proteins could also act as active metabolites of the innate immune system to affect microbial behaviors. Evidently, Aβ oligomers can bind to carbohydrates of microbial cell walls in a heparin-binding domain, thus endowing antimicrobial activity against fungal and bacterial infections in cell, nematode, and mouse models of AD.33 Similarly, hIAPP was also found to hinder Staphylococcus aureus and Escherichia coli bacteria growth.34 PAP248-286 in human semen can form amyloid-like aggregates (termed Semen Enhancer of Viral Infection) for enhancing HIV infection.35 These studies demonstrate the cross-seeding between amyloid proteins and bacterial amyloids through amyloid-like mimicry of β-sheet associations.
:10 molar ratio in vitro and in a zebrafish model.26 Additionally, amyloid proteins could also act as active metabolites of the innate immune system to affect microbial behaviors. Evidently, Aβ oligomers can bind to carbohydrates of microbial cell walls in a heparin-binding domain, thus endowing antimicrobial activity against fungal and bacterial infections in cell, nematode, and mouse models of AD.33 Similarly, hIAPP was also found to hinder Staphylococcus aureus and Escherichia coli bacteria growth.34 PAP248-286 in human semen can form amyloid-like aggregates (termed Semen Enhancer of Viral Infection) for enhancing HIV infection.35 These studies demonstrate the cross-seeding between amyloid proteins and bacterial amyloids through amyloid-like mimicry of β-sheet associations.
Different from single-targeted amyloid inhibitors with relatively poor marginal benefits from bench to bedside applications and beyond, antimicrobial peptides could serve as new amyloid inhibitors with multi-target inhibition activity against the aggregation of different amyloid proteins and the infection of different microbes simultaneously. If successful, antimicrobial peptides are expected to block different pathological pathways of amyloid aggregation and microbial infection towards the onset and progression of PMDs. Conceptually, the discovery or repurposing of antibacterial peptides, with intrinsic antimicrobial activity, as amyloid inhibitors is a plausible strategy to achieve both anti-neuroinflammation and anti-amyloid functions. Particularly, some antibacterial peptides (α-defensins,36,37 plantaricin A,38 uperin 3.5,39 and dermaseptin S9 (ref. 40)) have been found to possess amyloid-like properties in terms of their aggregation kinetics and β-rich structures, while other amyloid proteins (Aβ,41,42 hIAPP,43 GNNQQNY,44,45 and KLVFFGAIL46) have been recognized for their antibacterial and antifungal activities47–49 against different microorganisms including Candida albicans, Escherichia coli, Enterococcus faecalis, and Staphylococcus epidermidis.41,43,49–54 Given mutual resemblances in aggregation and structural characteristics between amyloid proteins and antimicrobial peptides, here, we discovered and repurposed two intestinal defensins, human α-defensin 6 (HD-6) and human β-defensin 1 (HBD-1), as amyloid inhibitors with dual anti-neuroinflammation and anti-amyloid functions. Selection of HD-6 and HBD-1 is largely attributed to their existence in intestinal Paneth and epithelial cells, as well as their important roles in regulating the gut microbial population/composition/physiology, and intestinal secretion/viability.55 From a structural viewpoint, both HD-6 and HBD-1 as cationic peptides contain six cysteine residues to form three intramolecular disulfide bridges for stabilizing a largely β-sheet structure, which serves as a critical structural motif to cross-seed with amyloid peptides to induce new amyloid inhibition functions (Fig. 1a & S5a†). Our collective results showed that both defensins were able to cross-seed with three different amyloid proteins, Aβ, hIAPP, and hCT, at sub-stoichiometric molar ratios in different bulk, surfactant, cellular, and bacterial environments. Such cross-seeding capacity of both defensins led to three distinct functions: (i) preventing amyloid formation of Aβ, hIAPP, and hCT; (ii) protecting different cells (SH-SY5Y and RIN-m5F) from the respective amyloid-induced toxicity; and (iii) exhibiting high antimicrobial activities against four bacterial strains. Given the close pathological links between amyloid aggregation and microbial infection and between AD, T2D, and MTC, we demonstrate a new repurposing strategy to discover antimicrobial peptides (beyond intestinal defensins) as multiple-target, dual function inhibitors to alleviate risk factors of amyloid aggregation, cellular dysfunction, and microbial infection, which contribute to the progressive pathogenesis and severity of neurodegenerative diseases.
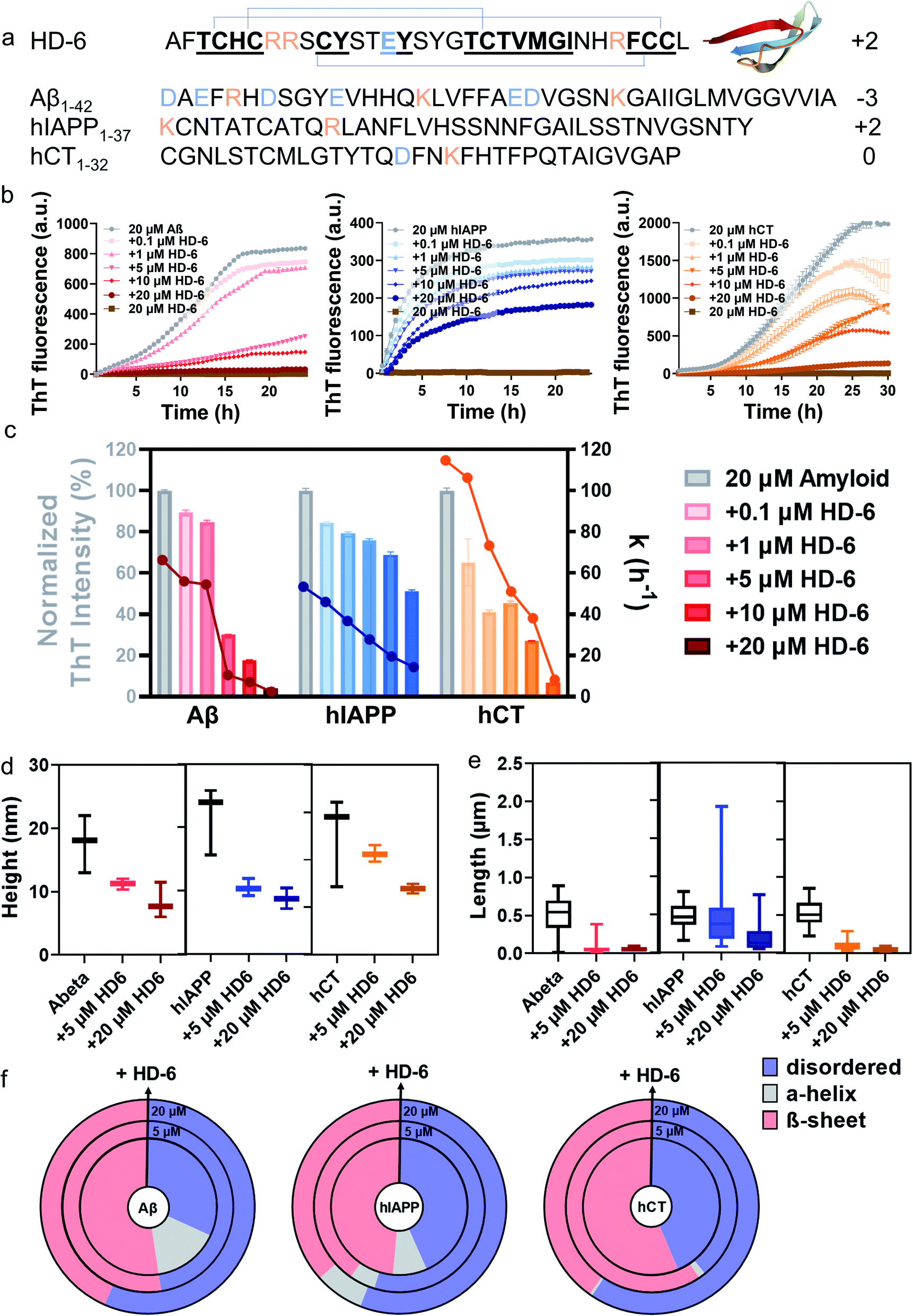 | ||
| Fig. 1 HD-6 exhibits a general cross-seeding-induced inhibition capacity against different amyloid aggregation pathways. (a) Sequence and structure of HD-6, Aβ, hIAPP, and hCT with β-rich structure. Color ID: positively charged residues (orange letters), negatively charged residues (blue letters), and β-structure fragments (bold & underline). (b) ThT aggregation kinetic profiles and (c) quantitative analysis of ThT profiles to show the dose-dependent inhibition effects of HD-6 on Aβ, hIAPP, and hCT aggregation at different amyloid:HD-6 ratios (1:0.005–1:1). Inhibition efficiency of HD-6 against amyloid aggregation is determined by the relative final fluorescence intensity (%) as normalized by that of pure amyloid aggregation (left axis). Error bar represents the standard deviation (s.d.) of triplicate measurements. The corresponding aggregation rate constant (k) is determined by a time point of t1/2, at which the fluorescence intensity reaches half of the maximum fluorescence intensity between the baseline and the plateau (right axis). (d) Secondary structure distributions of Aβ, hIAPP, or hCT (20 μM) in the presence of HD-6 of 0 μM (inner circle), 5 μM (middle circle), and 20 μM (outer circle) concentrations, obtained from circular dichroism (CD) spectra using the BestSel program. Morphological characterization of cross-seeding aggregates formed by 20 μM amyloid (Aβ, hIAPP, or hCT) and 5–20 μM HD-6 in (e) heights and (f) lengths, obtained from the AFM images in Fig. S4.† The upper, middle, and lower bars define the maximum, mean, and minimum values of aggregate heights and lengths, respectively. | ||
2. Results and discussion
2.1. General cross-seeding-induced inhibitory ability of defensins against different amyloid aggregation pathways
Our and other previous studies36,56 have shown that defensins and amyloids can form conformationally similar β-sheet-rich fibrils. Here, we aim to test the “like-interact-like” hypothesis by examining the cross-seeding possibility between two defensins (HD-6 and HBD-1) and three amyloid peptides (Aβ, hIAPP, and hCT) and how such cross-seeding, if occurs, affects amyloid aggregation. To this end, we examined a total of 6 cross-seeding systems by co-incubating Aβ, hIAPP, or hCT (20 μM) with HD-6 or HBD-1 (0.1–20 μM) separately at different molar ratios (1:0.005–1:1) at 37 °C for 24–30 h. ThT aggregation profiles in Fig. 1b show that Aβ, hIAPP, or hCT alone exhibit typical sigmoidal nucleation–polymerization curves, each starting with a short lag phase of 0.5–5 h, followed by a rapid growth phase of 1–25 h, finally reaching a saturated phase with the highest ThT intensities of 716, 435, and 1305 a.u., respectively. For comparison, we first used HD-6 defensin (0.1–20 μM) as a proof-of-concept example to co-incubate with Aβ, hIAPP, or hCT (20 μM) and to study their possible cross-seeding effects. At first glance of Fig. 1c, HD-6 exhibits significant inhibition effects on amyloid aggregation of the three amyloid peptides (Aβ, hIAPP, or hCT) at sub-stoichiometric concentrations (≤equimolar ratio) in a concentration-dependent manner, demonstrating that HD-6 can interact with the three different amyloid peptides to inhibit their aggregation. Specifically, at an equimolar ratio, HD-6 almost completely suppressed both amyloid fibril formation and the aggregation rate constant (k) of Aβ by 82% and 96%, hIAPP by 32% and 71%, and hCT by 73% and 92%, respectively. Even at an amyloid:HD-6 ratio of 1:0.25, HD-6 can still largely reduce Aβ fibrils by 70%, hIAPP fibrils by 25%, and hCT fibrils by 55%, respectively. To validate the ThT results, AFM images were recorded at different time points of kinetics experiments. AFM images of pure amyloid samples (20 μM) showed a large quantity of fibrils being formed at 24 h incubation, with the average height/length of 13/508 nm by Aβ, 15/483 nm by hIAPP, and 16/477 nm by hCT. The co-incubated samples of HD-6 with the three amyloid peptides, each with an equimolar ratio, almost completely suppressed amyloid fibril formation, as evidenced by the observation that there was no presence of large mature fibers, instead many discrete oligomers or short protofibrils. Analysis of these aggregates in AFM images revealed a significant reduction in the average height/length of 11/60 nm for HD-6–Aβ, 7/197 nm for HD-6–hIAPP, and 7/52 nm for HD-6–hCT systems, respectively (Fig. 1d and e). The inhibition of amyloid formation by HD-6 and large structural variance of HD-6–amyloid aggregates as observed with TEM were corroborated by ThT measurements.
From a structure-transition viewpoint, the secondary structure distribution of HD-6–amyloid systems (Fig. 1f and S3†), obtained from CD spectra (Fig. S1 & S2†), showed that upon 24–72 h incubation, (i) pure Aβ, hIAPP, and hCT (20 μM) adopted 52.4%, 48.5, and 56.4% of β-structure, while HD-6 (20 μM) retained 52.5% of its dominant disordered structure; (ii) the co-incubation of HD-6 (20 μM) with the three different amyloid peptides (20 μM) enabled the β-structure content of Aβ aggregates to be reduced to 43.4%, hIAPP aggregates to 36.6%, and hCT aggregates to 40.2%, respectively. The declining β-structure content of amyloid aggregates for HD-6 confirms the “like-interact-like” mechanism that HD-6 likely binds to conformationally similar β-structure-rich amyloid peptides so as to prevent the conformational changes from random coils to β-structures of amyloid aggregates, thus explaining the general inhibitory capacity of HD-6 for preventing the amyloid fibrillation of Aβ, hIAPP, and hCT.
To further explore the fundamental correlation between antimicrobial and amyloid peptides, we examined another defensin–amyloid system (i.e., HBD-1–Aβ, HBD-1–hIAPP, and HBD-1–hCT) by studying the cross-seeding of HBD-1 with different amyloid peptides (Aβ, hIAPP, and hCT) using the same aggregation assays. Consistently, co-incubation of HBD-1 (0.1–20 μM) with amyloid peptides (20 μM) showed a similar cross-seeding-induced inhibition effect of HBD-1 on the amyloid aggregation of Aβ, hIAPP, and hCT (Fig. S5†). Evidently, at all molar ratios of HBD-1:amyloids tested, HBD-1 enabled the final ThT intensity (an indicator of amyloid fibril amounts) to be reduced by 44–93%, the aggregation rate (half-time of aggregation) to be decreased by 71–96%, the structural transition towards β-structures to be delayed, the β-structure content to be lowered by 4–28%, and the longer and thicker mature fibrils to be disassembled into shorter and thinner ones. Taken together, collective results from ThT, AFM, and CD analyses demonstrate the cross-seeding interactions between the two defensins (HD-6 and HBD-1) and three amyloid peptides (Aβ, hIAPP, and hCT), and cross-seeding interactions enable amyloid aggregation pathways and kinetics to be altered by breaking their β-sheets and preventing their aggregation towards final fibril formation, confirming defensins to be effective and general inhibitors of amyloid peptides.
2.2. Molecular interactions of defensins with different amyloid proteins and seeds
To gain better insights into amyloid inhibition mechanisms of defensins, we quantified the binding affinity between 20 μM defensins (HD-6 and HBD-1) and 2.5–20 μM amyloid peptides (Aβ, hIAPP, and hCT) using surface plasmon resonance (SPR). To this end, defensins of HD-6 or HBD-1 were first covalently immobilized on carboxymethylated SPR chips via EDC/NHS coupling, followed by the flow of amyloid solutions through the defensin-coated channels for determining their specific binding affinity between immobilized defensins and amyloid aggregates in a PBS flow (Fig. 2a). At first glance of Fig. S6† & 2b–d, immobilized HD-6 did not adsorb any lysozyme (as a negative control), but exhibited a general, specific binding ability to the three different amyloid monomers in a concentration-dependent manner. In all three HD-6–amyloid systems, the HD-6-coated surfaces adsorbed more amyloid monomers as their concentrations increased from 2.5 to 20 μM, as evidenced by the increased protein adsorption (PA) from 3.2 to 71 ng cm−2 for Aβ monomers, from 2.4 to 35 ng cm−2 for hIAPP monomers, and from 6.3 to 49 ng cm−2 for hCT monomers. Additionally, the equilibrium dissociation constant (KD) obtained from binding constant linearization is in agreement with the specific binding preference for different amyloid proteins as indicated by different KD values, i.e., HD-6 exhibited a decreasing order of binding affinity: Aβ with a KD of 0.356 μM > hCT with a KD of 3.774 μM > hIAPP with a KD of 6.206 μM (Fig. S7†).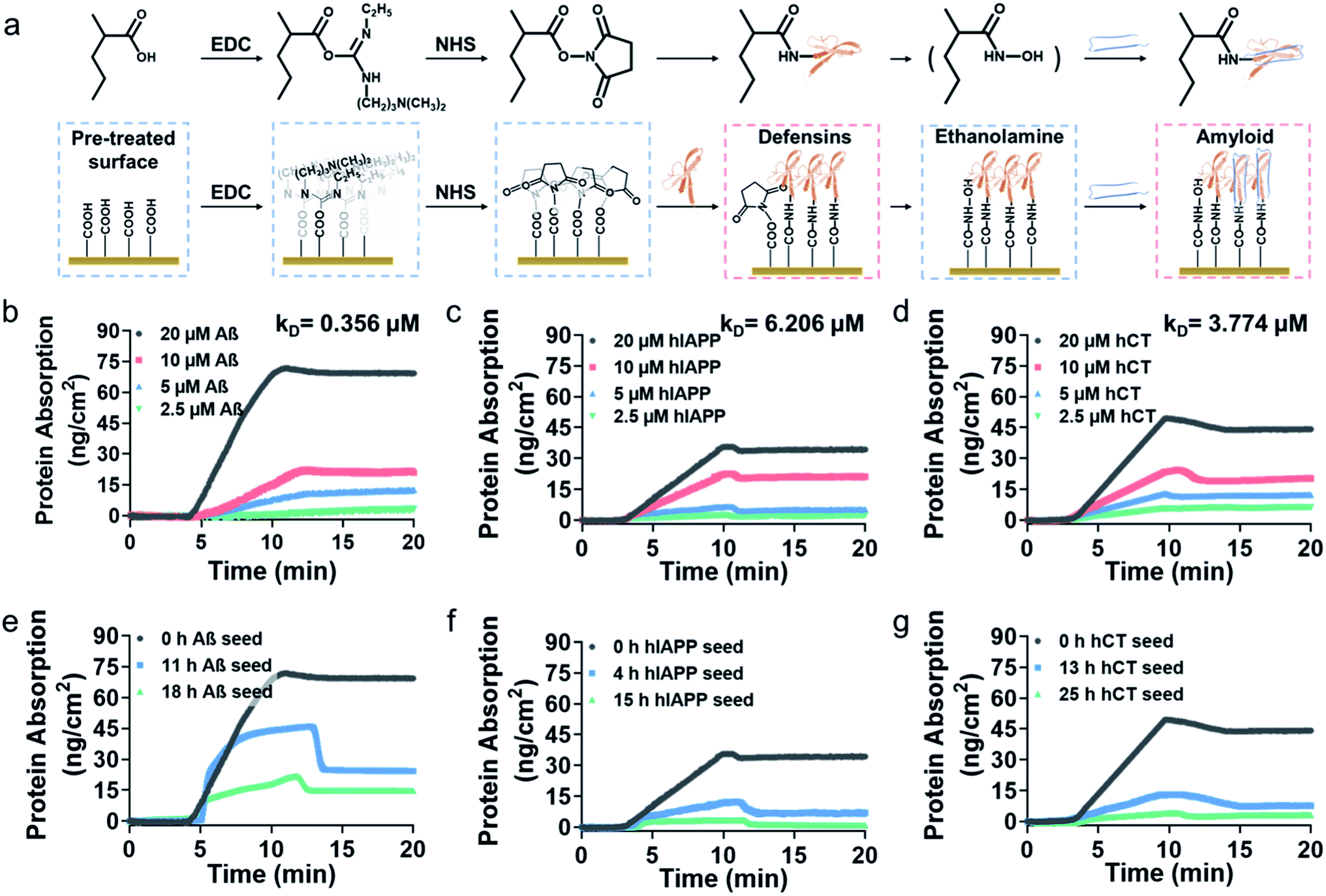 | ||
| Fig. 2 Binding affinity of HD-6 to amyloid aggregates of different concentrations and sizes by SPR. (a) Schematic workflow for immobilizing defensins on the SPR chip for amyloid binding. SPR sensorgrams to show the adsorption amount (binding preference) of (b) Aβ, (c) hIAPP, and (d) hCT monomers of varied concentrations of 2.5–20 μM on the HD-6 coated SPR surface. SPR sensorgrams to show the adsorption amount (binding preference) of 20 μM (e) Aβ, (f) hIAPP, and (g) hCT monomers (black), oligomers (blue), and fibrillar species (green) on the HD-6 coated SPR surface. | ||
In parallel to the binding of defensins to amyloid monomers, we also prepared different preformed amyloid seeds, which were incubated for different hours (ranging from 0 h to 25 h) to represent amyloid aggregates at the lag, growth, and equilibrium stages, to examine whether defensins have preferential interactions with some specific amyloid seeds. SPR sensorgrams in Fig. 2e show that upon flowing three different Aβ seed solutions through the HD-6-coated channels, the adsorption amount of Aβ on the immobilized HD-6 surfaces decreased with the aging of Aβ species. Quantitatively, immobilized HD-6 adsorbed 71 ng cm−2 of Aβ monomers, 45 ng cm−2 of 11 h Aβ seeds, and 21 ng cm−2 of 18 h Aβ seeds, respectively. This confirms the general binding ability of HD-6 to all of the three Aβ seeds in a seed-dependent way. In the cases of HD-6–hIAPP and HD-6–hCT systems (Fig. 2f and g), it appears that HD-6 exhibited very weak binding affinities to hIAPP seeds (PA,4h-hIAPP = 12 ng cm−2 and PA,15h-hIAPP = 3.4 ng cm−2) and hCT seeds (PA,4h-hCT = 13 ng cm−2 and PA,15h-hCT = 4.2 ng cm−2), but a strong binding affinity to hIAPP and hCT monomers (PA,hIAPP monomer = 35 ng cm−2 and PA,hCT monomer = 49 ng cm−2). In the case of SPR studies for HBD-1–amyloid systems, similar trends of concentration- and seed-dependent binding abilities of HBD-1 to different amyloid proteins were observed (Fig. S8†), i.e., HBD-1 interacts more preferentially and strongly with (i) amyloid monomers of higher concentrations than those of lower concentrations and (ii) amyloid monomers than amyloid oligomeric and fibrillar aggregates.
To validate SPR results, we performed the parallel ThT seeding experiments by adding 20 μM defensins (HD-6 or HBD-1) to 20 μM amyloid seed solutions (Aβ, hIAPP, and hCT) pre-incubated for different times. Any signal change in ThT curves before and after defensin addition will indicate the occurrence and efficiency of cross-seeding. Time points to add defensins were selected from Fig. 1b and S5b,† corresponding to different aggregation species of Aβ, hIAPP, and hCT in the oligomeric state (i.e., 11 h-incubated Aβ, 4 h-incubated hIAPP, and 13 h-incubated hCT) and the fibrillar state (18 h-incubated Aβ, 15 h-incubated hIAPP, and 25 h-incubated hCT). In the case of adding HD-6 (20 μM) to amyloid seed solutions at an equimolar ratio, the three HD-6–amyloid systems exhibited similar ThT kinetic curves in Fig. 3a–c, as indicated by several observable facts: (i) defensins can almost completely suppress Aβ and hCT fibrillization by 92–96% and largely decrease hIAPP fibrillization by 53% from their monomer aggregation (red line); (ii) defensins enabled the aggregation and growth of amyloid oligomers into mature fibrils to be immediately slowed down by reducing their final ThT intensities by 27–56% (blue line); (iii) defensins failed to stop amyloid fibrillation or disassemble existing amyloid fibrils, as shown by the two almost overlapped ThT curves after adding defensins to amyloid fibrils in the equivalent state (green line).
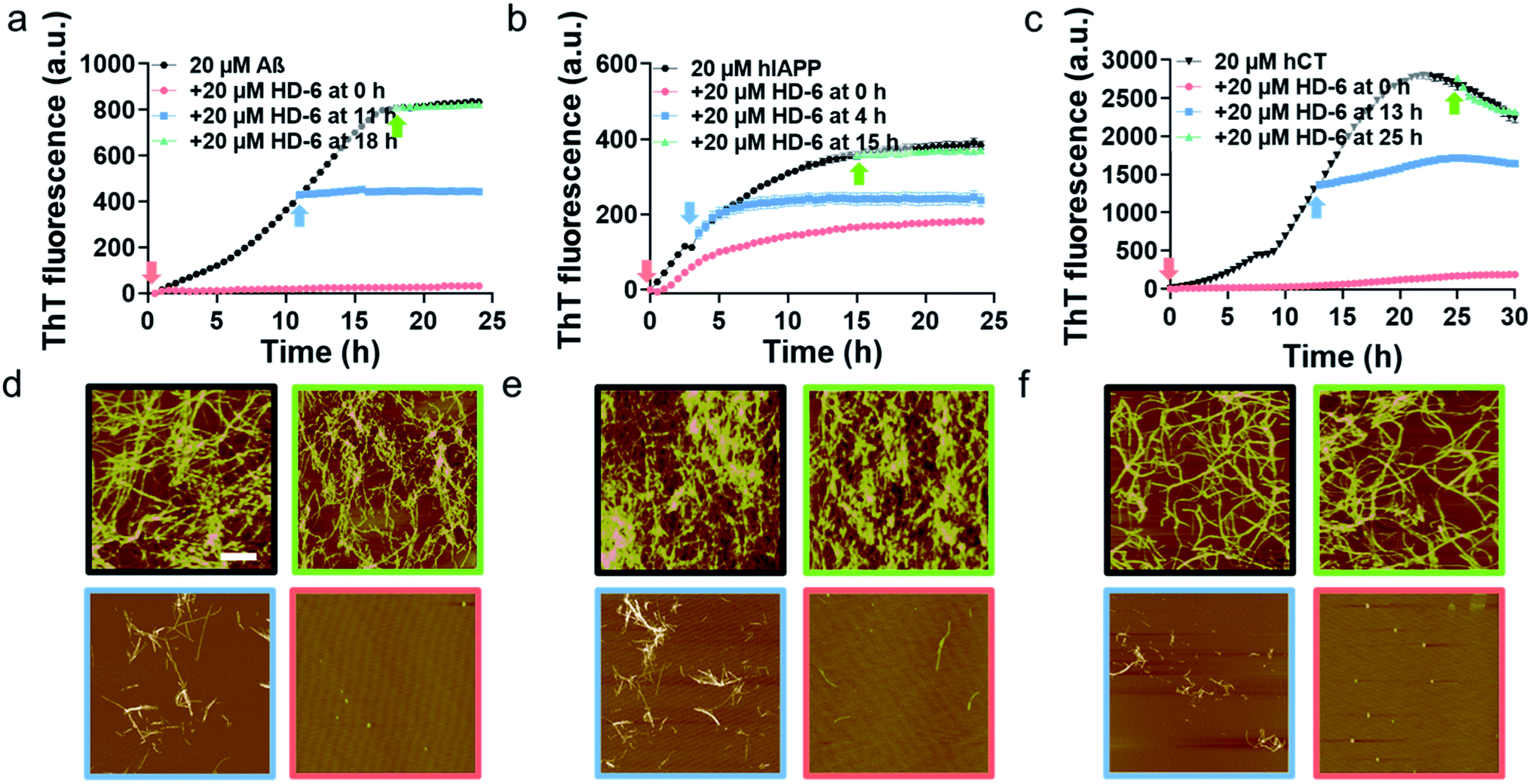 | ||
| Fig. 3 HD-6 interacts with different amyloid seeds to inhibit their aggregation to different extents. Time dependent (a–c) ThT fluorescence profiles and (d–f) representative AFM images for pure amyloid proteins (black) and the addition of 20 μM HD-6 to 20 μM (a, d) Aβ, (b, e) hIAPP, and (c, f) hCT seeds preformed in the monomeric (red), oligomeric (blue), and fibrillar (green) states. Time points for HD-6 to different amyloid seed solution are indicated by arrows in ThT profiles, while scale bars are 1 μm in AFM images. | ||
Consistently, additional lines of evidence from AFM images (Fig. 3d–f) at 24 h and 36 h again confirm the various cross-seeding efficiencies between different amyloid seeds and defensins. As compared to 20 μM pure amyloid fibrils (black box) with the average height/length of 13–16/477–508 nm, the introduction of equimolar HD-6 into amyloid monomer solutions completely eliminated amyloid fibril formation (red box), but such inhibition efficiency of HD6 decreased with the aging of amyloid seeds. Specifically, the addition of equimolar HD-6 (20 μM) to amyloid oligomers significantly retarded amyloid fibril formation, as observed from some short fibrils with the average height/length of 10/320 nm for Aβ, 12/248 nm for hIAPP, and 9/102 nm for hCT, together with very few, small amorphous aggregates (blue box). However, HD-6 had no influence on either aggregation or disassembly of the preformed amyloid fibrils (green box). Further, HDB-1 showed similar cross-seeding-induced inhibition efficacy to amyloid monomeric, oligomeric, and fibrillar aggregates (Fig. S9†).
Taken together, side-by-side result comparison between pure amyloids and amyloid–defensin systems using ThT, CD, AFM, and SPR data reveals the dose- and sequence-dependent inhibition effects of defensins on amyloid aggregation. Specifically, both HD-6 and HBD-1 exhibit sequence-dependent inhibition efficiency against amyloid aggregation in a decreasing order of Aβ > hCT > hIAPP. Such differences are likely attributed to different electrostatic interactions between defensins (+2e for HD-6 and +3e for HBD-1) and amyloid peptides (−3e for Aβ, 0e for hCT, and +2e for hIAPP). Fundamentally, the inhibition capacity of both HD-6 and HBD-1 mainly stems from their effective interaction with amyloid monomers and oligomers to alter their structural transitions and aggregation pathways at the early aggregation stages, but not amyloid fibrils probably because they are too stable to be disrupted. Different binding affinities of defensins to amyloid proteins, depending on their sequences, concentrations, and seeds of amyloids, clearly indicate the existence of different cross-seeding barriers and competitive interactions between defensins and amyloids, again supporting the defensin binding-induced (cross-seeding-induced) amyloid inhibition mechanism.
2.3. Defensins alleviate amyloid-induced cell toxicity
It is generally accepted that amyloid aggregates are key pathological species that cause cell degeneration and death associated with PMDs.57,58 Thus, inhibition of amyloid aggregation is considered as a critical and promising strategy for the prevention and treatment of PMDs. On the other hand, inhibition of amyloid aggregation does not necessarily lead to the reduction of amyloid toxicity, because amyloid oligomerization and fibrillization could be different pathological pathways. Here, upon demonstrating the amyloid inhibition properties of defensins, we continued to explore whether such inhibition performance of defensins can rescue cells from amyloid-mediated cytotoxicity. Given the three different amyloid peptides to be tested, different cell lines were chosen and used in MTT, LDH, and live/dead assays to study amyloid-induced cytotoxicity, i.e., the human neuroblastoma SH-SY5Y cell line for Aβ- and hCT-involved systems, while the rat pancreatic insulinoma RIN-m5F cell line for hIAPP-involved systems. As a control, both defensins of 1 μM were non-toxic to SH-SY5Y and RIN-m5F cells as evidenced by 97–99% cell viability and 0.5–2.6% cytotoxicity, while defensins of 20 μM were non-toxic to RIN-m5F (97–99% of cell viability and 1.1–1.2% of cytotoxicity), but little toxic to SH-SY5Y (76–79% of cell viability and 15–17% of cytotoxicity). This indicates that pure defensins show concentration-dependent and cell-specific cytotoxicity. In sharp contrast, pure Aβ, hIAPP, and hCT were highly toxic to cells by decreasing their cell viability to 31%, 55%, and 67% and increasing cell toxicity to 33%, 25%, and 35%, respectively (Fig. 4a–c & S10a–c†).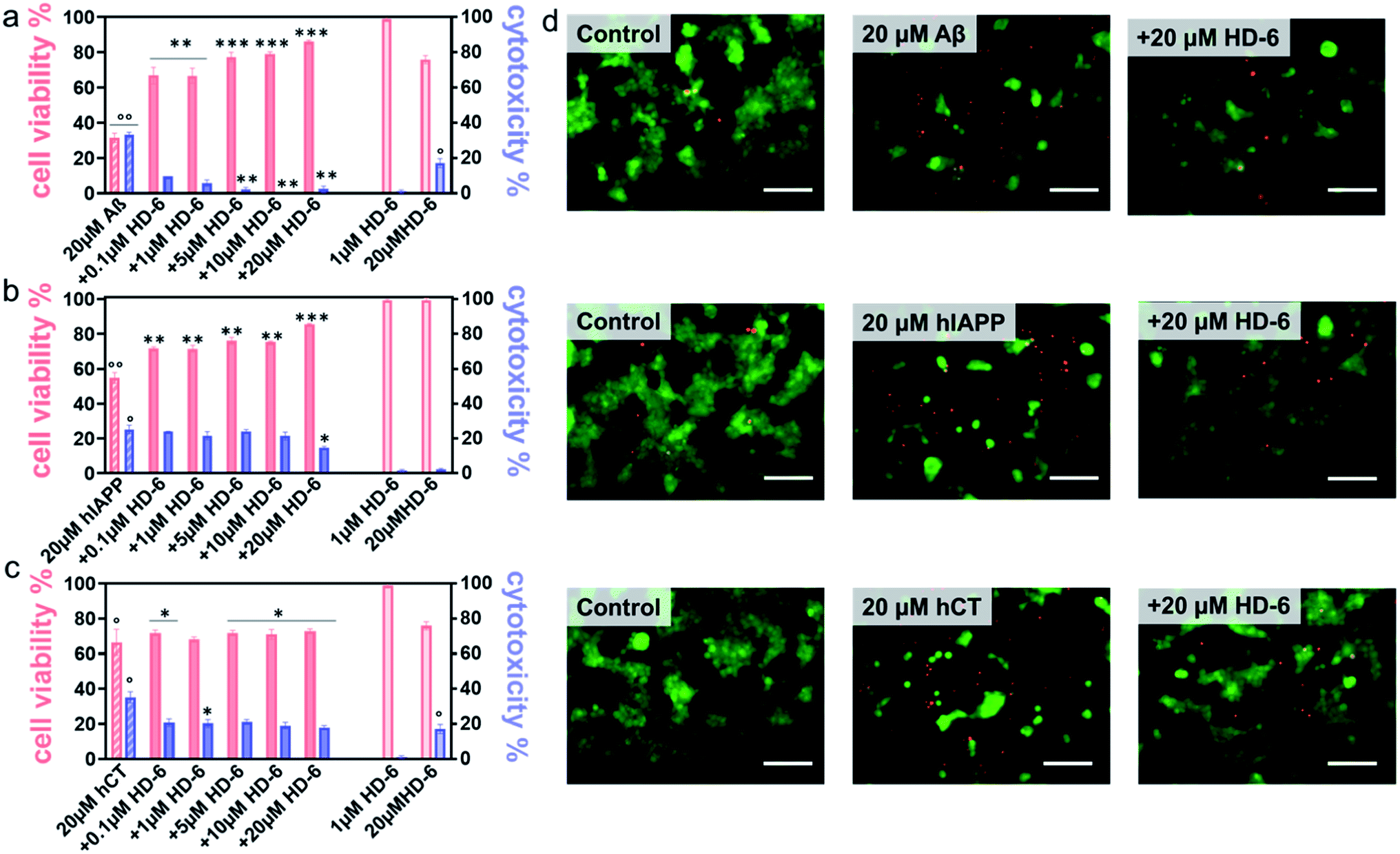 | ||
| Fig. 4 HD-6 rescues cells from amyloid-induced cytotoxicity. Dose-dependent protection role of HD-6 in (a) Aβ-, (b) hIAPP-, and (c) hCT-mediated cytotoxicity using cell viability from the MTT assay (red bar, left axis) and cell cytotoxicity from the LDH assay (blue bar, right axis), in which SH-SY5Y cells or RIN-m5F cells are treated with amyloid (20 μM) with and without 0.1–20 μM HD-6 for 24 h. Statistical analysis (n = 3) was conducted for cells treated with HD-6 or amyloid proteins alone relative to control (°, p < 0.05; °°, p < 0.01; °°°, p < 0.001), as well as cells treated with both HD-6 and amyloid proteins relative to cells treated with amyloid proteins alone (*, p < 0.05; **, p < 0.01; ***, p < 0.001). (d) Representative fluorescence microscopy images of cells upon treatment with 20μM HD-6 (1st column), 20μM amyloid proteins (2nd column), and their cross-seeds (3rd column) for 24 h. Scale bars = 180μm. | ||
In parallel, when co-incubating defensins with amyloid-treated cells, all of the six defensin–amyloid systems exhibited an increase of cell viability and a decrease of cytotoxicity to some extent as compared to the corresponding amyloid systems alone. Specifically, for the HD-6–Aβ systems, 0.1 μM of HD-6 enabled SH-SY5Y cells to be rescued from Aβ-induced toxicity by improving cell viability by 36% and reducing cell cytotoxicity by 24%, respectively. On the other hand, defensin concentration is a double-edged sword, i.e., while the increase of defensin concentration can be more effective in preventing amyloid aggregation, it also caused more cell death due to its intrinsic toxicity to some cell types. Evidently, HD-6 of 10 μM improved the cell viability by 48% and decreased the cell toxicity by 33% in the presence of Aβ and cells, but doubling the concentration of HD-6 to 20 μM only increased the cell viability by 55% and decreased the cell toxicity by 30% (Fig. 4a). In the case of HD-6–hCT systems, HD-6 of different concentrations between 0.1 and 20 μM exhibited a similar protection effect on hCT-induced cell death, as indicated by increasing cell viability by 3–7% and decreasing cell toxicity by 15–18% (Fig. 4c). For the HD-6–hIAPP system, thanks to the non-toxicity of defensins for RIN-m5F cells, HD-6 of different concentrations (0.1–20 μM) exhibited cell protection effects on RIN-m5F cells. As defensin concentrations increased from 0.1 to 20 μM, HD-6 increased the cell viability from 54% to 85% and decreased the cell toxicity from 25% to 14% (Fig. 4b).
Similarly, HBD-1 exhibited concentration-dependent cell protection effects on the HBD-1–Aβ system and HBD-1–hCT system, but not on the HBD-1–hIAPP system. Specifically, HBD-1 can protect SH-SY5Y cells from Aβ-induced toxicity by improving the cell viability by 35–51% and reducing the cell cytotoxicity by 24–25% (Fig. S10a†) as the HBD-1 concentration increased from 0.1 to 10 μM. The use of 20 μM of HBD-1 weakens its cell protection effect, as indicated by a cell viability of 42% and cell cytotoxicity of 15%. For HBD-1 and hCT systems, HBD-1 showed an optimal concentration of 1 μM to achieve its maximal cell protection with 84% of cell viability and 19% of cytotoxicity. Further increase of HBD-1 concentration to 20 μM led to a gradual decrease of cell viability to 61% and an increase of cytotoxicity to 20% (Fig. S10c†). This again indicates that there exists a balance between defensin-inhibition-induced cell protection and defensin-concentration-induced cell death for SH-SY5Y cells. In the case of HBD-1–hIAPP systems, HBD-1 showed a dose-dependent cell protection effect on RIN-m5F cells, in which the increase of HBD-1 concentration from 0.1 to 20 μM led to an increase of cell viability from 54% to 86% and a decrease of cell toxicity from 25% to 9% (Fig. S10b†).
Seeing is understanding. A live/dead cell assay was performed by staining live cells with Calcein AM (green) and dead cells with ethidium homodimer-1 (red). Upon overlaying both the green and red fluorescent images on the top of each other, it can be clearly seen in Fig. 4d & S10d† that defensins were almost non-cytotoxic to cells, while amyloids alone caused a significant extent of dead cells. Meanwhile, the same stoichiometry used in MTT assays that exhibits the best protection efficiency was also used in live/dead assays, i.e., 20 μM defensins were used in HD-6–Aβ, HD-6–hIAPP, HD-6–hCT, and HBD-1–hIAPP systems, 10 μM defensins in the HBD-1–hIAPP system, and 1 μM defensins in the HBD-1–hCT system. The results showed an appreciable reduction in the number of dead cells, which reinforces the protection role of defensins in amyloid-induced cell toxicity.
2.4. Cross-seeds of defensins-amyloids retain antimicrobial properties
Considering that the “amyloid cascade” and “microbial infection cascade” are two of the main mechanisms accounting for the pathogenesis of protein misfolding diseases, here we further tested the antimicrobial activity of defensins, amyloid peptides, and their heterocomplexes against both representative Gram-negative bacterial strains of Escherichia coli (E. coli) and P. aeruginosa (P.A.) and Gram-positive bacterial strains of S. aureus (S.A.) and S. epidermidis (S.E.) (Fig. S11†). As summarized in Fig. 5a–d and S12,† both defensins and amyloid peptides demonstrated some broad-spectrum antimicrobial activities against the four bacterial strains to different extents, with the best case showing 0.54-log reduction in bacterial growth. Both defensins and amyloid peptides displayed their antimicrobial activities against the four different bacterial strains in a descending order of S.E. > E. coli > S.A. > P.A. As compared to Gram positive pathogens of S.A. and S.E., Gram-negative pathogens of E. coli and P.A. possess an outer cell membrane and powerful molecular efflux pumps that prevent the cell entry of many categories of antimicrobial molecules.59,60 Moreover, it is not surprising to observe that both defensins, due to their intrinsic bacteria-killing ability, have higher antimicrobial activity than amyloid peptides. Between them, HD-6 was more effective in killing bacteria and inhibiting bacterial growth than HBD-1. Such common and broad-spectrum antimicrobial ability of defensins and amyloid peptides is mainly contributed by their cell membrane disruption capacity.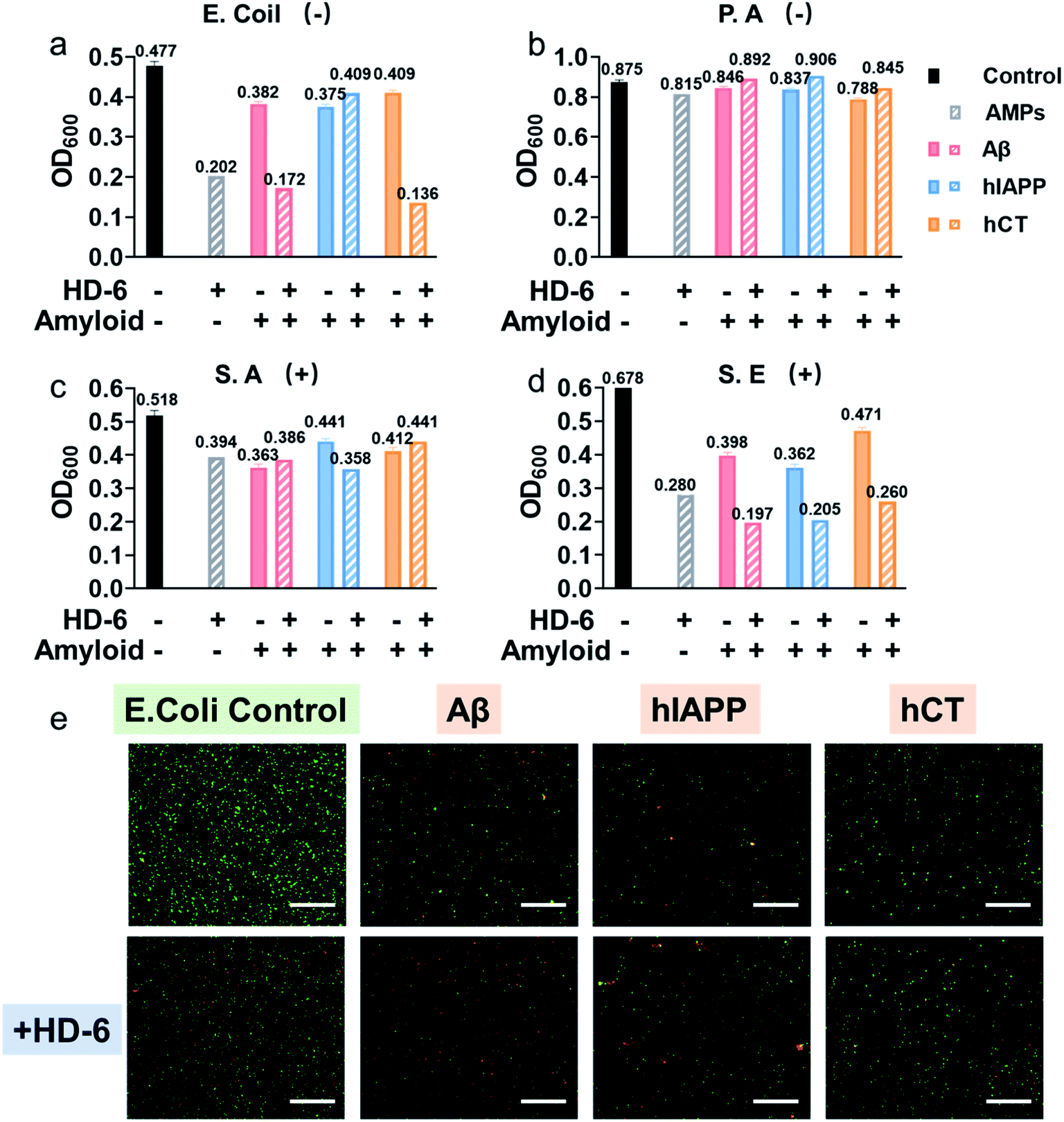 | ||
| Fig. 5 HD-6–amyloid heterocomplex retains a broad-spectrum antimicrobial activity. Antimicrobial activity of 20 μM HD-6, 20 μM amyloid peptides, and cross-species of HD-6–amyloid peptides against Gram-negative (a) E. Coli and (b) P. aeruginosa, and Gram-positive (c) S. aureus and (d) S. epidermidis quantified by the final bacterial density. Bacterial density is determined by OD600. (e) Representative fluorescence microscopy images of E. coli treated with freshly prepared amyloid peptides (20μM) in the absence and presence of 20μM HD-6. Red fluorescent propidium iodide and green fluorescent SYTO 9 are used to identify dead bacteria with damaged membranes and live bacteria with intact membranes, respectively. Scale bars = 180 μm. | ||
Upon demonstrating the effective antimicrobial activity of pure defensins or amyloid peptides, it is more interesting to examine whether and how defensin–amyloid heterocomplexes could alter their antimicrobial activity as compared to their pure counterparts. To achieve this goal, we co-incubated defensins and amyloid peptides (20 μM) at an equimolar ratio for 1 h and then added it into the bacterial solution. The OD600 values in Fig. S11† clearly show that apart from a few exceptions, the co-assemblies of defensins with the three different amyloid peptides can improve/retain their antimicrobial activities, as compared to their pure amyloid peptides or defensins. In the best cases, the co-incubation of HD-6 and amyloid peptides significantly enhanced their antimicrobial activities by 27–33% as compared to pure HD-6 and by 43–67% as compared to pure amyloid peptides. Similarly, such improved antimicrobial capacity can also be observed in the case of HBD-1–amyloid systems, as evidenced by the 1.8–15% and 7–36% higher antimicrobial capacities of HBD-1–amyloid cross-seeds than those of pure amyloid and pure HBD-1, respectively.
In principle, molecular interactions between antimicrobial molecules (including defensin–amyloid cross-seeds) and bacteria and the permeation of such molecules into bacteria have a major impact on the susceptibility of microorganism to these molecules. In this study, due to the complicated cell walls or outer membranes of bacteria and the presence of different defensin–amyloid heterocomplexes, it is not surprising to observe different antimicrobial activities of defensin–amyloid heterocomplexes. Among different bacterial strains, P.A. is the most stubborn bacterial strain, and its outer membrane permeability to small antibiotics is approximately 10 times smaller than that of E. coli.61 Therefore, all of the defensin–amyloid heterocomplexes appeared to have weak antimicrobial activity against P.A. (Fig. 5b and 12b). In contrast, Gram-positive S.E. can be readily killed by pure amyloids and defensins, as evidenced by 31–43% reduction in live bacteria. More importantly, amyloids and defensins can work together to induce synergistic effects on preventing the growth of S.E. bacteria, which leads to a further S.E. reduction of 45–71% (Fig. 5d and 12d). In the case of E. coli and S.A., defensin–amyloid heterocomplexes generally present a comparable or even higher antimicrobial activity than their corresponding defensins and amyloids alone. However, several exceptions were also observed presumably due to the natural antimicrobial/antibiotic resistance of E. coli and S.A. For instance, HD-6–hIAPP (14%, Fig. 5a) and HBD-1–Aβ (5.4%, Fig. S12a†) heterocomplexes exhibited a much weaker antimicrobial activity against E. coli than pure HD-6 (58%) and HBD-1 (14%). hCT–HD-6 (15%, Fig. 5c) and hIAPP–HBD-1 (15%, Fig. S12c†) heterocomplexes show weaker antimicrobial activity against S.A., as compared to HD-6 (24%) and HBD-1 (16%)-induced antimicrobial activity against S.A.
Additionally, Live/Dead staining was further performed to distinguish the live (green) bacteria and dead bacteria (red). As shown in Fig. 5e & S13,† all the bacterial strains without any treatment displayed massive green fluorescent signals, an indicator of normal bacterial growth. In contrast, all the samples treated with defensin and/or amyloids exhibited significant antimicrobial properties as evidenced by decreased bacterial density (i.e., less fluorescent signal) and live/dead ratio of bacteria (i.e., green/red ratio), thus again confirming that defensin–amyloid assemblies still retain bacterial killing properties.
3. Outlook and conclusions
The amyloid cascade caused by amyloid aggregation and neuroinflammation caused by microbial infection are considered as the two interconnected, pathological risk factors for initiating and promoting the onset and progression of neurodegenerative diseases. Due to the complex, multifactorial causes of neurodegenerative diseases, the cross-seeding (cross-interactions) of different amyloid proteins, together with the co-occurrence of different neurodegenerative diseases, indicates that single-target amyloid inhibitors against specific single amyloid proteins (Aβ, hIAPP, or hCT) are inefficient to modulate the co-aggregation pathways of different amyloid proteins, while the interlinked pathological risk factors between amyloid formation and neuroinflammation suggest that a single prevention mechanism is very unlikely to stop amyloid propagation and spreading via the brain–gut–microbiota axis. Different from the conventional amyloid inhibitors of small molecules,62,63 nanoparticles,64 polymers,65 and peptides66,67 with single-target or single-function properties (Fig. 6), it is highly desirable, but has proved to be very challenging, to develop new amyloid inhibitors capable of preventing amyloid formation and microbial infection simultaneously. Conceptually, antimicrobial peptides, due to their intrinsic antimicrobial activity, could serve as a huge and native source, but are much less explored, for repurposing them as potential and effective amyloid inhibitors. More importantly, some amyloid and antimicrobial peptides have been found to share certain structural and functional properties, i.e., several amyloid proteins are recognized for their antimicrobial activity, while some antimicrobial peptides possess amyloid-like properties to self-aggregate into β-structure rich fibrils. These lines of evidence raise the possibilities of co-aggregation and cross-seeding between amyloid proteins and antimicrobial peptides. Thus, it is hypothesized that the cross-seeding of antimicrobial peptides with amyloid proteins could deliver both anti-amyloid and antimicrobial properties against the two pathological pathways of neurodegenerative diseases.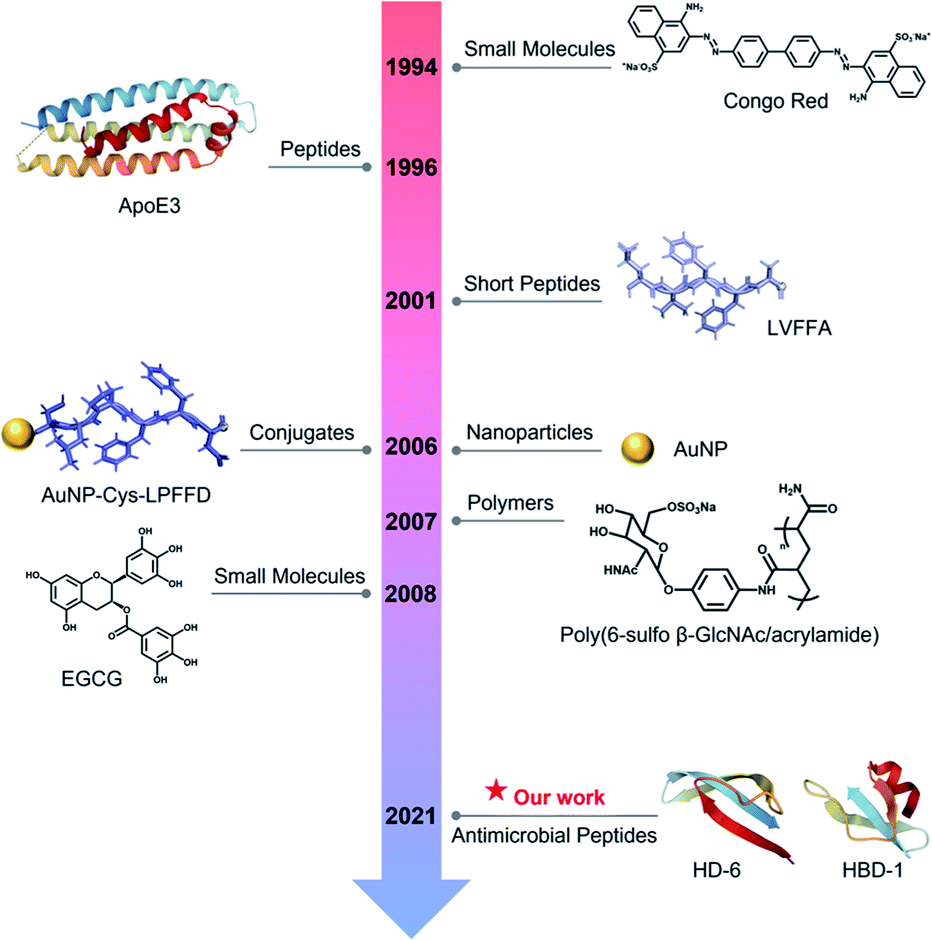 | ||
| Fig. 6 Historical literature review of the first-discovered or important amyloid inhibitors of different types. | ||
To test this hypothesis, we discovered and repurposed two intestinal defensins, HD-6 and HBD-1, as multiple-target, dual-function, amyloid inhibitors beyond few available today by demonstrating their (i) cross-seeding capacity with three different amyloid proteins, Aβ, hIAPP, and hCT and (ii) cross-seeding-induced anti-amyloid and antibacterial functions. Evidently, both defensins exhibited a general inhibition ability to prevent amyloid formation of Aβ, hIAPP, and hCT, but with different inhibition efficiencies that depend on amyloid sequences, concentrations, and aggregate states. Both defensins can significantly prevent amyloid formation from their monomers by up to 49–93% and from their oligomers by 27–56%, but not from their fibrillar species. Such differences in inhibition efficiency indicate the existence of cross-seeding barriers between intestinal defensins and amyloid proteins/aggregates. At the cellular level, both defensins enabled SH-SY5Y and RIN-m5F cells to be rescued from amyloid-induced cytotoxicity by increasing ∼35% of cell viability and reducing ∼24% of cytotoxicity. Apart from the amyloid inhibition function, defensin–amyloid assemblies can possess similar or even better antibacterial activities against both Gram-positive and Gram-negative bacteria as compared to pure defensins alone.
From a mechanistic viewpoint, the addition of defensins can act as a protective coating to either stabilize the native folded structures of amyloid monomers to prevent them from further misfolding and aggregation or bind to amyloid oligomers to disrupt their active aggregation sites including the β-sheet structure, specific residues, and peptide associations68 (Fig. 7a). Our cell assay results showed that as compared to highly toxic amyloid oligomers that often disrupt the structure and function of cell membranes, defensin–amyloid cross-seeds at the expense of amyloid homo-seeds not only showed much less cell cytotoxicity, but also reduced amyloid homo-seed-induced cell toxicity69 (Fig. 7b). Further, due to the presence of defensins in cross-seeds, they are still able to retain their intrinsic antimicrobial activity to some extent. Given that both defensins are rich in both hydrophobic and positively charged residues, the cationic residues (Lys or Arg) have a strong tendency to interact with negatively charged phospholipid head groups of the cell membrane for “anchoring” the cross-seeds at the membrane interface. Upon initial and loose membrane binding, hydrophobic residues, particularly ring-containing residues (Trp or Tyr), drive the cross-seeds to be partially or fully penetrated into the hydrocarbon core of lipid bilayers. As a result, depending on complex peptide–membrane interactions, insertion of cross-seeds or pore formation by cross-seeds is likely to occur, where both peptides and lipids can mutually adjust their conformations for accommodating membrane insertion and pore formation scenarios70 (Fig. 7c). Of note, the non/less toxicity of amyloid–defensin cross-seeds is likely attributed to several factors: (i) bacterial cell membranes are more negatively charged than host cell membranes, thus exerting stronger binding affinities to bacterial membranes than host cell membranes;71 (ii) cholesterols in host cell membranes act as a membrane stabilizing agent for protecting host cells from attack by amyloid–defensin cross-seeds;72 and (iii) the transmembrane potential of bacteria is much higher than that of host cells, thus rendering higher steric barriers for peptides to transport through the bacterial membranes.73 Taken together, our findings provide a new proof-of-concept strategy to repurpose antimicrobial peptides as amyloid inhibitors beyond few available today, which will greatly expand potential therapeutic drugs for amyloid diseases associated with the amyloid formation and microbial infection cascades. From a broader view, parallel efforts should be made to apply the data-driven, machine-learning approaches to computationally screen and identify some antimicrobial peptides as new amyloid inhibitors with built-in bacterial killing and amyloid inhibition functions from a huge dataset of existing antimicrobial peptides. Presumably, these repurposed antimicrobial peptides could serve as another pool or platform for researchers to explore their new structures and functions (e.g., biosensing, bioimaging, and tissue engineering) via different chemical or physical modifications (e.g., conjugation with nanoparticles and polymers), which has not been achieved before.
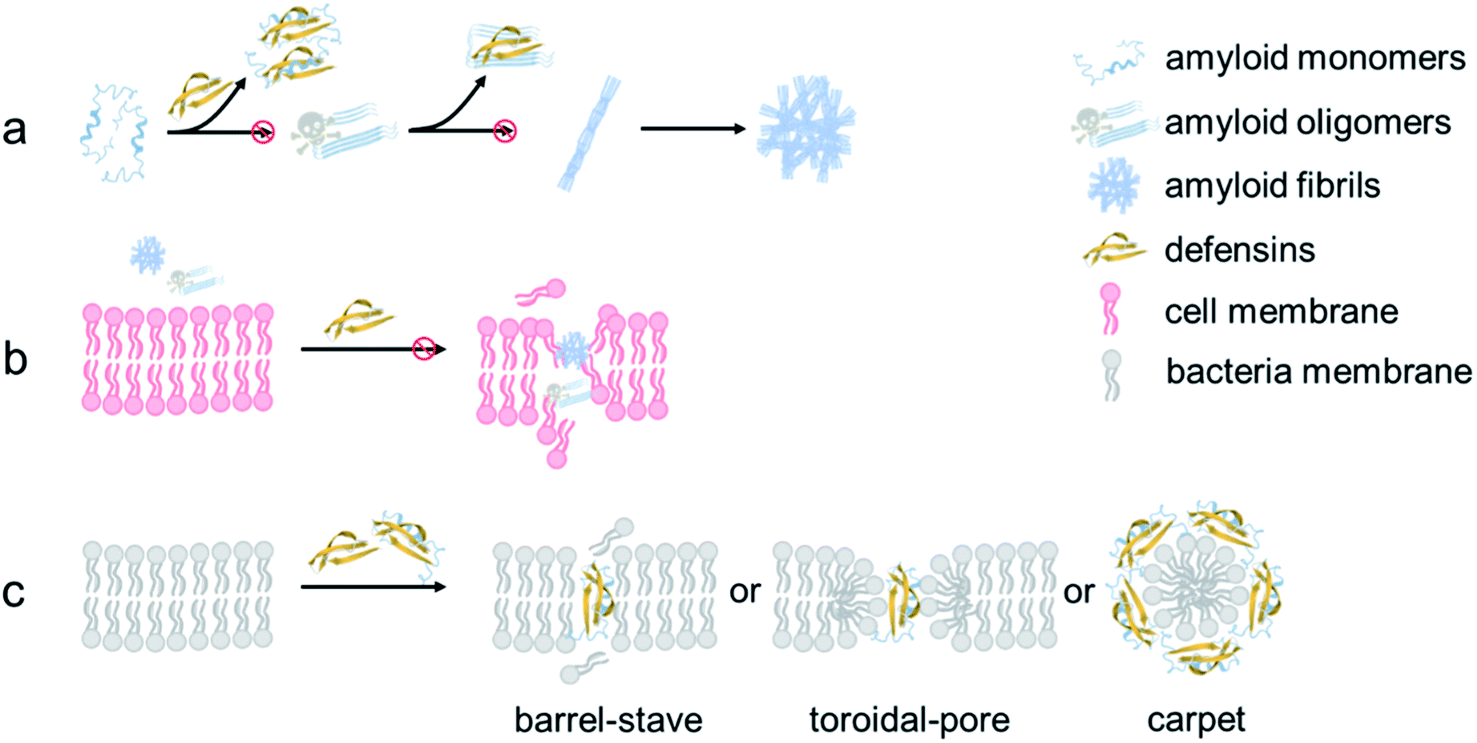 | ||
| Fig. 7 Hypothetical inhibition mechanisms of defensins against amyloid aggregation and toxicity via (a) anti-amyloid aggregation, (b) cell-protection, and (c) antimicrobial activity. | ||
Data availability
All data are available in the main text or the ESI.†Author contributions
Y. Tang designed this research, performed the characterization, assays and analysis. D. Zhang and X. Gong assisted to perform part of the SPR and antibacterial experiments. Y. Tang and J. Zheng wrote the manuscript. J. Zheng concepted the project and supervised this research. All authors contributed to the analysis and discussion of the results and have given approval to the final version of the manuscript.Conflicts of interest
There is no conflicts to declare.Acknowledgements
We acknowledge the financial support from NSF-CBET-2107619 and the Faculty Summer Research Fellowship from the University of Akron. We also trained three K12 students: Bowen Zheng from Copley High School, Alice Xu from Hudson High School, and Keven Gong from Western Reserve Academy via this project.References
- C. Soto and S. Pritzkow, Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases, Nat. Neurosci., 2018, 21(10), 1332 CrossRef CAS PubMed
.
- J. Gandhi, A. C. Antonelli, A. Afridi, S. Vatsia, G. Joshi, V. Romanov, I. V. Murray and S. A. Khan, Protein misfolding and aggregation in neurodegenerative diseases: a review of pathogeneses, novel detection strategies, and potential therapeutics, Rev. Neurosci., 2019, 30(4), 339 CrossRef PubMed
- A. Gámez, P. Yuste-Checa, S. Brasil, Á. Briso-Montiano, L. R. Desviat, M. Ugarte, C. Pérez-Cerdá and B. Pérez, Protein misfolding diseases: prospects of pharmacological treatment, Clin. Genet., 2018, 93(3), 450 CrossRef PubMed
- M. Leri, H. Chaudhary, I. A. Iashchishyn, J. Pansieri, Z. e. M. Svedružić, S. Gómez Alcalde, G. Musteikyte, V. Smirnovas, M. Stefani and M. Bucciantini, Natural compound from olive oil inhibits S100A9 amyloid formation and cytotoxicity: Implications for preventing Alzheimer's disease, ACS Chem. Neurosci., 2021, 12(11), 1905 CrossRef CAS PubMed
- P. K. Mandal, J. W. Pettegrew, E. Masliah, R. L. Hamilton and R. Mandal, Interaction between Aβ peptide and α synuclein: molecular mechanisms in overlapping pathology of Alzheimer's and Parkinson's in dementia with Lewy body disease, Neurochem. Res., 2006, 31(9), 1153 CrossRef CAS PubMed
- I. C. Stancu, B. Vasconcelos, D. Terwel and I. Dewachter, Models of β-amyloid induced Tau-pathology: the long and “folded” road to understand the mechanism, Mol. Neurodegener., 2014, 9(1), 1 CrossRef PubMed
- X. Li, X. Zhang, A. R. A. Ladiwala, D. Du, J. K. Yadav, P. M. Tessier, P. E. Wright, J. W. Kelly and J. N. Buxbaum, Mechanisms of transthyretin inhibition of β-amyloid aggregation in vitro, J. Neurosci., 2013, 33(50), 19423 CrossRef CAS PubMed
- P. Liu, S. Zhang, M. S. Chen, Q. Liu, C. Wang, C. Wang, Y. M. Li, F. Besenbacher and M. Dong, Co-assembly of human islet amyloid polypeptide (hIAPP)/insulin, Chem. Commun., 2012, 48(2), 191 RSC
- M. Mucibabic, P. Steneberg, E. Lidh, J. Straseviciene, A. Ziolkowska, U. Dahl, E. Lindahl and H. Edlund, α-Synuclein promotes IAPP fibril formation in vitro and β-cell amyloid formation in vivo in mice, Sci. Rep., 2020, 10(1), 1 CrossRef PubMed
- B. Ma and R. Nussinov, Selective molecular recognition in amyloid growth and transmission and cross-species barriers, J. Mol. Biol., 2012, 421(2–3), 172 CrossRef CAS PubMed
- B. Ren, Y. Zhang, M. Zhang, Y. Liu, D. Zhang, X. Gong, Z. Feng, J. Tang, Y. Chang and J. Zheng, Fundamentals of cross-seeding of amyloid proteins: an introduction, J. Mater. Chem. B, 2019, 7(46), 7267 RSC
- K. Lundmark, G. T. Westermark, A. Olsén and P. Westermark, Protein fibrils in nature can enhance amyloid protein A amyloidosis in mice: Cross-seeding as a disease mechanism, Proc. Natl. Acad. Sci. U. S. A., 2005, 02(17), 6098 CrossRef PubMed
- Y. Tang, D. Zhang, X. Gong and J. Zheng, A mechanistic survey of Alzheimer's disease, Biophys. Chem., 2022, 281, 106735 CrossRef CAS PubMed
- D. D. Li Puma, R. Piacentini, L. Leone, K. Gironi, M. E. Marcocci, G. De Chiara, A. T. Palamara and C. Grassi, Herpes Simplex Virus Type-1 Infection Impairs Adult Hippocampal Neurogenesis via Amyloid-β Protein Accumulation, Stem Cells, 2019, 37(11), 1467 CrossRef CAS PubMed
- M. Ortega and B. M. Ances, Role of HIV in amyloid metabolism, Journal of Neuroimmune Pharmacology, 2014, 9(4), 483 CrossRef PubMed
- D. Bortolotti, V. Gentili, A. Rotola, E. Caselli and R. Rizzo, HHV-6A infection induces amyloid-beta expression and activation of microglial cells, Alzheimer's Res. Ther., 2019, 11(1), 1 CrossRef PubMed
- F. Pistollato, S. Sumalla Cano, I. Elio, M. Masias Vergara, F. Giampieri and M. Battino, Role of gut microbiota and nutrients in amyloid formation and pathogenesis of Alzheimer disease, Nutr. Rev., 2016, 74(10), 624 CrossRef PubMed
- C. Roubaud-Baudron, P. Krolak-Salmon, I. Quadrio, F. Mégraud and N. Salles, Impact of chronic Helicobacter pylori infection on Alzheimer's disease: preliminary results, Neurobiol. Aging, 2012, 33(5), 1009 CrossRef PubMed
- C. J. Hammond, L. R. Hallock, R. J. Howanski, D. M. Appelt, C. S. Little and B. J. Balin, Immunohistological detection of Chlamydia pneumoniae in the Alzheimer's disease brain, BMC Neurosci., 2010, 11(1), 1 CrossRef PubMed
- D. Vigasova, M. Nemergut, B. Liskova and J. Damborsky, Multi-pathogen infections and Alzheimer's disease, Microb. Cell Fact., 2021, 20(1), 1 CrossRef PubMed
- E. A. Newcombe, J. Camats-Perna, M. L. Silva, N. Valmas, T. J. Huat and R. Medeiros, Inflammation: the link between comorbidities, genetics, and Alzheimer's disease, J. Neuroinflammation, 2018, 15(1), 1 CrossRef PubMed
- K. Kowalski and A. Mulak, Brain-Gut-Microbiota Axis in Alzheimer's Disease, Neurogastroenterol. Motil., 2019, 25(1), 48 CrossRef PubMed
- I. Blasko, R. Veerhuis, M. Stampfer-Kountchev, M. Saurwein-Teissl, P. Eikelenboom and B. Grubeck-Loebenstein, Costimulatory effects of interferon-γ and interleukin-1β or tumor necrosis factor α on the synthesis of Aβ1-40 and Aβ1-42 by human astrocytes, Neurobiol. Dis., 2000, 7(6), 682 CrossRef CAS PubMed
- J. W. Kinney, S. M. Bemiller, A. S. Murtishaw, A. M. Leisgang, A. M. Salazar and B. T. Lamb, Inflammation as a central mechanism in Alzheimer's disease, Alzheimer's & Dementia: Translational Research & Clinical Interventions, 2018, 4, 575 Search PubMed
- Z. Wang, P. Vilekar, J. Huang and D. F. Weaver, Furosemide as a Probe Molecule for the Treatment of Neuroinflammation in Alzheimer's Disease, ACS Chem. Neurosci., 2020, 11(24), 4152 CrossRef CAS PubMed
- I. Javed, Z. Zhang, J. Adamcik, N. Andrikopoulos, Y. Li, D. E. Otzen, S. Lin, R. Mezzenga, T. P. Davis and F. Ding, Accelerated amyloid beta pathogenesis by bacterial amyloid FapC, Adv. Sci., 2020, 7(18), 2001299 CrossRef CAS PubMed
- K. Kowalski and A. Mulak, Brain-gut-microbiota axis in Alzheimer's disease, Neurogastroenterol. Motil., 2019, 25(1), 48 CrossRef PubMed
- X. Zhan, B. Stamova, L.-W. Jin, C. DeCarli, B. Phinney and F. R. Sharp, Gram-negative bacterial molecules associate with Alzheimer disease pathology, Neurology, 2016, 87(22), 2324 CrossRef CAS PubMed
- D. C. Emery, D. K. Shoemark, T. E. Batstone, C. M. Waterfall, J. A. Coghill, T. L. Cerajewska, M. Davies, N. X. West and S. J. Allen, 16S rRNA next generation sequencing analysis shows bacteria in Alzheimer's post-mortem brain, Front. Aging Neurosci., 2017, 9, 195 CrossRef PubMed
- E. Y. Lee, Y. Srinivasan, J. De Anda, L. K. Nicastro, Ç. Tükel and G. C. Wong, Functional reciprocity of amyloids and antimicrobial peptides: rethinking the role of supramolecular assembly in host defense, immune activation, and inflammation, Front. Immunol., 2020, 11, 1629 CrossRef CAS PubMed
- T. R. Sampson, C. Challis, N. Jain, A. Moiseyenko, M. S. Ladinsky, G. G. Shastri, T. Thron, B. D. Needham, I. Horvath and J. W. Debelius, A gut bacterial amyloid promotes α-synuclein aggregation and motor impairment in mice, Elife, 2020, 9, e53111 CrossRef CAS PubMed
- M. L. Evans, E. Chorell, J. D. Taylor, J. Åden, A. Götheson, F. Li, M. Koch, L. Sefer, S. J. Matthews and P. Wittung-Stafshede, The bacterial curli system possesses a potent and selective inhibitor of amyloid formation, Molecular Cell, 2015, 57(3), 445 CrossRef CAS PubMed
- D. K. V. Kumar, S. H. Choi, K. J. Washicosky, W. A. Eimer, S. Tucker, J. Ghofrani, A. Lefkowitz, G. McColl, L. E. Goldstein and R. E. Tanzi, Amyloid-β peptide protects against microbial infection in mouse and worm models of Alzheimer's disease, Sci. Transl. Med., 2016, 8(340), 340ra72 Search PubMed
- L. Wang, Q. Liu, J.-C. Chen, Y.-X. Cui, B. Zhou, Y.-X. Chen, Y.-F. Zhao and Y.-M. Li, Antimicrobial activity of human islet amyloid polypeptides: an insight into amyloid peptides' connection with antimicrobial peptides, Biol. Chem., 2012, 393(7), 641 CrossRef CAS PubMed
- A. Röcker, N. R. Roan, J. K. Yadav, M. Fändrich and J. Münch, Structure, function and antagonism of semen amyloids, Chem. Commun., 2018, 54(55), 7557 RSC
- Y. Zhang, Y. Liu, Y. Tang, D. Zhang, H. He, J. Wu and J. Zheng, Antimicrobial α-defensins as Multi-target Inhibitors Against Amyloid Formation and Microbial Infection, Chem. Sci., 2021, 12, 9124 RSC
- Y. Zhang, Y. Liu, Y. Tang, D. Zhang, H. He, J. Wu and J. Zheng, Antimicrobial α-defensins as multi-target inhibitors against amyloid formation and microbial infection, Chem. Sci., 2021, 12(26), 9124 RSC
- H. Zhao, R. Sood, A. Jutila, S. Bose, G. Fimland, J. Nissen-Meyer and P. K. J. Kinnunen, Interaction of the antimicrobial peptide pheromone Plantaricin A with model membranes: Implications for a novel mechanism of action, Biochim. Biophys. Acta, Biomembr., 2006, 1758(9), 1461 CrossRef CAS PubMed
- A. N. Calabrese, Y. Liu, T. Wang, I. F. Musgrave, T. L. Pukala, R. F. Tabor, L. L. Martin, J. A. Carver and J. H. Bowie, The amyloid fibril-forming properties of the amphibian antimicrobial peptide uperin 3.5, ChemBioChem, 2016, 17(3), 239 CrossRef CAS PubMed
- L. Caillon, J. A. Killian, O. Lequin and L. Khemtémourian, Biophysical investigation of the membrane-disrupting mechanism of the antimicrobial and amyloid-like peptide dermaseptin S9, PLoS One, 2013, 8(10), e75528 CrossRef CAS PubMed
- S. J. Soscia, J. E. Kirby, K. J. Washicosky, S. M. Tucker, M. Ingelsson, B. Hyman, M. A. Burton, L. E. Goldstein, S. Duong and R. E. Tanzi,
et al., The Alzheimer's disease-associated amyloid beta-protein is an antimicrobial peptide, PLoS One, 2010, 5(3), e9505 CrossRef PubMed
- M. L. Gosztyla, H. M. Brothers and S. R. Robinson, Alzheimer's Amyloid-β is an Antimicrobial Peptide: A Review of the Evidence, J. Alzheimer's Dis., 2018, 62, 1495 CAS
- N. B. Last and A. D. Miranker, Common mechanism unites membrane poration by amyloid and antimicrobial peptides, Proc. Natl. Acad. Sci. U. S. A., 2013, 110(16), 6382 CrossRef CAS PubMed
- Y. Zhang, M. Zhang, Y. Liu, D. Zhang, Y. Tang, B. Ren and J. Zheng, Dual Amyloid Cross-Seeding Reveals Steric Zipper-Facilitated Fibrillization and Pathological Links between Protein Misfolding Diseases, J. Mater. Chem. B, 2021, 9, 3300 RSC
- M. Zhang, J. Zhao and J. Zheng, Molecular understanding of a potential functional link between antimicrobial and amyloid peptides, Soft Matter, 2014, 10(38), 7425 RSC
- Y. Zhang, Y. Tang, Y. Liu, D. Zhang and J. Zheng, Design and Engineering of Amyloid Aggregation-Prone Fragments and Their Antimicrobial Conjugates with Multi-Target Functionality, Adv. Funct. Mater., 2021, 31, 2102978 CrossRef CAS
- M. R. White, R. Kandel, S. Tripathi, D. Condon, L. Qi, J. Taubenberger and K. L. Hartshorn, Alzheimer's Associated β-Amyloid Protein Inhibits Influenza A Virus and Modulates Viral Interactions with Phagocytes, PLoS One, 2014, 9(7), e101364 CrossRef PubMed
- W. A. Eimer, D. K. Vijaya Kumar, N. K. Navalpur Shanmugam, A. S. Rodriguez, T. Mitchell, K. J. Washicosky, B. György, X. O. Breakefield, R. E. Tanzi and R. D. Moir, Alzheimer's Disease-Associated β-Amyloid Is Rapidly Seeded by Herpesviridae to Protect against Brain Infection, Neuron, 2018, 99(1), 56 CrossRef CAS PubMed
- B. L. Kagan, H. Jang, R. Capone, F. Teran Arce, S. Ramachandran, R. Lal and R. Nussinov, Antimicrobial properties of amyloid peptides, Mol. Pharm., 2012, 9(4), 708 CrossRef CAS PubMed
- Y. Hirakura, I. Carreras, J. D. Sipe and B. L. Kagan, Channel formation by serum amyloid A: a potential mechanism for amyloid pathogenesis and host defense, Amyloid, 2002, 9(1), 13 CrossRef CAS PubMed
- M. Pasupuleti, M. Roupe, V. Rydengård, K. Surewicz, W. K. Surewicz, A. Chalupka, M. Malmsten, O. E. Sörensen and A. Schmidtchen, Antimicrobial activity of human prion protein is mediated by its N-terminal region, PLoS One, 2009, 4(10), e7358 CrossRef PubMed
- H. Jang, F. T. Arce, M. Mustata, S. Ramachandran, R. Capone, R. Nussinov and R. Lal, Antimicrobial protegrin-1 forms amyloid-like fibrils with rapid kinetics suggesting a functional link, Biophys. J., 2011, 100(7), 1775 CrossRef CAS PubMed
- H. Chu, M. Pazgier, G. Jung, S.-P. Nuccio, P. A. Castillo, M. F. de Jong, M. G. Winter, S. E. Winter, J. Wehkamp and B. Shen,
et al., Human α-defensin 6 promotes mucosal innate immunity through self-assembled peptide nanonets, Science, 2012, 337(6093), 477 CrossRef CAS PubMed
- C. Code, Y. Domanov, A. Jutila and P. K. J. Kinnunen, Amyloid-type fiber formation in control of enzyme action: interfacial activation of phospholipase A2, Biophys. J., 2008, 95(1), 215 CrossRef CAS PubMed
- P. Kesika, N. Suganthy, B. S. Sivamaruthi and C. Chaiyasut, Role of gut-brain axis, gut microbial composition, and probiotic intervention in Alzheimer's disease, Life Sci., 2021, 264, 118627 CrossRef CAS PubMed
- K. De Smet and R. Contreras, Human antimicrobial peptides: defensins, cathelicidins and histatins, Biotechnol. Lett, 2005, 27(18), 1337 CrossRef CAS PubMed
- M. Zaman, A. N. Khan, S. M. Zakariya and R. H. Khan, Protein misfolding, aggregation and mechanism of amyloid cytotoxicity: an overview and therapeutic strategies to inhibit aggregation, Int. J. Biol. Macromol., 2019, 134, 1022 CrossRef CAS PubMed
- K. E. Marshall, R. Marchante, W.-F. Xue and L. C. Serpell, The relationship between amyloid structure and cytotoxicity, Prion, 2014, 8(2), 192 CrossRef CAS PubMed
- J. Costerton, J. Ingram and K. Cheng, Structure and function of the cell envelope of gram-negative bacteria, Bacteriol. Rev., 1974, 38(1), 87 CrossRef CAS PubMed
- S. Santajit and N. Indrawattana, Mechanisms of antimicrobial resistance in ESKAPE pathogens, BioMed Res. Int., 2016, 2016, 2475067 Search PubMed
- E. B. Breidenstein, C. de la Fuente-Núñez and R. E. Hancock, Pseudomonas aeruginosa: all roads lead to resistance, Trends Microbiol., 2011, 19(8), 419 CrossRef CAS PubMed
- A. Lorenzo and B. A. Yankner, Beta-amyloid neurotoxicity requires fibril formation and is inhibited by congo red, Proc. Natl. Acad. Sci. U. S. A., 1994, 91(25), 12243 CrossRef CAS PubMed
- D. E. Ehrnhoefer, J. Bieschke, A. Boeddrich, M. Herbst, L. Masino, R. Lurz, S. Engemann, A. Pastore and E. E. Wanker, EGCG redirects amyloidogenic polypeptides into unstructured, off-pathway oligomers, Nat. Struct. Mol. Biol., 2008, 15(6), 558 CrossRef CAS PubMed
- M. J. Kogan, N. G. Bastus, R. Amigo, D. Grillo-Bosch, E. Araya, A. Turiel, A. Labarta, E. Giralt and V. F. Puntes, Nanoparticle-mediated local and remote manipulation of protein aggregation, Nano Lett., 2006, 6(1), 110 CrossRef CAS PubMed
- Y. Miura, K. Yasuda, K. Yamamoto, M. Koike, Y. Nishida and K. Kobayashi, Inhibition of alzheimer amyloid aggregation with sulfated glycopolymers, Biomacromolecules, 2007, 8(7), 2129 CrossRef CAS PubMed
- K. C. Evans, E. P. Berger, C.-G. Cho, K. H. Weisgraber and P. T. Lansbury, Apolipoprotein E is a kinetic but not a thermodynamic inhibitor of amyloid formation: implications for the pathogenesis and treatment of Alzheimer disease, Proc. Natl. Acad. Sci. U. S. A., 1995, 92(3), 763 CrossRef CAS PubMed
- M. A. Findeis, J.-J. Lee, M. Kelley, J. D. Wakefield, M.-H. Zhang, J. Chin, W. Kubasek and S. M. Molineaux, Characterization of cholyl-leu-val-phe-phe-ala-OH as an inhibitor of amyloid beta-peptide polymerization, Amyloid, 2001, 8(4), 231 CrossRef CAS PubMed
- Y. Tang, D. Zhang, Y. Zhang, Y. Liu, X. Gong, Y. Chang, B. Ren and J. Zheng, Introduction and Fundamentals of Human Islet Amyloid Polypeptide Inhibitors, ACS Appl. Bio Mater., 2020, 3(12), 8286 CrossRef CAS PubMed
-
J. R. Harris and J. Marles-Wright, Macromolecular Protein Complexes II: Structure and Function, Springer, 2019 Search PubMed
- J. Lei, L. Sun, S. Huang, C. Zhu, P. Li, J. He, V. Mackey, D. H. Coy and Q. He, The antimicrobial peptides and their potential clinical applications, Am. J. Transl. Res., 2019, 11(7), 3919 CAS
- R. E. Hancock and H.-G. Sahl, Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies, Nat. Biotechnol., 2006, 24(12), 1551 CrossRef CAS PubMed
- M. Zasloff, Antimicrobial peptides of multicellular organisms, Nature, 2002, 415(6870), 389 CrossRef CAS PubMed
- K. Matsuzaki, K. Sugishita, N. Fujii and K. Miyajima, Molecular basis for membrane selectivity of an antimicrobial peptide, magainin 2, Biochemistry, 1995, 34(10), 3423 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc01447e |
| This journal is © The Royal Society of Chemistry 2022 |